
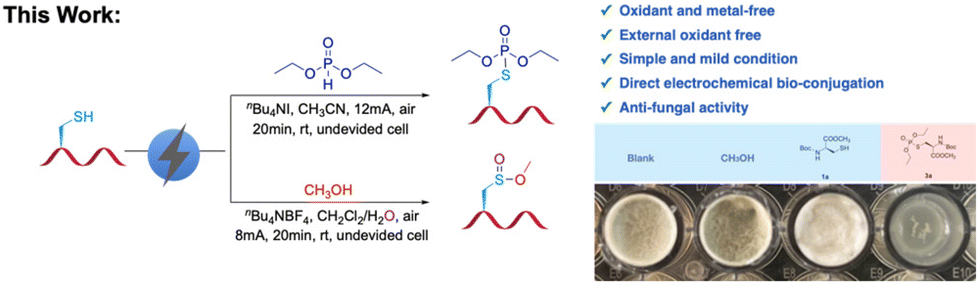
Electrochemical diversification of cysteine derivatives and cysteine-containing peptides to phosphorothioates and sulfinates†
Longyu
Xiao
a,
Yifan
Li
ac,
Jun
Huang
a,
Li
Pan
b,
Pan
Wu
b and
Yue
Weng
 *a
*a
aHubei Key Laboratory of Precision Manufacturing for Small-molecular Active Pharmaceutical Ingredients, School of Chemistry and Chemical Engineering, Hubei University, Wuhan, 430062, P. R. China. E-mail: wengyue@hubu.edu.cn
bState Key Laboratory of Biocatalysis and Enzyme Engineering, School of Life Sciences, Hubei University, Wuhan, 430062, P. R. China
cThe Key Laboratory for Chemical Biology of Fujian Province, The MOE Key Laboratory of Spectrochemical Analysis & Instrumentation, and Department of Chemical Biology, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China
First published on 1st October 2024
Abstract
This study explores the application of modified cysteine in oligopeptides to overcome challenges in amino acid residue modification, with a focus on cysteine. Novel strategies for the functionalization of biomolecules are presented, with mechanistic investigations indicating the involvement of free radical processes, structural substitution, and nucleophilic displacement. Notably, the gram-scale synthesis of modified cysteine is achieved with high yields, highlighting its potential as a novel anti-fungus agent in biomedical and agrochemical research.
Introduction
In the fields of chemical biology, biomedicine, and pesticide science, mild chemical reactions have gradually become key tools for modifying biomolecules.1–3 These reactions typically exhibit high efficiency, strong specificity, and mild conditions, allowing them to proceed in complex biological environments without disrupting the natural structure and function of biomolecules.4,5 For example, click chemistry reactions such as CuAAC (copper-catalyzed azide–alkyne cycloaddition)6 and SPAAC (strain-promoted azide–alkyne cycloaddition)7 have been widely used for the labeling and modification of macromolecules such as proteins and nucleic acids.8,9 These methods not only enhance the targeting and stability of drugs but also provide new means for studying the functional mechanisms of biomolecules.10 Modification of amino acid residues plays a crucial role in regulating protein functions and biological processes.11 Through chemical modifications, the structure and activity of proteins can be precisely controlled to investigate their functional mechanisms.12 Common modifications include phosphorylation,13 acetylation,14 methylation,15 and ubiquitination,16 which can affect protein stability, localization, interactions, and enzymatic activity. Currently, mild chemical reactions, such as bio-orthogonal chemistry reactions,17 have been widely applied to the specific modification of amino acid residues.18 These reactions can proceed in complex biological environments without disrupting the natural structure of proteins.19 For example, the use of 4-nitrophenyl phosphate allows for the specific modification of serine or threonine residues.20 These studies not only provide new tools for protein engineering but also offer new perspectives for disease treatment, such as the development of novel drugs21 that target and modify pathological proteins. The modification of amino acid residues22 has become an important research direction in chemical biology and drug development.23The modification of cysteine24 holds significant importance in protein function regulation and biomedical research. Cysteine contains a reactive thiol group (–SH) that can participate in various crucial biochemical reactions.25,26 By modifying cysteine, the structure, stability, and function of proteins could be regulated.27,28 For instance, the formation of disulfide bonds involving cysteine plays a key role in protein folding and stability.29,30 Additionally, cysteine modifications can regulate enzyme activity,31,32 signal transduction, and the cell cycle.33 Currently, mild chemical reactions are widely used for the specific modification of cysteine,34,35 employing reagents such as maleimide and iodoacetamide. These methods not only provide powerful tools for studying protein function and interactions but also show great potential in developing novel therapeutic approaches, such as designing targeted anticancer drugs through cysteine modification (Fig. 1).
 | ||
| Fig. 1 Electrocatalytic S–P and S(O)–O bonding reaction. | ||
In summary, cysteine modification plays an indispensable role in modern biochemistry and drug development. In recent years, electrochemical methods have made remarkable progress in the field of biomolecule modification. These methods utilize the efficiency and precision of electrochemical reactions to achieve specific modifications of biomolecules such as peptides and proteins under mild conditions. For instance, electrochemical redox reactions can selectively modify serine and tyrosine residues containing hydroxyl groups. Such modifications not only regulate the function and structure of biomolecules but also introduce functional tags for detection and imaging. The advancement of these technologies provides powerful tools for biomolecule functional research and drug development, pushing the frontiers of chemical biology, biomedicine, and pesticide science. Therefore, electrochemical strategies and bioconjugation processes have been designed to achieve highly selective and efficient modification of cysteine residues.
Results and discussion
Here, two novel strategies are presented for the bioconjugation of cysteine-containing biomolecules, employing electrochemical synthetic chemistry under simple, mild, and environmentally friendly conditions. This straightforward method involves the anodic oxidation of cysteine to generate free radicals, followed by cross-coupling with diethyl phosphite or methanol to form S–P or S(O)–O bonds, resulting in the formation of phosphorothioates and sulfinates. Next, the optimal reaction conditions for Boc-Cys-OMe (1a) with phosphite (2a) were explored. As shown in Table 1, using platinum electrodes for both the anode and cathode, with nBu4NI as the electrolyte, and conducting the reaction at a current of 12 mA for 1.5 hours, the amino acid-modified product 3a was obtained with an 87% yield (Table 1, entry 1). The effect of current intensity was also investigated, revealing that maintaining a steady current is crucial for the reaction (Table 1, entry 15). Both higher and lower currents resulted in reduced yields, indicating that 12 mA is the optimal current (Table 1, entries 2 and 3). The impact of different electrolytes on the yield was then examined (Table 1, entries 4–7), revealing that nBu4NBr reduced the yield, while nBu4NBF4 led to a significant decline. Neither KI nor the absence of an electrolyte resulted in any reaction. Choosing an appropriate solvent is also critical. Using methanol or dichloromethane significantly lowered the yield (Table 1, entries 8 and 9). Among the electrodes tested, the dual platinum electrode system provided the best results (Table 1, entries 10–12). The effect of the reaction atmosphere is also noteworthy; yields dropped significantly under nitrogen or oxygen (Table 1, entries 13 and 14). Interestingly, TLC analysis indicated the formation of an “unexpected” compound when methanol was used as the solvent (Table 1, entry 8). The initially low yield was attributed to competing side reactions. Upon reacting Boc-Cys-OMe with methanol, an unforeseen product was obtained with a yield of 78% (Table S1,† entry 7). The optimization of the reaction conditions for Boc-Cys-OMe and methanol is summarized in Table S1.†|
|
||
|---|---|---|
| Entry | Variation from the standard conditionsa | Yield (%) |
| a Reaction conditions: undivided cell, Pt anode, Pt cathode, 1a (0.2 mmol), 2a (0.4 mmol), nBu4NI (0.2 mmol), MeCN (8 mL), air, room temperature, 12 mA, 1.5 h. Yield of isolated products. N.D = not detected, N.R = not reaction. | ||
| 1 | None | 87 |
| 2 | 9 mA instead of 12 mA | 74 |
| 3 | 15 mA instead of 12 mA | 48 |
| 4 | n Bu4NBF4 instead of nBu4NI | 16 |
| 5 | n Bu4NBr instead of nBu4NI | 72 |
| 6 | KI instead of nBu4NI | N.D |
| 7 | Without nBu4NI | N.R |
| 8 | CH3OH instead of CH3CN | Trace |
| 9 | CH2Cl2 instead of CH3CN | 53 |
| 10 | C(+), Pt(−) as the electrodes | 64 |
| 11 | C(+), C(−) as the electrodes | 51 |
| 12 | Pt(+), Ni(−) as the electrodes | 73 |
| 13 | Under N2 | 43 |
| 14 | Under O2 | 34 |
| 15 | No electricity | N.R |
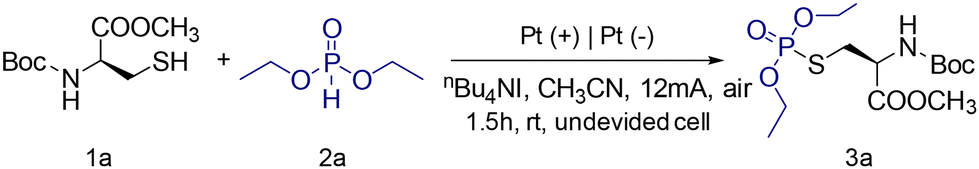
The applicability of the reaction with various phosphites or alcohols was subsequently explored. It was observed that phosphites with shorter carbon chains in the ester group led to phosphorothioates with higher yields (Scheme 1, 3a–3b). When using phosphites with longer carbon chains, the yields decreased (Scheme 1, 3c–3d). However, when the ester group contained a relatively stable conjugated structure, the target product was obtained with higher yields (Scheme 1, 3e). The effect of electron-withdrawing groups on the reaction was also investigated. It was found that product 3f was obtained in low yields when bis(2,2,2-trifluoroethyl) phosphonate was utilized in this protocol (Scheme 1, 3f). Additionally, pentaacetyl-5-thio-D-glucose, containing a thiol group, was reacted with diethyl phosphite, resulting in a considerable yield (Scheme 1, 3g). However, when alcohols reacted with Boc-Cys-OMe, the spatial structure had a significant impact on the reaction. When the carbon atoms in the main chain of the alcohol exceeded two, the alcohol could not react with Boc-Cys-OMe. Nevertheless, when using alcohols with shorter carbon chains, the sulfinates was obtained with moderate to high yields (Scheme 1, 6a–6c). It is noteworthy that all the electrochemical reactions had excellent isolated yields, with no residual self-coupled cystine.
 | ||
Scheme 1 Substrate scope of alcohols and phosphites. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction conditions: undivided cell, Pt anode, Pt cathode, 1a (0.2 mmol), phosphite ester (0.4 mmol), nBu4NI (0.2 mmol), MeCN (8 mL), air, room temperature, 12 mA, 1.5 h. bReact 1 h. cReaction condition: undivided cell, Pt anode, Pt cathode, 1a (0.2 mmol), alcohol (5.0 mmol), nBu4NBF4 (0.2 mmol), CH2Cl2 (6 mL), H2O (10 μL), air, room temperature, 8 mA, 4.0 h. Yield of isolated products. Reaction conditions: undivided cell, Pt anode, Pt cathode, 1a (0.2 mmol), phosphite ester (0.4 mmol), nBu4NI (0.2 mmol), MeCN (8 mL), air, room temperature, 12 mA, 1.5 h. bReact 1 h. cReaction condition: undivided cell, Pt anode, Pt cathode, 1a (0.2 mmol), alcohol (5.0 mmol), nBu4NBF4 (0.2 mmol), CH2Cl2 (6 mL), H2O (10 μL), air, room temperature, 8 mA, 4.0 h. Yield of isolated products. | ||
Furthermore, to investigate the selectivity of Boc-Cys-OMe labeling and its tolerance towards polypeptide labeling, various dipeptides containing Boc-Cys-OMe were introduced into the reaction system. These dipeptides can serve as protein components present in various tissues and cells. Subsequently, diethyl phosphite or methanol was used as a coupling reagent to evaluate the selectivity and tolerance of the Boc-Cys-OMe reaction on dipeptides. Excitingly, these dipeptides met the requirements for modifying the target molecules onto the peptide chains (Scheme 2, 4a–4j, and 7a–7f). Surprisingly, dipeptides formed with relatively stable amino acids such as glycine, alanine, valine, leucine, and isoleucine, resulted in the target products with relatively low yields (Scheme 2, 4a, 4c, 4d, 4i, 7a, 7c, 7d). In contrast, dipeptides formed with amino acids containing reactive groups, such as phenylalanine (containing a phenyl group), lysine (containing an amino group), glutamic acid (containing a carboxyl group), methionine (containing a methylthio group), and tryptophan (containing an indole structure), exhibited better selectivity and group tolerance (Scheme 2, 4b, 4e, 4f, 4g, 4h, 4j, 7b, 7e, 7f). Electrocatalytic bioconjugation often exhibits better selectivity and tolerance to reactive groups, especially the bioconjugation of dipeptides containing reactive groups with high selectivity and group tolerance is of great significance for electrochemical bioconjugation.
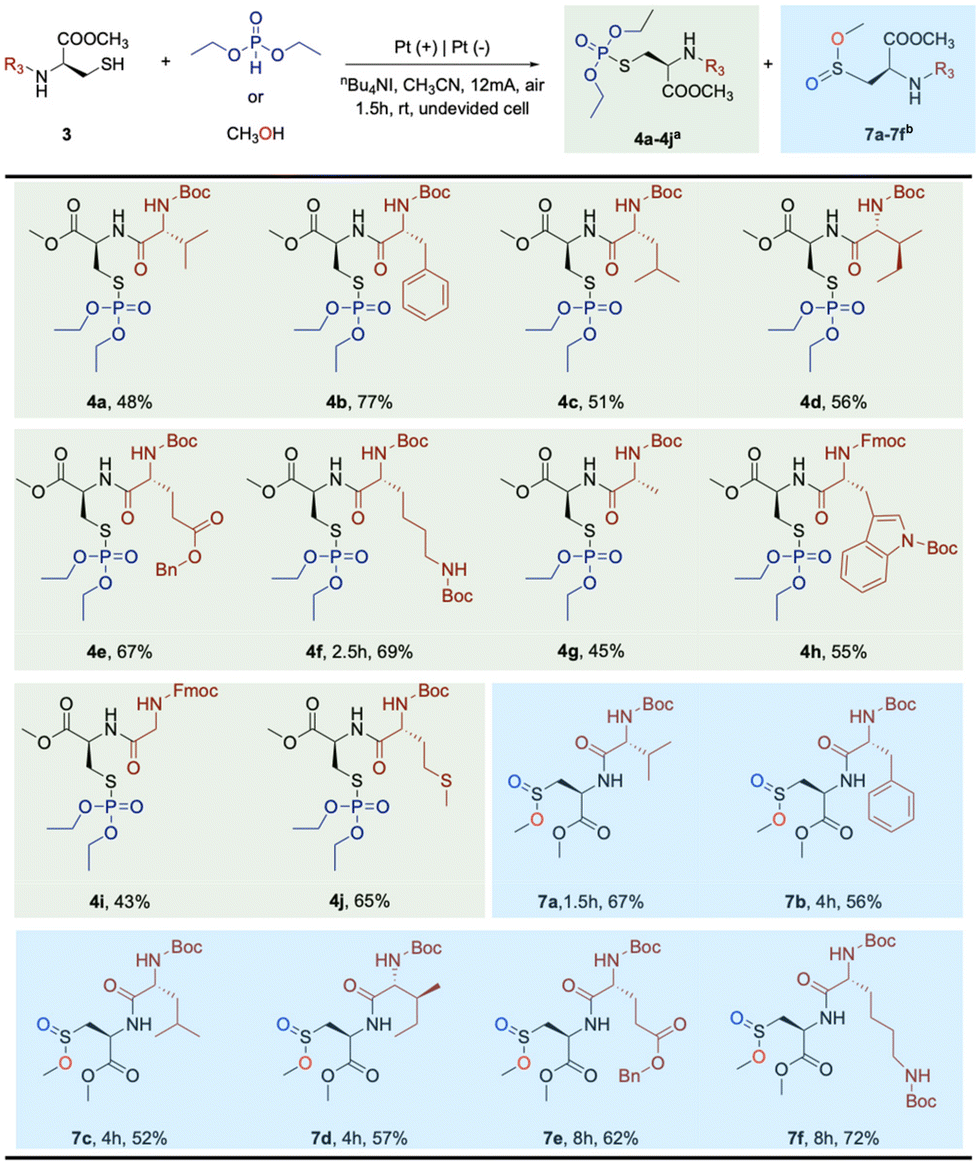 | ||
| Scheme 2 Substrate scope of dipeptides. aReaction conditions: undivided cell, Pt anode, Pt cathode, 3 (0.2 mmol), diethyl phosphite (0.4 mmol), nBu4NI (0.2 mmol), MeCN (8 mL), air, rt, 12 mA, 1.5 h. bReaction conditions: undivided cell, Pt anode, Pt cathode, 3 (0.2 mmol), methyl alcohol (5.0 mmol), nBu4NBF4 (0.2 mmol), CH2Cl2 (6 mL), H2O (10 μL), air, rt, 8 mA, 1.5–8.0 h. Yield of isolated products. | ||
To synthesize biomolecules, particularly polypeptides, via electrochemical methods, the applicability of electrocatalytic synthesis to polypeptides must be assessed. Building on the electrochemical synthesis of dipeptides, further research was initiated, beginning with the electrolysis of annexin inhibitory peptides with exposed amino and hydroxyl groups, cell adhesion peptides with exposed carboxyl, hydroxyl, and imidazole groups, and HPV16 E7-derived peptides with exposed hydroxyl groups. These peptides contain cysteine at the N-terminus or C-terminus and have certain biological activities. It is worth noting that some of these endogenous peptides containing cysteine successfully reacted to produce phosphorothioates and sulfinates (Scheme 3, 8a–8d). The successful labeling of these endogenous peptides in a short period of time indicates that the reaction has good site selectivity and functional group tolerance.
 | ||
| Scheme 3 Electrochemical synthesis of phosphorothioates and sulfinates of endogenous peptides. | ||
To demonstrate the industrial application value of this reaction, conducted gram-scale reactions: these included the reactions of Boc-Cys-OMe with diethyl phosphite and Boc-Cys-OMe with methanol (Scheme 4). A total of 1.48 grams of the target product 3a was obtained with an 80% yield, and 1.10 grams of the target product 6a was produced with a 78% yield. This experiment highlights the great potential of cysteine modification method in subsequent anti-fungus research and new drug synthesis.
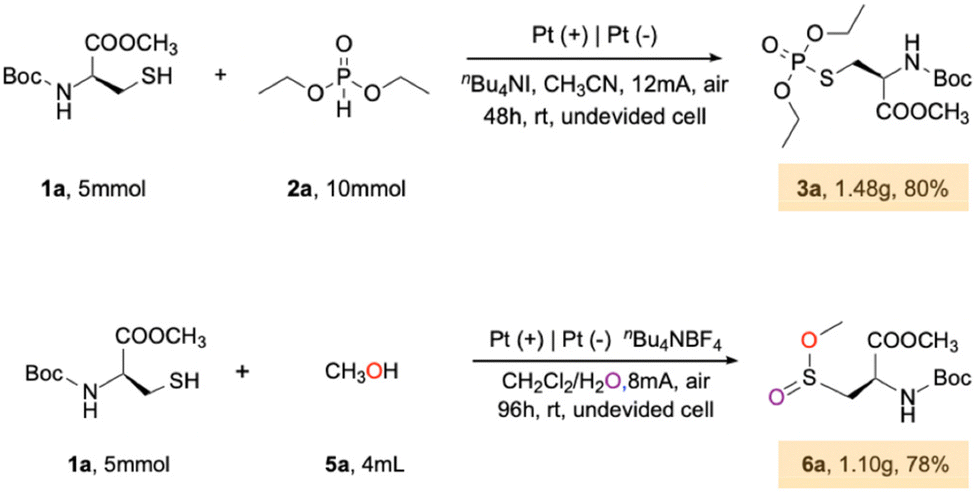 | ||
| Scheme 4 Gram-scale reaction. | ||
The antifungal efficacy of compounds 1a and 3a against the pathogenic fungus Magnaporthe grisea was also investigated (Scheme 5). To ensure the activity and consistency of the experimental materials (Scheme 5a), the target strain was first activated and cultured in LB liquid medium for 48 hours to reach the logarithmic growth phase. The activated Magnaporthe grisea strains were then inoculated into 100 mL of molten potato dextrose agar (PDA) medium, ensuring uniform distribution through thorough mixing. Using standardized 96-well plates as the experimental platform, three parallel samples were prepared for each group to enhance data reliability and reproducibility. Each well was filled with 200 μL of PDA solid medium containing Magnaporthe grisea. To assess the inhibitory effects of the compounds on fungal growth, 20 μL of the antibacterial solutions of 1a and 3a were introduced into specific wells, while negative control groups using methanol and pure water were included to rule out solvent effects. All treated plates were incubated at a constant temperature of 28 °C for 3–5 days to fully observe the potential effects of each anti-fungus substance. Then, the optical density at 600 nm (OD600) of each sample was determined (Scheme 5b). The experimental results show that Boc-Cys-Ome promotes the growth of Magnaporthe grisea, while the electrochemical reaction product 3a demonstrates a notable inhibitory effect on the fungus. These findings have potential implications for the development of bioactive molecules with fungicidal properties in agricultural chemistry.
 | ||
| Scheme 5 (a) Anti-fungus experiment; (b) optical density of treated fungal patches at 600 nm (OD600). | ||
To gain deeper insight into the electrochemical reaction mechanism, a series of control experiments were designed. Under the established conditions, Boc-Cys-OMe was allowed to react with diethyl phosphite for 30 minutes, yielding the intermediate cystine E with a 78% yield, along with a 6% yield of the target product. The reaction was then continued for an additional hour, during which intermediate E reacted with diethyl phosphite, producing the product 3a with a 73% yield. This preliminary analysis suggests that the reaction proceeds by first forming cystine from Boc-Cys-OMe, which subsequently reacts with diethyl phosphite to yield the product 3a (Scheme 6A). When the radical scavenger TEMPO was introduced into the reaction system under the same conditions, a significant reduction in yield was observed, indicating the involvement of radical intermediates in the reaction.
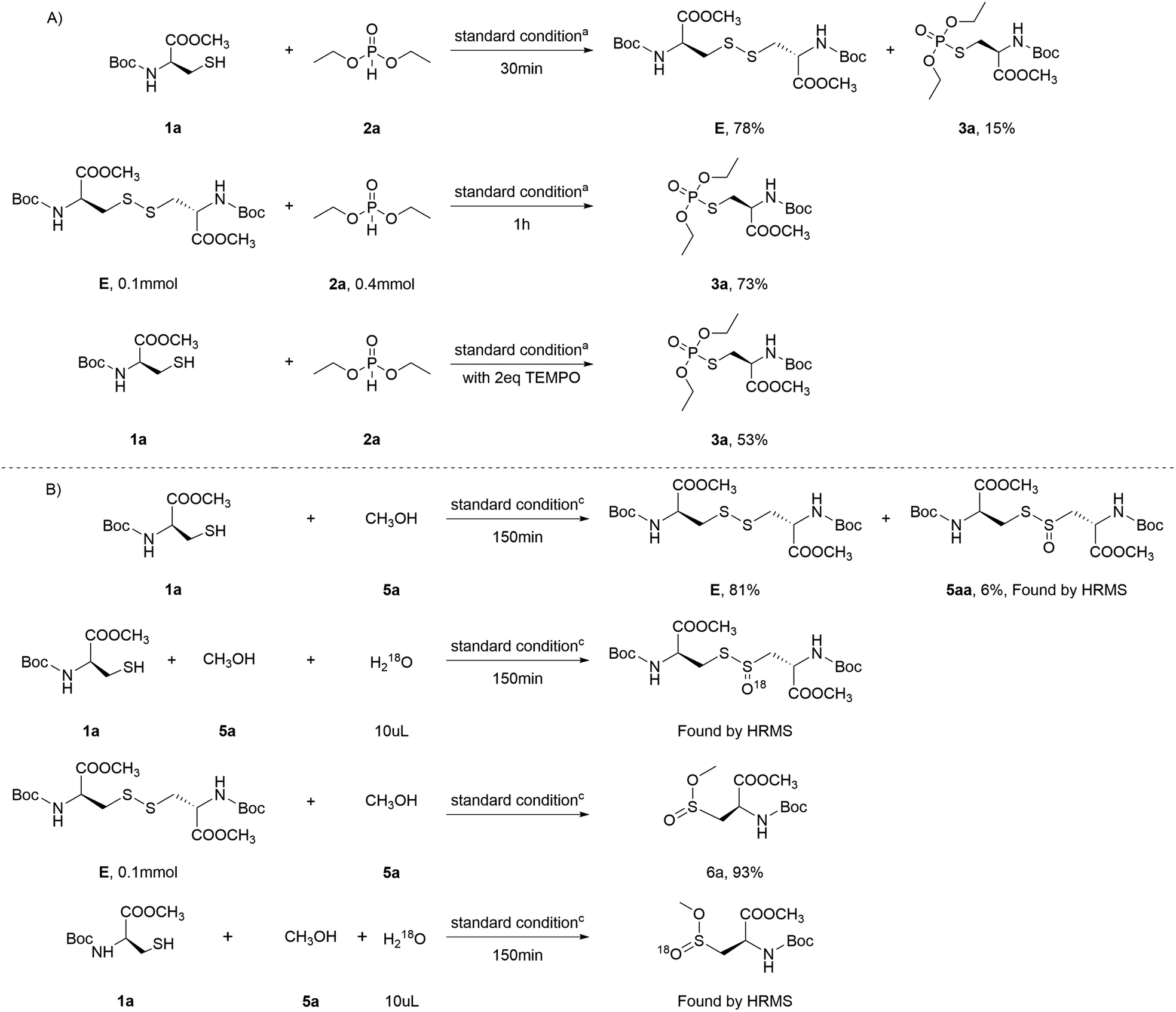 | ||
| Scheme 6 Control reaction. | ||
Furthermore, Boc-Cys-OMe was reacted with methanol for 150 minutes, again producing the cystine intermediate with a high yield, along with another intermediate, 5aa. This observation explains the reduced yields of products 3a and 6a when Boc-Cys-OMe, methanol, and diethyl phosphite are present in the same system. Similarly, when intermediate E was reacted with methanol for 4 hours, the product 6a was obtained with a 93% yield. Isotope labeling experiments were also conducted, confirming that the oxygen in the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond originates from water (Scheme 6B).
O bond originates from water (Scheme 6B).
Based on controlled experiments, a more in-depth investigation of the reaction mechanism was conducted using cyclic voltammetry. As shown in Fig. 2, nBu4NI is oxidized at 0.481 V and 1.062 V, and reduced at −1.102 V (Fig. 2 (1a), red curve). This corresponds to the oxidation of I− in nBu4NI to iodine radicals, which subsequently convert to I2, followed by a disproportionation reaction to form I− and I+. Boc-Cys-OMe is oxidized at 1.308 V (Fig. 2 (1a), blue curve), corresponding to the formation of Boc-Cys-OMe radicals. A mixture of Boc-Cys-OMe and diethyl phosphite exhibits a pronounced oxidation peak at 1.287 V, positioned between the oxidation peaks of nBu4NI and cysteine, indicating chemical interactions among the three compounds. To further confirm the presence of radical intermediates in the reaction, electron paramagnetic resonance (EPR) experiments were conducted (Fig. 2 (2)). The results revealed signals corresponding to the radicals of nBu4NI and Boc-Cys-OMe.
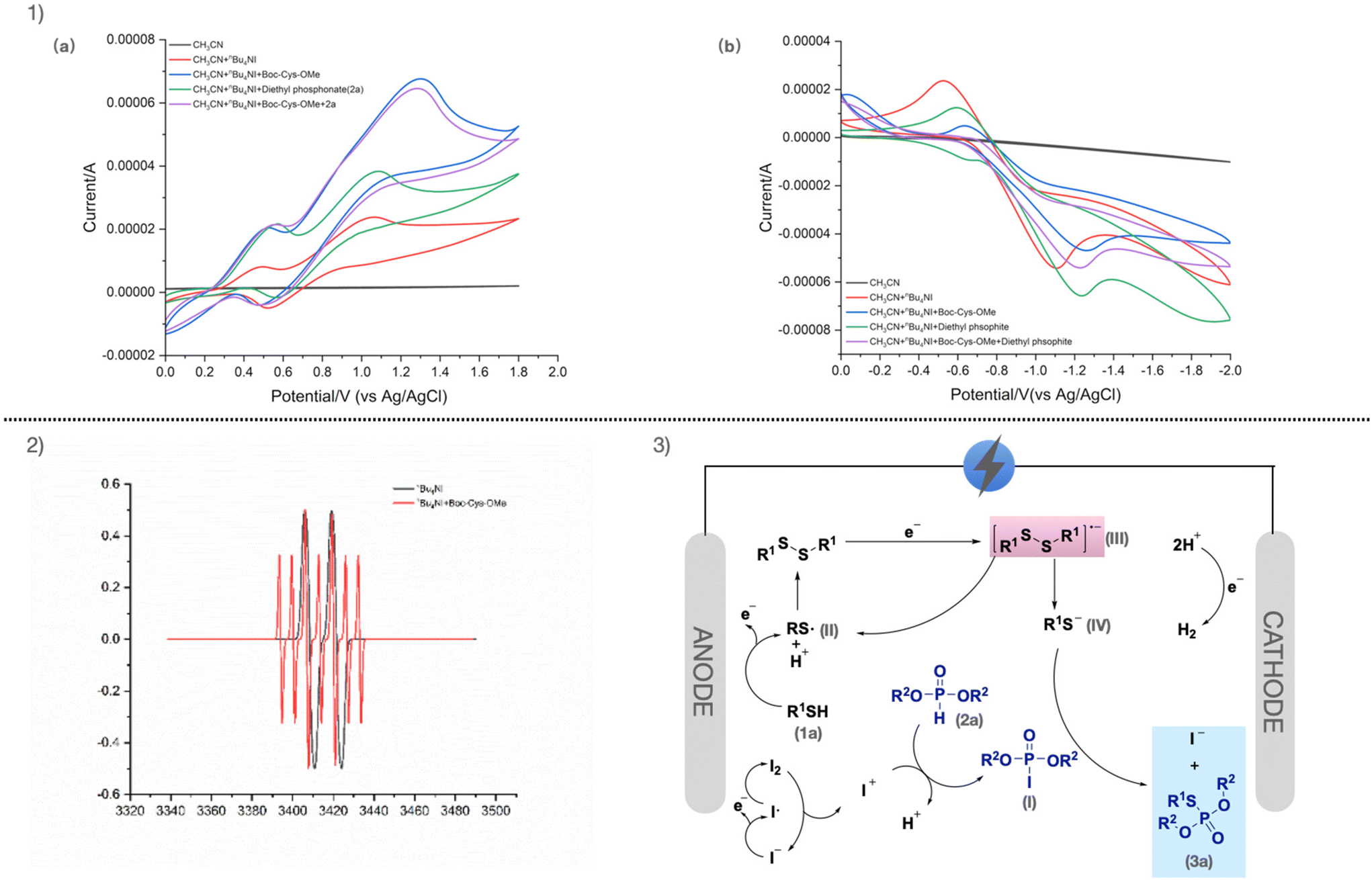 | ||
| Fig. 2 (1) Cyclic voltammograms of reactants and the mixtures in 0.1 M nBu4NI/CH3CN using a glassy carbon working electrode (diameter, 3 mm), oxidation peak (a), reduction peak (b). Pt electrode as counter electrode; Ag/AgCl as reference electrode, at 100 mV s−1 scan rate. Boc-Cys-OMe (8 mmol L−1), diethyl phosphite (16 mmol L−1). (2) Electron paramagnetic resonance (EPR) experiments of Boc-Cys-OMe and nBu4NI. (3) Proposed mechanism. | ||
Based on the aforementioned study and previous reports,36–38 a possible mechanism for the phosphorylation of cysteine is presented (Fig. 2 (3)). Firstly, I− in nBu4NI is oxidized to iodine radical at the anode. Iodine radical dimerizes to form I2, which disproportionates to yield I− and I+. I+ then undergoes electrophilic substitution with diethyl phosphite (2a) to produce diethyl iodophosphate (I). Simultaneously, cysteine is oxidized at the anode to generate cysteine radical (II). The cysteine radical dimerizes to form cystine, which is subsequently reduced at the cathode to form cystine radical anion (III). These cystine radical anion then convert back to cysteine radical and cysteine anion (IV). The cysteine anion then undergoes nucleophilic reaction with diethyl iodophosphate (I), ultimately yielding 3a.
Conclusions
In conclusion, a mild and efficient method for the modification and labeling of cysteine and cysteine-containing peptides via electrocatalysis has been developed. This approach offers excellent selectivity and functional group tolerance while maintaining operational simplicity. Importantly, it avoids the use of metals, strong acids, or bases, making it environmentally friendly. Through a combination of control experiments, isotope labeling, cyclic voltammetry, and electron paramagnetic resonance (EPR) studies, a plausible reaction mechanism has been proposed. This green modification strategy significantly broadens the scope of cysteine modifications. Additionally, the modified products demonstrated notable antifungal activity against the pathogenic fungus Magnaporthe grisea. Looking ahead, this methodology is expected to contribute to advancements in antifungal and agrochemical research.Author contributions
There are no conflicts to declare.Data availability
All relevant data are within the manuscript and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Mr Heming Qi College of Resources and Environmental Science, South-Central University for Nationalities for the help of EPR experiment. National Natural Science Foundation of China (Grant No. 22301069, 31900051); National Natural Science Foundation of China (Grant No. 22271085).References
- Y.-C. Song, S. Ingram, R. E. Arbon, D. O. Topping, D. R. Glowacki and J. P. Reid, Transient cavity dynamics and divergence from the Stokes–Einstein equation in organic aerosol, Chem. Sci., 2020, 11, 2999–3006 RSC.
- C. Wan, R. Sun, W. Xia, H. Jiang, B.-X. Chen, P.-C. Kuo, W.-R. Zhang, G. Yang, D. Li, C.-W. Chiang and Y. Weng, Electrochemical Bioconjugation of Tryptophan Residues: A Strategy for Peptide Modification, Org. Lett., 2024, 26, 5447–5452 CrossRef CAS PubMed.
- D. Alvarez-Dorta, C. Thobie-Gautier, M. Croyal, M. Bouzelha, M. Mével, D. Deniaud, M. Boujtita and S. G. Gouin, Electrochemically Promoted Tyrosine-Click-Chemistry for Protein Labeling, J. Am. Chem. Soc., 2018, 140, 17120–17126 CrossRef CAS PubMed.
- P. P. Singh, P. K. Singh and V. Srivastava, Visible light metallaphotoredox catalysis in the late-stage functionalization of pharmaceutically potent compounds, Org. Chem. Front., 2023, 10, 216–236 RSC.
- Y. Weng, C. Song, C.-W. Chiang and A. Lei, Single electron transfer-based peptide/protein bioconjugations driven by biocompatible energy input, Commun. Chem., 2020, 3, 171 CrossRef CAS PubMed.
- M. Z. C. Hatit, L. F. Reichenbach, J. M. Tobin, F. Vilela, G. A. Burley and A. J. B. Watson, A flow platform for degradation-free CuAAC bioconjugation, Nat. Commun., 2018, 9, 4021 CrossRef PubMed.
- Y. Xie, T. L. Lopez-Silva and J. P. Schneider, Hydrophilic Azide-Containing Amino Acid to Enhance the Solubility of Peptides for SPAAC Reactions, Org. Lett., 2022, 24, 7378–7382 CrossRef CAS PubMed.
- C. S. Swenson, A. Velusamy, H. S. Argueta-Gonzalez and J. M. Heemstra, Bilingual Peptide Nucleic Acids: Encoding the Languages of Nucleic Acids and Proteins in a Single Self-Assembling Biopolymer, J. Am. Chem. Soc., 2019, 141, 19038–19047 CrossRef CAS PubMed.
- C. Loynd, S. J. Singha Roy, V. J. Ovalle, S. E. Canarelli, A. Mondal, D. Jewel, E. D. Ficaretta, E. Weerapana and A. Chatterjee, Electrochemical labelling of hydroxyindoles with chemoselectivity for site-specific protein bioconjugation, Nat. Chem., 2024, 16, 389–397 CrossRef CAS PubMed.
- S. Jiang, L. Xiao, L. Pan, Q. Huang, F. Huo, M. Gao, C. Lu, P. Wu and Y. Weng, Electro-induced O–S bonding reaction targeting biological macromolecules, Org. Chem. Front., 2024, 11, 1090–1096 RSC.
- R. Bag and N. K. Sharma, Pd-catalyzed site-selective C(sp2)–H chalcogenation of amino acids and peptides using a picolinamide auxiliary, Org. Chem. Front., 2023, 10, 1252–1262 RSC.
- C.-F. Lee, R. Bai, K.-C. Liu, Z.-W. Chen, A. Gurjar and S. S. Badsara, Cs2CO3-Mediated synthetic strategy for iprobenfos derivatives via thiophilicaddition of H-phosphites on in situ generated thioaldehydes, ARKIVOC, 2024, 2, 202312055 Search PubMed.
- B. Huang, Y. Liu, H. Yao and Y. Zhao, NMR-based investigation into protein phosphorylation, Int. J. Biol. Macromol., 2020, 145, 53–63 CrossRef CAS PubMed.
- J. Baeza, M. J. Smallegan and J. M. Denu, Mechanisms and Dynamics of Protein Acetylation in Mitochondria, Trends Biochem. Sci., 2016, 41, 231–244 CrossRef CAS PubMed.
- M. E. Jakobsson, Enzymology and significance of protein histidine methylation, J. Biol. Chem., 2021, 297, 101130 CrossRef CAS PubMed.
- W. Gui, G. A. Davidson and Z. Zhuang, Chemical methods for protein site-specific ubiquitination, RSC Chem. Biol., 2021, 2, 450–467 RSC.
- J. Li and P. R. Chen, Development and application of bond cleavage reactions in bioorthogonal chemistry, Nat. Chem. Biol., 2016, 12, 129–137 CrossRef CAS PubMed.
- Y. Zheng, W. Lu, T. Ma and S. Huang, Recent advances in photochemical and electrochemical strategies for the synthesis of sulfonyl fluorides, Org. Chem. Front., 2024, 11, 217–235 RSC.
- Z. Jiang, N. Liu, D. Hu, G. Dong, Z. Miao, J. Yao, H. He, Y. Jiang, W. Zhang, Y. Wang and C. Sheng, The discovery of novel antifungal scaffolds by structural simplification of the natural product sampangine, Chem. Commun., 2015, 51, 14648–14651 RSC.
- S. Mukherjee, Y.-H. Hao and K. Orth, A newly discovered post-translational modification – the acetylation of serine and threonine residues, Trends Biochem. Sci., 2007, 32, 210–216 CrossRef CAS PubMed.
- B. Permanne, A. Sand, S. Ousson, M. Nény, J. Hantson, R. Schubert, C. Wiessner, A. Quattropani and D. Beher, O-GlcNAcase Inhibitor ASN90 is a Multimodal Drug Candidate for Tau and α-Synuclein Proteinopathies, ACS Chem. Neurosci., 2022, 13, 1296–1314 CrossRef CAS PubMed.
- M. N. Maas, J. C. J. Hintzen and J. Mecinović, Probing lysine posttranslational modifications by unnatural amino acids, Chem. Commun., 2022, 58, 7216–7231 RSC.
- Z.-W. Chen, A. Pratheepkumar, R. Bai, Y. Hu, S. S. Badsara, K.-W. Huang and C.-F. Lee, Cesium carbonate-catalyzed synthesis of phosphorothioates via S-phosphination of thioketones, Chem. Commun., 2022, 58, 11001–11004 RSC.
- S. A. Byrne, M. J. Bedding, L. Corcilius, D. J. Ford, Y. Zhong, C. Franck, M. Larance, J. P. Mackay and R. J. Payne, Late-stage modification of peptides and proteins at cysteine with diaryliodonium salts, Chem. Sci., 2021, 12, 14159–14166 RSC.
- K. Jiang, Y. Liu, Y. Yan, S. Wang, L. Liu and W. Yang, Combined chain- and step-growth dispersion polymerization toward PSt particles with soft, clickable patches, Polym. Chem., 2017, 8, 1404–1416 RSC.
- M. H. Asim, I. Nazir, A. Jalil, F. Laffleur, B. Matuszczak and A. Bernkop-Schnürch, Per-6-Thiolated Cyclodextrins: A Novel Type of Permeation Enhancing Excipients for BCS Class IV Drugs, ACS Appl. Mater., 2020, 12, 7942–7950 CrossRef CAS PubMed.
- P. C. Roehm and J. M. Berg, Selectivity of Methylation of Metal-Bound Cysteinates and Its Consequences, J. Am. Chem. Soc., 1998, 120, 13083–13087 CrossRef CAS.
- H. Seki, S. J. Walsh, J. D. Bargh, J. S. Parker, J. Carroll and D. R. Spring, Rapid and robust cysteine bioconjugation with vinylheteroarenes, Chem. Sci., 2021, 12, 9060–9068 RSC.
- R. Chang, M. Gruebele and D. E. Leckband, Protein Folding Stability and Kinetics in Alginate Hydrogels, Biomacromolecules, 2023, 24, 5245–5254 CrossRef CAS PubMed.
- N. G. Jayaprakash and A. Surolia, Role of glycosylation in nucleating protein folding and stability, Biochem. J., 2017, 474, 2333–2347 CrossRef CAS PubMed.
- Y. Zhang, Y. Deng, C. Wang, L. Li, L. Xu, Y. Yu and X. Su, Probing and regulating the activity of cellular enzymes by using DNA tetrahedron nanostructures, Chem. Sci., 2019, 10, 5959–5966 RSC.
- T. Kurakawa, N. Ueda, M. Maekawa, K. Kobayashi, M. Kojima, Y. Nagato, H. Sakakibara and J. Kyozuka, Direct control of shoot meristem activity by a cytokinin-activating enzyme, Nature, 2007, 445, 652–655 CrossRef CAS PubMed.
- E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Organometallic palladium reagents for cysteine bioconjugation, Nature, 2015, 526, 687–691 CrossRef CAS PubMed.
- J. Yu, X. Yang, Y. Sun and Z. Yin, Highly Reactive and Tracelessly Cleavable Cysteine-Specific Modification of Proteins via 4-Substituted Cyclopentenone, Angew. Chem., Int. Ed., 2018, 57, 11598–11602 CrossRef CAS PubMed.
- Y.-C. Liu and C.-F. Lee, N-Chlorosuccinimide-promoted synthesis of thiophosphates from thiols and phosphonates under mild conditions, Green Chem., 2014, 16, 357–364 RSC.
- C.-Y. Li, Y.-C. Liu, Y.-X. Li, D. M. Reddy and C.-F. Lee, Electrochemical Dehydrogenative Phosphorylation of Thiols, Org. Lett., 2019, 21, 7833–7836 CrossRef CAS PubMed.
- L. Deng, Y. Wang, H. Mei, Y. Pan and J. Han, Electrochemical Dehydrogenative Phosphorylation of Alcohols for the Synthesis of Organophosphinates, J. Org. Chem., 2019, 84, 949–956 CrossRef CAS PubMed.
- F. Xu, Y.-J. Li, C. Huang and H.-C. Xu, Ruthenium-Catalyzed Electrochemical Dehydrogenative Alkyne Annulation, ACS Catal., 2018, 8, 3820–3824 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qo01502a |
| This journal is © the Partner Organisations 2024 |