
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew pyrazolo[3,4-d]pyrimidine derivatives as EGFR-TK inhibitors: design, green synthesis, potential anti-proliferative activity and P-glycoprotein inhibition†
Aya I. Hassaballah *a,
Asmaa M. AboulMagd*b,
Magdy M. Hemdana,
Mohamed H. Hekala,
Amira A. El-Sayeda and
Paula S. Faraga
*a,
Asmaa M. AboulMagd*b,
Magdy M. Hemdana,
Mohamed H. Hekala,
Amira A. El-Sayeda and
Paula S. Faraga
aChemistry Department, Faculty of Science, Ain Shams University, Abbassia, 11566, Cairo, Egypt. E-mail: ayaibrahim@sci.asu.edu.eg
bPharmaceutical Chemistry Department, Faculty of Pharmacy, Nahda University (NUB), Beni-Suef, Egypt. E-mail: asmaa.aboulmaged@nub.edu.eg
First published on 9th January 2024
Abstract
In this study, four series of new pyrazolo[3,4-d]pyrimidine derivatives were designed and synthesized with both green and conventional methods. All the synthesized candidates were chemically confirmed using spectroscopic methods, and the DFT of the reaction mechanism was illustrated. The anti-proliferative activity of the synthesized compounds was evaluated against NCI 60 cancer cell lines. Two compounds (15 & 16) exhibited excellent broad-spectrum cytotoxic activity in NCI 5-log dose assays against the full 60-cell panel with GI50 values ranging from 0.018 to 9.98 μM. Moreover, the enzymatic assessment of the most active derivatives 4, 15, and 16 against EGFR tyrosine kinase showed significant inhibitory activities with IC50 of 0.054, 0.135, and 0.034 μM, respectively. The quantitative real-time PCR for the P-glycoprotein effect of compounds 15 and 16 was examined and illustrated the ability to inhibit the P-glycoprotein by 0.301 and 0.449 fold in comparison to the control. Mechanistic study using reversal activity in MDA-MB-468 cell line revealed the effect of both compounds 15 and 16 cytotoxicity against DOX/MDA-MB-468 with IC50 = 0.267 and 0.844 μM, respectively. Additionally, compound 16 was found to induce cell cycle arrest at the S phase with a subsequent increase in pre-G cell population in MDA-MB-468 cell line. It also increased the percentage of apoptotic cells in a time-dependent manner. Moreover, a molecular docking study was carried out to explain the target compounds' potent inhibitory activity within the EGFR binding site.
1. Introduction
Protein Tyrosine Kinases (PTKs) represent a large family of proteins that play a crucial role in many cellular activities including growth, differentiation, metabolism, adhesion, motility, and death.1–4 Numerous pathological diseases, including cancer are influenced by the aberrant catalytic activity of many PTKs caused by mutation or over-expression.5–7 The trans-membrane growth factor receptor PTK family includes many members, of these are the epidermal growth factor receptor (EGFR) family. The EGFR family has four members: HER2 (human epidermal growth factor receptor-2; also known as erbB2), HER1 (epidermal growth factor receptor; EGFR), HER3, and HER4. Numerous cancers of epithelial origin, including colon, breast, ovarian, and NSC lung cancer, have been linked to EGFR overexpression.8 So, to design novel anticancer therapies that work by competing with ATP for binding at the catalytic domain of their target enzyme, EGFR inhibition constitutes a logical strategy.9 In cancer clinical trials, a variety of small molecule EGFR kinase inhibitors have been evaluated. For instance, erlotinib10 (I) and gefitinib11 (II) are anilinoquinazoline-containing drugs that have been authorized for the chemotherapeutic treatment of individuals with advanced non-small lung cancer. Lapatinib (III) was also given the go-ahead to treat breast cancer12,13 (Fig. 1). Investigation of the clinically used EGFR-TK inhibitors showed that they should possess four pharmacophoric features to bind efficiently within the ATP binding pocket of EGFR-TK.14,15 The first feature is a flat heteroaromatic ring that can occupy the adenine binding pocket. The second one is a terminal hydrophobic tail to be embedded in the first hydrophobic groove of EGFR. The third pharmacophoric feature is a spacer that can bind via hydrogen bond interactions. Finally, one more hydrophobic tail occupies the hydrophobic II region of ATP.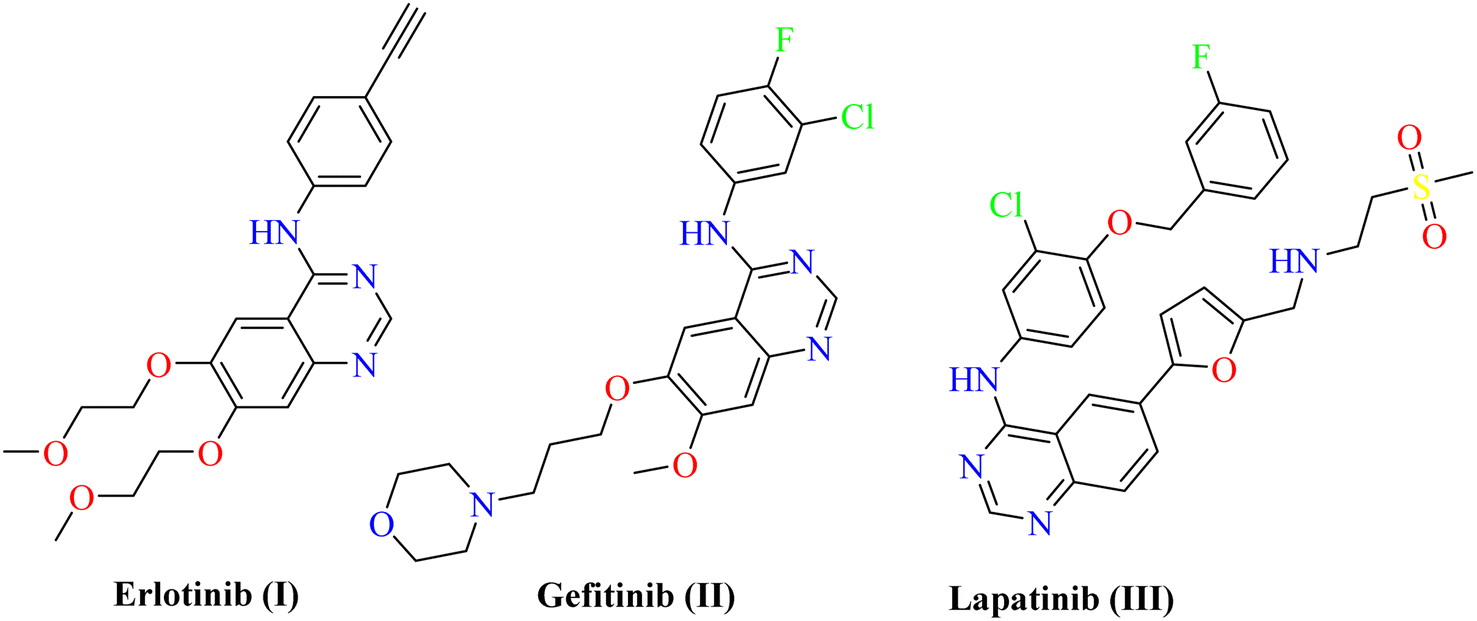 | ||
| Fig. 1 FDA-approved EGFR-TK inhibitors. | ||
Pyrazolo[3,4-d]pyrimidin-4(5H)-one is a bioisostere naturally occurring purine nucleoside heterocyclic base that is thought to be a main skeleton in some biologically active compounds. Differently substituted pyrazolo[3,4-d]pyrimidin-4(5H)-ones exhibit a wide range of pharmacological properties, including anti-inflammatory,16 anti-bacterial,17 anti-fungal,18 anti-viral,19 anti-microbial,20 CNS-modulating,21 anti-oxidant,22 and cardiovascular agents.23 Other pyrazoloprymidinone derivatives are also used to manage type II diabetes24 and insomnia.25 Moreover, the pyrazolo[3,4-d]pyrimidin-4(5H)- derivatives are considered as potential pharmacophores of several analogs that display anti-cancer activity against human colon (HCT-116),26 human leukemia (HL-60),27 non-small cell lung cancer (NCL-H 460), CNS cancer (SF- 268),28 and skin cancer cell lines (G-361).29 Additionally, hybrid molecules that built up via joining more than one pharmacophore are suggested to exhibit better activity than the individual activity of each isolated one. Thus, sunitinib (IV) and toceranib (V) are indoline-2-one derivatives that have been approved by the US FDA for cancer therapy. Also, compound VI was reported as an ATP-competitive inhibitor exerting EGFR inhibitory effect with nanomolar concentration.30 Compound VII is another 1H-pyrazolo [3,4-d]pyrimidine derivative with significant anti-EGFR activity31 (Fig. 2). Based on the mentioned evidences considering the potential EGFR as an anticancer target, the resistance arise against the FDA approved anticancer drugs, and the attractiveness of 1H-pyrazolo[3,4-d]pyrimidine moiety, it was interesting to design and synthesize new 1H-pyrazolo[3,4-d]pyrimidine derivatives that may exhibit possible anti-proliferative and EGFR inhibitory activity. Accordingly, the synthesized compounds were designed to have the essential pharmacophoric features of EGFR inhibitors.
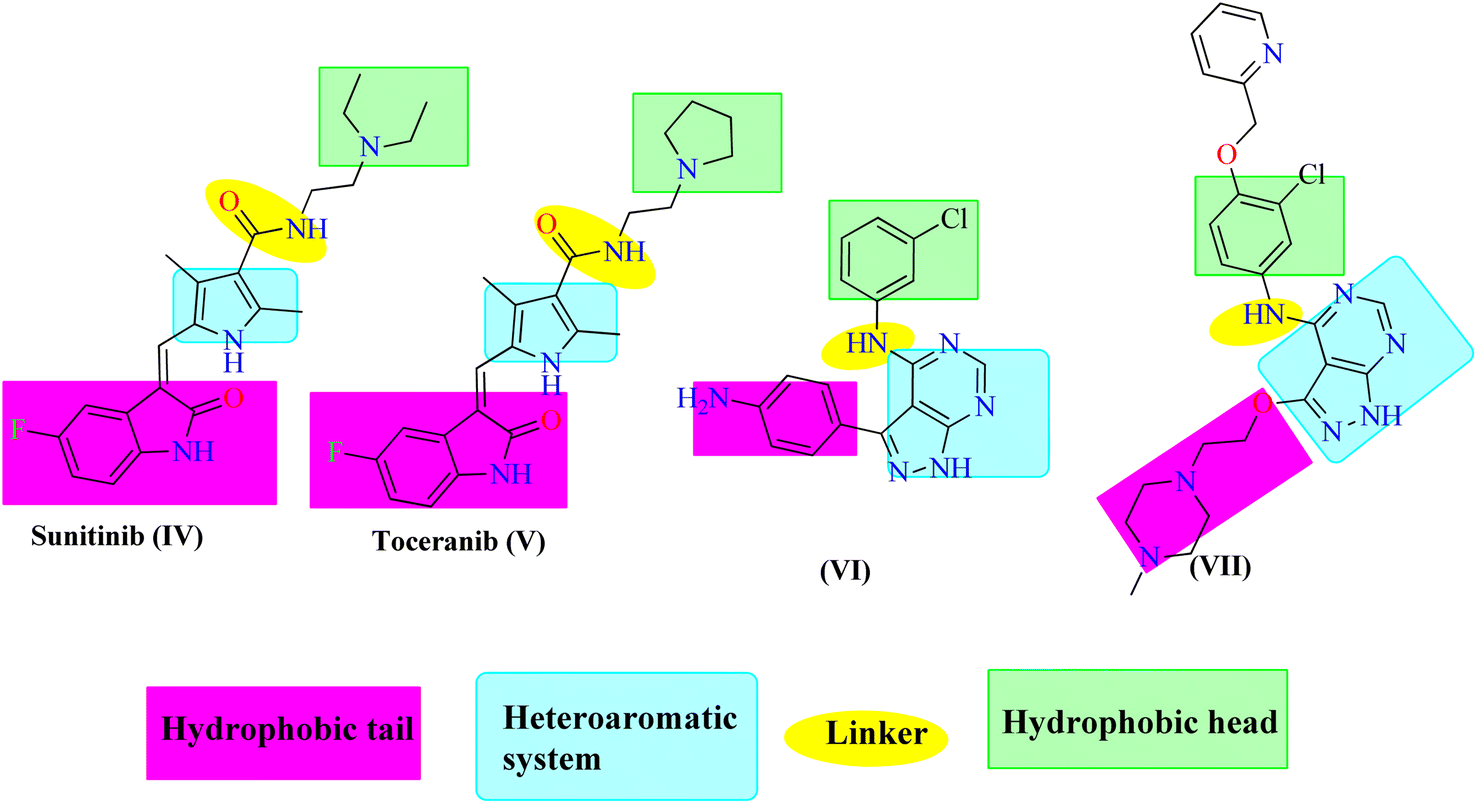 | ||
| Fig. 2 EGFR inhibitors and their pharmacophoric features. | ||
1.1. Rational of the work
Based on the previous results, regarding the potential anticancer activity of pyrazolo[3,4-d]pyrimidines, we herein, report the design and synthesis of a new series of pyrazolo[3,4-d]pyrimidine derivatives to evaluate their possible anti-proliferative activity and EGFR-TK inhibitory activity. In this work, 1H-pyrazolo[3,4-d]pyrimidine moiety was used as a heteroaromatic ring system occupying the adenine binding region.32,33 Also, phenyl ring was utilized as a hydrophobic moiety to occupy the hydrophobic region I of the ATP-binding site. The linker was reserved to make the essential binding interactions. Different moieties include substituted phenyl ring, tricyclic rings and aliphatic substituents were used to occupy the hydrophobic region II of the ATP-binding site to discover their possible cytotoxic activity. Correlation between the structure–activity relationship of the synthesized compounds and their anti-proliferative activity targeting EGFR was discussed due to these modifications (Fig. 3).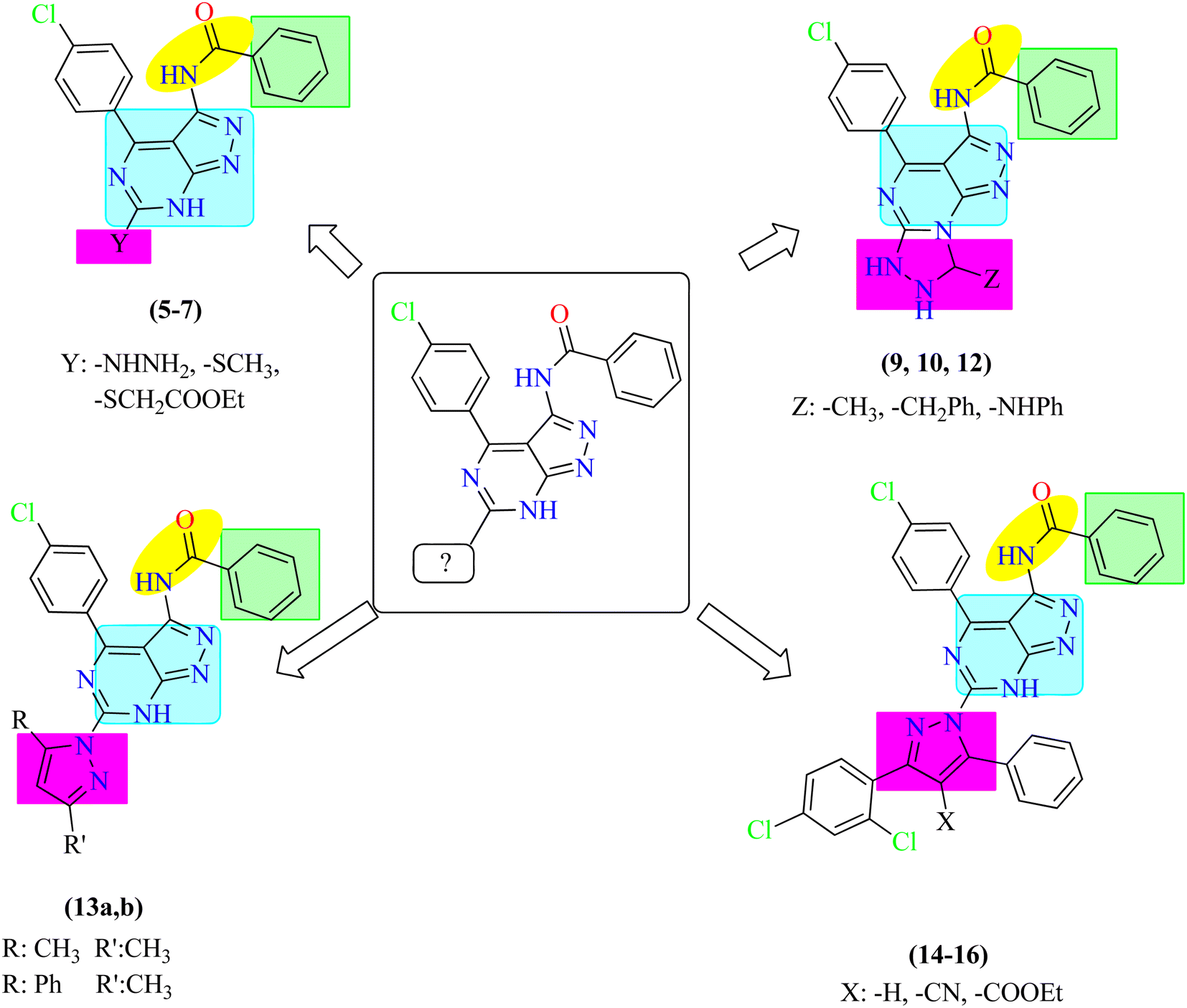 | ||
| Fig. 3 Rationale of molecular design of the new proposed EGFR-TK inhibitors. | ||
2. Results and discussion
2.1. Chemistry
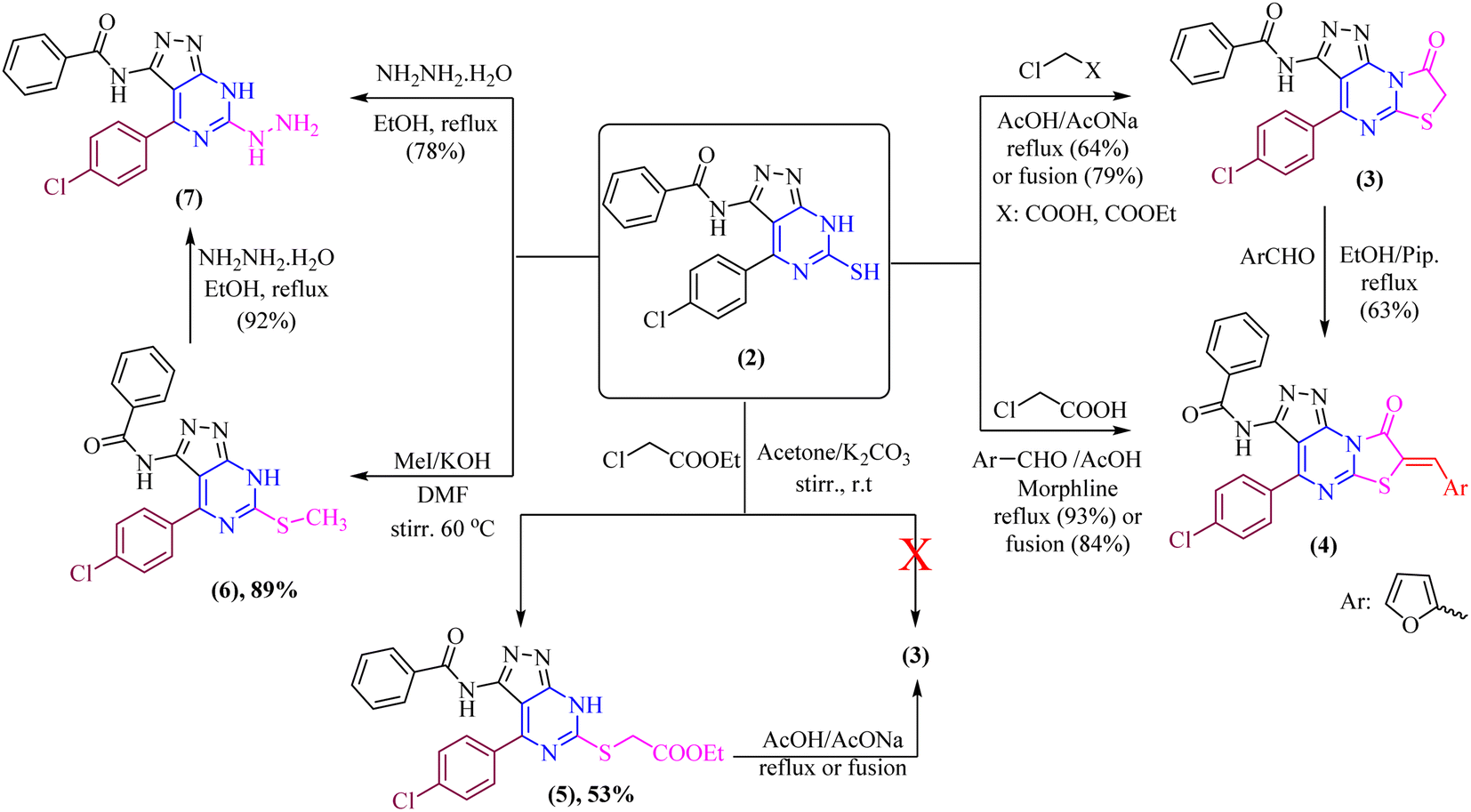
Our current research is concerned with the utility of N-(4-(4-chlorobenzylidene)-4H-pyrazol-3-yl)benzamide (1) in the synthesis of new substituted fused pyrimidine derivatives under conventional and green conditions as part of our interest in the synthesis of a wide range of heterocyclic systems with biological applications.34–37The starting chemical, N-(4-(4-chlorobenzylidene)-5-oxo-4,5-dihydro-1H-pyrazol-3-yl)benzamide (1), was synthesized according to an earlier reported procedure38 under different chemical reaction conditions. Thus, refluxing of compound 1 with ethanolic solution of thiourea in the presence of catalytic amount of sodium ethoxide or under free solvent fusion technique to afford the target compound N-(4-(4-chlorophenyl)-6-thioxo-6,7-dihydro-5H-pyrazolo[3,4-d]pyrimidin-3-yl)benzamide (2) with a beneficial yield, which was used as a key starting material as illustrated in (Scheme 1).
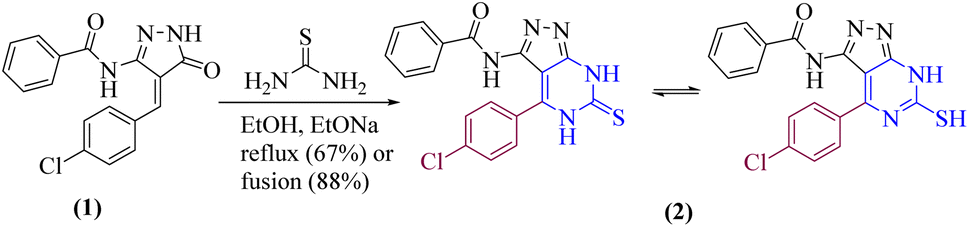 | ||
| Scheme 1 Synthesis of pyrimidinthione derivatives 2 under both conventional and green conditions. | ||
The structure of compound 2 was determined using spectroscopic data. Their IR spectra showed absorption bands corresponding to the –NH and –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups at υ 3197 br and 1681 cm−1, respectively as well as the presence of –NH groups at υ 11.46, 11.86 and 12.40 ppm in the 1H-NMR spectrum of compound 2 which agree well with the suggested structure.
O groups at υ 3197 br and 1681 cm−1, respectively as well as the presence of –NH groups at υ 11.46, 11.86 and 12.40 ppm in the 1H-NMR spectrum of compound 2 which agree well with the suggested structure.
Moreover, we illustrate the reaction of a secondary amine in absolute ethanol with the presence of a basic medium and a nitrosating agent. It was crucial to emphasize that, in these conditions, the absence of acidic conditions mitigates the risk of N-nitrosamine formation.39,40
The di-heterocyclic compound 2 was established and used as a mediator to synthesize tri-heterocyclic compounds linked with pyrazolopyrimidine moiety. Thus, refluxing of thiol derivative 2 with chloroacetic acid and/or ethyl chloroacetate in glacial acetic acid with sodium acetate anhydrous afforded thiazolo[3,2-a]pyrimidine derivative 3 as the sole product as shown in (Scheme 2). Compound 3 was also obtained by free solvent fusion condition for 75 min. Treatment of thiazolo[3,2-a]pyrimidine derivative, compound 3, with 2-furancarboxaldehyde yielded the corresponding condensation product, which surprisingly in situ undergoes knoevenagel condensation by presence of piperidine catalyst to give N-(4-(4-chlorophenyl)-7-(furan-2-ylmethylene)-8-oxo-7,8-dihydropyrazolo[4,3-e]thiazolo[3,2-a]pyrimidin-3-yl)benzamide, compound 4 as the sole product (one spot in TLC), which also obtained by fusion technique under solvent-free condition (Scheme 2). Likewise, compound 4 was obtained via one-pot multicomponent condensation reactions of 2, chloroacetic acid, and furfural, with morphline as a catalytic base to afford the corresponding thiazolo[3,2-a]pyrimidine derivative 4.
 | ||
| Scheme 2 Reaction of thiol derivative 2 with different electrophiles and nucleophiles. | ||
On another hand, by stirring of thiol derivative 2 at room temperature with ethyl chloroacetate in the presence of a catalytic amount of anhydrous potassium carbonate in dry acetone afforded uncyclized S-alkylated product ethyl 2-((3-benzamido-4-(4-chlorophenyl)-7H-pyrazolo[3,4-d]pyrimidin-6-yl)thio)acetate (5) as demonstrated in (Scheme 2) with no evidence was verified for the cyclized structure 3. Likewise, cyclization of S-alkylated product 5 via one-pot multicomponent condensation reactions of 5 with acetic acid in the presence of sodium acetate afforded the corresponding thiazolo[3,2-a]pyrimidine derivative 3 as the sole product. The absorption bands for the –CO ester group at υ 1717 cm−1 in the IR spectra verified the uncyclized S-alkylation. Furthermore, the 1H-NMR spectrum of compound 5 revealed the presence of a triplet at δ 2.06 ppm and a quartet at δ 4.35 ppm with equal coupling constants typical of –CH2CH3 protons, as well as a singlet signal at 5.44 ppm for 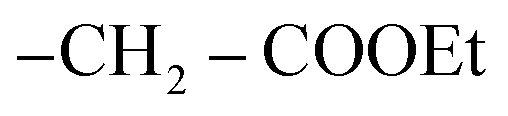 protons, all of which are completely consistent with the uncyclized structure, compound 5 with ruling out the cyclized product 3. Furthermore, the 13C-NMR spectrum of structure 5 matches the hypothesized structure well.
protons, all of which are completely consistent with the uncyclized structure, compound 5 with ruling out the cyclized product 3. Furthermore, the 13C-NMR spectrum of structure 5 matches the hypothesized structure well.
Moreover, methylthio derivative 6 was produced in 89% yield when thiol derivative 2 was alkylated by methyl iodide in dry N,N-dimethylformamide and in the presence of potassium hydroxide was stirred at 60 °C for 2.5 hours. The spectral data and the elemental analyses of the synthesized compound 6 were in accordance with the proposed structures of compound 6 (Scheme 2). The 1H-NMR spectrum showed the signal of the –CH3 group at δ 3.57 ppm. In addition, 13C-NMR spectra were consistent with the proposed structure.
Furthermore, the refluxing of ethanolic solution of thiol derivative 2 with hydrazine hydrate afforded hydrazineyl-7H-pyrazolo[3,4-d]pyrimidin derivative 7. Additionally, hydrazinolysis of the methylthio derivative 6 with a molar ratio [1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1] in absolute ethanol was heated under refluxing (one spot in TLC) (Scheme 2). The IR spectrum of compound 7 showed the appearance of –NH and –NH2 groups at υ 3424, 3309, and 3224 cm−1, respectively. Additionally, the structure of compound 7 is supported by the 1H-NMR spectrum [DMSO-d6], which exhibited signals for –NH2 group at δ 7.27 ppm as well as –NH protons in the downfield region that might be exchanged with D2O. Furthermore, the 13C-NMR spectrum of structure 7 matches the hypothesized structure well.
:1] in absolute ethanol was heated under refluxing (one spot in TLC) (Scheme 2). The IR spectrum of compound 7 showed the appearance of –NH and –NH2 groups at υ 3424, 3309, and 3224 cm−1, respectively. Additionally, the structure of compound 7 is supported by the 1H-NMR spectrum [DMSO-d6], which exhibited signals for –NH2 group at δ 7.27 ppm as well as –NH protons in the downfield region that might be exchanged with D2O. Furthermore, the 13C-NMR spectrum of structure 7 matches the hypothesized structure well.
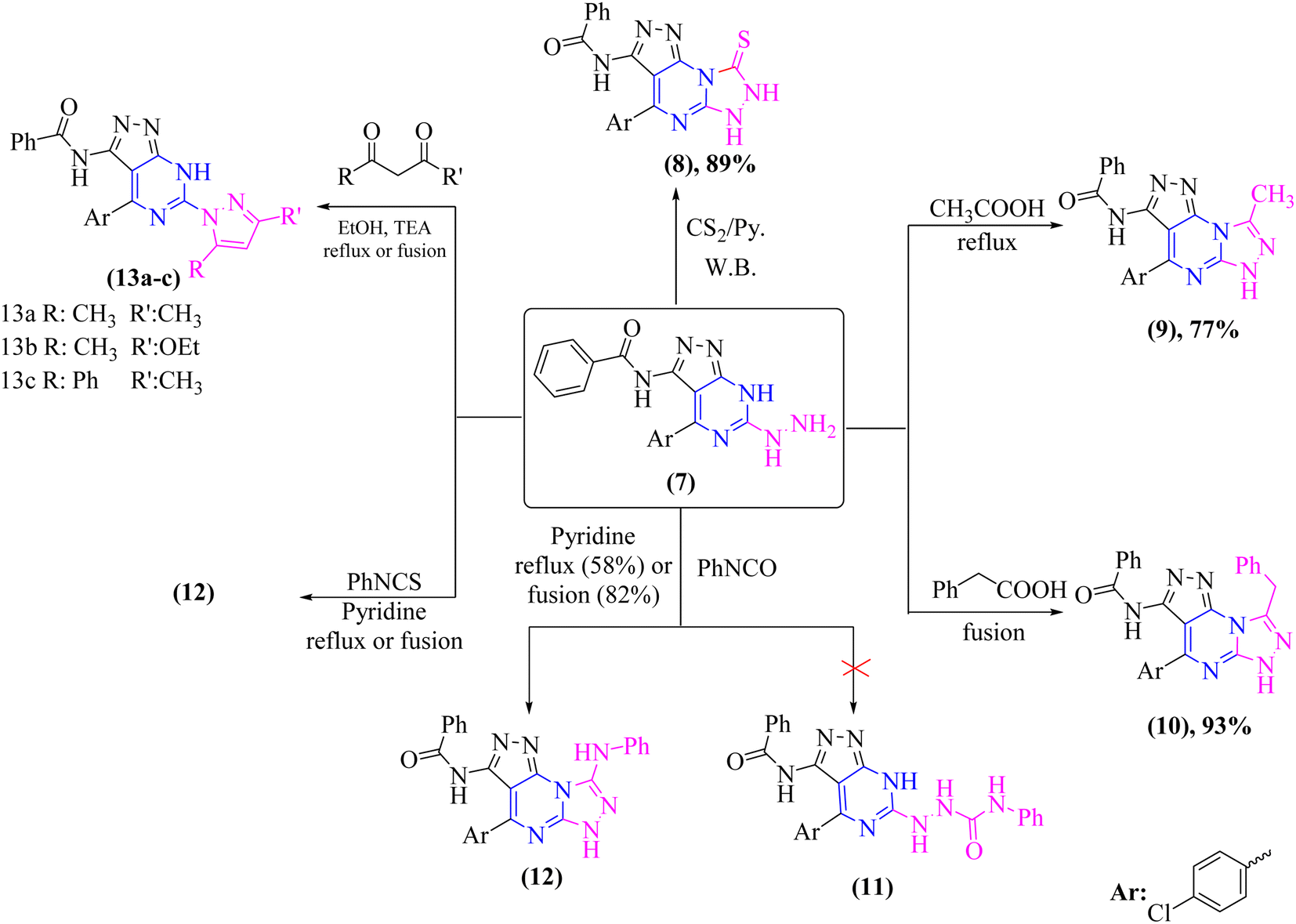
Chemical cyclization of hydrazine derivative 7 into distinct heterocyclic compounds having fused pyrazolo[3,4-d]pyrimidin scaffold has been explored under various chemical reaction circumstances, as shown in (Scheme 3). Thus, heating of compound 7 in water-bath with carbon disulfide in dry pyridine afforded thioxo-pyrazolo[4,3-e][1,2,4]triazolo[4,3-a]pyrimidin derivative, compound 8 in a rationally good yield (89%). Compound 8 lacked in its IR spectrum, the strong absorption band for –NH groups at υ 3180 and 3164 cm−1 with disappearing –NH2 group of compound 7. Furthermore, 1H-NMR spectrum of compound 8 displayed D2O exchangeable protons of –NH groups at δ 8.25, 11.44, and 13.39 ppm. In addition, the 13C-NMR spectrum of structure 8 agrees with the hypothesized structure well.
 | ||
| Scheme 3 Cyclization of hydrazine derivative 7 to afford fused tri-heterocyclic compounds. | ||
In addition, refluxing of compound 7 in glacial acetic acid for 3 h afforded N-(4-(4-chlorophenyl)-8-methyl-6H-pyrazolo[4,3-e][1,2,4]triazolo[4,3-a]pyrimidin-3-yl)benzamide (9) (Scheme 3). Moreover, compound 7 was fused under solvent-free condition with phenyl acetic acid afforded pyrazolo[4,3-e][1,2,4]triazolo[4,3-a]pyrimidin derivative 10 as the sole product in a good yield (93%). The proper elemental analysis and spectroscopic data provided sufficient proof for the chemical structure of compounds 9 and 10. Both compounds' 1H-NMR spectra [DMSO-d6] revealed signals corresponding to methyl and methylene protons of 9 and 10, respectively, and –NH protons in the down field area that could be exchanged with D2O. Furthermore, the 13C-NMR spectra of structures 9 and 10 revealed signals that matched the hypothesized structure (cf. experimental).
Moreover, treatment of hydrazine derivative 7 with phenyl isocyanate in refluxing pyridine for 15 hours yielded the unexpected pyrazolo[4,3-e][1,2,4]triazolo[4,3-a]pyrimidine derivative 11 rather than the uncyclized N-phenylhydrazine-1-carboxamide derivative 12 as illustrated in (Scheme 3).
Furthermore, refluxing compound 7 with phenyl isothiocyanate in dry pyridine for 15 hours or fusing it under free solvent condition for 2 hours afforded a sulfur-free product known as N-(4-(4-chlorophenyl)-8-(phenylamino)-6H-pyrazolo[4,3-e][1,2,4]triazolo[4,3-a]pyrimidin-3-yl)benzamide (12) (Scheme 3). The proper elemental analysis and spectroscopic data provided sufficient proof for the chemical structure of compound 12.
The fused di-heterocyclic compound 7 was employed as a starting material to synthesize tri-heterocyclic compounds with attached the pyrazolopyrimidine moiety. As indicated in (Scheme 3), treatment of compound 7 under both conventional and solvent free fusion conditions with different diketone derivatives such as acetyl acetone, ethyl acetoacetate, and/or benzoyl acetone yielded (pyrazol-1-yl)-7H-pyrazolo[3,4-d]pyrimidin derivatives 13a–c. The elemental analysis and spectral data IR and NMR of the produced compounds were compatible with the predicted structures of compounds 13a–c. The IR spectrum of compounds 13a–c revealed the elimination of the –NH2 group. Moreover, the 1H-NMR and 13C-NMR spectra were in concurrence with the proposed structures 13a–c.
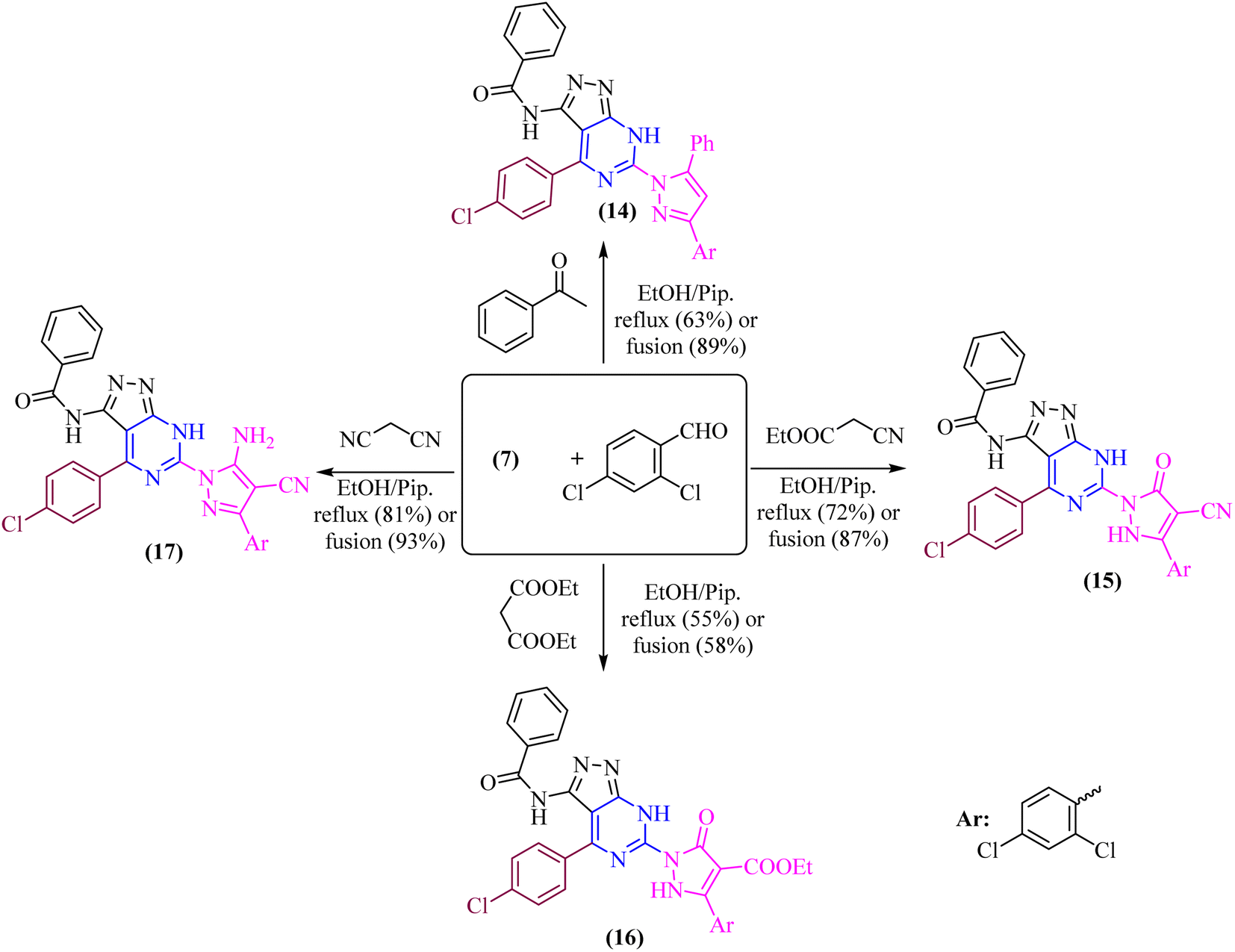
In both techniques, conventional and green conditions, one-pot multicomponent reactions of 7, and 2,4-dichlorobenzaldehyde in the presence of acetophenone, ethyl cyanoacetate, diethyl malonate, and/or malononitrile yielded the corresponding (1H-pyrazol-1-yl)-4-(4-chlorophenyl)-7H-pyrazolo[3,4-d]pyrimidin derivatives 14–17 as shown in (Scheme 4). The reaction mixture is refluxed or fused under solvent free conditions in absolute ethanol with a catalytic quantity of piperidine to produce arylidene derivatives (I) in situ as illustrated in (Scheme 5), resulting in Michael adducts that undergo intramolecular cyclization (II) to give the target derivatives 14–17 as the suggested mechanism (Scheme 5).
 | ||
| Scheme 4 One-pot multicomponent condensation reactions 7, and 2,4-dichlorobenzaldehyde with different active methylene. | ||
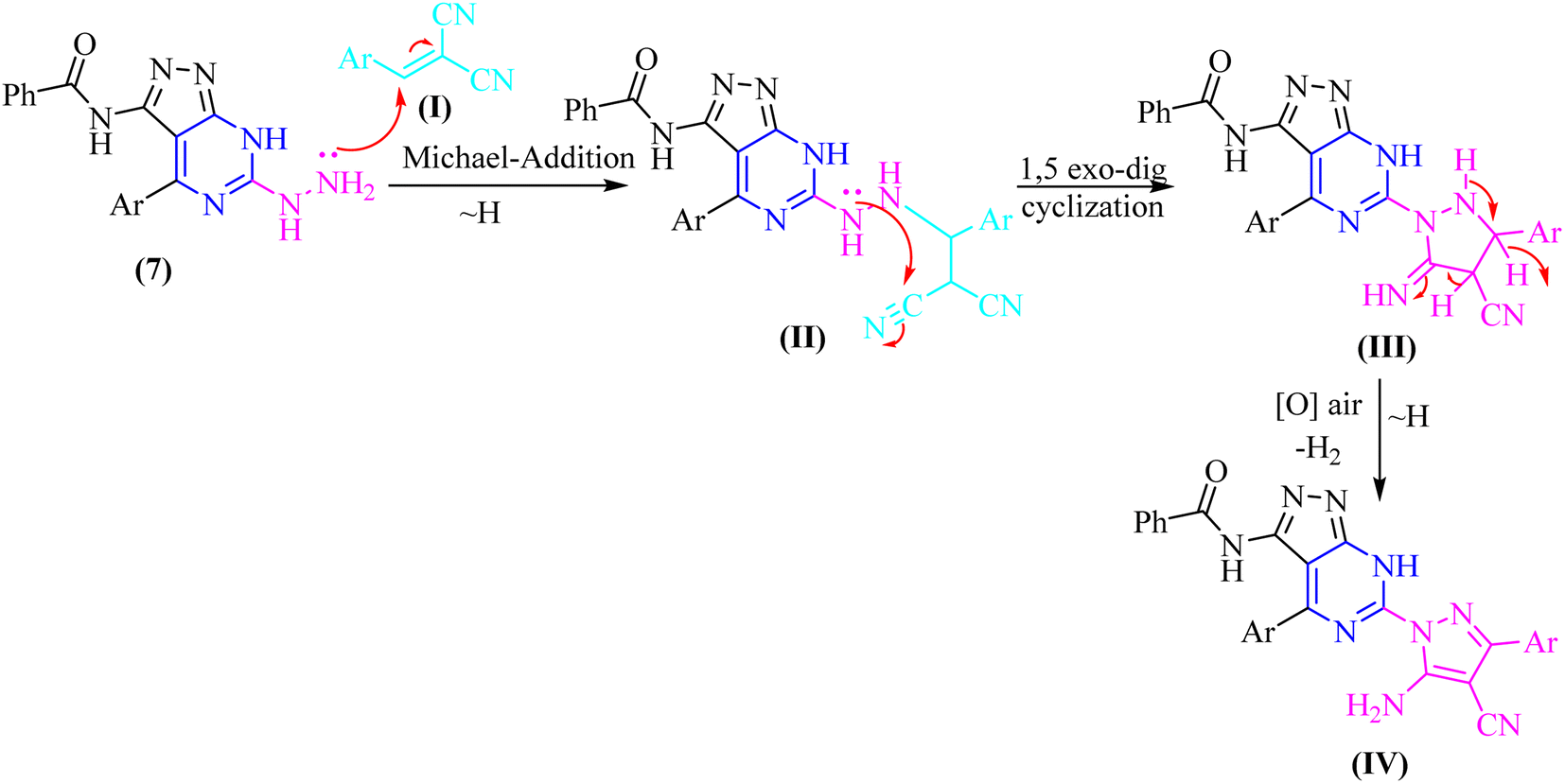 | ||
| Scheme 5 Suggested mechanism for the formation of compounds 14–17. | ||
Microanalytical and spectral data corroborated the structure of compounds 14–17. The IR spectra of compound 14 exhibit absorption at υ 3177 and 3062 cm−1, corresponding to the –NH and phenyl groups, respectively. The 1H-NMR spectra of these compounds indicate the phenyl and aromatic protons, in addition to the strongly deshielded exchangeable –NH proton at δ 14.14 ppm.
Moreover, the IR spectra of compounds 15 and 16 reveal absorption at υ 2215 and 1715 cm−1, which corresponds to the –CN and –CO ester groups of 15 and 16, respectively. The 1H-NMR spectrum of these compounds also exhibits the strongly deshielded exchangeable –NH protons at δ 13.37 and 14.18 ppm of 15 and 16, respectively as well as the aliphatic protons of –C2H5 of compound 16.
Furthermore, the IR spectrum revealed absorption bands in the range of υ 3210 and 3149 cm−1 for –NH2 and –NH, respectively, and absorption band at υ 2222 and 1657 cm−1 for –CN and amidic –CO groups, respectively. The exchangeable wide signals of –NH2 and amidic –NH in compound 17, and the aromatic protons in the expected region, were all visible in the 1H-NMR spectrum. Singlet signals were seen in the 13C-NMR spectrum for the CO, CN, and CC groups of compound 17.
2.2. DFT study of the reaction mechanism
To gain further insight into the reaction mechanisms and their possible pathways, theoretical calculations were carried out using compounds I and I′ as the model reactants (Fig. 1).First, DFT calculations suggested that the reactants 2 or its tautomer 2′ and CA can be converted into I or its tautomer I′ via the transition state 2-CA or 2′-CA with an activation energy of 25.53 kcal mol−1 (Fig. 4 and 5). At this point of the reaction, I or I′ exhibited intramolecular nucleophilic–electrophilic reaction between one of the –NH and –CO groups to form either II or II′ accordingly. Transition state TS I is more energetically favored than the competing TS I′ transition states by 7.36 kcal mol−1. In addition, the ΔG of the final step afforded II was lower than that afforded II′ by 2.13 kcal mol−1 (Fig. 5). Hence, the formation of II is significantly more preferred than II′.
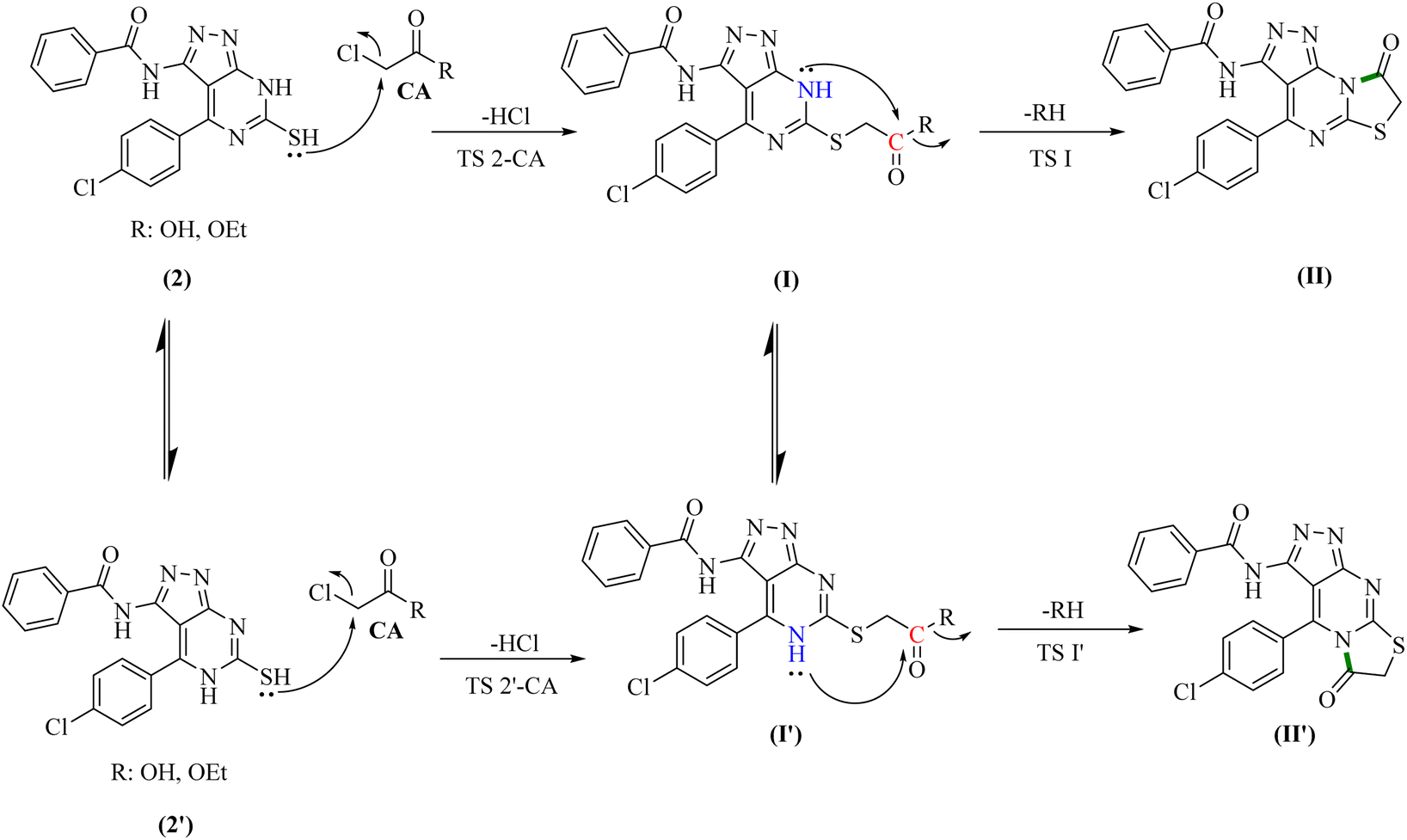 | ||
| Fig. 4 Two proposed reaction pathways of for the first reaction. | ||
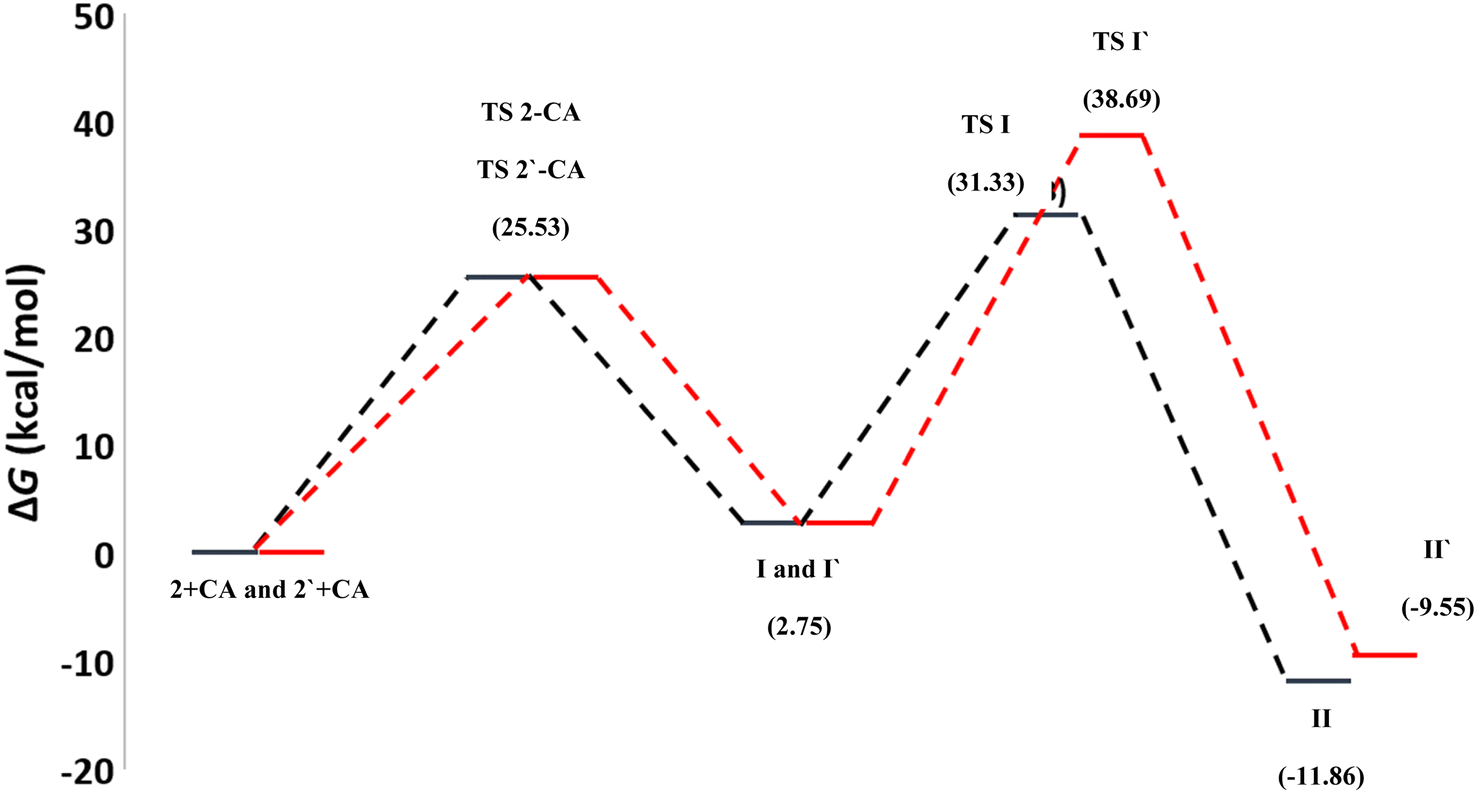 | ||
| Fig. 5 Potential energy curves for the reaction pathways of the first reaction. | ||
The second reaction showed, to some extent, similar findings, where DFT calculations showed that the reactants 7 or its tautomer 7′ and CD formed the intermediate III or its tautomer III′ via the transition state (7-CA and 7′-CA, respectively) with an activation energy of 42.58 kcal mol−1 (Fig. 6). At this stage of the reaction, III or III′ went into an intramolecular nucleophilic–electrophilic reaction between one of the –NH and –CS groups to form either IV or IV′ as unstable intermediates. Transition state TS III is very slightly energetically favored over TS III′ transition states by only 0.64 kcal mol−1. Finally, both IV and IV′ afforded D and D′, respectively via the transition states TS C and TS C′ with convergent ΔG values of −10.36 and −10.18 kcal mol−1, respectively, and hence, the formation of the final products V and V′ can occur with a slight preference to V.
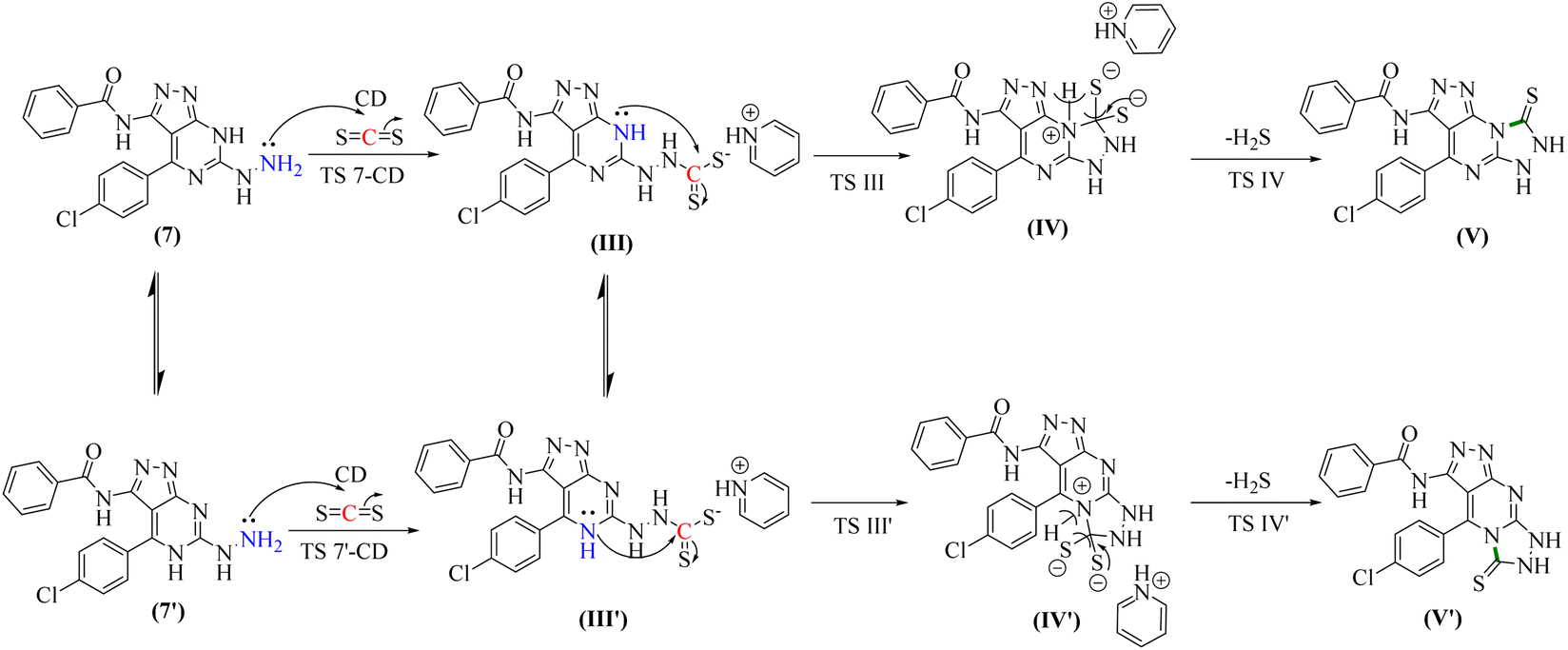 | ||
| Fig. 6 Two proposed reaction pathways for the second reaction. | ||
To support the previous findings from the frontier molecular orbital (FMO) perspective, we calculated the LUMO–HUMO energy gap for I and I′ in each reaction. I and I′ are the key intermediates in each reaction determining which final product will be formed.
The reacting orbital of the –NH is HOMO-1, while of the –CO or –CS group it is LUMO (Fig. 8). In the first reaction, the energy gap between the LUMO and HOMO-1 is 7.875 eV for I, while for I′, the corresponding energy gap between the two reacting orbitals is 8.174 eV, which is 0.299 eV larger than that in I. Therefore, the formation of II becomes favored over II′ (Fig. 7A).
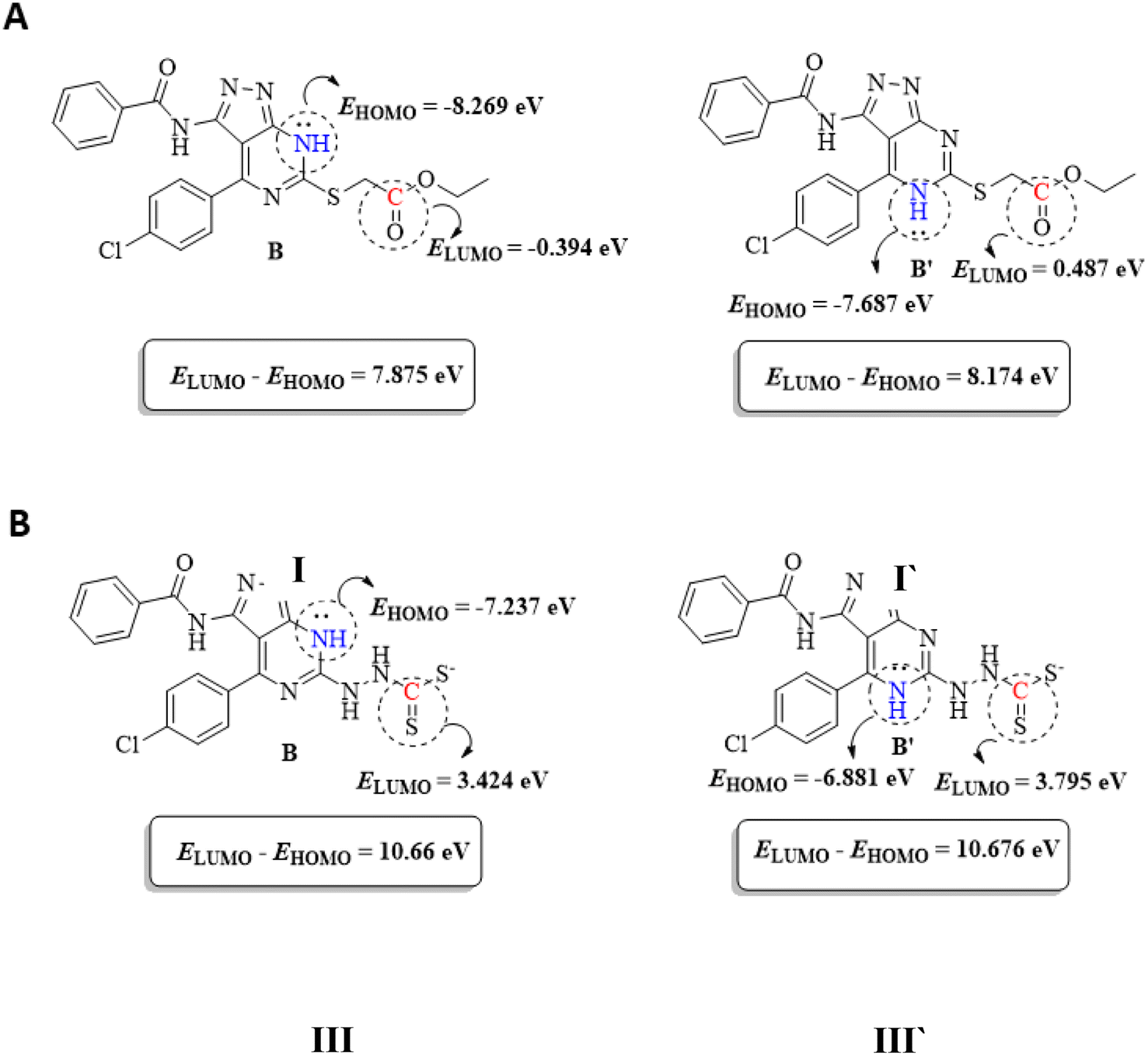 | ||
| Fig. 7 FMOs of B and B′ for the first and second reactions (A and B, respectively). | ||
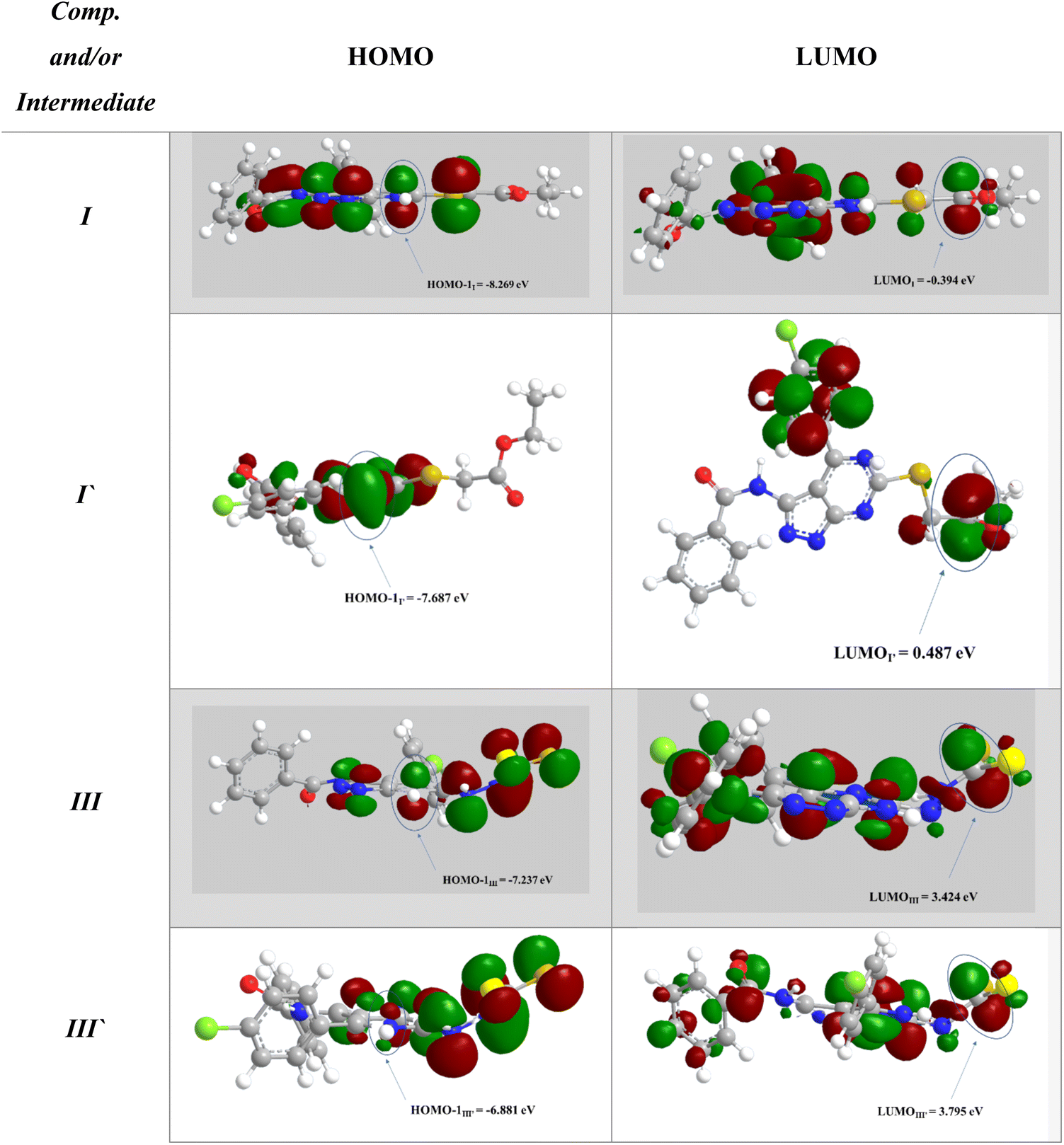 | ||
| Fig. 8 The frontier molecular orbital (FMO) of HOMO–LUMO. | ||
In the second reaction, the LUMO–HOMO-1 energy gap is 10.66 eV for III, while it is 10.676 eV for III′. Hence, the reaction is slightly favored toward the III formation over III′ formation (Fig. 7B).
2.3. Biological activity
Also, compound (4) showed moderate cell growth inhibition activity among most of the cancer cell lines. Surprisingly, candidate (6) revealed poor antiproliferative activity except on HOP-92 which revealed over 100% growth inhibition which may contribute to possible selectivity on HOP-92 (non – small cell lung cancer cell line).
Study of the SAR of the new derivatives showed certain findings. The presence of substituted imidazole ring occupying the hydrophobic pocket II remarkably increases the anticancer activity. Accordingly, compounds 15 and 16, have the most cytotoxic activity than other analogs either with tricyclic rings or aliphatic tail. This result is consistent with the significance of a terminal hydrophobic ring for the effectiveness of EGFR TK anticancer drugs, previously described. Also, the substitution with a phenyl ring in the tricyclic analog 4 contributes to its moderate cytotoxic activity. Conversely, the presence of tricyclic rings or aliphatic tails showed weak cytotoxic activity.
| Cpd no. | EGFR ± SD IC50 (μM) |
|---|---|
| 4 | 0.054 ± 0.003 |
| 15 | 0.135 ± 0.006 |
| 16 | 0.034 ± 0.002 |
| Lapatinib | 0.074 ± 0.003 |
| Cpd no. | MDA-MB-468 P-glycoprotein fold change |
|---|---|
| 15 | 0.301 |
| 16 | 0.449 |
| Control | 1 |
| Cpd no. | DOX/MDA-MB-468 (μM) | ±SD |
|---|---|---|
| 15 | 0.267 | 0.001 |
| 16 | 0.844 | 0.003 |
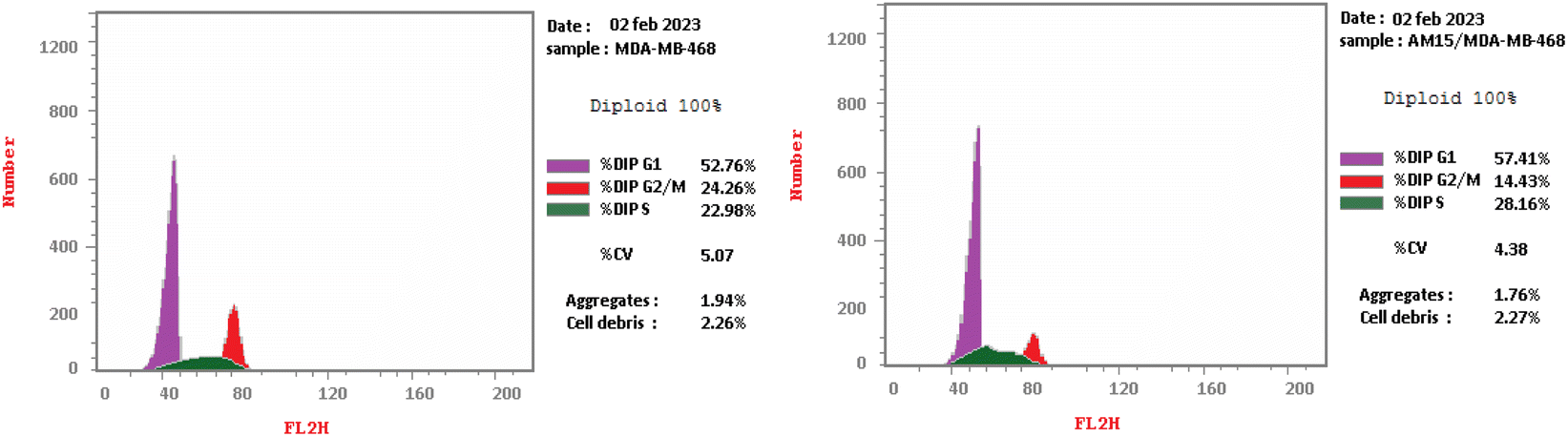 | ||
| Fig. 9 Flow cytometric analysis of cell cycle phases after treatment with compound 16. | ||
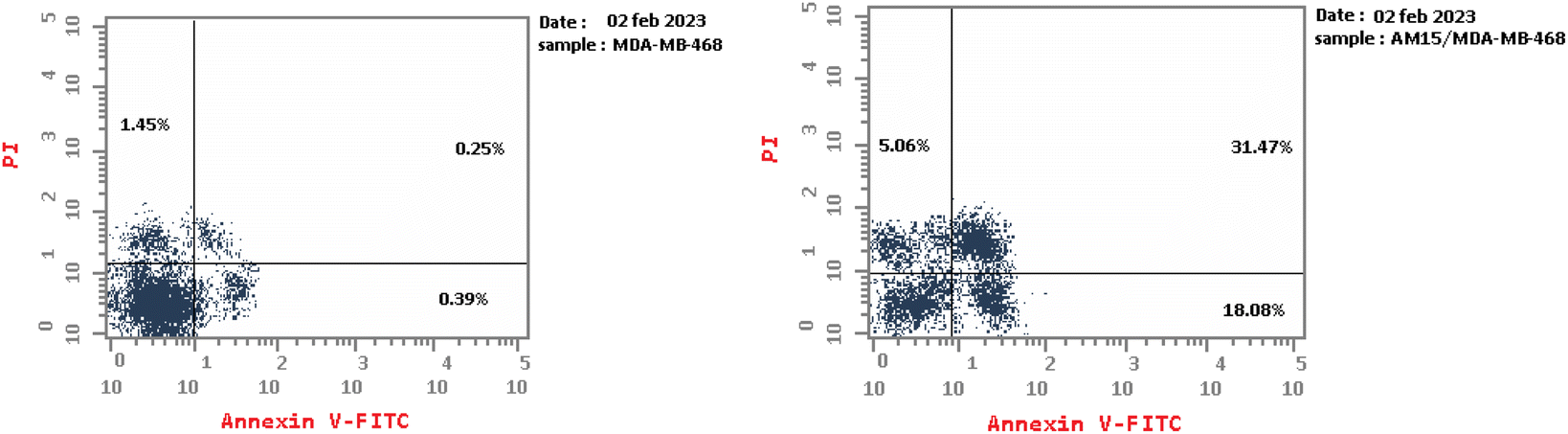 | ||
| Fig. 10 Flow cytometric analysis of apoptosis in MDA-MB-468 cells exposed to compound 16. | ||
The co-crystallized ligand of EGF showed a binding score of −7.03 kcal mol−1 and its binding mode was represented in Fig. 11. The heterocyclic pyrimidine moiety was oriented into the adenine pocket forming two hydrophobic interactions with Gly796, and Leu718. The phenyl moiety occupied the hydrophobic pocket I forming one hydrophobic interactions with Phe723. The terminal amine forms a hydrogen bond donor interaction with Phe795. Taking candidate 15 as one of the most active cytotoxic compounds, it showed a similar binding pattern to the ligand. Compound 15 displayed a binding score of −7.410 kcal mol−1. The 1H-pyrazolo[3,4-d]pyrimidin- 6-amine moiety oriented into the adenine pocket to form one hydrogen bond interaction with Met793. Also, it formed one hydrophobic interaction with Gly796. The dichlorophenyl moiety occupied the hydrophobic tail forming one hydrophobic interaction with Lys728. The phenyl ring connected to the amide linker occupied the hydrophobic head forming one hydrophobic interactions with Lys745, Fig. 12.
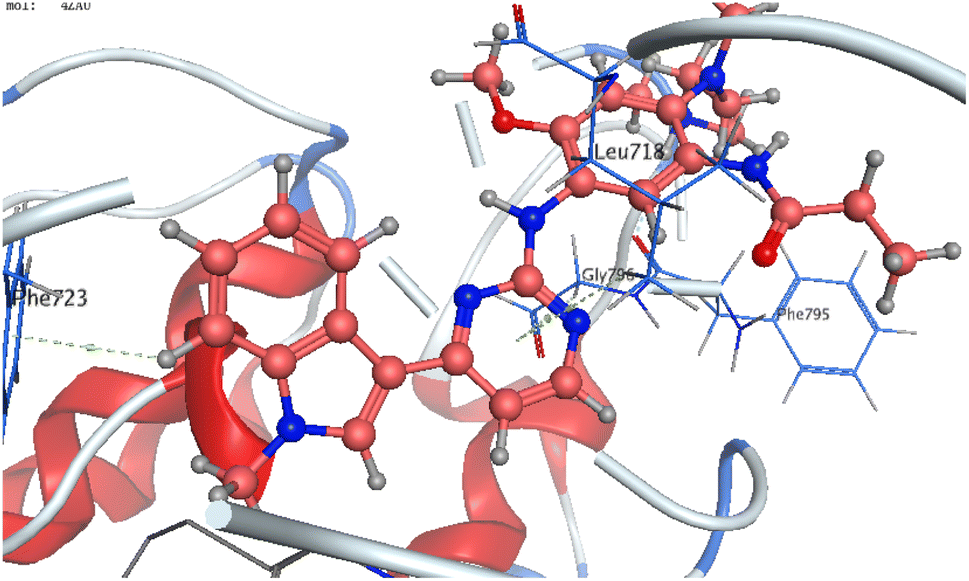 | ||
| Fig. 11 Three-dimensional binding modes of YY3 inside the binding pocket of EGFR-TK. | ||
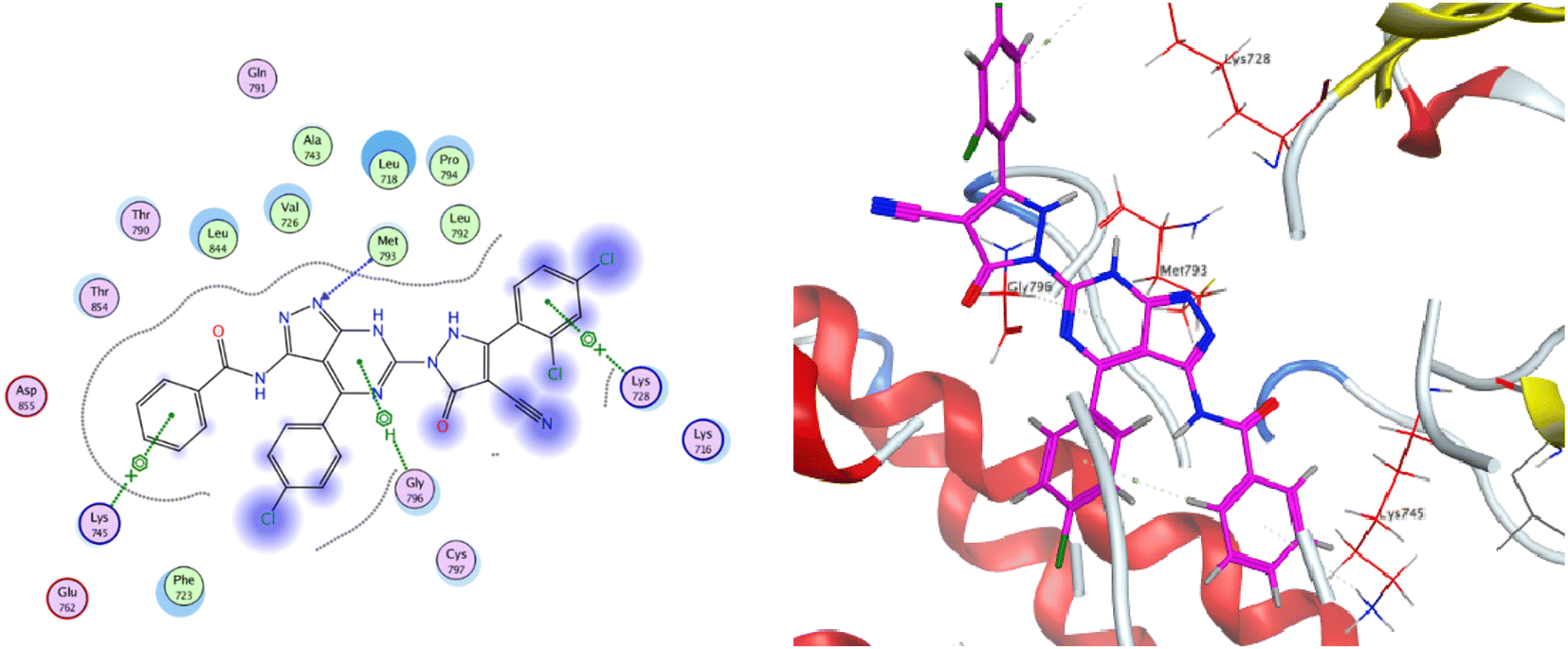 | ||
| Fig. 12 2D and 3D binding mode of compound 15 inside the binding pocket of EGFR. | ||
As for the newly designed pyrazolo[3,4-d]pyrimidine analogue 16, the key amino acid residue Met793 interacted via a hydrogen bond with the nitrogen atom of the pyrazole ring. The binding mode of compound 16 presented three other essential binding interactions with the binding site of EGFR-TK. The pyrimidine ring formed two hydrophobic interactions with the key residues Leu718 and Gly796. Additionally, the terminal phenyl ring showed another arene interaction with Lys745. As a summary, it is noticeable that the pyrazolo[3,4-d]pyrimidine moiety of the target compounds oriented into the adenine binding site, forming an essential binding interaction with the key residue Gly796. The aryl ring linker to the amide linker and the substituted phenyl rings occupied hydrophobic tail and head, forming additional hydrophobic interactions with a number of amino acid residues, Fig. 13.
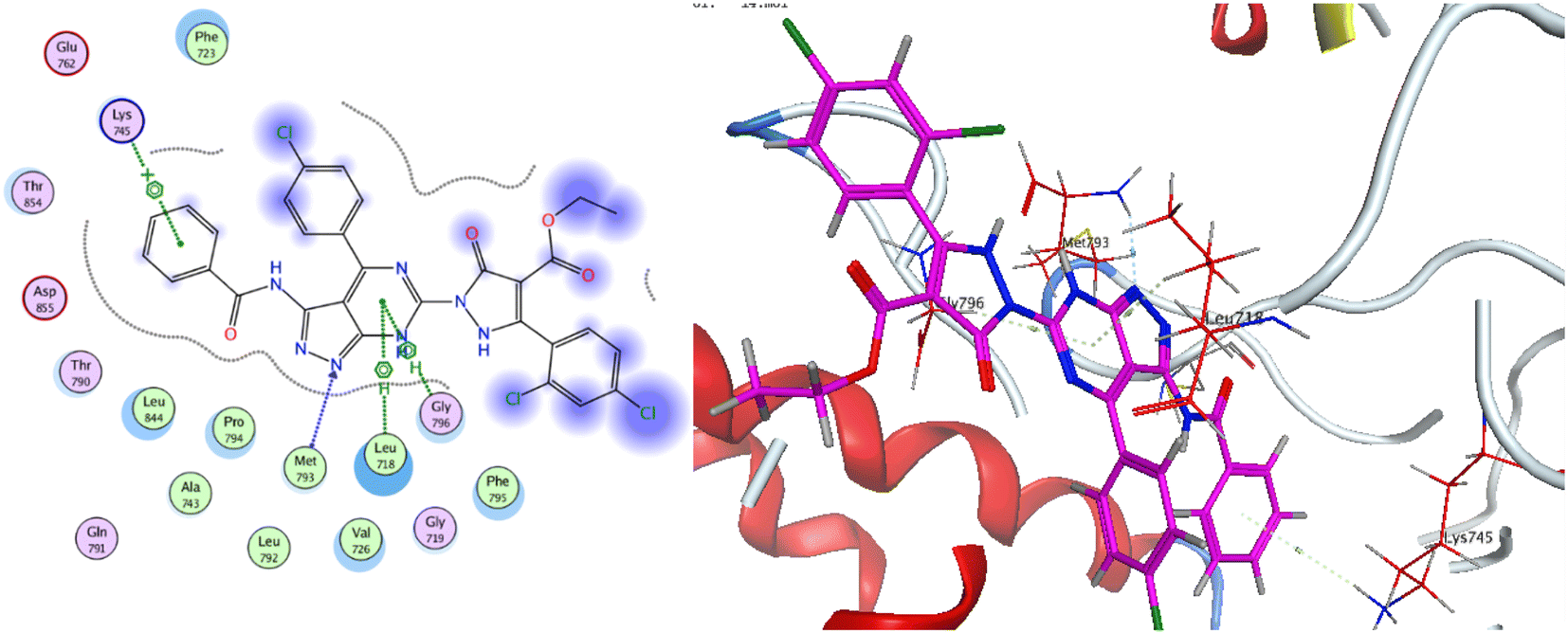 | ||
| Fig. 13 2D and 3D binding mode of compound 16 inside the binding pocket of EGFR. | ||
The results showed that both compounds had considerable oral absorption values. However, derivative 16 showed better water solubility value. The study revealed that both compounds showed high cellular permeability, particularly for intestinal cells (89.689%, and 83.764%, respectively) (Table 4).
| Property | Model name | Unit | 15 | 16 |
|---|---|---|---|---|
| Absorption | Water solubility | Numeric (log mol L−1) | −3.003 | −2.902 |
| Caco2 permeability | Numeric (log Papp in 10−6 cm s−1) | 0.632 | 0 | |
| Intestinal absorption (human) | Numeric (% absorbed) | 89.689 | 83.764 | |
| P-glycoprotein substrate | Categorical (yes/no) | Yes | Yes | |
| P-glycoprotein I inhibitor | Categorical (yes/no) | Yes | Yes | |
| P-glycoprotein II inhibitor | Categorical (yes/no) | Yes | Yes | |
| Distribution | Volume of distribution (human) | Numeric (log L kg−1) | −1.133 | −0.169 |
| Fraction unbound (human) | Numeric (Fu) | 0.049 | 0.099 | |
| BBB permeability | Numeric (log BB) | −0.2.833 | −0.2602 | |
| CNS permeability | Numeric (log PS) | −2.428 | −3.529 | |
| Metabolism | CYP2D6 substrate | Categorical (yes/no) | Yes | No |
| CYP3A4 substrate | Categorical (yes/no) | Yes | Yes | |
| CYP1A2 inhibitor | Categorical (yes/no) | No | No | |
| CYP2C19 inhibitor | Categorical (yes/no) | Yes | No | |
| CYP2C9 inhibitor | Categorical (yes/no) | No | Yes | |
| CYP2D6 inhibitor | Categorical (yes/no) | No | No | |
| CYP3A4 inhibitor | Categorical (yes/no) | Yes | No | |
| Excretion | Total clearance | Numeric (log ml min−1 kg−1) | 0.055 | 0.131 |
| Renal OCT2 substrate | Categorical (yes/no) | No | No | |
| Toxicity | AMES toxicity | Categorical (yes/no) | No | No |
| Max. tolerated dose (human) | Numeric (log mg kg−1 per day) | 0.626 | 0.642 | |
| hERG I inhibitor | Categorical (yes/no) | No | No | |
| hERG II inhibitor | Categorical (yes/no) | Yes | Yes | |
| Oral rat acute toxicity (LD50) | Numeric (mol kg−1) | 2.836 | 2.507 | |
| Oral rat chronic toxicity (LOAEL) | Numeric (log mg kg−1_bw per day) | 4.144 | 3.314 | |
| Hepatotoxicity | Categorical (yes/no) | Yes | No | |
| Skin sensitisation | Categorical (yes/no) | No | No | |
| T. pyriformis toxicity | Numeric (log ug L−1) | 0.285 | 0.285 | |
| Minnow toxicity | Numeric (log mM) | 1.932 | −0.635 |
Compound 16 showed the higher BBB permeability and unbound fraction than its analog, in accordance to distribution predication. Also, the parameters related to metabolism and excretion did not exhibit considerable variations among the examined derivatives. Moreover, the toxicological properties of compounds 15 and 16 were examined. The tested analogues showed no toxicity to AMES, and they showed minimum values for oral rat acute toxicity (LD50) and Tetrahymena pyriformis toxicity test. As for hERG I inhibition, both compounds predicted didn't inhibit hERG I enzyme but they inhibit hERG II which may help in decreasing side effects of heart including ventricular arrhythmia. These results represented might predict that both compounds 15 and 16 could be safe for human use.
2.4. Toxicological study
Initially, the doses 55, 90 and 100 mg kg−1 of compound 16 does not produce any significant effect on the animals. However, at the doses 1000 mg kg−1, the rats showed the abdominal muscle contractions and ataxia signs after intrapretonial injection. Then, the animals showed labored breathing, gasping and finally death within 7 days. The percentage of animals that died at each dose was then transformed to probit using Finney's method. LD50 of compound 16 after intraperitoneal injection in rats determined in the present study was 100.3 mg kg−1.3. Conclusion
Four series of pyrazolo[3,4-d]pyrimidine were designed, synthesized as anti-proliferative agents exhibited potent anti-proliferative activity and apoptosis inducers in tumor cells. Two candidates exhibited significant broad spectrum anticancer activity in NCI 5-log dose screening against different cancer cell panel with GI50 values ranging from 2.53 to 9.63 μM, especially on leukemia, renal, melanoma and breast cancer cell lines. Accordingly, compound 16a was chosen to further investigate the potential underlying mechanisms that induced cancer cell death. It was found that it has the ability to induce cell cycle arrest with concurrent increase in G1 and S population as well as increase the apoptotic cell population in time dependent manner. Moreover, the most significant anti-proliferative derivatives were evaluated for their EGFR inhibitory activity, that revealed the significant inhibition of the candidate (4) or cyano pyrazole derivative and its ethyl ester analogue (15 and 16) with IC50 of 0.054, 0.135 and 0.034 μM respectively. Finally, molecular docking study of the active compounds in EGFR kinase active site revealed their ability to form the essential H bond interactions with Met793, Gly796 and Lys745 key residues in the EGFR active site.4. Experimental
4.1. Chemistry
An uncorrected Griffin and George melting-point equipment was used to calculate all melting points (Griffin & George Ltd., Wembley, Middlesex, UK). The purity of the chemicals produced was controlled using aluminum sheet silica gel F254 (Merck) thin layer chromatography (TLC). IR-spectra were acquired using the KBr wafer technique on a Pye Unicam SP1200 spectrophotometer (Pye Unicam Ltd., Cambridge, UK). The 1H-NMR spectra were obtained using a Bruker Avance (III) with a Varian-Gemini 400 MHz and an internal standard of Tetramethyl silane (chemical shifts in scale ppm), while the 13C-NMR spectra were obtained using a Bruker Avance (III) with a Varian-Gemini 400 MHz and an internal standard of Tetramethyl silane (chemical shifts in scale ppm). In deuterated Dimethylsulphoxide, TMS was utilized as an internal standard (DMSO-d6). Elemental investigations were conducted out using a PerkinElmer 2400 CHN elemental analyzer at the Microanalytical Unit, Faculty of Science, Ain Shams University, and good analytical data (0.4) were obtained for all chemicals.Method (II): A mixture of arylidene derivative 1 (1.6 g, 5 mmol) and thiourea (0.38 g, 5 mmol) with base catalytic amount of sodium ethoxide was fused for 50 min at 180–185 °C in sand-bath. The reaction mixture was followed by TLC. The formed product was washed with boiling dioxane as (3.35 g, 88%). (Identity M.P., TLC, and IR).
Gold crystals; M.P. > 300 °C; IR (KBr) (υmax, cm−1): 3197 (NHbr), 3099, 3057 (CH aromatic), 1681 (COamide), 1656 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 6.75–6.76 (m, 5H, ArH), 7.69 (d, J = 3.44 Hz, 2H, ArHp-chloro), 8.03 (d, J = 3.45 Hz, 2H, ArHp-chloro), 11.46 (br s, 1H, 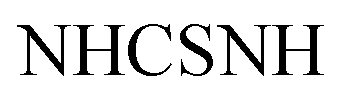 , exchangeable with D2O), 11.86 (br s, 1H,
, exchangeable with D2O), 11.86 (br s, 1H, 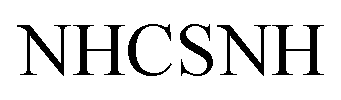 , exchangeable with D2O), 12.40 (br s, 1H,
, exchangeable with D2O), 12.40 (br s, 1H, 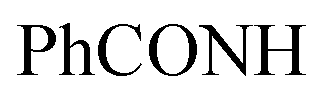 , exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 90.19, 112.88, 117.58, 124.37, 145.94, 146.48 (2C), 148.01 (2C), 150.07, 156.08, 156.79, 159.63, 170.94, 170.8, 171.55, 173.70, 187.64. Anal. Calcd for C18H12ClN5OS (381.84)%: C, 56.62; H, 3.17; N, 18.34; found: C, 56.64; H, 3.20; N, 18.31.
, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 90.19, 112.88, 117.58, 124.37, 145.94, 146.48 (2C), 148.01 (2C), 150.07, 156.08, 156.79, 159.63, 170.94, 170.8, 171.55, 173.70, 187.64. Anal. Calcd for C18H12ClN5OS (381.84)%: C, 56.62; H, 3.17; N, 18.34; found: C, 56.64; H, 3.20; N, 18.31.
:1] as (2.69 g, 64%).Method (II): A mixture of pyrazolo[3,4-d]pyrimidin 2 (1.9 g, 5 mmol) and chloroacetic acid (0.47 g, 5 mmol) and/or ethyl chloroacetate (0.61 mL, 5 mmol) with catalytic amount of sodium acetate anhydrous was fused for 75 min at 180–185 °C in sand-bath. TLC was used to monitor the reaction's progress. The formed product was washed with boiling ethanol as (3.32 g, 79%). (Identity M.P., TLC, and IR).
Red crystals; M.P. 284–286 °C; IR (KBr) (υmax, cm−1): 3270, 3129 (NH), 3091, 3006 (CH aromatic), 2962, 2924 (CH aliphatic), 1724 (COketone), 1698 (COamide), 1650, 1618 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 3.94 (s, 2H, CH2), 6.67–6.71 (m, 2H, ArH), 7.11–7.19 (m, 2H, ArH), 7.95–8.00 (m, 3H, ArH), 8.01–8.3 (m, 2H, ArH), 12.91 (br s, 1H, 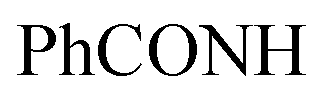 , exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 22.83, 116.04, 117.41, 120.44, 123.33, 123.69, 126.08, 128.56, 129.98, 131.66, 134.43, 140.38, 152.45, 158.87 (2C), 159.51 (2C), 166.93, 168.85, 169.66. Anal. Calcd for C20H12ClN5O2S (421.86)%: C, 56.94; H, 2.87; N, 16.60; found: C, 56.64; H, 3.20; N, 18.31.
, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 22.83, 116.04, 117.41, 120.44, 123.33, 123.69, 126.08, 128.56, 129.98, 131.66, 134.43, 140.38, 152.45, 158.87 (2C), 159.51 (2C), 166.93, 168.85, 169.66. Anal. Calcd for C20H12ClN5O2S (421.86)%: C, 56.94; H, 2.87; N, 16.60; found: C, 56.64; H, 3.20; N, 18.31.
Method (II): An ethanolic solution of compound 3 (2.1 g, 5 mmol) and furfural (0.48 mL, 5 mmol) with a catalytic amount of piperidine was refluxed for 3 h. TLC was used to monitor the reaction's progress. The compound 4 was produced by filtering the heated product and washed it from hot ethanol as (2.8 g, 63%).
Method (III): A mixture of compound 3 (2.1 g, 5 mmol) and furfural (0.48 mL, 5 mmol) with a catalytic amount of piperidine was fused under free solvent for 3 h. TLC was used to monitor the reaction's progress. The compound 4 was washed with boiling ethanol as (3.0 g, 84%).
Orange crystals; M.P. > 300 °C; IR (KBr) (υmax, cm−1): 3204, 3165 (NH), 3065, 3006 (CH aromatic), 1656 br (2C = Oamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 5.86 (s, 1H, =CHolefinic), 7.35–7.37 (m, 2H, ArH), 7.52–7.56 (m, 3H, ArH), 7.67 (t, J = 7.99 Hz, 1H, ArH), 7.97–7.99 (m, 3H, ArH), 8.17–8.19 (m, 3H, ArH), 12.41 (br s, 1H, 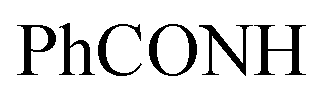 , exchangeable with D2O). Anal. Calcd for C25H14ClN5O3S (499.93)%: C, 60.06; H, 2.82; N, 14.01; found: C, 60.09; H, 2.79; N, 14.05.
, exchangeable with D2O). Anal. Calcd for C25H14ClN5O3S (499.93)%: C, 60.06; H, 2.82; N, 14.01; found: C, 60.09; H, 2.79; N, 14.05.
:1] giving the pure compound 5 as yellow crystals (2.43 g, 53%); M.P. 188–190 °C; IR (KBr) (υmax, cm−1): 3308, 3160 (NH), 3061, 3002 (CH aromatic), 2930 (CH aliphatic), 1717 (COester), 1661 (COamide), 1600 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 2.06 (t, J = 8.25 Hz, 3H, 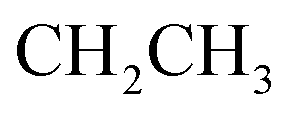 ), 4.35–4.44 (m, 2H,
), 4.35–4.44 (m, 2H, 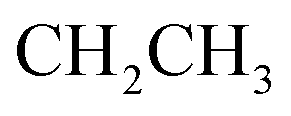 ), 5.44 (s, 2H,
), 5.44 (s, 2H, 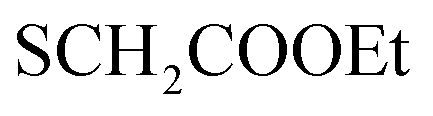 ), 7.03–7.05 (m, 2H, ArH), 7.34–7.38 (m, 2H, ArH), 7.56–7.64 (m, 5H, ArH), 11.28 (br s, 1H,
), 7.03–7.05 (m, 2H, ArH), 7.34–7.38 (m, 2H, ArH), 7.56–7.64 (m, 5H, ArH), 11.28 (br s, 1H, 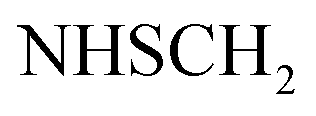 , exchangeable with D2O), 12.40 (br s, 1H,
, exchangeable with D2O), 12.40 (br s, 1H, 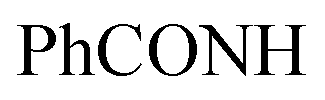 , exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 21, 23, 55, 112, 114 (2C), 118, 127 (2C), 128, 129 (2C), 137, 141, 146, 152, 159, 166 (2C), 168 (2C), 174. Anal. Calcd for C22H18ClN5O3S (467.93)%: C, 56.47; H, 3.88; N, 14.97; found: C, 56.45; H, 3.91; N, 14.75.Oamide), 1656 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 3.57 (s, 3H, SCH3), 6.77–6.78 (m, 3H, ArH), 7.79–7.80 (m, 2H, ArH), 8.04–8.05 (m, 4H, ArH), 11.78 (br s, 1H,
, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 21, 23, 55, 112, 114 (2C), 118, 127 (2C), 128, 129 (2C), 137, 141, 146, 152, 159, 166 (2C), 168 (2C), 174. Anal. Calcd for C22H18ClN5O3S (467.93)%: C, 56.47; H, 3.88; N, 14.97; found: C, 56.45; H, 3.91; N, 14.75.Oamide), 1656 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 3.57 (s, 3H, SCH3), 6.77–6.78 (m, 3H, ArH), 7.79–7.80 (m, 2H, ArH), 8.04–8.05 (m, 4H, ArH), 11.78 (br s, 1H, 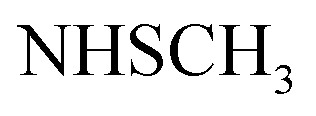 , exchangeable with D2O), 13.63 (br s, 1H,
, exchangeable with D2O), 13.63 (br s, 1H, 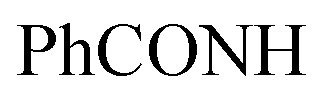 , exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 15, 113, 116, 121, 124, 128 (2C), 131 (2C), 133 (2C), 141, 144 (2C), 145, 148 (2C), 167, 168. Anal. Calcd for C19H14ClN5OS (395.87)%: C, 57.65; H, 3.56; N, 17.69; found: C, 57.68; H, 3.54; N, 17.72.
, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 15, 113, 116, 121, 124, 128 (2C), 131 (2C), 133 (2C), 141, 144 (2C), 145, 148 (2C), 167, 168. Anal. Calcd for C19H14ClN5OS (395.87)%: C, 57.65; H, 3.56; N, 17.69; found: C, 57.68; H, 3.54; N, 17.72.Method (II): An ethanolic solution of compound 6 (1.95 g, 5 mmol) and hydrazine hydrate (0.5 mL, 10 mmol) was refluxed for 2.5 h. TLC was used to monitor the reaction's progress. The compound 7 was produced by filtering the heated product and washed it from boiling ethanol as (3.5 g, 92%).
Gray crystals; M.P. > 300 °C; IR (KBr) (υmax, cm−1): 3424, 3309 (NH2), 3224, 3154 (NH), 3063 (CH aromatic), 1680 (COamide), 1604 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 6.75–6.76 (m, 3H, ArH), 7.27 (br s, 2H, 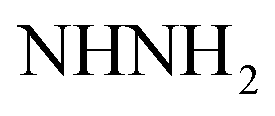 , exchangeable with D2O), 7.69–7.0 (m, 3H, ArH), 7.78 (br s, 1H,
, exchangeable with D2O), 7.69–7.0 (m, 3H, ArH), 7.78 (br s, 1H, 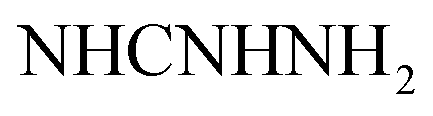 , exchangeable with D2O), 8.03–8.05 (m, 3H, ArH), 12.85 (br s, 1H,
, exchangeable with D2O), 8.03–8.05 (m, 3H, ArH), 12.85 (br s, 1H, 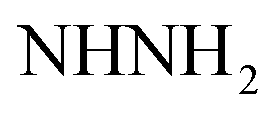 , exchangeable with D2O), 13.69 (br s, 1H,
, exchangeable with D2O), 13.69 (br s, 1H,  , exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 112.88 (2C), 117.61 (2C), 123.33, 123.69, 126.08, 131.66, 134.43, 140.38, 145.45, 148.04 (2C), 156.27, 159.08, 160.28, 169.81 (2C). Anal. Calcd for C18H14ClN7O (379.81)%: C, 56.92; H, 3.72; N, 25.82; found: C, 56.94; H, 3.75; N, 25.85.
, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 112.88 (2C), 117.61 (2C), 123.33, 123.69, 126.08, 131.66, 134.43, 140.38, 145.45, 148.04 (2C), 156.27, 159.08, 160.28, 169.81 (2C). Anal. Calcd for C18H14ClN7O (379.81)%: C, 56.92; H, 3.72; N, 25.82; found: C, 56.94; H, 3.75; N, 25.85.
Oamide), 1626 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 6.75–6.77 (m, 2H, ArH), 7.64–7.66 (m, 2H, ArH), 7.710–7.719 (m, 2H, ArH), 8.04–8.06 (m, 3H, ArH), 8.25 (br s, 1H,  , exchangeable with D2O), 11.44 (br s, 1H,
, exchangeable with D2O), 11.44 (br s, 1H,  , exchangeable with D2O), 13.39 (br s, 1H,
, exchangeable with D2O), 13.39 (br s, 1H, 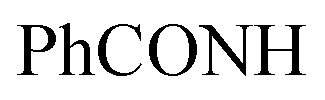 , exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 112.94, 115.85, 117.75, 117.93, 129.74 (2C), 131.93 (2C), 132.04, 136.85 (2C), 146.12, 146.39 (2C), 148.22, 156.91, 159.10, 160.49, 171.52. Anal. Calcd for C19H12ClN7OS (421.86)%: C, 54.10; H, 2.87; N, 23.24; found: C, 54.07; H, 2.89; N, 23.27.Oamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 1.49 (s, 3H, CH3), 7.32–7.34 (m, 3H, ArH), 7.68–7.77 (m, 2H, ArH), 7.94–7.95 (m, 2H, ArH), 8.04–8.09 (m, 2H, ArH), 11.43 (br s, 1H,
, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 112.94, 115.85, 117.75, 117.93, 129.74 (2C), 131.93 (2C), 132.04, 136.85 (2C), 146.12, 146.39 (2C), 148.22, 156.91, 159.10, 160.49, 171.52. Anal. Calcd for C19H12ClN7OS (421.86)%: C, 54.10; H, 2.87; N, 23.24; found: C, 54.07; H, 2.89; N, 23.27.Oamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 1.49 (s, 3H, CH3), 7.32–7.34 (m, 3H, ArH), 7.68–7.77 (m, 2H, ArH), 7.94–7.95 (m, 2H, ArH), 8.04–8.09 (m, 2H, ArH), 11.43 (br s, 1H,  , exchangeable with D2O), 13.33 (br s, 1H,
, exchangeable with D2O), 13.33 (br s, 1H, 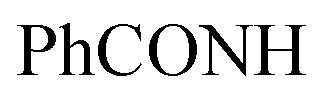 , exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 22.64, 113.03 (2C), 116.20, 118.26 (2C), 125.48 (2C), 128.94 (2C), 134.88, 136.99 (2C), 137.88 (2C), 141.22, 148.49, 159.46, 167.95 (2C). Anal. Calcd for C20H14ClN7O (403.83) C, 59.49; H, 3.49; N, 24.28; found: C, 59.53; H, 3.51; N, 24.30.Oamide), 1614 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 4.64 (s, 2H,
, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 22.64, 113.03 (2C), 116.20, 118.26 (2C), 125.48 (2C), 128.94 (2C), 134.88, 136.99 (2C), 137.88 (2C), 141.22, 148.49, 159.46, 167.95 (2C). Anal. Calcd for C20H14ClN7O (403.83) C, 59.49; H, 3.49; N, 24.28; found: C, 59.53; H, 3.51; N, 24.30.Oamide), 1614 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 4.64 (s, 2H, 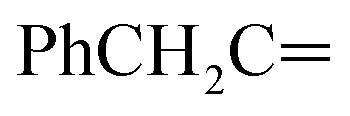 ), 7.56–7.64 (m, 6H, ArH), 7.81–7.86 (m, 3H, ArH), 8.10–8.17 (m, 3H, ArH), 11.97 (br s, 1H,
), 7.56–7.64 (m, 6H, ArH), 7.81–7.86 (m, 3H, ArH), 8.10–8.17 (m, 3H, ArH), 11.97 (br s, 1H,  , exchangeable with D2O), 13.66 (br s, 1H,
, exchangeable with D2O), 13.66 (br s, 1H, 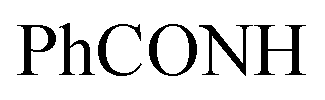 , exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 21.13, 61.78, 68.28, 115.85 (2C), 116.66 (2C), 120.90, 123.02, 123.13, 125.59 (2C), 126.03 (2C), 127.77 (2C), 128.11, 128.46, 128.55, 129.89 (2C), 135.19, 146.74, 151.71, 170.62, 170.77. Anal. Calcd for C26H18ClN7O (479.93): C, 65.07; H, 3.78; N, 20.43; found: C, 65.09; H, 3.80; N, 20.40.
, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 21.13, 61.78, 68.28, 115.85 (2C), 116.66 (2C), 120.90, 123.02, 123.13, 125.59 (2C), 126.03 (2C), 127.77 (2C), 128.11, 128.46, 128.55, 129.89 (2C), 135.19, 146.74, 151.71, 170.62, 170.77. Anal. Calcd for C26H18ClN7O (479.93): C, 65.07; H, 3.78; N, 20.43; found: C, 65.09; H, 3.80; N, 20.40.Method (II): A mixture of hydrazine derivative 6 (1.9 g, 5 mmol) and phenyl isothiocyanate (0.67 mL, 5 mmol) was fused for 2 h at 180–185 °C in sand-bath. TLC was used to monitor the reaction's progress. The formed product was washed with boiling dioxane to afford the pure compound 12 as (3.93 g, 82%). (Identity M.P., TLC, and IR).
Yellow crystals; M.P. > 300 °C; IR (KBr) (υmax, cm−1): 3240, 3197 (NH), 3060, 3035 (CH aromatic), 1686 (COamide), 1605 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 7.41–7.64 (m, 5H, ArH), 7.68–7.74 (m, 3H, ArH), 7.79–7.82 (m, 2H, ArH), 7.86–7.92 (m, 2H, ArH), 8.03–8.05 (m, 3H, ArH and  , exchangeable with D2O), 13.56 (br s, 1H,
, exchangeable with D2O), 13.56 (br s, 1H,  , exchangeable with D2O), 14.23 (br s, 1H,
, exchangeable with D2O), 14.23 (br s, 1H, 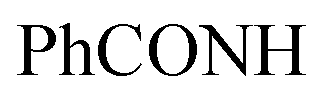 , exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 124.27, 125.54, 127.48, 127.82, 128.11, 128.36, 129.21, 129.39, 130.04, 130.44, 131.08, 131.61, 132.14, 132.68, 133.12, 133.93, 136.45, 138.10, 139.00, 148.15, 151.51, 163.29, 166.36, 166.40, 173.87. Anal. Calcd for C25H17ClN8O (480.92) C, 62.44; H, 3.56; N, 23.30; found: C, 62.48; H, 3.59; N, 23.33.
, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 124.27, 125.54, 127.48, 127.82, 128.11, 128.36, 129.21, 129.39, 130.04, 130.44, 131.08, 131.61, 132.14, 132.68, 133.12, 133.93, 136.45, 138.10, 139.00, 148.15, 151.51, 163.29, 166.36, 166.40, 173.87. Anal. Calcd for C25H17ClN8O (480.92) C, 62.44; H, 3.56; N, 23.30; found: C, 62.48; H, 3.59; N, 23.33.
Method (II): A mixture of compound 7 (1.9 g, 5 mmol) and the appropriate diketone derivatives namely; acetylacetone (0.50 mL, 5 mmol), ethyl acetoacetate (0.65 mL, 5 mmol) and/or benzoyl acetone (1.93 mL, 5 mmol) in the presence of a catalytic amount of triethylamine was fused under free solvent at 180–185 °C in sand-bath for one hour. The precipitated crystals were washed with boiling ethanol to afford the pure compounds 13a–c as (2.46 g, 56%), (2.94 g, 67%), and (3.35 g, 67%), respectively.
Oamide), 1637 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 1.85 (s, 3H, CH3), 1.89 (s, 3H, CH3), 5.30 (s, 1H, CHolefinic), 6.67–6.70 (m, 3H, ArH), 7.11–7.18 (m, 2H, ArH), 7.62–7.63 (m, 4H, ArH), 7.98 (br s, 1H,  , exchangeable with D2O), 9.59 (br s, 1H,
, exchangeable with D2O), 9.59 (br s, 1H, 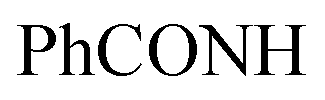 , exchangeable with D2O). Anal. Calcd for C23H18ClN7O (443.90): C, 62.23; H, 4.09; N, 22.09; found: C, 62.20; H, 4.12; N, 22.07.Oamide), 1659, 1617 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 2.11 (s, 3H, CH3), 5.83 (s, 1H, CHolefinic), 7.53–7.57 (m, 3H, ArH), 7.65–7.71 (m, 3H, ArH), 7.98–8.02 (m, 3H, ArH), 11.28 (s br, 1H,
, exchangeable with D2O). Anal. Calcd for C23H18ClN7O (443.90): C, 62.23; H, 4.09; N, 22.09; found: C, 62.20; H, 4.12; N, 22.07.Oamide), 1659, 1617 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 2.11 (s, 3H, CH3), 5.83 (s, 1H, CHolefinic), 7.53–7.57 (m, 3H, ArH), 7.65–7.71 (m, 3H, ArH), 7.98–8.02 (m, 3H, ArH), 11.28 (s br, 1H,  , exchangeable with D2O), 11.58 (br s, 1H,
, exchangeable with D2O), 11.58 (br s, 1H,  , exchangeable with D2O), 12.40 (br s, 1H,
, exchangeable with D2O), 12.40 (br s, 1H, 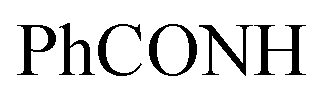 , exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 37.97, 68.28, 70.01, 120.90, 123.02, 123.13, 125.59 (2C), 126.03, 126.71, 127.77, 128.11, 128.46, 128.55, 129.80, 129.89, 135.19, 151.71, 152.70, 152.94, 161.70, 167.67. Anal. Calcd for C22H16ClN7O2 (445.87): C, 59.26; H, 3.62; N, 21.99; found: C, 59.28; H, 3.65; N, 22.03.Oamide), 1639 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 1.96 (s, 3H, CH3), 4.89 (s, 1H, CHolefinic), 7.18–7.22 (m, 2H, ArH), 7.36–7.39 (m, 2H, ArH), 7.61–7.65 (m, 6H, ArH), 7.99–8.01 (m, 2H, ArH), 8.64–8.66 (m, 2H, ArH), 9.05 (s br, 1H,
, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 37.97, 68.28, 70.01, 120.90, 123.02, 123.13, 125.59 (2C), 126.03, 126.71, 127.77, 128.11, 128.46, 128.55, 129.80, 129.89, 135.19, 151.71, 152.70, 152.94, 161.70, 167.67. Anal. Calcd for C22H16ClN7O2 (445.87): C, 59.26; H, 3.62; N, 21.99; found: C, 59.28; H, 3.65; N, 22.03.Oamide), 1639 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 1.96 (s, 3H, CH3), 4.89 (s, 1H, CHolefinic), 7.18–7.22 (m, 2H, ArH), 7.36–7.39 (m, 2H, ArH), 7.61–7.65 (m, 6H, ArH), 7.99–8.01 (m, 2H, ArH), 8.64–8.66 (m, 2H, ArH), 9.05 (s br, 1H,  , exchangeable with D2O), 13.66 (br s, 1H,
, exchangeable with D2O), 13.66 (br s, 1H, 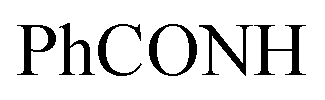 , exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 21.01, 21.13, 115.85, 116.66, 120.90, 123.02, 123.13, 125, 126.03, 126.71, 127.77 (2C), 128.11 (2C), 128.46 (2C), 128.46 (2C), 128.55 (2C), 129.80 (2C), 129.89 (2C), 146.74, 151.71, 161.70, 167.67. Anal. Calcd for C28H20ClN7O (505.97): C, 66.47; H, 3.98; N, 19.38; found: C, 66.49; H, 4.01; N, 19.40.
, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 21.01, 21.13, 115.85, 116.66, 120.90, 123.02, 123.13, 125, 126.03, 126.71, 127.77 (2C), 128.11 (2C), 128.46 (2C), 128.46 (2C), 128.55 (2C), 129.80 (2C), 129.89 (2C), 146.74, 151.71, 161.70, 167.67. Anal. Calcd for C28H20ClN7O (505.97): C, 66.47; H, 3.98; N, 19.38; found: C, 66.49; H, 4.01; N, 19.40.Method (II): A mixture of compound 7 (1.9 g, 5 mmol) with 2,4-dichlorobenzaldehyde (0.87 g, 5 mmol) and the appropriate active methylene derivatives namely; acetophenone (0.60 mL, 5 mmol), ethyl cyanoacetate (0.56 mL, 5 mmol), diethyl malonate (0.80 mL, 5 mmol) and/or malononitrile (0.33 mL, 5 mmol) in the presence of a catalytic amount of piperidine was fused under free solvent at 180–185 °C in sand-bath for 65–80 min. The precipitated crystals were washed with boiling ethanol to afford pure compounds 14–17 as (5.60 g, 89%), (5.22 g, 87%), (3.71 g, 58%), and (5.58 g, 93%), respectively.
Oamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 5.35 (s, 1H, CHolefinic), 7.55–7.57 (m, 3H, ArH), 7.64–7.72 (m, 3H, ArH), 7.79–7.83 (m, 1H, ArH), 7.97–8.08 (m, 6H, ArH), 8.13–8.18 (m, 4H, ArH), 8.95 (s br, 1H,  , exchangeable with D2O), 14.14 (br s, 1H,
, exchangeable with D2O), 14.14 (br s, 1H, 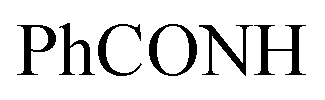 , exchangeable with D2O). Anal. Calcd for C33H20Cl3N7O (636.92): C, 62.23; H, 3.17; N, 15.39; found: C, 62.20; H, 3.19; N, 15.37.
, exchangeable with D2O). Anal. Calcd for C33H20Cl3N7O (636.92): C, 62.23; H, 3.17; N, 15.39; found: C, 62.20; H, 3.19; N, 15.37.![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1627 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 7.57–7.60 (m, 3H, ArH), 7.67–7.71 (m, 3H, ArH), 8.04–8.07 (m, 3H, ArH), 8.14–8.16 (m, 3H, ArH), 8.30 (s br, 1H,
N), 1627 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 7.57–7.60 (m, 3H, ArH), 7.67–7.71 (m, 3H, ArH), 8.04–8.07 (m, 3H, ArH), 8.14–8.16 (m, 3H, ArH), 8.30 (s br, 1H,  , exchangeable with D2O), 10.90 (s br, 1H,
, exchangeable with D2O), 10.90 (s br, 1H, 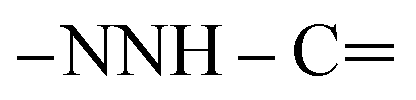 , exchangeable with D2O), 13.37 (br s, 1H,
, exchangeable with D2O), 13.37 (br s, 1H, 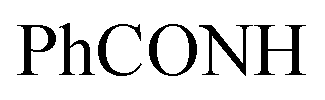 , exchangeable with D2O). Anal. Calcd for C28H15Cl3N8O2 (601.83): C, 55.88; H, 2.51; N, 18.62; found: C, 55.85; H, 2.54; N, 18.59. Purity calculated is 69.69%.Oester), 1666 (COamide), 1606 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 1.24 (t, J = 7.16 Hz, 3H,
, exchangeable with D2O). Anal. Calcd for C28H15Cl3N8O2 (601.83): C, 55.88; H, 2.51; N, 18.62; found: C, 55.85; H, 2.54; N, 18.59. Purity calculated is 69.69%.Oester), 1666 (COamide), 1606 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 1.24 (t, J = 7.16 Hz, 3H, 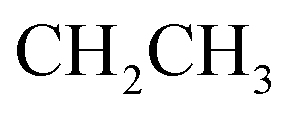 ), 4.17 (q, J = 7.08 Hz, 2H,
), 4.17 (q, J = 7.08 Hz, 2H, 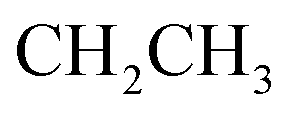 ), 7.55–7.59 (m, 5H, ArH), 7.66–7.70 (m, 5H, ArH), 8.07–8.12 (m, 2H, ArH), 10.65 (s br, 1H,
), 7.55–7.59 (m, 5H, ArH), 7.66–7.70 (m, 5H, ArH), 8.07–8.12 (m, 2H, ArH), 10.65 (s br, 1H, 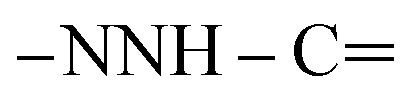 , exchangeable with D2O), 12.89 (s br, 1H,
, exchangeable with D2O), 12.89 (s br, 1H,  , exchangeable with D2O), 14.18 (br s, 1H,
, exchangeable with D2O), 14.18 (br s, 1H, 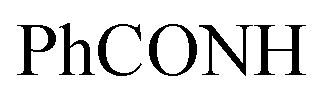 , exchangeable with D2O). Anal. Calcd for C30H20Cl3N7O4 (648.89): C, 55.53; H, 3.11; N, 15.11; found: C, 55.58; H, 3.15; N, 15.13. Purity calculated is 90.92%.N), 1657 (COamide), 1620 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 6.88 (br s, 2H, NH2, exchangeable with D2O), 7.48–7.49 (m, 2H, CH3), 7.52–7.56 (m, 4H, ArH), 7.65–7.69 (m, 2H, ArH), 7.97–7.99 (m, 4H, ArH), 8.20 (s br, 1H,
, exchangeable with D2O). Anal. Calcd for C30H20Cl3N7O4 (648.89): C, 55.53; H, 3.11; N, 15.11; found: C, 55.58; H, 3.15; N, 15.13. Purity calculated is 90.92%.N), 1657 (COamide), 1620 (CN). 1H-NMR (400 MHz, DMSO-d6) δppm: 6.88 (br s, 2H, NH2, exchangeable with D2O), 7.48–7.49 (m, 2H, CH3), 7.52–7.56 (m, 4H, ArH), 7.65–7.69 (m, 2H, ArH), 7.97–7.99 (m, 4H, ArH), 8.20 (s br, 1H,  , exchangeable with D2O), 11.82 (br s, 1H,
, exchangeable with D2O), 11.82 (br s, 1H, 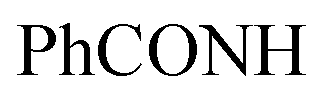 , exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 68.90, 100.05 (2C), 109.42, 116.06, 117.04 (2C), 120.52 (2C), 123.23 (2C), 123.86, 126.10 (2C), 128.60 (2C), 130.00 (2C), 131.15, 131.68, 134.65, 139.74, 140.38, 152.43, 159.67 (2C), 166.99, 169.64. Anal. Calcd for C28H16Cl3N9O (600.85): C, 55.97; H, 2.68; N, 20.98; found: C, 56.00; H, 2.71; N, 21.02.
, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 68.90, 100.05 (2C), 109.42, 116.06, 117.04 (2C), 120.52 (2C), 123.23 (2C), 123.86, 126.10 (2C), 128.60 (2C), 130.00 (2C), 131.15, 131.68, 134.65, 139.74, 140.38, 152.43, 159.67 (2C), 166.99, 169.64. Anal. Calcd for C28H16Cl3N9O (600.85): C, 55.97; H, 2.68; N, 20.98; found: C, 56.00; H, 2.71; N, 21.02.4.2. DFT studies
All calculations were carried out with Orca software.43 All geometrical optimizations were carried out at the B3LYP/6-311+G(d,p) level.44 The effect of were computed using UAHF (united atom topological model for Hartree–Fock) radii and a CPCM (conductor polarized continuum model).45,46 All the calculated energies are the Gibbs free energies in toluene at 298 K.4.3. Biological evaluation
4.3.1.1. Assay protocol. The human tumor cell lines of the cancer-screening panel were grown in RPMI 1640 medium containing 5% fetal bovine serum and 2 μM L-glutamine. For a typical screening experiment, cells are inoculated into 96 well microtiter plates in 100 mL at plating densities ranging from 5000 to 40
000 cells per well depending on the doubling time of individual cell lines. After cell inoculation, the microtiter plates are incubated at 37 °C, 5% CO2, 95% air and 100% relative humidity for 24 h prior to addition of experimental drugs. After 24 h, two plates of each cell line are fixed in situ with TCA, to represent a measurement of the cell population for each cell line at the time of drug addition (Tz). Experimental drugs are dissolved in dimethyl sulfoxide at 400-fold the desired final maximum test concentration and stored frozen prior to use. At the time of drug addition, an aliquot of frozen concentrate is thawed and diluted to twice the desired final maximum test concentration with complete medium containing 50 mg mL−1 Gentamicin. Additional four, 10-fold or 1/2log serial dilutions are made to provide a total of five drug concentrations plus control. Aliquots of 100 mL of these different drug dilutions are added to the appropriate microtiter wells already containing 100 mL of medium, resulting in the required final drug concentrations. Following drug addition, the plates are incubated for an additional 48 h at 37 °C, 5% CO2, 95% air, and 100% relative humidity. For adherent cells, the assay is terminated by the addition of cold TCA. Cells are fixed in situ by the gentle addition of 50 mL of cold 50% (w/v) TCA (final concentration, 10% TCA) and incubated for 60 min at 4 °C. The supernatant is discarded, and the plates are washed five times with tap water and air dried. Sulforhodamine B (SRB) solution (100 mL) at 0.4% (w/v) in 1% acetic acid is added to each well, and plates are incubated for 10 min at room temperature. After staining, unbound dye is removed by washing five times with 1% acetic acid and the plates are air dried. Bound stain is subsequently dissolved with 10 μM trizma base, and the absorbance is read on an automated plate reader at a wavelength of 515 nm. For suspension cells, the methodology is the same except that the assay is terminated by fixing settled cells at the bottom of the wells by gently adding 50 mL of 80% TCA (final concentration, 16% TCA).
4.3.1.2. Data analysis. Using the seven absorbance measurements [time zero, (Tz), control growth, (C), and test growth in the presence of drug at the five concentration levels (Ti)], the percentage growth is calculated at each of the drug concentrations levels. Percentage growth inhibition is calculated as: [(Ti − Tz)/(C − Tz)] × 100 for concentrations for which Ti>/1/4Tz and [(Ti − Tz)/Tz] × 100 for concentrations for which Ti < Tz.
Three dose response parameters are calculated for each experimental agent. Growth inhibition of 50% (GI50) is calculated from [(Ti − Tz)/(C − Tz)] × 100 = 50, which is the drug concentration resulting in a 50% reduction in the net protein increase (as measured by SRB staining) in control cells during the drug incubation. The drug concentration resulting in total growth inhibition (TGI) is calculated from Ti 1/4 Tz. The LC50 (concentration of drug resulting in a 50% reduction in the measured protein at the end of the drug treatment as compared to that at the beginning) indicating a net loss of cells following treatment is calculated from [(Ti − Tz)/Tz] × 100 = −50. Values are calculated for each of these three parameters if the level of activity is reached; however, if the effect is not reached or is exceeded, the value for that parameter is expressed as greater or less than the maximum or minimum concentration tested.
:1, Glu:Tyr) (Sigma#P7244) served as the standardized substrate & Kinase-Glo Plus Luminescence kinase assay kit (Promega#V3772). The IC50 determination was carried out where quality control testing is routinely performed on each of the targets to insure compliance to acceptable standards. 33P-ATP was purchased from PerkinElmer and ADP-Glo™ was purchased from Promega. All other materials were of standard laboratory grade.
4.3.2.1. Assay protocols. The EGFR tyrosine kinase activity was performed using Kinase-Glo Plus luminescence kinase assay kit (Promega). It measures kinase activity by quantitating the amount of ATP remaining in solution following a kinase reaction. The luminescent signal from the assay is correlated with the amount of ATP present and is inversely correlated with the amount of kinase activity. The compounds were diluted to 100 μM in 10% DMSO and 5 mL of the dilution was added to a 50 mL reaction so that the final concentration of DMSO is 1% in all of reactions. All of the enzymatic reactions were conducted at 30 °C for 40 min. The 50 μl reaction mixture contains 40 μM Tris, pH 7.4, 10 μM MgCl2, 0.1 mg mL−1 BSA, 0.2 mg mL−1 Poly (Glu, Tyr) substrate, 10 μM ATP and EGFR. After the enzymatic reaction, 50 mL of Kinase-GloPlus Luminescence kinase assay solution (Promega) was added to each reaction and incubate the plate for 5 min at room temperature. Luminescence signal was measured using a BioTek Synergy 2 microplate reader. The protein kinase assays used to determine IC50 value were performed using ADP-GloTM assay kit from Promega which measures the generation of ADP by the protein kinase. Generation of ADP by the protein kinase reaction leads to an increase in luminescence signal in the presence of ADP-GloTM assay kit. The assay was started by incubating the reaction mixture in a 96-well plate at 30 °C for 30 min. After the 30 min incubation period, the assay was terminated by the addition of 25 mL of ADP-GloTM Reagent (Promega). The 96 well plate was shaken and then incubated for 40 min at ambient temperature. 50 mL of Kinase detection reagent was added, the 96-well reaction plate was then read using the ADP-Glo Luminescences Protocol on a GloMax plate reader. Blank control was set up that included all the assay components except the addition of appropriate substrate (replace with equal volume of kinase assay buffer). The corrected activity for each protein kinase target was determined by removing the blank control value.
4.3.2.2. Data analysis. EGFR activity assays were performed in duplicate at each concentration. The luminescence data were analyzed using the computer software, Graphpad Prism. The difference between luminescence intensities in the absence of EGFR (Lut) and in the presence of EGFR (Luc) was defined as 100% activity (Lut − Luc). Using luminescence signal (Lu) in the presence of the compound, % activity was calculated as: % activity = (Lut − Lu)/(Lut − Luc) × 100%, where Lu = the luminescence intensity in the presence of the compound (all percent activities below zero were set to 0%). % Inhibition was calculated as: % inhibition = 100 (%) − % activity. IC50 determination for inhibitor against EGFR was estimated by generating a graph of log inhibitor vs. normalized response with variable using the Prism software.
4.4. Calculation of LD50
Twenty-five healthy adult male Wistar rats (weighing about 200 ± 20 g) were obtained from Animal House in Nahda University, Beni Suef, Egypt. All the animals were allowed to acclimatize to the experimental conditions for a period of 7 days. All the rats were housed in polyacrylic cages, not more than three animals per cage and maintained under standard laboratory conditions (natural light/dark cycle, room temperature 22 ± 3 °C). The experimental protocol was approved by the Ethical Committee in the university. Rats were fasted for 18 h prior to dosing. Compound 16 was administered once orally to the rats, using 22-gauze oral feeding needle. The volume of the dose depends on the size and weight of the animals. In rodents it should not exceed 1 mL/100 g body weight. In this study, compound 16 was dissolved in 0.5 mL of DMSO. An approximate LD50 can be determined by the so-called “up and down” or the “staircase method” using two animals and increasing the doses of compound 16. Five doses were chosen which were given orally to five groups of rats, five rats in each group, for the determination of LD50 of compound 16 from 0% mortality to 100% mortality. After 24 h, the numbers of deceased rats in each group were counted, and percentage of mortality was calculated using the graphical method of Miller and Tainter.Ethical approval
All animal procedures were performed in accordance with the guidelines for care and use of Animal House of Nahda University and approved by committee of ethics of scientific research on living organisms at Faculty of Pharmacy, Nahda University, Beni-Suef, Egypt under reference number NUB-018-023.Conflicts of interest
The authors declare no conflict of interest.References
- A. Detsi, M. Majdalani, C. A. Kontogiorgis, D. Hadjipavlou-Litina and P. Kefalas, Natural and synthetic 2'-hydroxy-chalcones and aurones: synthesis, characterization and evaluation of the antioxidant and soybean lipoxygenase inhibitory activity, Bioorg. Med. Chem., 2009, 17, 8073–8085, DOI:10.1016/j.bmc.2009.10.002.
- P. Traxler and P. Furet, Strategies toward the design of novel and selective protein tyrosine kinase inhibitors, Pharmacol. Ther., 1999, 82, 195–206, DOI:10.1016/S0163-7258(98)00044-8.
- P. S. Sharma, R. Sharma and T. Tyagi, Receptor tyrosine kinase inhibitors as potent weapons in war against cancers, Curr. Pharm. Des., 2009, 15, 758–776, DOI:10.2174/138161209787582219.
- A. Ocana, R. Serrano, R. Calero and A. Pandiella, Novel tyrosine kinase inhibitors in the treatment of cancer, Curr. Drug Targets, 2009, 10, 575–576, DOI:10.2174/138945009788488378.
- S. Vogel, M. Barbic, G. Jurgenliemk and J. Heilmann, Synthesis, cytotoxicity, anti-oxidative and anti-inflammatory activity of chalcones and influence of A-ring modifications on the pharmacological effect, Eur. J. Med. Chem., 2010, 45, 2206–2213, DOI:10.1016/j.ejmech.2010.01.060.
- D. G. Piotrowska, M. Cieslak, K. Krolewska and A. E. Wroblewski, Design, synthesis, and cytotoxicity of a new series of isoxazolidines derived from substituted chalcones, Eur. J. Med. Chem., 2011, 46, 1382–1389, DOI:10.1016/j.ejmech.2011.01.067.
- S. Shenvi, K. Kumar, K. S. Hatti, K. Rijesh, L. Diwakar and G. C. Reddy, Synthesis, anticancer and antioxidant activities of 2,4,5-trimethoxy chalcones and analogues from asaronaldehyde: structure activity relationship, Eur. J. Med. Chem., 2013, 62, 435–442, DOI:10.1016/j.ejmech.2013.01.018.
- J. H. Cheng, C. F. Hung, S. C. Yang, J. P. Wang, S. J. Won and C. N. Lin, Synthesis and cytotoxic, anti-inflammatory, and anti-oxidant activities of 2',5'-dialkoxylchalcones as cancer chemopreventive agents, Bioorg. Med. Chem., 2008, 16, 7270–7276, DOI:10.1016/j.bmc.2008.06.031.
- M. Cherry and D. H. Williams, Recent kinase, and kinase inhibitor X-ray structures: mechanisms of inhibition and selectivity insights, Curr. Med. Chem., 2004, 11, 663–673, DOI:10.2174/0929867043455792.
- J. Smith, Erlotinib: small-molecule targeted therapy in the treatment of non-small-cell lung cancer, Clin. Ther., 2005, 27, 1513–1534, DOI:10.1016/j.clinthera.2005.10.014.
- G. R. Simon, J. C. Ruckdeschel, C. Williams, A. Cantor, A. Chiappori, C. M. Rocha Lima, S. Antonia, E. Haura, H. Wagner, L. Robinson, E. Sommers, M. Alberts and G. Bepler, Gefitinib (ZD1839) in previously treated advanced non-small-cell lung cancer: experience from a single institution, Moffitt Cancer Center, 2003, 10, 388–395, DOI:10.1177/107327480301000506.
- A. J. Tevaarwerk and J. M. Kolesar, Lapatinib: a small-molecule inhibitor of epidermal growth factor receptor and human epidermal growth factor receptor-2 tyrosine kinases used in the treatment of breast cancer, Clin. Ther., 2009, 31(Pt 2), 2332–2348, DOI:10.1016/j.clinthera.2009.11.029.
- K. Bouchalova, M. Cizkova, K. Cwiertka, R. Trojanec, D. Friedecky and M. Hajduch, Lapatinib in breast cancer - the predictive significance of HER1 (EGFR), HER2, PTEN and PIK3CA genes and lapatinib plasma level assessment, Biomed. Pap. Med. Fac. Univ. Palacky. Olomouc. Czech. Repub., 2010, 154, 281–288, DOI:10.5507/bp.2010.043.
- P. Mahajan, N. Suri, R. Mehra, M. Gupta, A. Kumar and S. K. Singh, Med. Chem. Res., 2017, 26, 74, DOI:10.1007/s00044-016-1728-2.
- M. S. Abdelbaset, M. Abdel-Aziz, M. Ramadan, M. H. Abdelrahman, S. N. Abbas Bukhari, T. F. S. Ali and G. E. D. A. Abuo-Rahma, Bioorg. Med. Chem., 2019, 27, 1076, DOI:10.1016/j.bmc.2019.
- S. B. Yewale, S. B. Ganorkar, K. G. Baheti and R. U. Shelke, Novel 3-substituted-1-aryl-5- phenyl-6-anilinopyrazolo[3,4-d]pyrimidin-4-ones: docking, synthesis and pharmacological evaluation as a potential anti-inflammatory agents, Bioorg. Med. Chem. Lett., 2012, 22, 6616–6620, DOI:10.1016/j.bmcl.2012.08.119.
- M. Bakavoli, G. Bagherzadeh, M. Vaseghifar, A. Shiri, M. Pordel, M. Mashreghi, P. Pordeli and M. Araghi, Molecular iodine promoted synthesis of new pyrazolo[3,4- d]pyrimidine derivatives as potential antibacterial agents, Eur. J. Med. Chem., 2010, 45, 647–650, DOI:10.1016/j.ejmech.2009.10.051.
- B. S. Holla, M. Mahalinga, M. S. Karthikeyan, P. M. Akberali and N. S. Shetty, Synthesis of some novel pyrazolo[3,4-d]pyrimidine derivatives as potential antimicrobial agents, Bioorg. Med. Chem., 2006, 14, 2040–2047, DOI:10.1016/j.bmc.2005.10.053.
- J.-H. Chern, K.-S. Shia, T.-A. Hsu, C.-L. Tai, C.-C. Lee, Y.-C. Lee, C.-S. Chang, S.-N. Tseng and S.-R. Shih, Design, synthesis, and structure–activity relationships of pyrazolo[3,4-d]pyrimidines: a novel class of potent enterovirus inhibitors, Bioorg. Med. Chem. Lett., 2004, 14, 2519–2525, DOI:10.1016/j.bmcl.2004.02.092.
- A. Bazgir, M. M. Khanaposhtani and A. A. Soorki, One-pot synthesis and antibacterial activities of pyrazolo[4′,3′:5,6]pyrido[2,3-d]pyrimidine-dione derivatives, Bioorg. Med. Chem. Lett., 2008, 18, 5800–5803, DOI:10.1016/j.bmcl.2008.09.057.
- B. R. Neustadt, J. Hao, N. Lindo, W. J. Greenlee, A. W. Stamford, D. Tulshian, E. Ongini, J. Hunter, A. Monopoli, R. Bertorelli, C. Foster, L. Arik, J. Lachowicz, K. Ng and K.-I. Feng, Potent, selective, and orally active adenosine A2A receptor antagonists: arylpiperazine derivatives of pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidines, Bioorg. Med. Chem. Lett., 2007, 17, 1376–1380, DOI:10.1016/j.bmcl.2006.11.083.
- A. El-Mekabaty, Synthesis and Antioxidant Activity of Some New Heterocycles Incorporating the Pyrazolo[3,4-d]pyrimidin-4-one Moiety, Chem. Heterocycl. Compd., 2015, 50, 1698–1706, DOI:10.1007/s10593-015-1640-6.
- P. Gong, Y. F. Zhao and D. Wang, Synthesis and vasodilatory activities of new pyrazolo[3,4- d]pyrimidin-4-one derivatives, Chin. Chem. Lett., 2002, 13, 613–616 CAS.
- K. W. Weitzel, J. M. Wickman, S. G. Augustin and J. G. Strom, Zaleplon: a pyrazolopyrimidine sedative-hypnotic agent for the treatment of insomnia, Clin. Ther., 2018, 22, 1254–1267, DOI:10.1016/S0149-2918(00)83024-6.
- B. D. Manning, Challenges and Opportunities in Defining the Essential Cancer Kinome, Sci. Signaling, 2009, 2, pe15, DOI:10.1126/scisignal.263pe15.
- M. M. El-Enany, M. M. Kamel, O. M. Khalil and H. B. El-Nassan, Synthesis and antitumor activity of novel 6-aryl and 6-alkylpyrazolo[3,4-d]pyrimidin-4-one derivatives, Eur. J. Med. Chem., 2010, 45, 5286–5291, DOI:10.1016/j.ejmech.2010.08.048.
- X. J. Song, Y. Shao and X. G. Dong, Microwave-assisted synthesis of some novel fluorinated pyrazolo[3,4-d]pyrimidine derivatives containing 1,3,4-thiadiazole as potential antitumor agents, Chin. Chem. Lett., 2011, 22, 1036–1038, DOI:10.1016/j.cclet.2011.05.012.
- S. Gupta, L. M. Rodrigues, A. P. Esteves, A. M. F. Oliveira-Campos, M. S. J. Nascimento, N. Nazareth, H. Cidade, M. P. Neves, E. Fernandes, M. Pinto, N. M. F. S. A. Cerqueira and N. Brás, Synthesis of N-aryl-5-amino-4-cyanopyrazole derivatives as potent xanthine oxidase inhibitors, Eur. J. Med. Chem., 2008, 43, 771–780, DOI:10.1016/j.ejmech.2007.06.002.
- M. Devarakonda, R. Doonaboina, S. Vanga, J. Vemu, S. Boni and R. P. Mailavaram, Synthesis of novel 2-alkyl-4-substituted-amino-pyrazolo[3,4-d]pyrimidines as new leads for anti-bacterial and anti-cancer activity, Med. Chem. Res., 2013, 22, 1090–1101, DOI:10.1007/s00044-012-0084-0.
- P. Traxler, G. Bold, J. Frei, M. Lang, N. Lydon, H. Mett, E. Buchdunger, T. Meyer, M. Mueller and P. Furet, Use of a pharmacophore model for the design of EGF-R tyrosine kinase inhibitors: 4- (phenylamino) pyrazolo [3, 4-d] pyrimidines, J. Med. Chem., 1997, 40, 3601–3616, DOI:10.1021/jm970124v.
- A. A. Gaber, A. H. Bayoumi, A. M. El-Morsy, F. F. Sherbiny, A. B. M. Mehany and I. H. Eissa, Design, synthesis and anticancer evaluation of 1H-pyrazolo [3, 4-d] pyrimidine derivatives as potent EGFRWT and EGFRT790M inhibitors and apoptosis inducers, Bioorg. Chem., 2018, 80, 375–395, DOI:10.1016/j.bioorg.2018.06.017.
- V. K. Sharma, P. P. Nandekar, A. Sangamwar, H. P. Sánchez and S. M. Agarwal, Structure guided design and binding analysis of EGFR inhibiting analogues of erlotinib and AEE788 using ensemble docking, molecular dynamics and MM-GBSA, RSC Adv., 2016, 6, 65725–65735, 10.1039/C6RA08517B.
- P. Traxler and P. Furet, Strategies toward the design of novel and selective protein tyrosine kinase inhibitors, Pharmacol. Ther., 1999, 82, 195–206, DOI:10.1016/S0163-7258(98)00044-8.
- S. R. Atta-Allah, A. M. AboulMagd and P. S. Farag, Design, microwave assisted synthesis, and molecular modeling study of some new 1,3,4-thiadiazole derivatives as potent anticancer agents and potential VEGFR-2 inhibitors, Bioorg. Chem., 2021, 112, 104923, DOI:10.1016/j.bioorg.2021.104923.
- P. S. Farag, M. M. Hemdan and A. A. El-Sayed, Nano nickel [1,2,4]-triazole-3-thiones complex: design, sonochemical synthesis, and antimicrobial evaluation, J. Heterocycl. Chem., 2020, 57, 3428–3441, DOI:10.1002/jhet.4063.
- M. H. Hekal, P. S. Farag, M. M. Hemdan and W. M. El-Sayed, New N-(1,3,4-thiadiazol-2-yl)furan-2-carboxamide derivatives as potential inhibitors of the VEGFR-2, Bioorg. Chem., 2021, 115, 105176, DOI:10.1016/j.bioorg.2021.105176.
- P. S. Farag, M. M. Hemdan and A. I. Hassaballah, The Reformatsky analogous reaction for the synthesis of novel β-thioxoesters via using aroyl isothiocyanates under solvent-free ball milling and conventional conditions, RSC Adv., 2022, 12, 10204, 10.1039/D2RA01154A.
- P. S. Farag, A. M. AboulMagd, M. M. Hemdan and A. I. Hassaballah, Annulated pyrazole derivatives as a novel class of urokinase (uPA) inhibitors: green synthesis, anticancer activity, DNA-damage evaluation, and molecular modelling study, Bioorg. Chem., 2023, 130, 106231, DOI:10.1016/j.bioorg.2022.106231.
- T. Shaikh, A. Gosar and H. Sayyed, Nitrosamine Impurities in Drug Substances and Drug Products, J. Adv. Pharm. Pract., 2020, 2, 48–57, DOI:10.5281/zenodo.3629095.
- H. Akkaraju, R. Tatia, S. S. Mane, A. B. Khade and S. J. Dengale, A comprehensive review of sources of nitrosamine contamination of pharmaceutical substances and products, Regul. Toxicol. Pharmacol., 2023, 139, 105355, DOI:10.1016/j.yrtph.2023.105355.
- S. Pani, S. Mohapatra, A. Sahoo, B. Baral and P. R. Debata, Shifting of cell cycle arrest from the S-phase to G2/M phase and downregulation of EGFR expression by phytochemical combinations in HeLa cervical cancer cells, J. Biochem. Mol. Toxicol., 2022, 36, e22947, DOI:10.1002/jbt.22947.
- D. E. Pires, T. L. Blundell and D. B. Ascher, pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures, J. Med. Chem., 2015, 58(9), 4066–4072 CrossRef CAS PubMed.
- F. Neese, Software update: the ORCA program system, version 4.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 8, e1327, DOI:10.1002/wcms.1327.
- J. Antony and S. Grimme, Density functional theory including dispersion corrections for intermolecular interactions in a large benchmark set of biologically relevant molecules, Phys. Chem. Chem. Phys., 2006, 8, 5287–5293, 10.1039/B612585A.
- V. Barone and M. Cossi, Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model, J. Phys. Chem., 1998, 102, 1995–2001, DOI:10.1021/jp9716997.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model, J. Comput. Chem., 2003, 24, 669–681, DOI:10.1002/jcc.10189.
- MOE, Chemical Computing Group Inc., Montreal, https://www.chemcomp/com Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra05401b |
| This journal is © The Royal Society of Chemistry 2024 |