
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Shaping cycles with light: a regiodivergent approach to tetracyclic aza-aromatic compounds†
Clara
Mañas
 ab,
Belén
Ibarra
ab and
Estíbaliz
Merino
*ab
ab,
Belén
Ibarra
ab and
Estíbaliz
Merino
*ab
aUniversidad de Alcalá, Departamento de Química Orgánica y Química Inorgánica, Instituto de Investigación Andrés M. del Río (IQAR), Facultad de Farmacia, Alcalá de Henares, 28805 Madrid, Spain. E-mail: estibaliz.merino@uah.es
bInstituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), Ctra. de Colmenar Viejo, Km. 9.100, 28034 Madrid, Spain
First published on 28th October 2024
Abstract
The development of regiodivergent methods that allow access to different structures from a single substrate through intramolecular processes is crucial for accelerating new molecule discovery, as well as making processes more sustainable and efficient in terms of waste production and economy. In this study, we report a novel regiodivergent cyclization procedure to access two distinct azapolyaromatic regioisomers from 2-alkynylazobenzenes. The key to achieving this regiodivergence lies in the presence or absence of a gold catalyst. The irradiation with visible light of 2-alkynylazobenzenes in the presence of a Ir photocatalyst affords 11H-indolo[1,2-b]indazoles, whereas under similar conditions with AuCl3, indazolo[2,3-a]quinolines are produced. Control experiments and DFT calculations suggest that both transformations operate through different reaction mechanisms: the formation of 11H-indolo[1,2-b]indazoles involves a radical mechanism, whereas the formation of indazolo[2,3-a]quinolines appears to proceed predominantly through a polar mechanism. This transformation enables the one-step conversion of simple 2-alkynylazobenzenes into diverse azapolyaromatic structures via an intramolecular visible light-promoted process, holding significant potential for new nitrogenated heterocycles.
Introduction
Regiodivergent reactions represent a significant advancement in organic chemistry, offering the ability to produce different regioisomeric products from a single substrate under varying conditions. This versatility not only enhances the efficiency of synthetic routes by minimizing the need for multiple starting materials but also contributes to the sustainability of chemical processes by reducing waste and energy consumption. The ability to selectively access different structural isomers expands the chemical space available for new molecule discovery, facilitating the development of novel compounds with potential applications in pharmaceuticals, materials science, and agrochemicals. Thus, the development and understanding of regiodivergent reactions are crucial for advancing both the practical and theoretical aspects of modern organic synthesis.Fused polycyclic (hetero)aromatic scaffolds are an essential class of compounds in organic chemistry characterized by their complex and rigid structures. These scaffolds are highly valued for their unique electronic properties serving as core structures in a wide range of functional materials, including organic semiconductors,1 dyes,2 and light-emitting diodes3 and in a wide range of biologically active compounds,4 so that the exploration and synthesis of these scaffolds continue to drive innovation in both materials science and drug discovery, highlighting their broad applicability and importance.
11H-Indolo[1,2-b]indazoles and indazolo[2,3-a]quinolines are two distinct classes of azapolyaromatic compounds with unique structural properties and potential applications. Indolo[1,2-b]indazoles are potent inhibitors of DNA topoisomerases I and II5 but have been relatively unexplored. In contrast, indazoloquinolines, with their extended conjugated system, are used in fluorescence materials, such as OLED devices,6 or as photocatalysts.7 Quinolines are prevalent scaffolds in pharmacology, found in over 600 natural products, mainly alkaloids.8 Both natural and synthetic quinolines exhibit a wide range of biological activities, including antifungal, antibacterial, antioxidant, anticancer, and antimalarial properties.9 Despite their medicinal relevance and extensive investigation,10 the fusion of quinolines with the indazole core to obtain indazolo[2,3-a]quinolines has been scarcely reported.
Indolo[1,2-b]indazoles were first synthesized serendipitously by Wilshire in 197311 and later explored for their reactivity and bioactivity.12 Various methodologies, including palladium and copper-catalyzed intramolecular C–N bond formation13 at high temperatures and Rh(III)-catalyzed dehydrogenative annulation,14 have been developed to synthesize these compounds. Additionally, the synthesis of these compounds can be achieved through a 1,4-dilithio intermediate formed by reacting 2-phenyl-2H-indazole with n-BuLi and trapping it with an acyl chloride.15 Initially, indazolo[2,3-a]quinolines were synthesized in 1980 by Zurawel and Mitsuo.16 These compounds have been explored through various methodologies, including PPh3-mediated reductive cyclizations of 2-(o-nitrophenyl)quinolines,17 aryne [3 + 2] dipolar cycloadditions,18 the Pd-catalyzed tandem cross-coupling of 2-(o-iodophenyl)-2H-indazoles with organozinc reagents19 as well as Rh(III)-catalyzed regioselective C–H functionalization of 2-aryl-2H-indazoles with alkynes,20 α-diazo carbonyl compounds,21 cyclic enones22 or β-ketosulfoxonium ylides.23 Additionally, rhodium-catalyzed cyclizations of azobenzenes24 and 2-phenyl-2H-indazoles25 with vinylene carbonate via C–H bond activation have also been reported (Fig. 1a). These efforts have expanded the scope and synthetic accessibility of indazolo[2,3-a]quinolines, allowing for the exploration of new applications for these compounds. Dihydroindazolo[2,3-a]quinolines can be synthesized via the Povarov reaction and a tandem process involving visible light-promoted intramolecular N–N bond formation using choline chloride/CuCl26 or ruthenium complex (Fig. 1a).27
 | ||
| Fig. 1 (a) Approaches to synthesize 11H-indolo[1,2-b]indazoles, indazolo[2,3-a]quinolines and 5,6-dihydroindazolo[2,3-a]quinolines. (b) Photoreactivity of azobenzenes. (c) Photoreactivity of 2-alkynylazobenzenes. (d) Visible light-mediated regiodivergent synthesis of 11H-indolo[1,2-b]indazoles, indazolo[2,3-a]quinolines and 5,6-dihydroindazolo[2,3-a]quinolines from 2-alkynylazobenzenes. | ||
Azobenzenes are ubiquitous molecules that play a central role in both fundamental and applied science. Initially investigated as dyes28 their applications have expanded significantly into fields such as medicine, textiles, food, cosmetics as well as chemical sensing, organic transistors, and cell signaling.29 Nowadays, research on azobenzenes is primarily centered on their trans–cis photoisomerization and remarkable photostability.30,31 However, despite the growing interest in their photoisomerization properties, their application as synthetic intermediates in photo-induced reactions to form complex molecules remains underexplored. Since the 1960s, few examples have shown azobenzenes irradiated with UV or visible light to transform their scaffold. Most involve difunctionalization of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond without forming nitrogen-containing heterocycles.32 A few examples involve forming complex molecules as seven-membered 1,3,2-diazaborepin heterocycles33 or carbazoles34 (Fig. 1b).
N bond without forming nitrogen-containing heterocycles.32 A few examples involve forming complex molecules as seven-membered 1,3,2-diazaborepin heterocycles33 or carbazoles34 (Fig. 1b).
These examples highlight the potential of azobenzene reactivity under visible light irradiation for synthesizing nitrogen-containing heterocycles. Recently, our group reported a study on the heterodifunctionalization of 2-alkynylazobenzenes exclusively promoted by visible light, without the use of any transition metals or photocatalysts, describing oxyamination, sulfenoamination and diamination reactions (Fig. 1c).35 This process exhibited excellent regioselectivity across a broad range of substrates and was characterized by its broad scope, simple set up, and mild reaction conditions.
We hypothesized that 2-alkynylazobenzenes could be engaged in additional bond-forming events in the presence of metal transitions and/or photocatalysts, thereby expanding the synthetic utility of these visible light-promoted processes. Consequently, we aimed to design cascade reactions that would enable the highly controlled synthesis of azapolyaromatic structures from 2-alkynylazobenzenes (Fig. 1d). First, we decided to test the reactivity of a 2-alkynylazobenzene 1a by irradiating it with visible light in the presence of a photocatalyst. Our goal was to generate a reactive species of 2-alkynylazobenzene by the photocatalyst and visible light, to induce cyclization and form a 2H-indazole intermediate. This intermediate of radical nature could then undergo an additional cyclization with a phenyl ring through a subsequent C–C bond forming event.
Second, we speculated that the presence of a metal complex to activate the alkynyl moiety could lead to the formation of a 3-alkenyl-2H-indazole intermediate. This intermediate would then evolve to form an additional C–C bond, generating a new ring of different size, as previously observed for similar compounds.36
Herein, we present the realization of these concepts through a regiodivergent strategy for synthesizing 11H-indolo[1,2-b]indazoles and indazolo[2,3-a]quinolines from 2-alkynylazobenzenes via intramolecular processes under photoredox conditions (Fig. 1d). To elucidate the nature of the productive reaction intermediates involved in these transformations, we conducted control experiments, mechanistic probes, and DFT calculations, the results of which will be presented. Our mechanistic studies suggest that different pathways operate depending on the presence or absence of AuCl3 in the reaction medium. The ability to selectively synthesize these scaffolds from a common precursor demonstrates the versatility of modern synthetic methods. This provides valuable tools for synthesizing complex scaffolds from simple starting materials and expanding the catalogue of sustainable methodologies for organic synthesis that utilize visible light to promote transformations.
Results and discussion
Initially, (E)-1-(2-(hex-1-yn-1-yl)phenyl)-2-phenyldiazene (1a) was selected as a benchmark substrate to explore its photoreactivity. We began our investigation by studying the reactivity of this substrate using 5 mol% of AuCl3 and 2 mol% of Ir[dF(CF3)ppy]2(dtbpy)PF6 in MeCN at room temperature, irradiated with a blue LED (40 W) (Scheme 1). | ||
| Scheme 1 Initial experiment. | ||
Under these conditions, a mixture of four cyclization products was obtained: alongside indolo[1,2-b]indazole 2a, 5,6-dihydroindazolo[2,3-a]quinoline 3a, indazolo[2,3-a]quinoline 4a and 3-alkenyl-2H-indazole 5a were formed. 2a, 3a, and 4a result from the formation of a C–N bond and a C–C bond, generating two five-membered rings for 2a and one five-membered ring and one six-membered ring for compounds 3a and 4a in a single reaction step. Considering this result, we decided to optimize the synthesis of compounds 2a, 3a, and 4a to selectively obtain different azapolycycles from 2-alkynylazobenzenes.
Firstly, our focus shifted to optimizing the formation of compound 2a. A control experiment conducted without the gold(III) catalyst resulted in 2a being the major product, which was isolated by column chromatography with a 34% yield (Table 1, entry 1). Next, we investigated the effect of the photocatalyst by testing various organometallic and organic photocatalysts (Fig. 2). Among the Ir(III) organometallic photocatalysts, Ir(dFppy)3 offered a yield of 47% (Table 1, entry 2), while Ir(ppy)3 delivered a significantly improved yield of 62% (Table 1, entry 3). In contrast, photocatalysts derived from Cu(I) and Ru(II) resulted in lower yields (Table 1, entries 4 and 5). Organic photocatalysts from the xanthene family (Table S1†) as well as acridinium and 4CzIPN photocatalysts did not yield better results (Table 1, entries 6 and 7). Based on these findings, Ir(ppy)3 was selected for further optimization studies. Regarding the solvents, acetone provided a better outcome than MeCN, yielding a 72% yield (Table 1, entry 8). Using MeOH as a solvent favoured the formation of 3-(1-methoxypentyl)-2-phenyl-2H-indazole (A) with no indolo[1,2-b]indazole 2a detected (Table 1, entry 9).25 DCE emerged as the optimal solvent, achieving an 85% yield (Table 1, entry 10). Finally, the reaction was performed under irradiation with 50 or 100 W (Table 1, entries 11 and 12). No significant changes in the reaction were observed at 50 W and increasing the intensity to 100 W did not improve the yield or affect the reaction time.
 | ||
| Fig. 2 Some of the photocatalysts tested in this work. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Blue LED (W) | Photocatalyst | Solvent | Yielda (%) |
| a Isolated yields after column chromatography on silica gel. b A was isolated with 90% yield. | ||||
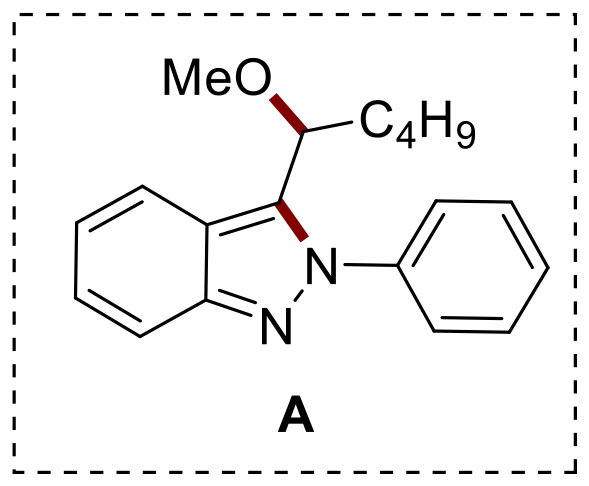
|
||||
| 1 | 40 | PC1 | MeCN | 34 |
| 2 | 40 | PC2 | MeCN | 47 |
| 3 | 40 | PC3 | MeCN | 62 |
| 4 | 40 | PC4 | MeCN | 33 |
| 5 | 40 | PC5 | MeCN | 15 |
| 6 | 40 | PC6 | MeCN | 34 |
| 7 | 40 | PC7 | MeCN | 24 |
| 8 | 40 | PC3 | Acetone | 72 |
| 9 | 40 | PC3 | MeOH | 0b |
| 10 | 40 | PC3 | DCE | 85 |
| 11 | 50 | PC3 | DCE | 85 |
| 12 | 100 | PC3 | DCE | 86 |

With the optimized reaction conditions in hand, a scale-up experiment was conducted, increasing the scale tenfold (1 mmol) with compound 1a. The corresponding product 2a was isolated with a similar yield after purification by column chromatography (Scheme 2).
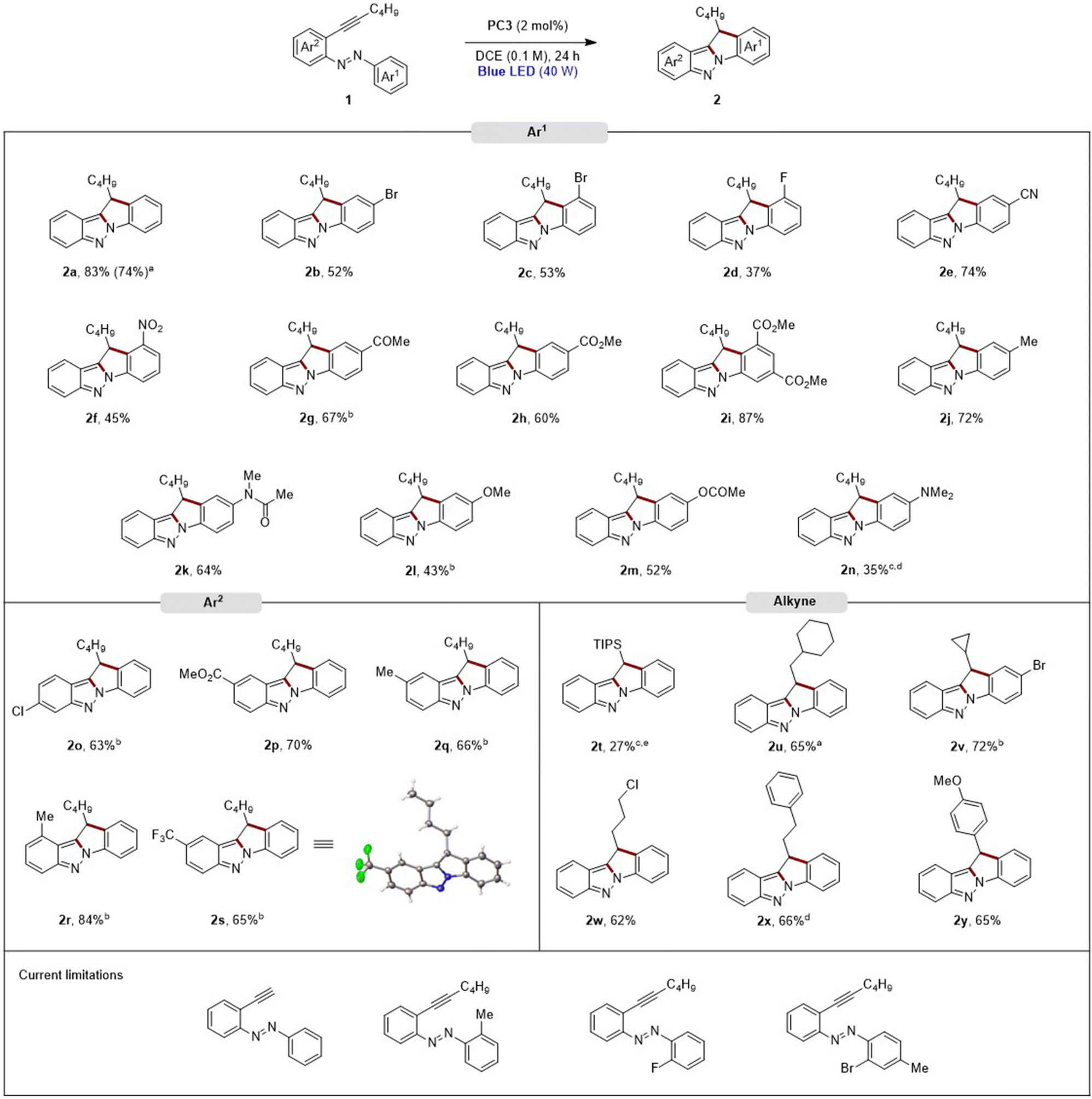 | ||
Scheme 2 Scope for the reaction from 2-alkynylazobenzenes 1 to indolo[1,2-b]indazoles 2. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction was carried out at 1 mmol scale. bReaction time: 48 h. cYield based on recovered starting material. dReaction time: 72 h. eReaction time: 7 days. Reaction was carried out at 1 mmol scale. bReaction time: 48 h. cYield based on recovered starting material. dReaction time: 72 h. eReaction time: 7 days. | ||
To demonstrate the versatility and robustness of our method for synthesizing indolo[1,2-b]indazoles 2via C–N and C–C bond formation from 2-alkynylazobenzenes 1, we set out to explore the substrate scope. A series of 2-alkynylazobenzenes 1, with different modifications on both aromatic rings and the alkynyl chain, were subjected to the optimized reaction conditions. The method achieved moderate to very good yields across a broad range of functional groups, with substitutions on both the para and the meta positions of Ar1 being well tolerated (Scheme 2). Different halogen atoms were introduced at these positions, resulting in moderate yields (2b–2d). For 2c and 2d, the formation of the C–C bond exclusively occurred at the position of the aromatic ring with the azo group and halogen in the ortho position. Nitrogenated electron-withdrawing groups were successfully incorporated: a cyano group in the para position yielded 2e with a 74%, and a nitro group in the meta position gave 2f with 45% yield. Carbonylated electron-withdrawing groups such as ketones and esters (2g and 2h), were introduced in the para position with good yields. A disubstituted substrate with ester groups delivered 2i with a very good yield. Electron-donating groups were also evaluated: a methyl group at the para position produced 2j successfully while an amide group led to a 64% yield of 2k. Strong and weak oxygenated electron-donating groups gave moderate yields for 2l and 2m, with lower yields generally due to the formation of byproducts 3a and 4a. Nitrogenated electron-donating groups at the para position were also tested, but the reaction with a dimethylamino group did not achieve complete conversion after 7 days, yielding 2n with a 35% yield. Further testing on the aromatic ring Ar2 and the alkynyl chain showed good results. A chlorine atom at position 8 yielded 2o with a 63% and an ester group at position 9 yielded 2p with a 70% yield. Methyl groups at positions 9 and 10 resulted in good and very good yields, respectively (2q and 2r), although with longer reaction times (48 hours). A trifluoromethyl group at position 9 gave 2s with a 65% yield and the structure was confirmed by X-ray diffraction.37 With a bulky protecting group as triisopropylsilyl group on the triple bond, the reaction achieved 70% conversion after 7 days isolating 2t with 27% yield. Deprotection of the triisopropylsilyl ether group with TBAF and subsequent cyclization under standard conditions led to decomposition, likely due to the low stability of (E)-1-(2-ethynylphenyl)-2-phenyldiazene and the extended reaction time. Introducing cycloalkyl groups, a cyclohexyl group (2u) yielded 65%, while a cyclopropyl group (2v) gave a 72% yield. A chlorine atom in the alkynyl chain was compatible, providing 2w with a 62% yield. Incorporating a phenyl group into the alkynyl chain or a p-methoxyphenyl group directly linked at the triple bond gave the corresponding products 2x and 2y, respectively, in similar good yields. With substrates containing substitutions in the ortho position of Ar1 with fluorine, bromo or methyl groups, the corresponding indolo[1,2-b]indazoles 2 were not obtained, instead starting material, decomposition and dehalogenation products were recovered.
To investigate the mechanism of this reaction, experiments were designed to produce 2a by deviating from standard conditions. When the reaction was carried out without a catalyst, a 33% yield of the product was observed. The role of light during the process was also evaluated. In the absence of light, the formation of 2a was not observed, indicating its crucial role in this transformation. The reaction proceeds exclusively under light irradiation (Fig. 3a). Next, the reaction was conducted in the presence of a radical quencher. Using 2,6-di-tert-butyl-4-methylphenol (BHT), the resulting radical intermediate formed after the C–N bond formation was trapped, and compound 6 was isolated with a 39% yield. This result indicates the likely involvement of radical species along the reaction pathway. When the reaction was carried out with (E)-1-(hex-1-yn-1-yl)-2-styrylbenzene, after 36 hours of irradiation, only isomerization of the double bond was observed, yielding a 1:3.5 E:Z ratio. This result suggests that the excitation of the azo group by the photocatalyst triggers the reaction (Fig. 3b).
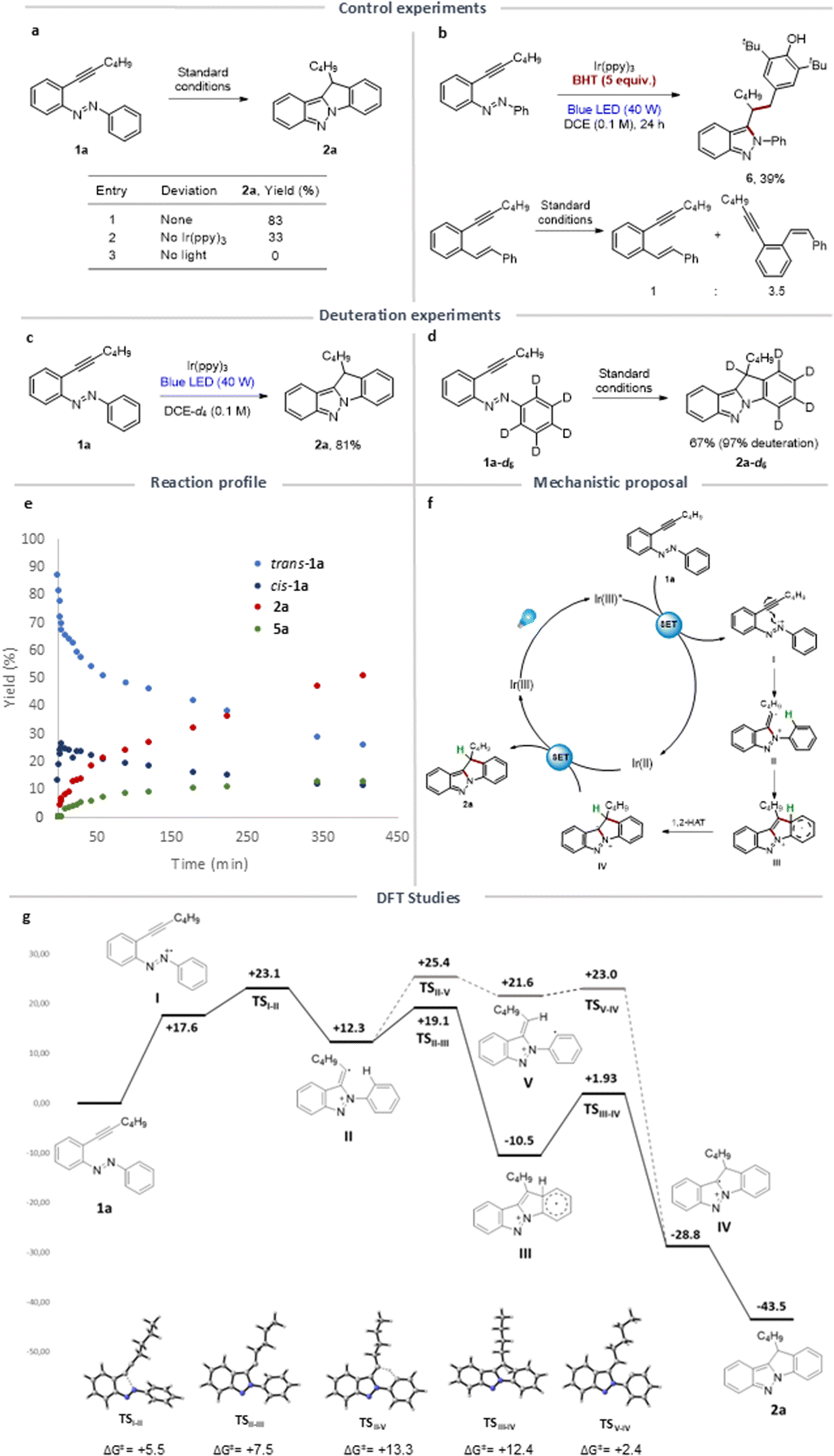 | ||
| Fig. 3 (a) and (b) Control experiments. (c) and (d) Deuteration experiments. (e) Kinetic study. (f) Mechanistic proposal. (g) Computed reaction coordinate profile of the reaction to synthesize indolo[1,2-b]indazole 2a from (E)-1-(2-(hex-1-yn-1-yl)phenyl)-2-phenyldiazene 1a. All calculations were conducted at B3LYP/6-31g+(d,p)/IEFPCM (solvent = DCE) level of theory (see ESI† for full details). The energy values are given in kcal mol−1. | ||
Next, two deuteration experiments were conducted. In the first, the reaction was performed using deuterated dichloroethane to determine whether deuterium incorporation into the product occurred via hydrogen transfer from the solvent to the substrate (Fig. 3c). In this case, product 2a was isolated with an 81% yield, and no deuterium incorporation was observed. Subsequently, (E)-1-[2-(hex-1-yn-1-yl)phenyl]-2-(phenyl-d5)-diazene (1a-d5) was synthesized, and under standard reaction conditions, the corresponding deuterated compound 2a-d5 was formed with a 67% yield and 97% deuteration (Fig. 3d). This suggests that during the reaction, a hydrogen atom from the aromatic ring Ar1 migrates to the five-membered ring.
Analysis of the reaction progress by 1H NMR revealed that (Z)-1a is generated during the reaction. The reaction profile, monitored by nuclear magnetic resonance spectroscopy, indicates that a photo-stationary equilibrium of the cis–trans isomers of 1a is reached at 10 minutes. The concentration of both isomers decreases during the reaction, while the product yield increases. After 7 hours, a 10% formation of indazole 5a was observed (Fig. 3e).
To gain deeper insight into the mechanism operating in this transformation, DFT calculations were conducted (Fig. 3g). The initial step, the formation of the 2-phenyl-2H-indazole intermediate II from the radical cation I generated by activating 1a with the Ir catalyst, is both kinetically and thermodynamically favorable (ΔG‡ = +5.5 kcal mol−1 and ΔG = −5.3 kcal mol−1). Subsequently, two distinct mechanistic pathways can be proposed. The first pathway involves cyclization to form the C–C bond, leading to intermediate III, followed by a 1,2-intramolecular hydrogen migration to furnish intermediate IV. The second pathway considers the 1,5-intramolecular hydrogen migration occurs before C–C bond formation. The formation of the C–C bond is favored by 6.3 kcal mol−1 over the 1,5-intramolecular hydrogen migration, making the first pathway more likely. The formation of intermediate III from IIvia C–C bond formation has a kinetic barrier of ΔG‡ = +6.8 kcal mol−1 and is highly thermodynamically favored (ΔG = −22.8 kcal mol−1). The subsequent 1,2-intramolecular hydrogen migration is the rate-determining step, with a kinetic barrier of ΔG‡ = +12.4 kcal mol−1.
Following the experimental and computational investigations above, we propose a plausible mechanism (Fig. 3f). The reaction likely begins with the activation of the Ir catalyst by visible light absorption, forming an excited Ir species. This is followed by a single-electron transfer (SET) process, where 2-alkynylazobenzene 1a transfers an electron to the Ir catalyst, generating an Ir(II) species and a radical cation I. The C–N bond is then formed by the attack of a nitrogen atom on a carbon of the alkyne group, producing the radical cation II of the indazole intermediate. Next, the C–C bond formation occurs to generate intermediate III, followed by a 1,2-hydrogen atom transfer. Finally, a second SET process closes the catalytic cycle, forming compound 2a.
The next goal was to optimize the reaction conditions for obtaining indazolo[2,3-a]quinolines 4. The benchmark substrate for optimization was again 1a. An experiment was conducted using 2-alkynylazobenzene 1a (0.2 mmol), with AuCl3 (5 mol%) and Ir[dF(CF3)ppy]2(dtbpy)PF6 (2 mol%) in MeCN (0.1 M), irradiated with blue light (40 W) for 24 hours (Table 2, entry 1). In this case, 2H-indazole 5a was the main product with a 45% yield, while 5,6-dihydroindazolo[2,3-a]quinoline 3a and indazolo[2,3-a]quinoline 4a were minor products, yielding 20% and a 5%, respectively, as determined by 1H NMR. Increasing the blue LED potency to 80 W (Table 2, entry 2) reduced the yield of 2H-indazole 5a to 27% yield, with a corresponding increase in the yields of 3a and 4a to 29% and 10%, respectively. When the irradiation was further increased to 100 W, the temperature rose to 70 °C (Table 2, entry 3). Under these conditions, no 2H-indazole 5a was observed. Instead, a 50% yield of 5,6-dihydroindazolo[2,3-a]quinoline 3a and a 30% yield of indazolo[2,3-a]quinoline 4a were detected by 1H NMR. Next, different gold(III) catalysts were tested. AuBr3 showed promising results, furnishing 49% yield of 3a and 24% yield of 4a (Table 2, entry 4). In contrast, Au(OAc)3 was less effective, providing only a 29% yield of 3a with negligible amounts of 4a (Table 2, entry 5). A copper(I) catalyst, Cu(MeCN)4BF4 was also evaluated (Table 2, entry 6). Despite achieving a 17% yield of 4a, the overall results did not show significant improvement. After testing several catalysts, no substantial improvements were observed.38 Copper (PC4) and ruthenium (PC8) photocatalysts predominantly produced 5a (Table 2, entries 7 and 8). In contrast, using 2DPAFIPN (PC9) shifted the major product to 3a (Table 2, entry 9). Other solvents were tested, DCE yielded only 30% yield of 3a and traces of 4a, whereas acetone produced a mixture of 3a and 4a with yields of 15% and traces, respectively (Table 2, entries 10 and 11). Notably, no presence of 5a was detected in both experiments.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Blue LED (W) | Catalyst | PC | Solvent | Oxidant (2 equiv.) | 3a (%) | 4a (%) | 5a (%) |
| a Yields calculated by 1H NMR, using dibromomethane as internal standard. b Reaction was performed in two steps: (i) AuCl3 (5 mol%), Ir[dF(CF3)ppy2](dtbpy)PF6 (2 mol%), MeCN (0.1 M), Blue LED (100 W), 70 °C, 24 h. (ii) DDQ (1.2 equiv.), 1,4-dioxane (0.1 M), Blue LED (80 W), 50 °C, 24 h. c Isolated yield (3a + 4a) by column chromatography. | ||||||||
| 1 | 40 | AuCl3 | PC1 | MeCN | — | 20 | 5 | 45 |
| 2 | 80 | AuCl3 | PC1 | MeCN | — | 29 | 10 | 27 |
| 3 | 100 | AuCl3 | PC1 | MeCN | — | 50 | 30 | — |
| 4 | 100 | AuBr3 | PC1 | MeCN | — | 49 | 24 | — |
| 5 | 100 | Au(OAc)3 | PC1 | MeCN | — | 29 | Traces | — |
| 6 | 100 | Cu(MeCN)4BF4 | PC1 | MeCN | — | 38 | 17 | — |
| 7 | 100 | AuCl3 | PC4 | MeCN | — | — | — | 35 |
| 8 | 100 | AuCl3 | PC8 | MeCN | — | 25 | Traces | 48 |
| 9 | 100 | AuCl3 | PC9 | MeCN | — | 38 | 5 | — |
| 10 | 100 | AuCl3 | PC1 | DCE | — | 30 | Traces | — |
| 11 | 100 | AuCl3 | PC1 | Acetone | — | 15 | Traces | — |
| 12 | 100 | AuCl3 | PC1 | MeCN | PhI(OAc)2 | 0 | 18 | — |
| 13 | 100 | AuCl3 | PC1 | MeCN | DDQ | 0 | 32 | — |
| 14b,c | 100 | AuCl3 | PC1 | MeCN | DDQ | 7 | 46 | — |
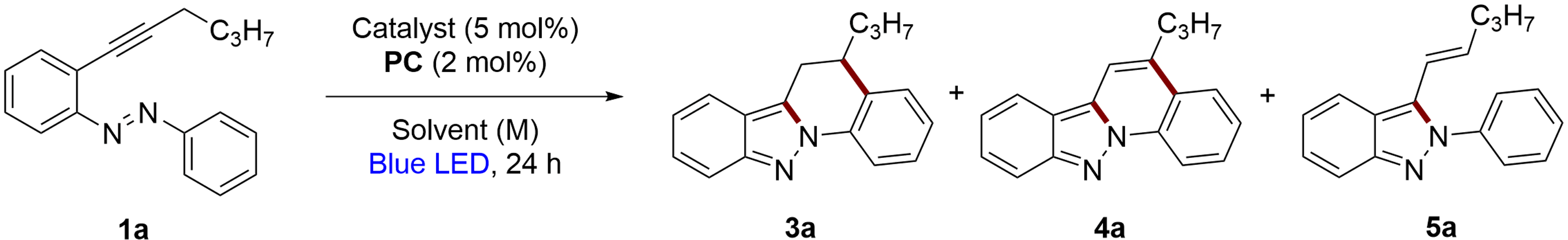
Recognizing that both 3-alkenyl-2H-indazole 5a and 5,6-dihydroindazolo[2,3-a]quinoline 3a are likely intermediates, further experiments with extended reaction times or enhanced blue LED power were conducted. These conditions achieved complete conversion, with longer reaction times favouring the formation of indazolo[2,3-a]quinoline 4a. However, increased reaction times also led to higher decomposition of the compounds. To improve the yield of 4a, the addition of an oxidant was tested to oxidize 5,6-dihydroindazolo[2,3-a]quinoline 3a into the corresponding indazolo[2,3-a]quinoline 4a. (Diacetoxyiodo)benzene initially yielded exclusively 4a but with low yield (Table 2, entry 12). Other oxidants as NBS, IBX, cyclooctene and MnO2 were tested but no improvement in the yields of 4a were obtained (Tables S3 and S4†). Given DDQ's effectiveness as an oxidant, we tried adding it after 24 hours of irradiation and filtering the mixture over silica. Although this resulted in complete oxidation to 4a, the yield was only 32% yield and the formation of decomposition byproducts (Table 2, entry 13). To minimize decomposition and promote oxidation, we added only 1.2 equivalents of DDQ, used 1,4-dioxane as the solvent for the second reaction step, and reduced the blue LED irradiation to 80 W. Under these conditions, 4a along with 3a were isolated by column chromatography with a yield of 53% (Table 2, entry 14).
Instances of photocatalyzed dehydrogenations have been documented through distinct mechanisms, including the generation of hydrogen gas, which increases the pressure in the reaction vessel.39 Our reaction conducted in a 5 mL pressure Schlenk, led to the hypothesis that the liberated H2 increased the pressure, potentially slowing the reaction rate. To address this, we modified the set up. Initially, the reaction was carried out in a round-bottom flask with a balloon punctured through the septum, resulting in significant hydrazine formation, indicating decomposition of the starting material. Conducting the reaction in a Schlenk tube improved the outcome, yielding a 3:10 ratio of 5,6-dihydroindazolo[2,3-a]quinoline 3a to indazolo[2,3-a]quinoline 4a, but trace hydrazine still indicated ongoing decomposition. We then used a pressure Schlenk for the first 24 hours, ensuring full conversion of the starting material to 3a and 4a, followed by transferring the mixture under argon to a Schlenk tube for another 24 hours of irradiation. This afforded only indazolo[2,3-a]quinoline 4a, but with a low 1H NMR yield of 27%, suggesting further decomposition.38 Finally, performing the entire reaction in a pressure Schlenk with periodic pressure releases resulted in a 5:4 ratio of 3a and 4a, indicating incomplete conversion. After optimizing the reaction conditions to obtain indazolo[2,3-a]quinoline 4a, the scope of the reaction was explored (Scheme 3). A variety of 2-alkynylazobenzenes 1, with modifications on the aromatic rings and alkynyl chain, were evaluated. The corresponding indazolo[2,3-a]quinolines 4 were obtained in synthetically useful yields. Products without substitution (3a/4a) or with halogens in Ar1, such as bromine in the para position (3b/4b) or fluorine in the ortho position (3e/4e), were isolated with yields ranging from 30–51%. The structure of 4b was confirmed by X-Ray diffraction.37 When a fluorine atom was present in the meta position, a mixture of the 2- and 3-substituted products (3d/4d and 3d′/4d′) was obtained with yields of 18 and 29%, respectively. Electron-donating groups were also assessed. A methyl group or N-methylacetamide gave the corresponding products in moderate yields (3k/4k and 3l/4l). Modifications on the aromatic ring Ar2 were also evaluated. Compounds with a chlorine atom in position 9 and a methyl group in position 7 were isolated with yields of 34 and 55%, respectively (3p/4p and 3s/4s). Modifications to the alkynyl chain were also explored. 2-Alkynylazobenzene 1x, with a chlorine atom at the end of the alkyl chain, delivered 3x/4x with a 42% yield.40 The introduction of an extra phenyl group directly conjugated with the indazolo[2,3-a]quinoline core enabled the exclusive isolation of the desired compound 4aa with a 41% yield. Due to this result, we investigated the scope using a benzyl substituent on the alkynyl moiety. With a fluorine substituent on Ar1 in the ortho position relative to the azo group, the tetracycle 4ab could be isolated in pure form, albeit with a low yield. The substrate 1ac, with the fluorine atom in the meta position relative to the azo group, under standard reaction conditions, led to a mixture of compounds 4ac and 4ac′ with 24% and 23% yields, respectively. Substrates with acetyl or methyl substituents on Ar1, or methyl groups in positions 7 and 8 on Ar2, resulted in moderate yields of 4ad–4ai. Given the exclusive selectivity for forming 4 with a benzyl substituent on the alkynyl fragment, we decided to study the variability of substituents on this aromatic ring. Substrates with electron-withdrawing groups like bromine and electron-donating groups like methyl and methoxy in different positions allowed the exclusive isolation of compounds 4aj–4an with yields between 15% and 26%. Selective isolation of compound 3j with a yield of 51% allowed confirmation of its structure by X-ray diffraction.37 This example, along with substrate 1ae, demonstrates that selective formation of 3 can be achieved by modifying the alkynyl fragment when the substrate presents two CO2Me groups on Ar1. Given these results, the synthesis of starting substrates with other electron-withdrawing groups in the meta position, such as nitro and bromo substituents was proposed to selectively obtain 5,6-dihydroindazolo[2,3-a]quinolines 3. However, the synthesis of these substrates was not successful. Surprisingly, when the aromatic ring Ar1 has a dimethylamino group, high yields of compounds 3m and 3ag were obtained regardless of whether the alkynyl fragment had an alkyl or aromatic group. The results from this scope study suggest that the reaction selectivity is strongly influenced by the electronic nature of the substituents.
 | ||
| Scheme 3 Scope for the reaction from 2-alkynylazobenzenes 1 to 5,6-dihydroindazolo[2,3-a]quinolines 3 and indazolo[2,3-a]quinolines 4. aConditions: AuCl3 (5 mol%), PC1 (2 mol%), MeCN (0.1 M), Blue LED (30 W), 25 °C, 24 h. bConditions: AuCl3 (5 mol%), PC1 (2 mol%), MeCN (0.1 M), Blue LED (100 W), 70 °C, 24 h. | ||
To gain a deeper understanding of the reaction, experiments were designed to synthesize 4a by deviating from standard conditions (Fig. 4a). When the reaction was performed without AuCl3, the yield was 18% for 3a and traces of 4a, with 34% of the mixture identified as indolo[1,2-b]indazole 2a. This outcome highlighted the lack of selectivity in the absence of gold(III). In a second experiment without the photocatalyst, the yield was 34% (3a + 4a), with no other products detected. These results suggest that the yield decreases primarily during the formation of the 3-alkenyl-2H-indazole 5, rather than during the subsequent C–C bond formation to generate the 5,6-dihydroindazolo[2,3-a]quinoline 3a. When the reaction was carried out in the dark conditions at room temperature, no conversion to the desired product was observed. Similarly, a reaction conducted in the dark at 90 °C yielded no traces of 3a or 4a but resulted in a 68% yield of 3-alkenyl-2H-indazole 5a. Next, a control experiment with a radical scavenger was performed by adding 5 equivalents of BHT to the standard reaction conditions for both steps. This resulted in a mixture of 3a and 4a in a 2.3:1 ratio with a 42% yield. A similar result was obtained when 1,3-pentadiene, a known triplet quencher, was used.41 Notably, no traces of 1a or 5a were detected, confirming the involvement of a polar mechanism for the first part of the reaction. However, the ratio of 3a to 4a suggested that the oxidation from 3a to 4a might involve a radical pathway.
 | ||
| Fig. 4 (a) Control experiments. (b) Reactions with 5a. (c) Deuteration experiment. | ||
To prove that 5a is an intermediate in the reaction pathway, 3-alkenyl-2H-indazole 5a (0.02 mmol), AuCl3 (5 mol%) and Ir[dF(CF3)ppy]2(dtbpy)PF6 (2 mol%) were dissolved in MeCN and irradiated with a blue LED (100 W) for 24 hours. This produced a 52% yield of N-heterocycles 3a and 4a. In absence of PC1, the yield and the 3a:4a ratio were similar to the previous experiment. Another reaction, without AuCl3 and PC1, produced 3a and 4a with a 55% yield and a 3:1 ratio. These results support the assumption that both Au(III) and Ir(III) catalysts promote the formation of 5a and that 5a is likely an intermediate in the reaction (Fig. 4b). They also indicate that the intramolecular cyclization to form the six-membered ring is exclusively promoted by visible light.
To determine whether the hydrogen at position 6 in 4a originated from the aromatic ring of 3a, (E)-1-[2-(hex-1-yn-1-yl)phenyl]-2-(phenyl-d5)-diazene (3a-d5) was subjected to the standard reaction conditions. After purification by column chromatography and analysis of the 1H NMR spectra and HRMS, a mixture of four products with the same polarity was observed: 5,6-dihydroindazolo[2,3-a]quinolines 3a-d4 and 3a-d5, and indazolo[2,3-a]quinolines 4a-d4 and 4a-d5 (Fig. 4c). This result suggests the possibility of simultaneous intramolecular and intermolecular hydrogen migrations from the aromatic ring to position 6 of 3a. Due to the overlapping signals in the 1H NMR and the inability to separate the products by column chromatography, the ratio of the products could not be determined. However, HPLC analysis provided approximate relative concentrations of each compound, with 4a-d4 being the major product and 3a-d4 and 3a-d5 as the minor ones, each with almost the same relative ratio.38
Based on the control experiments, it seems that two mechanisms are operating in the formation of indazolo[2,3-a]quinolines 4. DFT calculations were conducted to further investigate the proposed mechanism. A plausible mechanistic hypothesis has been proposed to rationalize the dual gold photoredox catalyzed transformation of 2-alkynylazobenzenes 1 into indazolo[2,3-a]quinolines 4. This hypothesis suggests an initial excitation of 1a generating the first singlet which by coordination of AuCl3 to the alkynyl moiety, generating intermediate I. Next, the formation of a C–N bond (TS not found) generates the indazole intermediate II. Next, a 1,2-hydride shift occurs, generating intermediate III, with a transition state of ΔG‡ = +20 kcal·mol−1.42 The elimination of AuCl3 produces 3-alkenyl-2H-indazole 5a, which can cyclize to 3a in the presence of light. Finally, DDQ facilitates the oxidation of 3a, yielding the desired indazolo[2,3-a]quinoline 4a. A secondary mechanism involving the Ir(III) catalyst may start with the coordination of AuCl3 to the alkynyl moiety, followed by a single-electron transfer (SET) from the Ir(III) catalyst to intermediate IV. This forms radical anion V and oxidizes Ir(III) to Ir(IV). Radical anion V can then react with the triple bond, forming the C–N bond and giving rise to radical intermediate VI through a low-barrier transition state (ΔG‡ = +3.2 kcal mol−1). A 1,2-hydride shift with a transition state energy of +12.6 kcal mol−1 delivers intermediate VII. This intermediate undergoes a SET process with the Ir(IV) catalyst, oxidizing to intermediate III and reducing the iridium catalyst, thus closing the photocatalytic cycle. Intermediate III can then evolve to 5a, closing the gold catalytic cycle (Fig. 5).
 | ||
| Fig. 5 Mechanistic proposal and DFT calculations. All calculations were conducted at M06/6-311g(d,p)/IEFPCM (solvent = MeCN)/Au (SDD) level of theory (see ESI† for full details). The energy values are given in kcal mol−1. | ||
Conclusions
In conclusion, this study demonstrates the efficacy of a novel regiodivergent cyclization procedure for transforming 2-alkynylazobenzenes into two distinct azapolyaromatic regioisomers. By manipulating the presence or absence of a gold catalyst and utilizing visible light irradiation, we achieved selective synthesis of 11H-indolo[1,2-b]indazoles and indazolo[2,3-a]quinolines. The mechanistic insights provided by control experiments and DFT calculations reveal that these transformations proceed through different pathways, with radical and polar mechanisms being predominant. This methodology not only enhances the structural diversity of azapolyaromatic compounds but also offers a more sustainable and efficient approach for the discovery and synthesis of new molecules.Author contributions
Conceptualization: E. M.; methodology: C. M. and E. M.; funding acquisition: E. M.; investigation: C. M., B. I. and E. M.; supervision: E. M.; writing-original draft: E. M.; writing: review & editing: C. M. and E. M.Data availability
Data for this article, including procedures, NMR spectra description, NMR spectra and DFT results are available at the ESI.†Conflicts of interest
There are no conflicts to declare.Note added after first publication
This article replaces the version published on the 28th of October 2024. The structure of Compound A was omitted below Table 1, and the reaction scheme shown at the top of Table 2 was included instead in error. The RSC apologises for any confusion.Acknowledgements
Financial support was provided by Comunidad de Madrid Research Talent Attraction Program (grant ref. 2018-T1/IND-10054 and 2022-5A/IND-24227), the Spanish Ministry of Science and Innovation (MICINN) (grant ref. CNS2022-135304 and TED2021-129634B-I00) and Universidad de Alcalá (grant ref. CCG20/CC-009 and IUAH21/CC-003). Computational resources for the DFT calculations were provided by the HERMES Cluster at the University of Zaragoza. We also gratefully acknowledge the computing time granted by the Spanish Supercomputing Network (grant ref. QH-2023-2-0005) and provided on CESGA (Galicia Supercomputing Center).References
- (a) W. Jiang, Y. Li and Z. Wang, Heteroarenes as high-performance organic semiconductors, Chem. Soc. Rev., 2013, 42, 6113–6127, 10.1039/c3cs60108k; (b) Z. Chen, W. Li, M. A. Sabuj, Y. Li, W. Zhu, M. Zeng, C. S. Sarap, M. M. Huda, X. Qiao, X. Peng, D. Ma, Y. Ma, N. Rai and F. Huang, Evolution of the electronic structure in open-shell donor-acceptor organic semiconductors, Nat. Commun., 2021, 12, 5889, DOI:10.1038/s41467-021-26173-3.
- (a) N. Benson, O. Suleiman, S. O. Odoh and Z. R. Woydziak, Pyrazole, imidazole, and isoindolone dipyrrinone analogues: pH-dependent fluorophores that red-shift emission frequencies in a basic solution, J. Org. Chem., 2019, 84, 11856–11862, DOI:10.1021/acs.joc.9b01708; (b) G. Li, M. Zhao, J. Xie, Y. Yao, L. Mou, X. Zhang, X. Guo, W. Sun, Z. Wang, J. Xu, J. Xue, T. Hu, M. Zhang, M. Li and L. Hong, Efficient synthesis of cyclic amidine-based fluorophores via 6π-electrocyclic ring closure, Chem. Sci., 2020, 11, 3586–3591, 10.1039/d0sc00798f.
- (a) Y. Yang, Y. Wang, Y. Xie, T. Xiong, Z. Yuan, Y. Zhang, S. Qian and Y. Xiao, Fused perylenebisimide–carbazole: new ladder chromophores with enhanced third-order nonlinear optical activities, Chem. Commun., 2011, 47, 10749–10751, 10.1039/c1cc14071j; (b) M. Stępień, E. Gońka, M. ZÌyła and N. Sprutta, Heterocyclic nanographenes and other polycyclic heteroaromatic compounds: synthetic routes, properties, and applications, Chem. Rev., 2017, 117, 3479–3716, DOI:10.1021/acs.chemrev.6b00076; (c) Z. Huang, Z. Bin, R. Su, F. Yang, J. Lan and J. You, Molecular design of non-doped OLEDs based on a twisted heptagonal acceptor: a delicate balance between rigidity and rotatability, Angew. Chem., Int. Ed., 2020, 59, 9992–9996, DOI:10.1002/anie.201915397.
- (a) J. Akhtar, A. A. Khan, Z. Ali, R. Haider and M. S. Yar, Structure-activity relationship (SAR) study and design strategies of nitrogen-containing heterocyclic moieties for their anticancer activities, Eur. J. Med. Chem., 2017, 125, 143–189, DOI:10.1016/j.ejmech.2016.09.023; (b) M. M. Heravi and V. Zadsirjan, Prescribed drugs containing nitrogen heterocycles: an overview, RSC Adv., 2020, 10, 44247–44311, 10.1039/d0ra09198g.
- K. Umemura, T. Mizushima, H. Katayama, Y. Kiryu, T. Yamori and T. Andoh, Inhibition of DNA topoisomerases II and/or I by pyrazolo[1,5-a]indole derivatives and their growth inhibitory activities, Mol. Pharmacol., 2002, 62, 873–880, DOI:10.1124/mol.62.4.873.
- (a) P. Stoessel, H. Heil, D. Joosten, C. Pflumm, A. Gerhard and E. Breuning, Organic electroluminescent materials and devices, WO2010086089A1, 2010 (International patent application); (b) P. Stoessel, D. Joosten, A. Gerhard and E. Breuning, Metal Complexes, WO2012007086A1, 2012 (International patent application); (c) S.-J. Jung, K.-Y. Kim, J.-M. Hong, S.-J. Eum, J.-D. Lee, J.-H. Jung, M.-J. Kim, Y.-S. No and S.-S. Chung, Polycyclic compound containing pyrazole and organic light-emitting device using same, WO2015034140A1, 2015 (International patent application).
- M. Vijay, S. V. Kumar, V. Satheesh, P. Ananthappan, H. K. Srivastava, S. Ellairaja, V. S. Vasantha and T. Punniyamurthy, Stereospecific assembly of fused imidazolidines via tandem ring opening/oxidative amination of aziridines with cyclic secondary amines using photoredox catalysis, Org. Lett., 2019, 21, 7649–7654, DOI:10.1021/acs.orglett.9b02957.
- J. P. Michael, Quinoline, quinazoline and acridone alkaloids, Nat. Prod. Rep., 2007, 24, 223–246, 10.1039/b509528j.
- A. Weyesaa and E. Mulugeta, Recent advances in the synthesis of biologically and pharmaceutically active quinoline and its analogues: a review, RSC Adv., 2020, 10, 20784–20793, 10.1039/d0ra03763j.
- (a) S. M. Prajapati, K. D. Patel, R. H. Vekariya, S. N. Panchala and H. D. Patel, Recent advances in the synthesis of quinolines: a review, RSC Adv., 2014, 4, 24463–24476, 10.1039/c4ra01814a; (b) V. F. Batista, D. C. G. A. Pinto and A. M. S. Silva, Synthesis of quinolines: a green perspective, ACS Sustainable Chem. Eng., 2016, 4, 4064–4078, DOI:10.1021/acssuschemeng.6b01010.
- D. J. Gale and J. F. K. Wilshire, Studies in the indazole series. Ring opening of some 1-Arylindazole-3-carboxylic acids during decarboxylation, Aust. J. Chem., 1973, 26, 2683–2695, DOI:10.1071/ch9732683.
- J. Rosevear and J. F. K. Wilshire, Cyclization reactions in azole chemistry: the reaction of some azoles with o-fluoroacetophenone, o-fluorobenzaldehyde and o-fluorobenzophenone, Aust. J. Chem., 1991, 44, 1097–1114, DOI:10.1071/ch9911097.
- (a) Y. Zhu, Y. Kiryu and H. Katayama, Intramolecular aromatic amination by a hydrazino group for the synthesis of indolo[1,2-b]indazole derivatives, Tetrahedron Lett., 2002, 43, 3577–3580, DOI:10.1016/s0040-4039(02)00545-2; (b) J. Chi, C. Hang, Y. Zhu and H. Katayama, Synthesis of indolo[1,2-b]indazole derivatives via copper(I)-catalyzed intramolecular amination reaction, Synth. Commun., 2010, 40, 1123–1133, DOI:10.1080/00397910903043017.
- C. Guo, B. Li, H. Liu, X. Zhang, X. Zhang and X. Fan, Synthesis of fused or spiro polyheterocyclic compounds via the dehydrogenative annulation reactions of 2-arylindazoles with maleimides, Org. Lett., 2019, 21, 7189–7193, DOI:10.1021/acs.orglett.9b01889.
- H. Zhang, S.-S. Di, X.-B. Huang, Y.-B. Zhou, M.-C. Liu and H.-Y. Wu, Direct dilithiation of N-aryl heterocycles for the construction of condensed N-heterocycles, Org. Chem. Front., 2022, 9, 2659–2663, 10.1039/d1qo01896e.
- (a) K. T. Potts, H. P. Youzwak and S. J. Zurawel, Nonclassical heterocycles. 6. Tri- and tetracyclic ring systems containing a “nonclassical” thiophene nucleus, J. Org. Chem., 1980, 45, 90–97, DOI:10.1021/jo01289a019; (b) Y. Yoshiro, H. Takashi and M. Mitsuo, Reactions of substituted pyridinium N-imines with benzyne: syntheses of pyrido[1,2-b]indazoles and related compounds, Chem. Lett., 1980, 9, 1133–1136, DOI:10.1246/cl.1980.1133.
- N. Shindoh, H. Tokuyama, Y. Takemoto and K. Takasu, Auto-tandem catalysis in the synthesis of substituted quinolines from aldimines and electron-rich olefins: cascade Povarov−hydrogen-transfer reaction, J. Org. Chem., 2008, 73, 7451–7456, DOI:10.1021/jo8009243.
- (a) J. Zhao, P. Li, C. Wu, H. Chen, W. Ai, R. Sun, H. Ren, R. C. Larock and F. Shi, Aryne [3 + 2] cycloaddition with N-sulfonylpyridinium imides and in situ generated N-sulfonylisoquinolinium imides: a potential route to pyrido[1,2-b]indazoles and indazolo[3,2-a]isoquinolines, Org. Biomol. Chem., 2012, 10, 1922–1930, 10.1039/c2ob06611d; (b) L. Li, H. Wang, X. Yang, L. Kong, F. Wang and X. Li, Rhodium-catalyzed oxidative synthesis of quinoline-fused sydnones via 2-fold C–H bond activation, J. Org. Chem., 2016, 81, 12038–12045, DOI:10.1021/acs.joc.6b02356.
- B. Haag, Z. Peng and P. Knochel, Preparation of polyfunctional indazoles and heteroarylazo compounds using highly functionalized zinc reagents, Org. Lett., 2009, 11, 4270–4273, DOI:10.1021/ol901585k.
- S. V. Kumar, S. Ellairaja, V. Satheesh, V. S. Vasantha and T. Punniyamurthy, Rh-catalyzed regioselective C–H activation and C–C bond formation: synthesis and photophysical studies of indazolo[2,3-a]quinolines, Org. Chem. Front., 2018, 5, 2630–2635, 10.1039/c8qo00557e.
- S. Guo, L. Sun, X. Li, X. Zhang and X. Fan, Selective synthesis of indazolo[2,3-a]quinolines via Rh(III)-catalyzed oxidant-free [4 + 2] or [5 + 1] annulation of 2-aryl-2H-indazoles with α-diazo carbonyl compounds, Adv. Synth. Catal., 2020, 362, 913–926, DOI:10.1002/adsc.201901422.
- Y. C. Devulapally, C. Ittamalla, S. Balasubramanian and B. V. S. Reddy, Rhodium(III)-catalyzed dehydrogenative annulation of 2-arylindazoles with cyclic enones, Eur. J. Org. Chem., 2021, 3083–3090, DOI:10.1002/ejoc.202100272.
- A. Chidrawar, Y. C. Devulapally, S. Balasubramanian and B. V. S. Reddy, Rh(III)-catalyzed oxidative annulation of 2-arylindazoles with β-ketosulfoxonium ylides, ChemistrySelect, 2021, 6, 13046–13050, DOI:10.1002/slct.202102982.
- W. Hu, C. Pi, D. Hu, X. Han, Y. Wu and X. Cui, Rh(III)-catalyzed synthesis of indazolo[2,3-a]quinolines: vinylene carbonate as C1 and C2 building blocks, Org. Lett., 2022, 24, 2613–2618, DOI:10.1021/acs.orglett.2c00580.
- K. Ghosh, Y. Nishii and M. Miura, Oxidative C-H/C-H annulation of imidazopyridines and indazoles through rhodium-catalyzed vinylene transfer, Org. Lett., 2020, 22, 3547–3550, DOI:10.1021/acs.orglett.0c00975.
- A. R. Hajipour, Z. Khorsandi and S. Bahri, An efficient and inexpensive visible light photoredox copper catalyst for N-N bond-forming reactions: the one-pot synthesis of indazolo[2,3-α]quinolines, J. Iran. Chem. Soc., 2018, 15, 981–986, DOI:10.1007/s13738-018-1295-1.
- W.-C. Lin and D.-Y. Yang, Visible light photoredox catalysis: synthesis of indazolo[2,3-a]quinolines from 2-(2-nitrophenyl)-1,2,3,4-tetrahydroquinolines, Org. Lett., 2013, 15, 4862–4865, DOI:10.1021/ol402286d.
- (a) P. F. Gordon and P. Gregory, Organic chemistry in colour, Springer, NY, 1983 CrossRef; (b) H. Zollinger, Color chemistry: syntheses, properties and applications of organic dyes and pigments, VCH, NY, 1987 Search PubMed; (c) K. Hunger, Industrial dyes: chemistry, properties, applications, ed. Wiley-VCH, Germany, 2003 Search PubMed.
- (a) T. Ikeda and O. Tsutsumi, Optical switching and image storage by means of azobenzene liquid-crystal films, Science, 1995, 268, 1873–1875, DOI:10.1126/science.268.5219.1873; (b) H. Murakami, A. Kawabuchi, K. Kotoo, M. Kutinake and N. Nakashima, A light-driven molecular shuttle based on a rotaxane, J. Am. Chem. Soc., 1997, 119, 7605–7606, DOI:10.1021/ja971438a; (c) J. C. Crano and R. Guglielmetti, Organic Photochromic and Thermochromic Compounds, Plenum Press, NY, 1999. DOI:10.1007/b114211; (d) W. Zhen, H. Han, M. Anguiano, C. Lemere, C.-G. Cho and P. T. Lansbury, Synthesis and amyloid binding properties of rhenium complexes: preliminary progress toward a reagent for SPECT imaging of Alzheimer's disease brain, J. Med. Chem., 1999, 42, 2805–2815, DOI:10.1021/jm990103w; (e) B. L. Feringa, R. A. Van Delden, N. Koumura and E. M. Geertsema, Chiroptical Molecular Switches, Chem. Rev., 2000, 100, 1789–1816, DOI:10.1021/cr9900228; (f) H. Finkelmann, E. Nishikawa, G. G. Pereira and M. Warner, A new opto-mechanical effect in solids, Phys. Rev. Lett., 2001, 87, 015501, DOI:10.1103/physrevlett.87.015501; (g) T. Hugel, N. B. Holland, A. Cattani, L. Moroder, M. Seitz and H. E. Gaub, Single-molecule optomechanical cycle, Science, 2002, 296, 1103–1106, DOI:10.1126/science.1069856; (h) I. A. Banerjee, L. Yu and H. Matsui, Application of host-guest chemistry in nanotube-based device fabrication: photochemically controlled immobilization of azobenzene nanotubes on patterned α-CD monolayer/Au substrates via molecular recognition, J. Am. Chem. Soc., 2003, 125, 9542–9543, DOI:10.1021/ja0344011; (i) A. Jain, Y. Gupta and S. K. Jain, Azo chemistry and its potential for colonic delivery, Crit. Rev. Ther. Drug Carrier Syst., 2006, 23, 349–400, DOI:10.1615/critrevtherdrugcarriersyst.v23.i5.10; (j) Smart light-responsive materials, ed. Y. Zhao and T. Ikeda, John Wiley & Sons, Hoboken, NJ, 2009 Search PubMed; (k) Molecular switches, ed. B. L. Feringa and W. R. Browne, Wiley-VCH, 2011 Search PubMed; (l) A. Polosukhina, J. Litt, I. Tochitsky, J. Nemargut, Y. Sychev, I. De Kouchkovsky, T. Huang, K. Borges, D. Trauner, R. N. Van Gelder and R. H. Kramer, Photochemical restoration of visual responses in blind mice, Neuron, 2012, 75, 271–282, DOI:10.1016/j.neuron.2012.05.022; (m) W. A. Velema, W. Szymanski and B. L. Feringa, Photopharmacology: beyond proof of principle, J. Am. Chem. Soc., 2014, 136, 2178–2191, DOI:10.1021/ja413063e; (n) J. Zhu, T. Guo, Z. Wang and Y. Zhao, Triggered azobenzene-based prodrugs and drug delivery systems, J. Controlled Release, 2022, 345, 475–493, DOI:10.1016/j.jconrel.2022.03.041; (o) C. Fedele, T. P. Ruoko, K. Kuntze, M. Virkki and A. Priimagi, New tricks and emerging applications from contemporary azobenzene research, Photochem. Photobiol. Sci., 2022, 21, 1719–1734, DOI:10.1007/s43630-022-00262-8.
- (a) N. Siampiringue, G. Guyot, S. Monti and P. Bortolus, The cis → trans photoisomerization of azobenzene: an experimental re-examination, J. Photochem., 1987, 37, 185–188, DOI:10.1016/0047-2670(87)85039-6; (b) A. Cembran, F. Bernardi, M. Garavelli, L. Gagliardi and G. Orlandi, On the mechanism of the cis–trans isomerization in the lowest electronic states of azobenzene: S0, S1, and T1, J. Am. Chem. Soc., 2004, 26, 3234–3243, DOI:10.1021/ja038327y; (c) E. Merino and M. Ribagorda, Control over molecular motion using the cis–trans photoisomerization of the azo group, Beilstein J. Org. Chem., 2012, 8, 1071–1090, DOI:10.3762/bjoc.8.119; (d) M. Quick, A. L. Dobryakov, M. Gerecke, C. Richter, F. Berndt, I. N. Ioffe, A. A. Granovsky, R. Mahrwald, N. P. Ernsting and S. A. Kovalenko, Photoisomerization dynamics and pathways of trans- and cis-azobenzene in solution from broadband femtosecond spectroscopies and calculations, J. Phys. Chem. B, 2014, 118, 8756–8771, DOI:10.1021/jp504999f; (e) I. C. D. Merritt, D. Jacquemin and M. Vacher, Cis-trans photoisomerisation of azobenzene: a fresh theoretical look, Phys. Chem. Chem. Phys., 2021, 23, 19155–19165, 10.1039/d1cp01873f.
- (a) A. M. Sánchez and R. H. de Rossi, Solvent effects on the thermal cis-trans isomerization and charge-transfer absorption of 4-(diethylamino)-4′-nitroazobenzene, J. Org. Chem., 1996, 61, 3446–3451, DOI:10.1021/jo00165a005; (b) M. Alemani, M. V. Peters, S. Hecht, K.-H. Rieder, F. Moresco and L. Grill, Electric field-induced isomerization of azobenzene by STM, J. Am. Chem. Soc., 2006, 128, 14446–14447, DOI:10.1021/ja065449s; (c) G. L. Hallett-Tapley, C. D'Alfonso, N. L. Pacioni, C. D. McTiernan, M. González-Béjar, O. Lanzalunga, E. I. Alarcona and J. C. Scaiano, Gold nanoparticle catalysis of the cis–trans isomerization of azobenzene, Chem. Commun., 2013, 49, 10073–10075, 10.1039/c3cc41669k; (d) S. K. Surampudi, H. R. Patel, G. Nagarjunaa and D. Venkataraman, Mechano-isomerization of azobenzene, Chem. Commun., 2013, 49, 7519–7521, 10.1039/c3cc43797c; (e) A. Goulet-Hanssens, M. Utecht, D. Mutruc, E. Titov, J. Schwar, L. Grubert, D. Bléger, P. Saalfrank and S. Hecht, Electrocatalytic Z → E isomerization of azobenzenes, J. Am. Chem. Soc., 2017, 139, 335–341, DOI:10.1021/jacs.6b10822.
- (a) G. E. Lewis and R. J. Mayfield, Photoinduced reaction of azobenzene with acetyl chloride, Tetrahedron Lett., 1966, 7, 269–271, DOI:10.1016/s0040-4039(00)70226-7; (b) X. Ke, S. Sarina, J. Zhao, X. Zhang, J. Changa and H. Zhu, Tuning the reduction power of supported gold nanoparticle photocatalysts for selective reductions by manipulating the wavelength of visible light irradiation, Chem. Commun., 2012, 48, 3509–3511, 10.1039/c2cc17977f; (c) F. Cumine, F. Palumbo and J. A. Murphy, Reduction of nitroarenes, azoarenes and hydrazine derivatives by an organic super electron donor, Tetrahedron, 2018, 74, 5539–5545, DOI:10.1016/j.tet.2018.04.069; (d) N. Xu, Y. Zhang, W. Chen, P. Li and L. Wang, Photoinduced N-methylation and N-sulfonylation of azobenzenes with DMSO under mild reaction conditions, Adv. Synth. Catal., 2018, 360, 1199–1208, DOI:10.1002/adsc.201701548; (e) X. Wang, X. Wang, C. Xia and L. Wu, Visible-light-promoted oxidative dehydrogenation of hydrazobenzenes and transfer hydrogenation of azobenzenes, Green Chem., 2019, 21, 4189–4193, 10.1039/c9gc01618j; (f) Y. Katsuma, L. Wu, Z. Lin, S. Akiyama and M. Yamashita, Reactivity of a tetra(o-tolyl)diborane(4) dianion as a diarylboryl anion equivalent, Angew. Chem., Int. Ed., 2019, 58, 317–321, DOI:10.1002/anie.201907400; (g) J. Yang, M. Song, H. Zhou, G. Wang, B. Ma, Y. Qi and C. Huo, Visible-light-mediated hydroacylation of azobenzenes with α-keto acids, Org. Lett., 2020, 22, 8407–8412, DOI:10.1021/acs.orglett.0c03039; (h) M. Song, H. Zhou, G. Wang, B. Ma, Y. Jiang, J. Yang, C. Huo and X.-C. Wang, Visible-light-promoted diboron-mediated transfer hydrogenation of azobenzenes to hydrazobenzenes, J. Org. Chem., 2021, 86, 4804–4811, DOI:10.1021/acs.joc.1c00394; (i) J. Yang, M. Song, H. Zhou, Y. Qi, B. Ma and X.-C. Wang, Visible-light-promoted decarboxylative addition cyclization of N-aryl glycines and azobenzenes to access 1,2,4-triazolidines, Green Chem., 2021, 23, 5806–5811, 10.1039/d1gc02272e; (j) J. Yang, C. Wang, H. Zhou, Q. Kong and W. Ma, Visible-light-driven hydrophosphorylation of azobenzenes enabled by trans-to-cis photoisomerization, Adv. Synth. Catal., 2022, 364, 4275–4280, DOI:10.1002/adsc.202201089; (k) Q. Li, Y. Luo, J. Chen and Y. Xia, Visible-light-promoted hydrogenation of azobenzenes to hydrazobenzenes with thioacetic acid as the reductant, J. Org. Chem., 2023, 88, 2443–2452, DOI:10.1021/acs.joc.2c02873; (l) J. Yang, C. Wang, B. Huang, H. Zhou, J. Li and X. Liu, Photoredox catalytic phosphine-mediated deoxygenative hydroacylation of azobenzenes with carboxylic acids, Org. Lett., 2024, 26, 498–502, DOI:10.1021/acs.orglett.3c03875.
- V. A. K. Adiraju and C. D. Martin, Isomer dependence on the reactivity of diazenes with pentaphenylborole, Chem. – Eur. J., 2017, 23, 11437–11444, DOI:10.1002/chem.201702539.
- W. Zhang, J. Bu, L. Wang, P. Li and H. Li, Sunlight-mediated [3 + 2] cycloaddition of azobenzenes with arynes: an approach toward the carbazole skeleton, Org. Chem. Front., 2021, 8, 5045–5051, 10.1039/d1qo00739d.
- C. Mañas and E. Merino, Visible light-mediated heterodifunctionalization of alkynylazobenzenes for 2H–indazole synthesis, Org. Lett., 2024, 26, 1868–1873, DOI:10.1021/acs.orglett.4c00097.
- (a) B. E. Moulton, H. Dong, C. T. O'Brien, S. B. Duckett, Z. Lin and I. J. S. Fairlamb, A natural light induced regioselective 6π-electrocyclisation–oxidative aromatisation reaction: experimental and theoretical insights, Org. Biomol. Chem., 2008, 6, 4523–4532, 10.1039/b811284c; (b) J. Xi, D. Wang, J. Hu, H. Shen, T. Wang and Z. Zhang, Rearrangement of 2-(benzofuran-2-yl)-3-phenylpyridines via photoinduced 6π-electrocyclization, Org. Biomol. Chem., 2023, 21, 7188–7193, 10.1039/d3ob00883e.
- Deposition numbers CCDC 2374941 (for 2s), by 2374943 (for 3j) and 2374942 (for 4b) contain the supporting crystallographic data for this paper.†.
- For more details, see ESI.†.
- (a) S. Kato, Y. Saga, M. Kojima, H. Fuse, S. Matsunaga, A. Fukatsu, M. Kondo, S. Masaoka and M. Kanai, Hybrid catalysis enabling room-temperature hydrogen gas release from N-heterocycles and tetrahydronaphthalenes, J. Am. Chem. Soc., 2017, 139, 2204–2207, DOI:10.1021/jacs.7b00253; (b) K.-H. He, F.-F. Tan, C.-Z. Zhou, G.-J. Zhou, X.-L. Yang and Y. Li, Acceptorless dehydrogenation of N-heterocycles by merging visible-light photoredox catalysis and cobalt catalysis, Angew. Chem., Int. Ed., 2017, 56, 3080–3084, DOI:10.1002/ange.201612486; (c) J. Fan, W. Zhang, W. Gao, T. Wang, W.-L. Duan, Y. Liang and Z. Zhang, Syntheses of benzofuranoquinolines and analogues via photoinduced acceptorless dehydrogenative annulation of o-phenylfuranylpyridines, Org. Lett., 2019, 21, 9183–9187, DOI:10.1021/acs.orglett.9b03556; (d) Y. Kang, W. Zhang, T. Wang, Y. Liang and Z. Zhang, Two-step synthesis of π-expanded maleimides from 3,4-diphenylfuran-2(5H)-ones, J. Org. Chem., 2019, 84, 12387–12398, DOI:10.1021/acs.joc.9b01792; (e) M.-J. Zhou, L. Zhang, G. Liu, C. Xu and Z. Huang, Site-selective acceptorless dehydrogenation of aliphatics enabled by organophotoredox/cobalt dual catalysis, J. Am. Chem. Soc., 2021, 143, 16470–16485, DOI:10.1021/jacs.1c05479.
- Even thought, traces of 5,6-dihydroindazolo[2,3-a]quinoline 3x could be detected by 1H NMR spectroscopy in the crude mixture, the two products could be purified by column chromatography.
- G. S. Hammond, J. Saltiel, A. A. Lamola, N. J. Turro, J. S. Bradshaw, D. O. Cowan, R. C. Counsell, V. Vogt and C. Dalton, Mechanisms of photochemical reactions in solution. XXII. Photochemical cis-trans isomerization, J. Am. Chem. Soc., 1964, 86, 3197–3217, DOI:10.1021/ja01070a002.
- (a) I. V. Seregin and V. Gevorgyan, Gold-Catalyzed 1,2-Migration of Silicon, Tin, and Germanium en Route to C-2 Substituted Fused Pyrrole-Containing Heterocycles, J. Am. Chem. Soc., 2006, 128, 12050–12051, DOI:10.1021/ja063278l; (b) I. V. Seregin, A. W. Schammel and V. Gevorgyan, Multisubstituted N-fused heterocycles via transition metal-catalyzed cycloisomerization protocols, Tetrahedron, 2008, 64, 6876–6883, DOI:10.1016/j.tet.2008.04.023.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2374941–2374943. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qo01606h |
| This journal is © the Partner Organisations 2024 |