
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePerformance and biocompatibility of OSTEMER 322 in cell-based microfluidic applications†
Petr Aubrecht *a,
Jiří Smejkala,
Petr Panuškaa,
Klára Španbauerováa,
Viktorie Neubertováa,
Pavel Kauleab,
Jindřich Matoušekc,
Stanislav Vinopala,
Michaela Liegertováa,
Marcel Štofika and
Jan Malý*a
*a,
Jiří Smejkala,
Petr Panuškaa,
Klára Španbauerováa,
Viktorie Neubertováa,
Pavel Kauleab,
Jindřich Matoušekc,
Stanislav Vinopala,
Michaela Liegertováa,
Marcel Štofika and
Jan Malý*a
aCentre for Nanomaterials and Biotechnology, Faculty of Science, Jan Evangelista Purkyně University in Ústí nad Labem, Pasteurova 3632/15, 400 96 Ústí nad Labem, Czech Republic. E-mail: petr.aubrecht@ujep.cz; malyjalga@seznam.cz
bDepartment of Chemistry, Faculty of Science, Jan Evangelista Purkyně University in Ústí nad Labem, Pasteurova 3632/15, 400 96 Ústí nad Labem, Czech Republic
cDepartment of Physics, Faculty of Science, Jan Evangelista Purkyně University in Ústí nad Labem, Pasteurova 3632/15, 400 96 Ústí nad Labem, Czech Republic
First published on 23rd January 2024
Abstract
The Off-Stoichiometry Thiol–ene and Epoxy (OSTE+) polymer technology has been increasingly utilised in the field of microfluidics and lab-on-a-chip applications. However, the impact of OSTEMER polymers, specifically the OSTEMER 322 formulation, on cell viability has remained limited. In this work, we thoroughly explored the biocompatibility of this commercial OSTEMER formulation, along with various surface modifications, through a broad range of cell types, from fibroblasts to epithelial cells. We employed cell viability and confluence assays to evaluate the performance of the material and its modified variants in cell culturing. The properties of the pristine and modified OSTEMER were also investigated using surface characterization methods including contact angle, zeta potential, and X-ray photoelectron spectroscopy. Mass spectrometry analysis confirmed the absence of leaching constituents from OSTEMER, indicating its safety for cell-based applications. Our findings demonstrated that cell viability on OSTEMER surfaces is sufficient for typical cell culture experiments, suggesting OSTEMER 322 is a suitable material for a variety of cell-based assays in microfluidic devices.
1 Introduction
The field of microfluidics has experienced consistent growth over the past two decades.1 Nevertheless, despite the overall advancements, the anticipated improvements have not been realised thus far,2 as many devices remain as proof-of-concept prototypes and are often not commercially utilised.1,3 Not respecting the end-user's needs and preferences together with a lack of consistency in terms of material, fabrication, and interfacing technology, might be the main reasons for a slow rate of commercialisation.4There are many materials and fabrication techniques that can be utilised for the preparation of microfluidic devices. Thus far, none of the materials have been specifically tailored for microfluidics in mind. In the past, there has been a reliance on hard materials such as silicon, quartz, and glass in conjunction with microfabrication technologies. However, these methods have proven to be costly, complicated, and inaccessible to the majority of researchers.5–7
The invention of polydimethylsiloxane (PDMS) and soft-lithography resulted in a major shift in the field.8 This elastomer rapidly gained popularity as the preferred material for the creation of proof-of-concept prototypes and microfluidic devices. Replica moulding with PDMS is a more straightforward process, often eliminating the need for costly clean room facilities.9 However, PDMS also has disadvantages such as high permeability to gases, swelling in organic solvents, hydrophobicity, and also moulding and casting processes are difficult to transfer to industrial production.
Other materials such as thermoplastics including poly(methyl methacrylate) (PMMA) and cyclic olefin polymers (COP) are more suitable for mass production via injection moulding and hot embossing.10–13 They are generally considered more appropriate for achieving the chemical resistance and mechanical strength of the final device.14 However, these materials are hard to modify and suffer from slow mould fabrication, which renders them not viable for rapid prototyping.15
The thiol–ene (TE) polymers consists of two monomers, each with at least two thiol or allyl (or ene) groups.16 Thiol–ene polymerisation belongs to so-called click–chemistry reactions characterised by high yields and conversion rates, reduced polymerisation shrinkage stress, and the absence of undesirable by-products.17,18 These characteristics, together with material properties such as enhanced resistance to acids and organic solvents and the availability of different multifunctional monomers, make them excellent candidates for the production of microfluidic devices.19
The first examples of the use of thiol–enes, like UV curing glue Norland Optical Adhesive, date back to 2007.20,21 These devices have already demonstrated advantages over PDMS, particularly in terms of enhanced resistance to organic solvents.21,22 Subsequently, there was a progression towards employing more customized formulations23 and the introduction of off-stoichiometric thiol–enes (OSTE) pioneered by Carlborg et al. In this approach, the monomers were mixed in non-stoichiometric ratios.19
The primary benefit of non-stoichiometric ratios is the presence of an excess of thiol or allyl groups on the surface. This surplus enables the direct grafting of molecules through click reactions24,25 or the direct bonding and sealing of the microfluidic device.19 The exceptional characteristics of OSTE as a novel microfluidic fabrication platform were demonstrated in various instances.19,26–28 Nevertheless, the native OSTE surface has certain limitations, such as restricted hydrophilicity. Additionally, there has been a concern regarding the leaching of unreacted monomers, as well as the requirement for a corresponding thiol- or ene-surface chemistry to facilitate bonding.29
In order to address these limitations, a superior dual-cure polymer based on the thiol–ene-epoxy formulation (OSTE+), was developed.29 The OSTE+ system is a ternary system consisting of three distinct monomers that undergo polymerisation through two separate stages. The first UV-initiated step produces an intermediate, soft-solid material with surface properties ready for bio/chemical modification and bonding. The second thermally-initiated step results in the formation of an inert polymer with thermoplastic-like properties.29
The OSTEMER 322 is part of the OSTE+ family, which was designed as a manufacturing platform, and specifically tailored for use in microfluidics. OSTE+ exhibits several advantages compared to previously examined materials. These include the ability to adjust mechanical features, tune surface properties, and facilitate manufacturing through UV-curing and photopatterning, which make it compatible with standard soft lithography processes for rapid prototyping.30 Another notable advantage is the ability to be heterogeneously bonded to other materials and itself,31–34 which is an important part of the final packaging.
Biocompatibility is of significant importance regarding materials utilised in microfluidics, particularly in the context of cell culture studies and the development of complex in vitro models such as organs-on-chips. It is defined as the ability of a device material to perform with an appropriate (biological) host response in a specific situation.35 In the context of this study, the term “biocompatibility” refers to the ability of cell cultures to grow in OSTE/OSTE+ devices while maintaining the viability and integrity of the cells. To our knowledge, there are only a few studies that examine the use of OSTE as a material for biological applications32,36–40 and even fewer examples that directly examine the biocompatibility of OSTE+/OSTEMER material,41,42 so no definite conclusions about its biocompatibility could have been drawn so far.
The performance of the TE material is significantly influenced by the selection of monomers and the quantity of active groups present on the surface. According to several reports, thiol-excess OSTE exhibits lower cell viability in comparison to allyl-excess OSTE43 and plasma treatment can improve cell viability.41 In the context of a TE-based NOA81, no instances of toxicity were reported, and the cells showed normal proliferation.44 There have been studies on cell morphology and cell differentiation that provide additional evidence supporting the biocompatibility of OSTE+,36,45 making it possible to categorise it as biocompatible based on the ISO 10993-5 standard.42 However, leaching monomers were identified as a potential source of cytotoxicity, although incubation in water alleviated the problem.42 In addition, the absorption of oxygen from the surrounding environment might also influence OSTE+ biocompatibility.46 For this reason, it is desirable to conduct additional biocompatibility tests.
The main objective of this study was to assess the biocompatibility of OSTEMER 322 Crystal Clear (oste322) and its various modifications to develop adhesive and nonadhesive surfaces for cell culture applications. Additionally, we aimed to investigate the surface properties of oste322-derived materials using several characterisation techniques, including contact angle measurement, X-ray photoelectron spectroscopy (XPS), and electrokinetic analysis (zeta potential). Furthermore, we conducted mass spectrometry (HPLC-MS) analyses to assess the potential leaching constituents. The biocompatibility of different formulations of oste322 was examined through an in vitro cell viability assay, specifically the CCK-8 assay, combined with a confluence assay conducted using confocal microscopy and time-lapse of attaching cells to the oste322 surfaces. Our primary focus was on testing the oste322 in multi-well plate inserts, assessing its biocompatibility and performance, and briefly acknowledging its potential in microfluidic systems.
2 Materials and methods
2.1 Fabrication, surface modification and biocompatibility evaluation of the oste322 inserts
 | ||
| Fig. 1 Schematic of the fabrication process. Workflow of the oste322 inserts and static microfluidic system preparation. | ||
Step 1: A 3D model of the master for casting (Fig. 2A; left) was created using the CAD software Fusion 360 (Autodesk, USA) and printed using the 3D printer Perfactory 4 LED (EnvisionTEC, Germany) from the photopolymer HTM series (EnvisionTEC, Germany). The printed master was sonicated with the ultrasonic bath SHE-UT 8031 EUK (Shesto, UK) in isopropanol A.G. (PENTA, Czech Republic) for two cycles and five minutes each before being exposed to 150 J cm−2 of UV light with a photolithographic system (Newport Oriel, USA) and a photolithography mercury lamp 350–450 nm (Newport Oriel, USA).
 | ||
| Fig. 2 Oste322 inserts and microfluidic system preparation and surface characterisation of the oste322 inserts. (A) Left: 3D model of the HTM master and a photograph of the PDMS mould for oste322 inserts preparation. Right: 3D model of the PDMS mould and photograph of the oste322 static microfluidic system. (B) Electrokinetic analysis (zeta potential) measurements. (C) Water contact angle measurement. (D) Surface free energy values calculated by the OWRK method. The data are presented as mean ± SD (n = 6) and were analysed using a 2-way ANOVA, comparing the pristine data set vs. pristine oste322 and plasma data set to a pristine data set. The symbol * represents a p-value ≤0.05, **p ≤ 0.01 and ***p ≤ 0.001. Data that lacks symbols are considered statistically insignificant. | ||
Step 2: PDMS Sylgard 184 (Dow Corning, USA) was mixed in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 w/w ratio (base elastomer to curing agent) and degassed in a desiccator under vacuum for 1 h. The mixture was poured into the HTM master form and left to cure at ambient temperature for 24 h, the PDMS replica was then released.
:1 w/w ratio (base elastomer to curing agent) and degassed in a desiccator under vacuum for 1 h. The mixture was poured into the HTM master form and left to cure at ambient temperature for 24 h, the PDMS replica was then released.
Step 3: The commercial polymer OSTEMER 322 Crystal Clear (Mercene Labs, Sweden) was mixed in a 1.09:1 w/w ratio (component A to component B), thoroughly stirred, and degassed in the desiccator for half an hour. The mixture was then poured into the replica PDMS mould and sealed with a PDMS lid.
Step 4: The mould containing oste322 was polymerized on the MA/BA Gen4 Series Mask and Bond Aligner (SÜSS MicroTec, Germany) through dispersion glass with a dose of 1400 mJ cm−2 of UV radiation (mercury light source with no filters).
Step 5: The PDMS mould with oste322 inserts was then heated to 100 °C in a laboratory oven LAC-LH 30/12 (LAC) by a programmable unit Ht Industry (HT8) for 1.5 h, followed by a gradual decrease in temperature. The completed Oste322 inserts were extracted from the mould.
Step 6: The oste322 inserts were stirred for 20 minutes in 200 mL of 96% ethanol A.G. (PENTA, Czech Republic) before being rapidly immersed in dH2O. The inserts were nitrogen-blow-dried after 20 minutes of additional stirring in 200 mL of dH2O. Using the PE-100 Benchtop Plasma System (Plasma Etch, USA), half of the inserts were then activated by oxygen plasma. The vacuum chamber was pumped down to a background pressure of 65 mTorr using a rotary vane mechanical pump RV12F (Edwards, UK). The RF power was set to 50 W for 30 s with negligible back-reflected power and a 20 sccm oxygen flow.
Step 7: The pristine and plasma-activated oste322 inserts were placed in a 96-well plate, Nunclon™ Delta Surface (Thermo Fisher Scientific) and 100 μL of a protein-containing solution was added to each well (Table 1): collagen type I 10 μg mL−1 in Phosphate Buffered Saline (PBS); collagen type II 5 μg mL−1 in PBS; collagen type IV 5 μg mL−1 in PBS; elastin 5 μg mL−1 in PBS; fibronectin 5 μg mL−1 in PBS; laminin 6 μg mL−1 in Hank's Balanced Salt Solution (HBSS); and poly-L-lysine 5 μg mL−1 in borate buffer. The plate was incubated in the CellCulture® CO2 incubator (Esco Micro, Singapore) at 37 °C. After incubation, the wells were washed five times with 100 μL of PBS and gently dried by a nitrogen stream for use in surface characterisation experiments and cell viability assays.
| Oste322 insert | Viability (CCK-8)/confluence (24 h) | Characterisation | |||
|---|---|---|---|---|---|
| HTB-177 | U-373MG | BJ | B14 | XPS, zeta, CA | |
| Oste322 | • | • | • | • | • |
| Fluo-ST2 | - | - | - | - | • |
| Collagen I | - | - | • | • | • |
| Collagen II | • | - | - | - | • |
| Collagen IV | • | • | - | - | • |
| Elastin | • | • | - | - | • |
| Fibronectin | - | • | • | • | • |
| Laminin | • | • | - | - | • |
| Poly-L-lysine | • | • | • | • | • |
| Oste322 system | Viability/confluence (72 h) | ||||
| HTB-177 | U-373 MG | BJ | B14 | B14 | |
| Channel | 1 | 2 | 3 | 4 | 5 |
| Modification | Laminin | Collagen IV | Fibronectin | Fibronectin | Fluo-ST2 |
Surface Free Energy (SFE) was calculated in Advance software employing the Owens, Wendt, Rabel, and Kaelble (OWRK) method. This involved analysing the contact angles of two liquids, namely water as a polar liquid and diiodomethane (CH2I2) as a nonpolar liquid. This model takes into account the surface tension phenomenon by considering both the polar and dispersive components.47–49 A total of six values from each liquid were utilized in the calculation of the surface free energy.
The mobile phase consisted entirely of acetonitrile, with a flow rate of 150 μL min−1. The measurement was conducted in the absence of a chromatographic column, utilising solely a pre-column equipped with a pre-filter. The autosampler extracted a complete loop from the HPLC vial, specifically 25 μL of the leach solution. This solution was then diluted in a 1:1 ratio by volume with 100 μL of the leach solution and 100 μL of acetonitrile. The resulting mixture was transferred into a glass insert with a capacity of 250 μL, which was subsequently placed inside the HPLC vial. The blank samples were prepared following a standardised procedure in which a mixture of ethanol and MiliQ water was combined with acetonitrile. Additionally, acetonitrile alone was measured as the third blank sample.
The mass spectra (MS) were obtained using a Thermo Scientific LCQ Fleet spectrometer equipped with a mass analyser operating based on the ion trap principle. The ionisation process was carried out at atmospheric pressure using water, nitrogen, and helium molecules (5.0 Messer, Czech Republic) as collision gases in the ion trap. The experimental setup involved a tube voltage of −125 V, a capillary voltage of −35.0 V, a capillary temperature of 275 °C, and the use of nitrogen as an inert sheath gas with a flow rate of 40 p.d.u. The screening of aqueous and ethanol extracts was conducted in both negative (APCI-N) and positive (APCI-P) modes. The chromatograms and MS APCI spectra obtained were analysed using XCalibur® software (T. E. Corp., 1998–2007).
000 cells/mL in the appropriate medium was introduced into each well. The plate was subsequently placed in the CellCulture® CO2 incubator (Esco Micro, Singapore) and incubated overnight at a temperature of 37 °C. The incubator maintained a humidified environment with a relative humidity of 95%, which included 5% CO2.In each well, a volume of 10 μL of the CCK-8 assay (Dojindo Molecular Technologies, USA) was introduced, followed by incubation of the entire plate at 37 °C for 2 h. The measurement of the viability assay was conducted using the GloMax® Explorer instrument (Promega, USA), with absorbance readings taken at 450 nm and a reference wavelength set to 600 nm. Following the completion of the measurement, the subsequent steps were carried out in Section 2.1.7 Oste322 inserts confluence Assay.
The cells attached to the oste322 inserts were visualised using a Leica SP8 confocal microscope (Leica, Germany), equipped with an environmental chamber (Okolab, Italy). The environmental chamber ensured a consistent temperature of 36.5 °C and provided humidified air with a 5% concentration of CO2. The images were captured utilising the HC PL FLUOTAR CS 10×/0.30 N.A. dry Leica objective. The Leica Las X Navigator feature was employed to capture images of specified wells on a 96-well plate. Typically, a set of 4 × 4 image tiles with a 10% overlap was captured for each well.
The acquisition of fluorescence images was performed utilising a 561 nm excitation laser, while the HyD detector range was configured to detect emissions within the 566–700 nm range. Concurrently, a wide-field image was obtained using a transmitted light detector. In order to maintain consistent focus throughout the imaging procedure, the Autofocus feature of LAS X Navigator was utilised. This involved implementing Contrast Based Method 1 on the fluorescence channel, with a capture range of 200 μm and 9–11 focusing steps performed before capturing each image. The tiles were merged using the Mosaic Merge function in LAS X software following the imaging process.
The data sets underwent processing using the FIJI ImageJ software. Initially, the analyse function was established. The specified measurements include area, mean grey value, area fraction, threshold limitation, and display label. The image underwent the application of the Gaussian Blur filter, where the value of Sigma (radius) was set to 2.00. The background subtraction was performed using the “subtract background” function, with a rolling ball radius of 30.0 pixels. The threshold was modified to include a majority of cells, thereby enabling the generation of an analysis measurement table for each image.
2.2 Fabrication, modification, and time-lapse imaging of cell attaching to the oste322 plate
Step 1: The frame for casting was 3D printed on a PRUSA MK 3 3D printer from PET material (Prusament PET, Prusa, Czech Republic). The PET frame was pressed onto the Silicon wafer with a spin coated thin layer of PDMS serving as an anti-adhesion layer.
Step 2: The glass component of the oste322 plate consisted of a glass slide (76 × 50 × 1 mm) with drilled holes (Corning, USA). Holes in the glass slides were fabricated by the powder micro-blasting technique (micro-blasting CNC lathe, Comco, USA). The abrasive Al2O3 particles (Comco, USA) with dimensions of 50 μm and the nozzle MB2520-18 Hi/Performance Accuflo Nozzle (Comco, USA) were used in the blasting process.
Step 3: The commercial polymer OSTEMER 322 Crystal Clear (Mercene Labs, Sweden) was mixed in a 1.09:1 w/w ratio (component A to component B), stirred thoroughly and degassed for half an hour in a desiccator. The mixture was then poured into the PET frame.
Step 4: The polymerisation of the mixture was carried out on the MA/BA Gen4 Series Mask and Bond Aligner (SÜSS MicroTec, Germany) using dispersion glass and UV radiation from a mercury light source without any filters. The UV dose applied was 500 mJ cm−2. After exposure, the frame was cut and peeled off and the machined glass from step 2 was pressed to the Oste322 surface and treated with an additional 1000 mJ cm−2 of UV radiation.
Step 5: The oste322 plate was heated on the hotplate to 100 °C for 1.5 h, with a gradual decrease in temperature after the end of the process.
Step 6: The plate was then sterilised by stirring in 200 mL of 96% ethanol A.G. (PENTA, Czech Republic) for 20 min, washed by dH2O and subsequently stirred in 200 mL of dH2O for 20 minutes. Finally, the oste322 plate was dried using a stream of nitrogen gas.
Step 7: To each well of the oste322 plate, 100 μL of a corresponding solution with protein was added in column triplicate (no treatment; fibronectin 5 μg mL−1; collagen type I 10 μg mL−1; no treatment; laminin 6 μg mL−1; and collagen IV 5 μg mL−1). The oste322 plate was subsequently placed in the CellCulture® CO2 incubator (Esco Micro, Singapore) and incubated at a temperature of 37 °C for 2 h. The solution was extracted from each well and rinsed by dH2O. To each well 100 μL of solution with cell B14 (first three columns) and HTB (last three columns) at a concentration of 40000 cells per mL in DMEM was added. Cells were counted by CellDrop™ FL (DeNovix Inc), stained with 0.33 nM CellTracker™ Orange CMTMR Dye (ThermoFisher Scientific) and incubated at a temperature of 37 °C for 20 min. Then they were centrifugated at 110 RCF for 10 min in a centrifuge Sigma 3-30KS (Sartorius, Germany) and resuspended in DMEM.
The acquisition of fluorescence images was performed utilising a 561 nm excitation laser, while the HyD detector range was configured to detect emissions within the 566–700 nm range. Concurrently, a wide-field image was obtained using a transmitted light detector. To maintain consistent focus throughout the imaging procedure, the manual focus after each imaging was utilised. A series of images was repeatedly taken after 15 min continuously for 6 h. After the time-lapse imaging, each well of the oste322 plate was rinsed with DMEM and a series of images were taken after the wash. The tiles were merged using the Mosaic Merge function in LAS X software following the imaging process.
The quantification of cell attachment on the time-lapse data set was performed using automated segmentation. The entire scan area was segmented, and cell quantification was performed using the generalist algorithm Cellpose (v. 2.0).50,51 The segmentation procedure was conducted on the wide-field channel, with additional training of the model on the data set. Each image was segmented, and a PNG cp map with segmentation and a zip file with ROI was generated. The map and ROI segments were imported into the FIJI ImageJ and the analyse function was used for the determination of the circularity (calculated using the formula: circularity = (4 π area)/perimeter2) and perimeter of each cell.
2.3 Fabrication, surface modification, and biocompatibility evaluation of the oste322 microfluidic system
Step 3: The commercial polymer OSTEMER 322 Crystal Clear (Mercene Labs, Sweden) was mixed in a 1.09:1 w/w ratio (component A to component B), stirred thoroughly, and degassed for half an hour within the desiccator. The mixture was then poured into the PDMS replica of the system and sealed with a Corning glass lid 75 × 50 × 1 mm (Dow Corning, USA).
Step 4: The polymerisation of the mould was carried out on the MA/BA Gen4 Series Mask and Bond Aligner (SÜSS MicroTec, Germany) using dispersion glass and UV radiation from a mercury light source without any filters. The UV dose applied was 1000 mJ cm−2. The bottom part, composed of a 75 × 50 × 1 mm Corning glass (Dow Corning, USA), was subjected to a polymerisation process with a thin layer of oste322 by a dose of 800 mJ cm−2 in order to enhance its reactivity. The two components were combined, compressed, and subjected to an additional polymerisation process with an exposure of 1400 mJ cm−2.
Step 5: Both parts of the oste322 system were then heated in a laboratory oven LAC-LH 30/12 (LAC) by a programmable unit Ht Industry (HT8) to 100 °C for 1.5 h, with a gradual decrease in temperature after the end of the process.
Step 6: The oste322 system was sterilised by stirring in 200 mL of 96% ethanol A.G. (PENTA, Czech Republic) for 20 minutes. The system underwent three rounds of washing with deionized water (dH2O) and was subsequently stirred in 200 mL of dH2O for 20 minutes. Finally, the system was dried using a stream of nitrogen gas.
Step 7: Channel 5, as depicted in Fig. 2A on the right, within the oste322 static microfluidic system underwent initial modification using Fluorosilane Fluo-ST2 (Emulseo, France). Subsequently, the entire system was heated to a temperature of 60 °C for 1.5 h. The remaining channels (refer to Table 1) were subjected to protein modifications, specifically Fibronectin at a concentration of 5 μg mL−1 in PBS, collagen type IV (5 μg mL−1) in PBS, and laminin (6 μg mL−1) in HBSS. The system was subsequently placed in the CellCulture® CO2 incubator (Esco Micro, Singapore) and incubated at a temperature of 37 °C for 2 h. Following the incubation period, each channel of the system was rinsed with 1 mL of PBS and then dried using a gentle stream of nitrogen.
The system was inspected using the Leica SP8 (Leica, Germany). The images were captured utilising the HC PL FLUOTAR CS 10×/0.30 N.A. dry objective (Leica, Germany). The Leica Las X Navigator function was employed to capture images of the specified region. Typically, a set of 4 × 6 image tiles with a 10% overlap was captured for each well. The scanning process was conducted sequentially. The fluorescence (Hoechst) images were acquired using a 405 nm laser for excitation and the PMT1 detector range was set to 410–500 nm. The fluorescence (Cell Tracker Orange) images were acquired using a 561 nm laser for excitation and the PMT2 detector range was set to 566–650 nm. Simultaneously, a wide-field image was acquired on a transmitted light detector. The fluorescence (propidium iodide) images were acquired using a 514 nm laser for excitation and the HyD detector range was set to 660–756 nm.
In order to maintain consistent focus throughout the imaging process, the Autofocus feature of the LAS X Navigator was utilised. Specifically, contrast-based method 1 was employed on fluorescence channel 2, which corresponds to the Cell Tracker Orange dye. The capture range was set at 70 μm, and before capturing each image, 5–7 focusing steps were performed. The tiles were merged using the Mosaic Merge function in LAS X software.
The quantification of viability data sets was performed using automated segmentation. The entire scan area was segmented, and cell quantification was performed using the StarDist plugin in FIJI ImageJ.52 The aforementioned procedure was conducted for both the Hoechst channel, with specifically labelled nuclei, and the Propidium Iodide channel, identifying dead cells. The default configuration of StarDist was employed, except for the PI counting parameter, which was adjusted to a higher probability/score threshold of 0.65. To enhance visual representation, the Gaussian Blur filter was implemented with a Sigma value (radius) of 2.00, followed by the adjustment of contrast.
2.4 Maintenance and cultivation of in vitro cell lines
Several model cell lines were used for the experiments. HTB-177 cell line (ATCC® NCI-H460, human lung carcinoma; ATCC) was cultivated in the RPMI-1640 medium (Thermo Fisher Scientific) supplemented with 10% (v/v) foetal bovine serum (FBS, Sigma-Aldrich), 0.1% (w/v) penicillin, and 0.1% (w/v) streptomycin (Sigma-Aldrich).U-373 cell line (Human glioblastoma astrocytoma, Sigma-Aldrich) was cultivated in DMEM medium (Thermo Fisher Scientific) supplemented with 10% (v/v) foetal bovine serum (FBS, Sigma-Aldrich), 0.1% (v/v) non-essential amino acids (Sigma-Aldrich), 0.1% (w/v) penicillin, and 0.1% (w/v) streptomycin (Sigma-Aldrich).
BJ cell line (ATCC® CRL-2522, human skin fibroblasts; ATCC) was cultivated in DMEM medium (Thermo Fisher Scientific) supplemented with 10% (v/v) foetal bovine serum (FBS, Sigma-Aldrich), 0.1% (w/v) penicillin, and 0.1% (w/v) streptomycin (Sigma-Aldrich).
Chinese Hamster B14 cell line (CCL-14.1, fibroblast, Cricetulus griseus, ATCC, Sigma-Aldrich) was cultivated in the high-glucose DMEM medium (Thermo Fisher Scientific) supplemented with 10% (v/v) foetal bovine serum (FBS, Sigma-Aldrich), 0.1% (w/v) penicillin, and 0.1% (w/v) streptomycin (Sigma-Aldrich).
All cell lines were maintained in the culture flasks (surface area 25 cm2, maximum volume 10 mL) and cultured in the CellCulture® CO2 incubator (Esco Micro, Singapore) at 37 °C in a humidified atmosphere (95% relative humidity), including 5% CO2.
The cells were harvested and utilised, once they reached a confluence level of 40–60%. Prior to cell passaging, the culture medium was removed and the cells were rinsed with 5 mL of PBS buffer (phosphate buffered saline, Sigma-Aldrich). The cells underwent trypsinisation using 0.5 mL of trypsin solution (trypsin–EDTA, Sigma-Aldrich, diluted 10-fold) for 5 min. The cells were subsequently resuspended in a 2 mL volume of the cell culture medium and mechanically dissociated through repetitive pipetting. The remaining cells in the flask were mixed with 5 mL of fresh medium and incubated. The quantification of viable cells was conducted using the trypan blue exclusion method on a CellDrop™ FL (DeNovix, USA).
2.5 Statistical analysis
The XPS measurement was performed as a series of three independent experiments. Two samples from the same batch were subjected to electrokinetic potential measurements in technical duplicate in two independent experiments. The contact angle was measured in two independent experiments in technical triplicates. The biological experiments involving oste322 inserts were measured in two independent experiments in technical triplicates. The time-lapse experiment was carried out in technical triplicates in three independent experiments. The static microfluidic oste322 system experiments were conducted as three independent experiments. Statistical significance was evaluated using either one-way or two-way analysis of variance (ANOVA) with Tukey's, Dunnett's, or Šídák's multiple comparisons test, employing Prism 8.0 GraphPad software (GraphPad Software, USA). A p-value <0.05 was considered statistically significant.3 Results and discussion
Material selection for microfluidic cell cultivation requires careful consideration of various factors such as chemistry, mechanics, and geometry.53 The chemistry of the surface is a crucial factor in determining the hydrophilicity of a material, as it is influenced by the presence of accessible chemical groups. These chemical groups directly impact the wettability of the material. The significance of surface charge extends to specific microfluidic applications and contributes to electrostatic interactions with cells.3.1 Oste322 surface optimisation for cell interactions
To optimise the oste322 surface, we investigated several surface-modifying methods. First, we utilised oxygen plasma treatment. This method is frequently used to enhance the properties of materials used in microfluidics.54–56 Additionally, we explored the Fluo-ST2 hydrophobic surface treatment, which is based on the use of a fluorosilane polymer. This particular coating forms a covalent bond with a range of surfaces commonly employed in microfluidics, such as PDMS and glass. It is used to improve the performance and stability of aqueous droplets within fluorinated oils. In theory, it is expected to possess an anti-fouling characteristic due to its fluorinated nature.57Oste322 has several limitations that are comparable to those of plastic culture vessels. Notably, it has the potential to induce alterations in cell shape, polarity, and morphology, as well as to promote increased cell proliferation and decreased differentiation.58 To address these effects and enhance cellular adhesion, we opted to modify the oste322 insert surface through the introduction of proteins, which are common components of the extracellular matrix found in the majority of mammalian tissues.
Collagens, elastin, and laminin have been selected for the oste322 surface modifications due to their status as the predominant proteins within the extracellular matrix of mammals.59 Collagen types I and II are prevalent as fibrillar collagens within connective tissue; collagen type IV is a network-forming collagen that comprises a significant portion of the basal lamina.60 In contrast to collagen, elastin does not undergo glycosylation of its amino acid residues. Fibronectin was selected due to its multifunctionality in mediating cell adhesion and migration. Additionally, it plays a role in the regulation of cellular morphology, cytoskeletal organization, haemostasis, and wound repair. Laminin constitutes a significant component of the basal lamina, acts as an adhesive agent, facilitating the cohesion between cells and the ECM (by binding collagen), and is known to exert regulatory effects on cellular processes such as cell growth, motility, and signalling pathways.58,61
In order to enhance cell adhesion on plastic surfaces, it is also possible to employ synthetic polymers. Poly(amino acids), such as poly-L-lysine, introduce a positive charge on plastic surfaces, thereby increasing the number of positively charged sites that are accessible for cellular binding.62 This phenomenon facilitates the electrostatic interaction between negatively charged ions present on the cell membrane and the positively charged surface of the culture substrate. Poly-L-lysine has been selected to complement the selected naturally occurring proteins.63
3.2 Material properties of oste322 inserts
Oste322 inserts, pristine or modified as described above, were comprehensively characterised using multiple techniques (Table 1). The chemical composition of their surfaces was analysed by XPS, (ESI Table S1†). The noted increase in oxygen content in plasma-treated oste322 suggests an occurrence of oxidation, a common reaction in polymers under such treatment.64–66 This modification can enhance the material's properties, such as improved wettability and surface roughness, which is beneficial for applications requiring effective cell adhesion. Furthermore, the formation of surface oxides might boost chemical stability, providing resistance to degradation while potentially influencing the material's biocompatibility, impacting its interaction with biological systems in biomedical applications.In the case of the Fluo-ST2 modification, the significant increase of fluorine in both the pristine and plasma pre-treated inserts of oste322 suggests the successful application of the Fluo-ST2 treatment, effectively modifying the surface characteristics of the material. The consequential reduction of other elements' levels indicates a prominent presence of the Fluo-ST2 coating. These changes could potentially enhance the material's anti-fouling properties, stability, and performance in microfluidic applications.
The increase in nitrogen content observed in some protein modifications (e.g. collagen II, fibronectin, and elastin) might indicate successful protein adsorption, given that nitrogen is used as a marker of protein presence.67 Silicon, which is detected on oste322 inserts, decreased in content when modifications were applied. Its presence likely stems from surface contamination during the heating step of PDMS masters. The presence of sodium and chlorine contaminants, presumably from PBS washing, calls for improved cleaning protocols to ensure the reliability of surface modifications and the preservation of desirable material properties.
The Electrokinetic analysis measures the charge on the material surface and can determine very small changes in its chemistry. The zeta potential of pristine oste322 was −30.0 ± 1.6 mV (Fig. 2B). This negative potential could be attributed to the presence of hydroxyl groups on the surface of the material. Certain protein modifications can lead to a noticeable reduction in the overall negative charge of the modified surface.68 Indeed, oste322 coating with collagen type II or IV resulted in reduced surface negativity (Fig. 2B). The charge status of collagen proteins in our experimental conditions (1 mM KCl) remains uncertain, as they can exhibit both positive and negative charges at pH 7.4, depending on the ionic strength ranging from 100 to 250 mM.69 As poly-L-lysine has a positive charge under physiological conditions due to the protonation of primary amino groups,70 this modification reduced the surface negativity the most (Fig. 2B).
The application of plasma treatment on pristine oste322 did not have a substantial impact on the surface charge (Fig. 2B). However, when the surface was pre-treated with plasma prior to further modification, it influenced the charge across all types of modification (Fig. 2B). This suggests that the surface plasma pre-treatment likely creates additional active functional groups, thereby enhancing the bonding between oste322 and surface-modifying substances.
The electrokinetic analysis results indicate that surface modifications to the oste322 material can significantly alter its surface charge, potentially impacting how cells and biomolecules interact with the material. A less negative or even positive zeta potential, as observed with some protein modifications and especially with poly-L-lysine, could potentially enhance cell adhesion due to increased electrostatic interactions. This may be particularly beneficial in applications involving cell cultivation, where enhanced cell–material interactions (adhesion or repulsion effects) are crucial.
The measurement of contact angle is influenced not only by the chemical composition of the surface but also by factors such as droplet size, surface roughness, and image analysis. Consequently, it becomes challenging to make direct comparisons between CA measurements reported in different publications and draw precise conclusions. Nevertheless, the majority of publications indicate that contact angles of OSTEMERs tend to exhibit a slight hydrophilic nature, primarily attributed to the occurrence of epoxy-ring opening polymerization, leading to the generation of hydroxyl groups.
In our experiment, the CA of deionized water on pristine oste322 inserts was 99 ± 3° (Fig. 2C). The measured angle exceeds the reported values of 67° for standard cured oste322 as published by Sandström et al.71 and 73 ± 2° (for oste327) and 70 ± 3° (for oste322) reported by Zhou et al.37
For reverse cured oste322,72 the CA was found to be approximately 80° (ranging from 73° to 85°) and 96° for OSTE-80.41 The increase in the contact angle measured in this study could be attributed to the alteration in the fabrication process of the oste322 inserts, which were subjected to a heating procedure before their removal from the PDMS mould.
The findings demonstrate that various protein modifications of pristine oste322 significantly affect the CA, resulting in increased hydrophilicity of the surface, with values ranging from approximately 80° to 50° (Fig. 2C). This observation provides further indirect evidence of protein binding to the surface of oste322. The application of oxygen plasma to the pristine oste322 resulted in a notable reduction in the contact angle (CA) to a value of 14 ± 4°. This decrease could be attributed to the increased presence of hydroxyl groups on the surface.41 Additionally, the application of plasma pre-treatment on protein-coated variants led to a further reduction in contact angle (CA), lowering it to 20°. The Fluo-ST2 modification did not result in a substantial alteration of CA in the case of pristine oste322. However, there was an observed increase to 114 ± 1° for plasma pre-treated Fluo-ST2. This suggests that the surface modification led to a higher level of saturation of the surface with Fluo-ST2 molecules.
In conclusion, the contact angle measurements in our study have shown that surface modifications of the oste322 material substantially influence its hydrophilicity, an important factor in mediating cell–material interactions.73–75 The pristine oste322 exhibited a higher contact angle than previously reported likely due to variations in the fabrication process, where the oste322 was heated inside of the PDMS moulds (this should be noted for future applications since the presence of traces of PDMS can be detrimental to some experiments). Protein modifications generally increased the hydrophilicity of the surface, suggesting successful protein binding. Moreover, oxygen plasma treatment substantially decreased the contact angle (Fig. 2C), indicating an enhanced presence of hydroxyl groups. Plasma pre-treatment further reduced the contact angle on protein-coated surfaces, hinting at a possible synergy between these treatments. In contrast, Fluo-ST2 modification did not significantly alter the contact angle of pristine oste322, but it did increase the angle for plasma pre-treated oste322 (Fig. 2C), implying greater surface saturation with Fluo-ST2 molecules.
The Owens, Wendt, Rabel and Kaelble (OWKR) method was employed for calculating the surface energy. The SFE of pristine oste322 was found to be 15.4 ± 3.2 mN m−1 (Fig. 2D) which falls within the range of low-energy polymers such as polytetrafluoroethylene, polydimethylsiloxane, and polypropylene, typically exhibiting SFE values in the range of 18 to 30 mN m−1.76 The application of Fluo-ST2 on the surface did not result in any significant alteration in surface energy. However, pre-treating with oxygen plasma led to a decrease in the SFE to a value of 7.1 ± 1.2 mN m−1.
The oxygen plasma treatment resulted in a significant increase in the surface energy, reaching a value of 77.8 ± 2.8 mN m−1. This increase was accompanied by a corresponding rise in the polar contribution (Fig. 2D). The observed changes in SFE suggest that the reactive plasma species interact with oste322, resulting in a transformation of its hydrophobic properties to hydrophilic. Except for poly-L-lysine, all protein modifications resulted in an increase in surface energy, ranging from 31.9 to 57.8 mN m−1. The impact of plasma pre-treatment on SFE exceeded that of proteins alone, resulting in increased values ranging from 57.2–75.4 mN m−1.
To summarise, the baseline SFE of oste322 resembles low-energy polymers, with Fluo-ST2 treatment having negligible effect and oxygen plasma pretreatment decreasing the SFE. Notably, plasma treatment dramatically increased the surface energy, transforming oste322 inserts from hydrophobic to hydrophilic. Most protein modifications increase surface energy, especially when paired with plasma pre-treatment. Importantly, the correlation between SFE and water contact angle measurements suggests that surfaces with higher contact angles, such as Fluo-ST2 treated ones, have lower surface energy, potentially hindering cell attachment. The Fluo-ST2 SFE values are comparable to Teflon coatings, which are regarded as anti-adhesive.77 Thus, strategic surface modifications can fine-tune oste322 characteristics to suit specific applications involving cell cultivation.
To assess the potential cytotoxic effects of leaching components from oste322 preparations, we performed a leaching test on pristine and plasma-treated oste322 inserts using MiliQ water or ethanol. We tested leaching times of 20 min, 24 h, and 72 h. The qualitative screening of the leached materials was conducted using HPLC-MS.
Instrument background peaks were consistently observed in all mass spectra that were measured using APCI-N (applies to blank samples as well). The masses with m/z values of 130.08 and 170.75 Da could potentially be inferred from the spectra obtained through APCI-P measurements. The identical masses were observed in the blank samples and thus omitted from the analyses. The only analyte present in the ethanolic extracts exhibited a mass of m/z 354.17 Da. This phenomenon was observed exclusively in APCI-P mode. Based on the information provided by the available sources, it was possible to suggest the corresponding analytes, as indicated in ESI Table S2.†
Several lines of evidence indicate possible leaching of detergents from various polymeric materials, including 3D printing resins.78,79 Based on the XPS spectra indicating the presence of silicon and the observed elevated contact angle of the pristine oste322, it was reasonable to consider PDMS as a likely source of contamination. However, it was not possible to determine the concentration of the leached compounds due to the limitations of mass spectrometry. Although PDMS contamination of the oste322 surface might have some impact on cell confluence and viability, cytotoxic effects would be rather unlikely, as judged by the properties of PDMS.80
It is important to understand the implications of the material properties of oste322 on the performance of the fabricated microfluidic devices. The robustness and flexibility, coupled with the adjustable hydrophilicity/hydrophobicity, make oste322 a versatile material for microfabrication. These properties ensure that the devices can withstand varying operational conditions and can be adapted to accommodate a diverse range of biomedical applications, thus expanding the usability and applicability of oste322 in microfluidic device fabrication.
3.3 Biocompatibility and performance of modified surfaces on oste322 inserts and oste322 plate
The evaluation of surface properties and their impact on cell viability is critical in cell-based assays and for cell cultivation in general. In order to evaluate the biocompatibility of pristine oste322 and its modified variants (Table 1), we performed a CCK-8 cell viability assay and a cell confluence assay using epithelial (HTB-177), glioblastoma (U-373 MG), skin (BJ), and animal fibroblast (B14) cells.Various cell lines have unique requirements for the extracellular matrix composition. Therefore, we always carefully choose a set of proteins that are considered appropriate for facilitating the growth of a given cell line. To optimise efficiency and accuracy, we selectively omitted the protein modifications that were considered irrelevant to the individual cell lines (based on the cell line provider information). This approach allowed us to identify the optimal combination of proteins for each cell line, which was used as the basis for the later experiments with the oste322 plate and system.
We have modified the oste322 surface with selected proteins to facilitate cell attachment to its surface. We were especially interested in the impact of oxygen plasma treatment of the oste322 surface since it could improve both protein adsorption41 and cell adhesion.81 Fig. 3–6 presents a comparison of the results obtained from viability and confluence assays. The figures emphasise the similarities observed in most instances between the standard cell culture substrate, namely the polystyrene culture plate with a physically modified surface suitable for cell cultivation (“tissue culture-treated”), and the oste322 substrate with its corresponding modifications.
 | ||
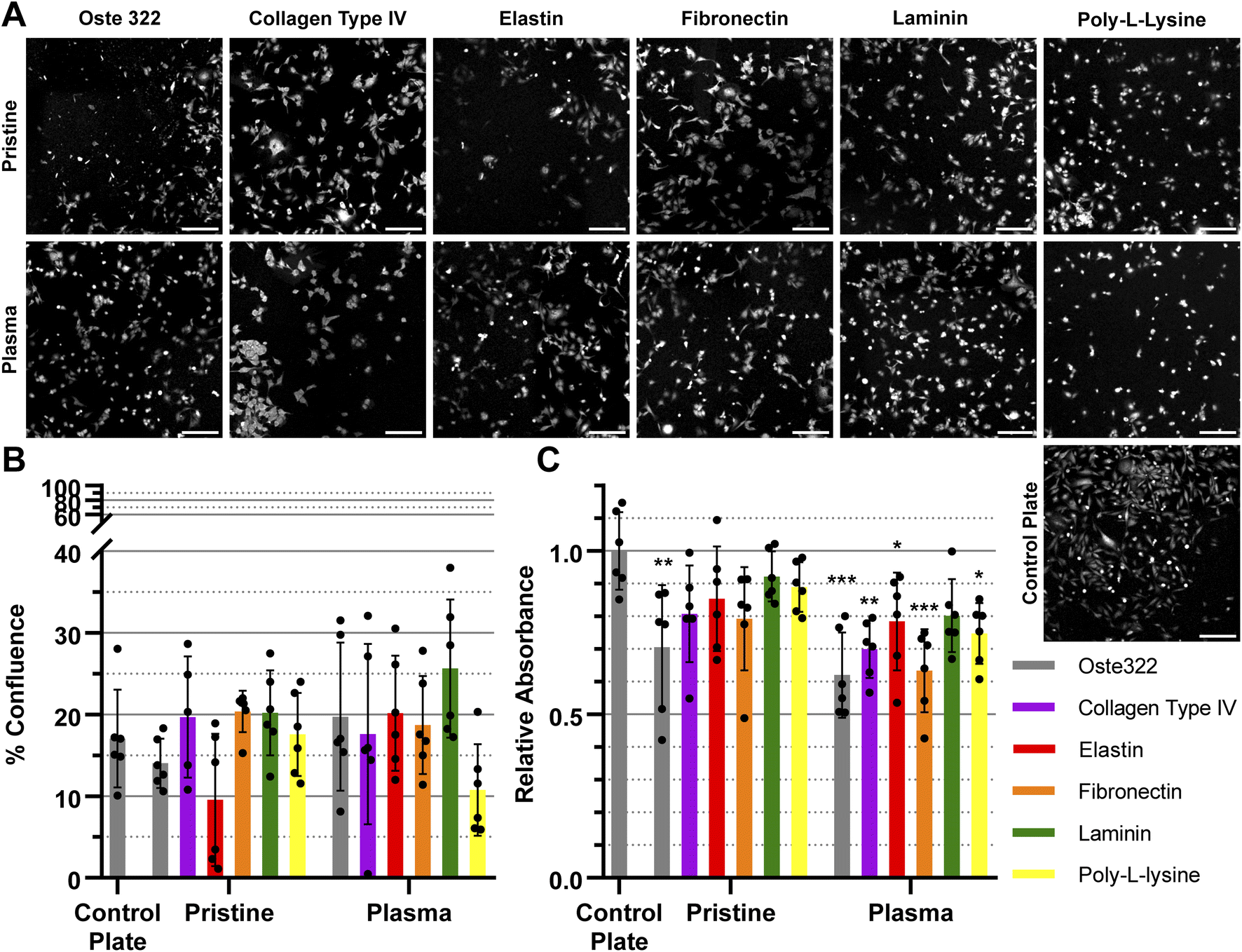
| Fig. 3 The confluence and viability of HTB-177 (epithelial) cells. Cells were evaluated after 24 h of incubation on the oste322 inserts and a control plate. (A) Representative tile scans of different modifications applied to pristine and plasma pre-treated oste322 inserts were obtained using confocal microscopy. The visualisation of cells was achieved by employing the CellTracker™ Orange dye. (B) The assessment of confluence was conducted using confocal images, measuring the percentage of the area covered by cells. (C) Cell viability was evaluated using the CCK-8 assay, and the data were normalised to the control. Values (shown as dots) are presented as the mean ± SD (n = 6). The data were analysed using a one-way ANOVA, comparing each data set with control plate, where * represents a p-value ≤0.05, **p ≤ 0.01, and ***p ≤ 0.001. Data with no symbols are statistically not significant. Scale bars = 200 μm. | ||
 | ||
| Fig. 4 The confluence and viability of U-373 MG (glioblastoma) cells. Cells were evaluated after 24 h of incubation on the oste322 inserts and a control plate. (A) Representative tile scans of different modifications applied to pristine and plasma pre-treated oste322 inserts were obtained using confocal microscopy. The visualisation of cells was achieved by employing the CellTracker™ Orange dye. (B) The assessment of confluence was conducted using confocal images, measuring the percentage of the area covered by cells. (C) Cell viability was evaluated using the CCK-8 assay, and the data were normalised to the control. Values (shown as dots) are presented as the mean ± SD (n = 6). The data were analysed using a one-way ANOVA, comparing each data set with control plate, where * represents a p-value ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001. Data with no symbols are statistically not significant. Scale bars = 200 μm. | ||
 | ||
| Fig. 5 The confluence and viability of CRL-2522 BJ (skin) cells were evaluated after 24 h of incubation on the oste322 inserts and a control plate. (A) Representative tile scans of different modifications applied to pristine and plasma pre-treated oste322 inserts were obtained using confocal microscopy. The visualisation of cells was achieved by employing the CellTracker™ Orange dye. (B) The assessment of confluence was conducted using confocal images, measuring the percentage of the area covered by cells. (C) Cell viability was evaluated using the CCK-8 assay, and the data were normalised to the control. Values (shown as dots) are presented as the mean ± SD (n = 6). The data were analysed using a one-way ANOVA, comparing each data set with control plate, where * represents a p-value ≤0.05, **p ≤ 0.01, and ***p ≤ 0.001. Data with no symbols are statistically not significant. Scale bars = 200 μm. | ||
 | ||
| Fig. 6 The confluence and viability of B14 (fibroblast) cells were evaluated after 24 h of incubation on the oste322 inserts and a control plate. (A) Representative tile scans of different modifications applied to pristine and plasma pre-treated oste322 inserts were obtained using confocal microscopy. The visualisation of cells was achieved by employing the CellTracker™ Orange dye. (B) The assessment of confluence was conducted using confocal images, measuring the percentage of the area covered by cells. (C) Cell viability was evaluated using the CCK-8 assay, and the data were normalised to the control. Values (shown as dots) are presented as the mean ± SD (n = 6). The data were analysed using a Brown-Forsythe and Welch ANOVA test, comparing each data set with control plate, where * represents a p-value ≤0.05. Data with no symbols are statistically not significant. Scale bars = 200 μm. | ||
The HTB-177 cell line exhibited the highest responsiveness to the various modifications of the oste322 surface (Fig. 3A). Based on the data obtained from the measured confluence, as depicted in Fig. 3B, it was evident that the modifications improved the cells' capacity to spread over the surface, reaching a level comparable to that of the control plate. This was not the case for pristine oste322 and the elastin-coated variant, as their confluence was lower compared to the control. From Fig. 3C, it was evident that the viability of cells on all of the modified variants, as well as on the pristine oste322, was significantly lower when compared to the control plate.
The confluence of U-373 MG cell line was generally similar to that of the control plate, as depicted in Fig. 4A and B. The viability of the modified variants, as indicated in Fig. 4C was found to be similar to that of the control plate, except for the pristine oste322, which exhibited lower viability. In the case of the plasma pre-treated variants, it appears that only the laminin modification exhibits a level of comparability to the control plate. All other variants have lower viability.
The BJ cell line demonstrated favourable performance when cultivated on oste322 surfaces. The confluence was comparable to the control plate in all variants, as depicted in Fig. 5A and B. The viability was similar or slightly lower across most of the modifications. The viability of cells on pristine oste322 exhibits the lowest value in comparison to the other variants.
The B14 cells were overall the best performing, where both confluence (Fig. 6A and B), and viability (Fig. 6C) were comparable to the plate control. The only exception was the plasma pre-treated poly-L-lysine variant of modification, where the viability was lower than that of the control.
In summary, the viability and confluence measurements suggested a rather favourable cell toxicity profile of oste322. Moreover, the majority of cell lines, specifically B14 and BJ, exhibited similar behaviour to that of the plate control. Nevertheless, the rate of cell growth is partially decreased on variously modified pristine oste322 surfaces (Fig. 3–5C), which correlates with the slight hydrophobic nature of inserts moulded from PDMS along with a low surface free energy (Fig. 2C and D).
Furthermore, the application of plasma treatment to the native oste322 surface led to increased confluence, particularly in HTB-177 cells. However, it should be noted that this does not apply to all cell lines. In addition, our results do not indicate an effect of the surface plasma pre-treatment on cell confluence before protein modification. This implies that the inherent chemistry of the native oste322 surface is capable of facilitating protein adhesion. This discovery simplifies the surface preparation process, as the plasma treatment of enclosed channels in microfluidic systems could be challenging.
To further understand the interactions of cells with oste322 surfaces, the time-lapse experiments focusing on cell attachment using the best (B14) and worst performing (HTB-177) cell lines were carried out. Two protein modifications performing best for the given cell line on oste322 inserts were used on the newly manufactured thin oste322 plate (see Methods Section 2.2.1). In this experiment, the circularity and cell perimeter were measured in 15 minute intervals for 6 h. Finally, the confluence was measured in the last time point. Then, the cells were washed, using cell culture medium and confluence was established after washing as well. From Fig. 7A–C it is evident that B14 cells growing either on pristine oste322 or Fibronectin-coated oste332 are flatter than the control. On the other hand, B14 growing on collagen I-coated oste332 exhibited lower attachment to the surface than the control. However, the wash-out experiments showed that the cell attachment strength was not affected per se (Fig. 7D), which suggests that other cellular mechanisms (e.g., kinetics of cell attachment, cell viability) might have been impacted by the oste332 surface. In the case of HTB-177 cells, the circularity (Fig. 7E and F) was overall comparable to the control. However, the perimeter (Fig. 7G) of cells growing on oste322 surfaces was significantly lower than the control for all oste322 variants. It suggests that the spreading of HTB-177 cells on oste322 and modified variants is hindered compared to control. It is also evident (Fig. 7H) that the confluence after washing remained the same also for HTB-177, further corroborating that the strength of the cell attachment is not negatively affected by oste322.
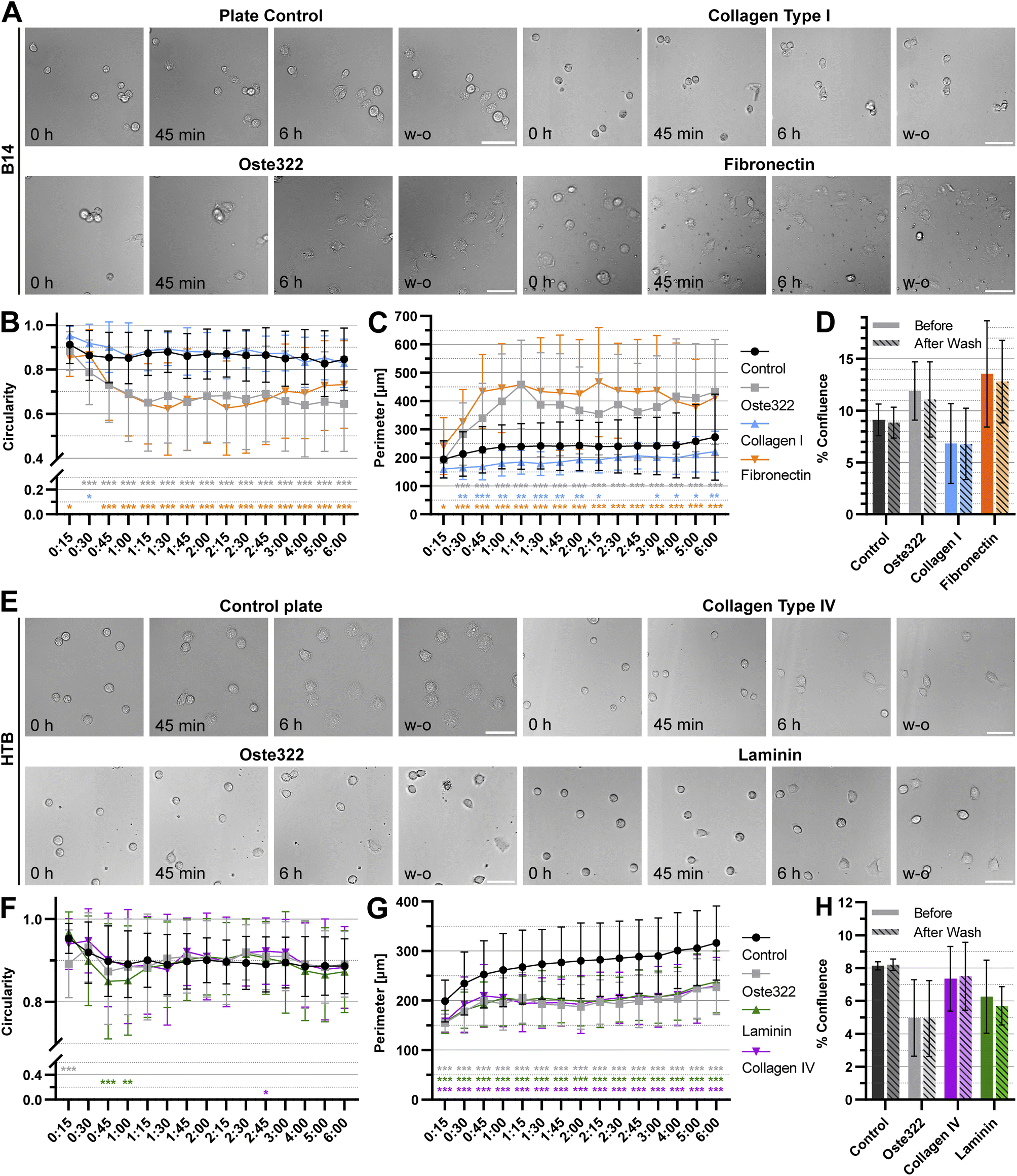 | ||
| Fig. 7 Time-lapse of cell B14 (fibroblast) and HTB-177 (epithelial) attachment. (A) Still images from time-lapse experiments with B14 cells with indicated times and surface treatment. (B) Circularity of B14 cells (1.0 represents a perfect circle). (C) Perimeter of B14 cells. (D) Confluence of B14 cells after 6 h of imaging and a washing step. (E) Still images from time-lapse experiments with HTB-177 cells (HTB) with indicated times and surface treatment. (F) Circularity of HTB-177 cells (1.0 represents a perfect circle). (G) Perimeter of HTB-177 cells. (H) Confluence of HTB-177 cells after 6 h of imaging and a washing step. Values represent the mean ± SD. The data were analysed using a two-way ANOVA, comparing each data set with control, where * represents a p-value ≤0.05, ** p-value ≤ 0.01 and ***p ≤ 0.001. Scale bars = 50 μm. | ||
The overall viability of the HTB-177 and U373MG cell lines on the oste322 appears to be consistently decreased, regardless of any modifications. Multiple factors may influence the cells cultivated on the oste322 substrate. In certain instances, particularly when the off-stoichiometric ratios are high, the OSTE polymers may undergo unreacted monomer leaching.42 It has the potential to adversely affect the viability of cells and their metabolic activity due to the potential cytotoxic effects introduced by the presence of unreacted monomers. However, our investigation using mass spectrometry did not detect any contaminants in the leachates that could be attributed to toxicity.
Although OSTE and OSTE+ polymers provide surface anchors for direct surface modification, it is important to consider that the modifications themselves may influence cell viability. The potential impact of modifications on various aspects, such as surface chemistry, hydrophobicity/hydrophilicity, and other properties, should be considered, as they can influence cellular behaviour and metabolic processes. The consideration of the distinct properties and characteristics of proteins employed for surface modification, along with their potential interaction with the OSTEMER material, holds significant importance. In the instance of the HTB-177 cell line, there is a possibility that the proteins selected for modifying the oste322 surface were not compatible with the specific needs of the cells, which could have contributed to the observed decrease in viability. Potential improvements in the compatibility of HTB-177 cells with oste322 could be achieved by optimising the surface modification process. This could involve adjusting the concentration or type of proteins used for surface modification or even investigating other surface modification methods that might be better suited for this particular cell line.
The mechanical properties of the oste322 should also be considered. The material's consistency can range from rubbery to thermoplastic-like, depending on the off-stoichiometry ratio. The sensitivity of cells to substrate stiffness has been previously examined.82 It is important to note that an unsuitable range of stiffness could potentially impact cellular metabolic activity by impeding cellular functions.
In conclusion, the evaluation of various oste322 surfaces, both pristine and modified, brought novel insights into the material's biocompatibility across different cell lines. While the viability and confluence of cells were mostly comparable to those observed on the conventional polystyrene culture plate, there were some notable differences depending on the cell line and surface modification. In particular, the HTB-177 cell line showed lower viability and confluence on the oste322 surfaces, while the B14 cells performed best. These findings underscore the need to carefully select and tailor surface modifications to the specific requirements of each cell line.
3.4 Biocompatibility and performance of oste322 in a static microfluidic system
To evaluate the prolonged impact of the oste322 material on the cultured cells, we fabricated a simple static microfluidic system (Fig. 1 and 2A, right). The cell viability and confluence experiments were conducted under static conditions with medium exchange every 24 h over a period of 72 h. The static system provided a controlled environment to evaluate the viability and proliferation of cells on a protein-modified surface within a setup simulating potential microfluidic applications. The proteins selected for modification of the system channels were based on the results from the previous screening, as indicated in Section 3.1 (please refer to Table 1 for the specific protein choices). We have also included the anti-fouling Fluo-ST2 since this coating could be particularly useful in instances, where a strong cell-substrate adhesion is not desired.Although the confluence of HTB-177, U-373 MG, and B14 appears to be lower than the control, this effect is not statistically significant, as seen in (Fig. 8A and C). In contrast, the confluence of B14 was drastically reduced on the Fluo-ST2 surface (Fig. 8A and C). The overall cell viability on tested surfaces was the same as the control (Fig. 8B and D). However, from Fig. 8E we can observe, that the number of attached cells per area is significantly lower in all cases, except for the BJ cell line. This indicates that the cell attachment was affected by the scale-down, which might reflect both cell adhesion issues and cell viability problems, since unfit and dying cells might have been removed during the washing steps after cell labelling.
 | ||
| Fig. 8 Confluence and viability evaluation after 72 h of cell incubation in the static microfluidic oste322 system and control plate. (A) Representative tile scans from confocal microscopy. Cell confluence visualised by CellTracker™ Orange dye within a channel of the oste322 system. (B) Cell viability in the oste322 system; cell nuclei (Hoechst32258, green) and non-viable cells (Propidium Iodide, magenta). The second row shows viability staining from the inside of a corresponding well of plate control. (C) Confluence was assessed using confocal images as a percentage of the area covered by cells. (D) Cell viability was assessed by counting all cells and the dead cell fraction, as shown on the graph as live/dead ratio. The data were normalised to the control. (E) The number of cells attached per area. Values (shown as dots) represent the mean ± SD (n = 3). The data were analysed using a 2-way ANOVA, comparing the data for each set vs. control, where ** represents a p-value ≤0.01 and ***p ≤ 0.001. Scale bar = 200 μm. | ||
In general, discussion on microfluidic cultures is challenging due to the fundamental shift in the physicochemical perspective caused by transitioning from well plate inserts to scaled-down cell-based microfluidic devices. The effect of this scale-down is apparent from Fig. 8E. Additional testing, preferably in microfluidic conditions and for longer time periods would be necessary for conclusions regarding the suitability of this material for use in applications like organs-on-chip or organoids.
The application of the anti-fouling Fluo-ST2 coating led to limited cellular adhesion to the surface, with a notable effect on the proliferation of the rapidly growing B-14 cell line. This implies that the surfaces of oste322 could be altered to inhibit cell adhesion and fouling, thereby preventing undesired interactions or blockages within microfluidic channels, even when cells are incubated in the system for an extended period of time. The presence of this anti-fouling property confers several benefits, including enhanced device performance and decreased maintenance needs. We showed that the silane modification chemistry commonly employed on glass or plasma-activated PDMS could also be applied to the pristine oste322 material.
Oste322 demonstrated the ability to efficiently produce complex structures, making it suitable for the fabrication of microfluidic devices. Its compatibility with a standard microfluidic workflow allows for convenient and cost-effective fabrication processes. Another notable advantage is the ability of oste322 to be bonded to several types of materials and to itself without the need for additional means such as adhesive or plasma activation. Moreover, the potential for utilisation of reactive UV injection moulding in the fabrication of microfluidic devices establishes this material as suitable not only for rapid prototyping but also for large-scale manufacturing.
The impact of our findings extends to the broader field of microfluidics, offering promising avenues for the development of more efficient and versatile lab-on-a-chip technologies. Oste322 unique properties, such as chemical stability, tunability, and biocompatibility, make it a good candidate for the fabrication of microfluidic devices designed for a wide array of applications, from chemical synthesis to biological assays.
4 Conclusions
Our study established the photosensitive thermoset polymer, OSTEMER 322 (oste322), as a promising material for bio-microfluidic device fabrication. Initial surface characterisation demonstrated oste322's versatile properties, enabling adaptability in terms of hydrophilicity/hydrophobicity, zeta potential, and surface chemistry.The viability and confluence of various cell types, including epithelial, skin, and fibroblast cells, on the protein-coated oste322 were still lower than the standard tissue-culture plastics. However, oste322 demonstrated minimal cytotoxicity after 24 h incubation. Moreover, a simple static microfluidic system fabricated from oste322 exhibited good biocompatibility for most of the cell lines even after a 72 h incubation period. However, some cells (like HTB-177) were notably affected by the scale-down. Therefore, additional testing and surface modifications, especially for prolonged incubation times, might be needed.
One of the unique aspects of the oste322 fabrication process is its two-step curing method, which combines the advantages of soft-lithography with those of solid thermosets. This, along with its slightly hydrophilic native surface, low water vapour permeability, and bonding properties (ability to be bonded to other materials and to itself), makes it an attractive choice for bio-microfluidic applications. Oste322 can be the material, that can serve the academic needs for rapid prototyping, while having the potential for mass production via UV reaction injection moulding. The practical benefits of this material are promising for future developments in bio-microfluidic devices.
Author contributions
Petr Aubrecht: conceptualisation, methodology, investigation, formal analysis, visualisation, writing – original draft, writing – review and editing. Jiří Smejkal: investigation, formal analysis, writing – review and editing. Petr Panuška: investigation. Klára Španbauerová: investigation. Viktorie Neubertová: investigation. Pavel Kaule: investigation. Jindřich Matoušek: investigation. Stanislav Vinopal: formal analysis, supervision, writing – review and editing. Michaela Liegertová: formal analysis, writing – review and editing. Marcel Štofik: conceptualisation, writing – review and editing. Jan Malý: conceptualisation, funding acquisition, supervision, writing – review and editing. All authors have approved the final version of the manuscript.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
This work was supported by the Internal Grant Agency of the University of Jana Evangelista Purkyně in Ústí nad Labem, grant No. UJEP-SGS-2022-53-005-3 and by the Czech Science Foundation (GACR No. 22-10536S). The authors acknowledge the assistance provided by the Research Infrastructure NanoEnviCz (Project No. LM2018124) and the project Pro-NanoEnviCz (Reg. No. CZ.02.1.01/0.0/0.0/16_013/0001821 and CZ.02.1.01/0.0/0.0/18_046/0015586), supported by the Ministry of Education, Youth and Sports.References
- N. Convery and N. Gadegaard, Micro Nano Eng., 2019, 2, 76–91 CrossRef.
- G. M. Whitesides, Nature, 2006, 442, 368–373 CrossRef CAS PubMed.
- D. Mark, S. Haeberle, G. Roth, F. Von Stetten and R. Zengerle, in Microfluidics Based Microsystems, ed. S. Kakaç, B. Kosoy, D. Li and A. Pramuanjaroenkij, Springer Netherlands, Dordrecht, 2010, pp. 305–376 Search PubMed.
- H. Becker, Lab Chip, 2010, 10, 271–273 RSC.
- D. J. Harrison, K. Fluri, K. Seiler, Z. Fan, C. S. Effenhauser and A. Manz, Science, 1993, 261, 895–897 CrossRef CAS PubMed.
- S. C. Jacobson, A. W. Moore and J. M. Ramsey, Anal. Chem., 1995, 67, 2059–2063 CrossRef CAS.
- A. W. Moore, S. C. Jacobson and J. M. Ramsey, Anal. Chem., 1995, 67, 4184–4189 CrossRef CAS.
- J. C. McDonald, D. C. Duffy, J. R. Anderson, D. T. Chiu, H. Wu, O. J. A. Schueller and G. M. Whitesides, Electrophoresis, 2000, 21, 27–40 CrossRef CAS.
- Y. Xia, J. J. McClelland, R. Gupta, D. Qin, X.-M. Zhao, L. L. Sohn, R. J. Celotta and G. M. Whitesides, Adv. Mater., 1997, 9, 147–149 CrossRef CAS.
- H. Becker and U. Heim, Sens. Actuators, A, 2000, 83, 130–135 CrossRef CAS.
- C.-S. Chen, S.-C. Chen, W.-H. Liao, R.-D. Chien and S.-H. Lin, Int. Commun. Heat Mass Transfer, 2010, 37, 1290–1294 CrossRef CAS.
- J. Narasimhan and I. Papautsky, J. Manuf. Syst., 2004, 14, 96–103 Search PubMed.
- T. S. Hansen, D. Selmeczi and N. B. Larsen, J. Micromech. Microeng., 2010, 20, 015020 CrossRef.
- K. Giri and C.-W. Tsao, Micromachines, 2022, 13, 486 CrossRef PubMed.
- S. Scott and Z. Ali, Micromachines, 2021, 12, 319 CrossRef.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301–5338 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS.
- T. O. Machado, C. Sayer and P. H. H. Araujo, Eur. Polym. J., 2017, 86, 200–215 CrossRef CAS.
- C. F. Carlborg, T. Haraldsson, K. Öberg, M. Malkoch and W. van der Wijngaart, Lab Chip, 2011, 11, 3136 RSC.
- S. H. Kim, Y. Yang, M. Kim, S.-W. Nam, K.-M. Lee, N. Y. Lee, Y. S. Kim and S. Park, Adv. Funct. Mater., 2007, 17, 3493–3498 CrossRef CAS.
- Ph. Wägli, A. Homsy and N. F. de Rooij, Sens. Actuators, B, 2011, 156, 994–1001 CrossRef.
- L.-H. Hung, R. Lin and A. P. Lee, Lab Chip, 2008, 8, 983 RSC.
- J. F. Ashley, N. B. Cramer, R. H. Davis and C. N. Bowman, Lab Chip, 2011, 11, 2772 RSC.
- S. Silvestrini, D. Ferraro, T. Tóth, M. Pierno, T. Carofiglio, G. Mistura and M. Maggini, Lab Chip, 2012, 12, 4041 RSC.
- N. A. Feidenhans’l, J. P. Lafleur, T. G. Jensen and J. P. Kutter, Electrophoresis, 2014, 35, 282–288 CrossRef PubMed.
- G. Pardon, T. Haraldsson and W. van der Wijngaart, in 2014 IEEE 27th International Conference on Micro Electro Mechanical Systems (MEMS), IEEE, San Francisco, CA, USA, 2014, pp. 96–99 Search PubMed.
- F. Saharil, C. F. Carlborg, T. Haraldsson and W. van der Wijngaart, Lab Chip, 2012, 12, 3032 RSC.
- J. M. Karlsson, F. Carlborg, F. Saharil, F. Forsberg, F. Niklaus, W. van der Wijngaart and T. Haraldsson, Chemistry and Life Sciences (microTAS, 2012), 2012, pp. 225–227 Search PubMed.
- T. Haraldsson, C. F. Carlborg and W. van der Wijngaart, Proc. SPIE, Microfluidics, BioMEMS, and Medical Microsystems XII, 2014, vol. 8976 Search PubMed.
- C. F. Carlborg, A. Vastesson, Y. Liu, W. van der Wijngaart, M. Johansson and T. Haraldsson, J. Polym. Sci. Part A: Polym. Chem., 2014, 52, 2604–2615 CrossRef CAS.
- L. El Fissi, R. Fernández, P. García, M. Calero, J. V. García, Y. Jiménez, A. Arnau and L. A. Francis, Sens. Actuators, A, 2019, 285, 511–518 CrossRef CAS.
- D. Sticker, R. Geczy, U. O. Häfeli and J. P. Kutter, ACS Appl. Mater. Interfaces, 2020, 12, 10080–10095 CrossRef CAS.
- Y.-J. Fan, H.-Y. Hsieh, S.-F. Tsai, C.-H. Wu, C.-M. Lee, Y.-T. Liu, C.-H. Lu, S.-W. Chang and B.-C. Chen, Lab Chip, 2021, 21, 344–354 RSC.
- D. M. D. Siu, K. C. M. Lee, B. M. F. Chung, J. S. J. Wong, G. Zheng and K. K. Tsia, Lab Chip, 2023, 23, 1011–1033 RSC.
- J. Black, Biological Performance of Materials: Fundamentals of Biocompatibility, CRC Taylor & Francis, Boca Raton, 4th edn, 2006 Search PubMed.
- D. Sticker, M. Rothbauer, S. Lechner, M.-T. Hehenberger and P. Ertl, Lab Chip, 2015, 15, 4542–4554 RSC.
- X. C. Zhou, R. Sjöberg, A. Druet, J. M. Schwenk, W. van der Wijngaart, T. Haraldsson and C. F. Carlborg, Lab Chip, 2017, 17, 3672–3681 RSC.
- R. P. S. de Campos, C. D. M. Campos, G. B. Almeida and J. A. F. da Silva, IEEE Trans. Biomed. Circuits Syst., 2017, 11, 1470–1477 Search PubMed.
- H. Liu, J. Usprech, Y. Sun and C. A. Simmons, Acta Biomater., 2016, 34, 113–124 CrossRef PubMed.
- I. Matthiesen, D. Voulgaris, P. Nikolakopoulou, T. E. Winkler and A. Herland, Small, 2021, 17, 2101785 CrossRef CAS PubMed.
- T. F. Chen, K. S. Siow, P. Y. Ng and B. Y. Majlis, Materials Science and Engineering: C, 2017, 79, 613–621 CrossRef CAS PubMed.
- F. Ejserholm, J. Stegmayr, P. Bauer, F. Johansson, L. Wallman, M. Bengtsson and S. Oredsson, Biomater. Res., 2015, 19, 19 CrossRef PubMed.
- W. Liu, Y. Li and X. Ding, Biofabrication, 2017, 9, 025006 CrossRef PubMed.
- R. Li, X. Lv, M. Hasan, J. Xu, Y. Xu, X. Zhang, K. Qin, J. Wang, D. Zhou and Y. Deng, J. Chromatogr. Sci., 2016, 54, 523–530 CAS.
- R. Rimsa, A. Galvanovskis, J. Plume, F. Rumnieks, K. Grindulis, G. Paidere, S. Erentraute, G. Mozolevskis and A. Abols, Micromachines, 2021, 12, 546 CrossRef PubMed.
- D. Sticker, M. Rothbauer, J. Ehgartner, C. Steininger, O. Liske, R. Liska, W. Neuhaus, T. Mayr, T. Haraldsson, J. P. Kutter and P. Ertl, ACS Appl. Mater. Interfaces, 2019, 11, 9730–9739 CrossRef CAS PubMed.
- D. K. Owens and R. C. Wendt, J. Appl. Polym. Sci., 1969, 13, 1741–1747 CrossRef CAS.
- W. Rabel, Farbe und Lack, 1971, 77, 997–1005 CAS.
- D. H. Kaelble, The Journal of Adhesion, 1970, 2, 66–81 CrossRef CAS.
- C. Stringer, T. Wang, M. Michaelos and M. Pachitariu, Nat. Methods, 2021, 18, 100–106 CrossRef CAS PubMed.
- M. Pachitariu and C. Stringer, Nat. Methods, 2022, 19, 1634–1641 CrossRef CAS PubMed.
- U. Schmidt, M. Weigert, C. Broaddus and G. Myers, in Medical Image Computing and Computer Assisted Intervention – MICCAI 2018, ed. A. F. Frangi, J. A. Schnabel, C. Davatzikos, C. Alberola-López and G. Fichtinger, Springer International Publishing, Cham, 2018, vol. 11071, pp. 265–273 Search PubMed.
- J. Kim and R. C. Hayward, Trends Biotechnol., 2012, 30, 426–439 CrossRef CAS PubMed.
- S. Roy, C. Y. Yue, Y. C. Lam, Z. Y. Wang and H. Hu, Sens. Actuators, B, 2010, 150, 537–549 CrossRef CAS.
- S. H. Tan, N.-T. Nguyen, Y. C. Chua and T. G. Kang, Biomicrofluidics, 2010, 4, 032204 CrossRef PubMed.
- B. Da Silva, M. Zhang, G. Schelcher, L. Winter, C. Guyon, P. Tabeling, D. Bonn and M. Tatoulian, Plasma Processes Polym., 2017, 14, 1600034 CrossRef.
- M. Lejars, A. Margaillan and C. Bressy, Chem. Rev., 2012, 112, 4347–4390 CrossRef CAS PubMed.
- H. K. Kleinman, L. Luckenbill-Edds, F. W. Cannon and G. C. Sephel, Anal. Biochem., 1987, 166, 1–13 CrossRef CAS PubMed.
- J. Halper and M. Kjaer, in Progress in Heritable Soft Connective Tissue Diseases, ed. J. Halper, Springer Netherlands, Dordrecht, 2014, vol. 802, pp. 31–47 Search PubMed.
- Molecular Biology of the Cell, ed. B. Alberts, Garland Science, New York, 4th edn, 2002 Search PubMed.
- D. M. Gamm, J. N. Melvan, R. L. Shearer, I. Pinilla, G. Sabat, C. N. Svendsen and L. S. Wright, Invest. Ophthalmol. Visual Sci., 2008, 49, 788 CrossRef PubMed.
- T. Saxena, L. Karumbaiah and C. M. Valmikinathan, in Natural and Synthetic Biomedical Polymers, Elsevier, 2014, pp. 43–65 Search PubMed.
- D. Mazia, G. Schatten and W. Sale, J. Cell Biol., 1975, 66, 198–200 CrossRef CAS PubMed.
- R. Foerch, J. Izawa and G. Spears, J. Adhes. Sci. Technol., 1991, 5, 549–564 CrossRef CAS.
- R. W. Paynter, Surf. Interface Anal., 1998, 26, 674–681 CrossRef CAS.
- A. Vesel, M. Mozetic and A. Zalar, Vacuum, 2007, 82, 248–251 CrossRef CAS.
- S. Ray and A. G. Shard, Anal. Chem., 2011, 83, 8659–8666 CrossRef CAS PubMed.
- N. L. Burns and K. Holmberg, in Trends in Colloid and Interface Science X, ed. C. Solans, M. R. Infante and M. J. García-Celma, Steinkopff, Darmstadt, 1996, vol. 100, pp. 271–275 Search PubMed.
- K. G. Wilcox, G. M. Kemerer and S. Morozova, J. Chem. Phys., 2023, 158, 044903 CrossRef CAS PubMed.
- M. Zheng, M. Pan, W. Zhang, H. Lin, S. Wu, C. Lu, S. Tang, D. Liu and J. Cai, Bioact. Mater., 2021, 6, 1878–1909 CAS.
- N. Sandström, R. Z. Shafagh, A. Vastesson, C. F. Carlborg, W. van der Wijngaart and T. Haraldsson, J. Micromech. Microeng., 2015, 25, 075002 CrossRef.
- L. Sønstevold, M. Yadav, N. B. Arnfinnsdottir, A. K. Herbjørnrød, G. U. Jensen, A. Aksnes and M. M. Mielnik, J. Micromech. Microeng., 2022, 32, 075008 CrossRef.
- A. S. Curtis, J. V. Forrester, C. McInnes and F. Lawrie, J. Cell Biol., 1983, 97, 1500–1506 CrossRef CAS PubMed.
- P. B. Van Wachem, A. H. Hogt, T. Beugeling, J. Feijen, A. Bantjes, J. P. Detmers and W. G. Van Aken, Biomaterials, 1987, 8, 323–328 CrossRef CAS PubMed.
- M. Lampin, R. Warocquier-Clérout, C. Legris, M. Degrange and M. F. Sigot-Luizard, J. Biomed. Mater. Res., 1997, 36, 99–108 CrossRef CAS PubMed.
- Solid Surface Energy Data (SFE) for Common Polymers, http://www.surface-tension.de/solid-surface-energy.htm, accessed June 13, 2023 Search PubMed.
- M. E. Callow and R. L. Fletcher, Int. Biodeterior. Biodegrad., 1994, 34, 333–348 CrossRef CAS.
- D. Lithner, I. Nordensvan and G. Dave, Environ. Sci. Pollut. Res., 2012, 19, 1763–1772 CrossRef CAS PubMed.
- Z. Nejedlá, D. Poustka, R. Herma, M. Liegertová, M. Štofik, J. Smejkal, V. Šícha, P. Kaule and J. Malý, RSC Adv., 2021, 11, 16252–16267 RSC.
- S. Halldorsson, E. Lucumi, R. Gómez-Sjöberg and R. M. T. Fleming, Biosens. Bioelectron., 2015, 63, 218–231 CrossRef CAS PubMed.
- N. Recek, M. Jaganjac, M. Kolar, L. Milkovic, M. Mozetič, K. Stana-Kleinschek and A. Vesel, Molecules, 2013, 18, 12441–12463 CrossRef CAS PubMed.
- R. W. Tilghman, C. R. Cowan, J. D. Mih, Y. Koryakina, D. Gioeli, J. K. Slack-Davis, B. R. Blackman, D. J. Tschumperlin and J. T. Parsons, PLoS One, 2010, 5, e12905 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra05789e |
| This journal is © The Royal Society of Chemistry 2024 |