DOI:
10.1039/D4QO01348D
(Review Article)
Org. Chem. Front., 2024,
11, 5306-5324
Recent advances in electrochemical difunctionalization of alkenes and alkynes for the synthesis of organohalides
Received
22nd July 2024
, Accepted 21st August 2024
First published on 22nd August 2024
Abstract
Organohalides are particularly important and versatile, as they are found not only in bioactive natural products, pharmaceuticals, agrochemicals, and materials but also serve as fundamental reagents in synthetic chemistry. The rapid development of electrosynthesis as an environmentally friendly and efficient technology provides solutions for the synthesis of organohalides that avoid the use of chemical oxidants and minimize the formation of by-products. This review summarizes recent advances in the electrochemical difunctionalization of alkenes and alkynes for the synthesis of organohalides, as well as their underlying mechanisms. We anticipate that this work will stimulate new strategies for the synthesis of organohalides.
1. Introduction
Organohalides are important structural units in natural products, pharmaceuticals, agrochemicals, and materials.1 They are also fundamental and widely used coupling reagents in synthetic chemistry.2 As a consequence, great efforts have been devoted to incorporating the halide atom in organic molecules. Among the numerous established methods,3 the difunctionalization of alkenes and alkynes represents an efficient and straightforward strategy for the synthesis of halide-containing compounds, partially because alkenes and alkynes are readily available substrates. Although the dihalogenation of alkenes is one of the earliest organic reactions,4 wherein molecular chlorine or bromine is used as the halogenating reagent, which has several severe drawbacks, including the operation of toxic and corrosive reagent, poor chemoselectivity, harsh reaction conditions, and the formation of hazardous HCl or HBr as a by-product. In this context, alternatives including a new generation of halogenation reagents,5 the combination of electrophilic halogenated reagent and halogen source6 or halogen anion and external oxidant,7 halogenation by metal catalysis8 and photocatalysis9 among others have been extensively developed to mitigate the toxic and corrosive issues. Furthermore, the halogenium ion intermediate in the electrophilic halogenation reaction would be readily trapped by other nucleophiles, resulting in the corresponding halofunctionalized products being endowed with halide and another functional group at the same time.10 In addition, radical based-halofunctionalization of alkene/alkynes provides another strategy for synthesizing halogenated compounds.11 However, these strategies present one or more disadvantages, which include: (1) the preparation of some reagents still involves the use of elemental halides or high acquisition costs; (2) the need for stoichiometric amounts of oxidants; (3) the need for expensive transition metals or photocatalysts; (4) the harsh reaction conditions. Therefore, synthetic organic chemists are constantly motivated to search for environmentally friendly and sustainable tactics in this field.
With the renaissance of electrosynthesis as an environmentally friendly and sustainable platform for organic synthesis,12 it also offers an alternative out of the above-mentioned dilemma. This benefits from the direct use of traceless and green electrons as redox reagents, thereby circumventing the reliance on stoichiometric chemical redox reagents. Despite a few reviews have been comprehensively summarized on the electrochemical difunctionalization of alkenes and alkynes,13 the relevant synthesis of organohalides was sporadically discussed. Recently, Deagostino and co-workers reviewed the progress in the synthesis of chlorinated compounds via electrocatalysis and photoredox catalysis.14 In this review, we focally showcase the progress in the electrochemical difunctionalization of alkenes and alkynes for the synthesis of organohalides, as well as their proposed mechanisms (Scheme 1). In addition, possible directions for further development in this field are suggested with the hope of stimulating further new strategies for the synthesis of diverse organohalides.
 |
| | Scheme 1 Electrochemical difunctionalization of alkenes and alkynes for the synthesis of organohalides. | |
2. Electrochemical dihalogenation of alkenes and alkynes
Dihalogenation of alkenes/alkynes provides an efficient and straightforward strategy to simultaneously incorporate two vicinal carbon–halogen bonds in one molecule. In recent years, some elegant sustainable electrocatalytic dihalogenation approaches using different halogen sources have been developed in this field. Accordingly, the following section discusses recent advances in the electrochemical dihalogenation of alkenes and alkynes.
In 2017, the Lin group reported the first Mn-catalyzed electrochemical dichlorination of alkenes (Scheme 2).15 This operationally simple and efficient protocol utilizes electricity as an energy input and MgCl2 as a safe chlorine source, enabling various vicinal dichlorides in excellent yields with good to excellent diastereomeric ratios. Notably, a series of compounds bearing oxidatively fragile functionalities, including alcohol, aldehyde, sulfide, and carboxylic acid, remain unaffected under the optimized conditions, offering the possibility of further derivatization. Control experiments demonstrated that the mechanism via electrochemical generation of Cl2 followed by its electrophilic addition to the alkene could be excluded. Combined with the CV studies, the authors proposed the plausible mechanism for dichlorination as outlined at the bottom of Scheme 2. First, the Cl-bound MnII is formed through ligand exchange, followed by anodic oxidation that generates MnIIICl species. The MnIIICl species serves as a vital atom-transfer reagent and adds to the alkene to form a carbon-centered radical. After the Mn-assisted second round of radical group transfer, the final dichlorinated product is formed.
 |
| | Scheme 2 Electrochemical vicinal dichlorination of alkenes.15 | |
Fluorinating alkyl groups as a class of important structural motifs have received considerable attention since their functions in improving bioactivity and pharmacokinetics. Oxidative difluorination of alkenes provides an expedient strategy for incorporating fluorine atoms in molecules to access vicinal difluorides. However, the traditional strategies suffer from toxicity, high cost, stoichiometric oxidants, and limited substrate scope with restricted to electron-poor ones.16 To tackle these shortcomings, the Lennox group developed an electrochemical method for the generation of p-tolyl difluoro λ3-iodane (Scheme 3),17 which was previously reported as an oxidant for the difluorination of alkenes.16e,f Under their “undivided cell” optimized conditions, a range of alkenes harboring electron-poor moiety could be difluorinated in good to excellent yields. However, the electron-rich substrates failed since they are more easily oxidized before iodotoluene, thereby hampering the key formation of p-tolyl difluoro λ3-iodane. By redevising the reaction carried out in a divided cell, a wide range of electron-rich substrates was tolerated and afforded the desired products in moderate to very good yields. Mechanistically, the key to success is the in situ electrochemical generation of the hypervalent iodine, which serves as an oxidant and enables the subsequent difluorination of alkenes (Scheme 3). Meanwhile, the generation of H2 as the sole byproduct at the cathode makes this method attractive and eco-friendly.
 |
| | Scheme 3 Electrochemical vicinal difluorination of alkenes.17 | |
In contrast to the above-mentioned oxidative dichlorination and difluorination of alkenes, in which the generation of H2 is a sacrificial half-reaction, Waldvogel and Morandi reported a consecutive paired electrolysis merged with a halogen shuttle reaction for the vicinal dibromination and dichlorination of alkenes (Scheme 4).18 This strategy employs 1,2-dihaloethane such as 1,2-dibromoethane or 1,1,1,2-tetrachloroethane, as a simple, easily available, and noncorrosive halide source thereby eluding the use of volatile and corrosive Br2 and Cl2. It is worth mentioning that the addition of small amounts of 1,1,1,3,3,3-hexafluoisopropanol (HFIP) facilitates the reduction of the halide donor and suppresses the undesirable side reaction, and the supporting electrolyte can be recycled by simple crystallization. Under their optimal conditions, various dibrominated and dichlorinated products were obtained in moderate to good yields. This scalable reaction, equipped with two inexpensive graphite electrodes, tolerates a range of functional groups including amide, free alcohol, aldehyde, ester, and imide. Remarkably, the previously worldwide used insecticide, lindane as a persistent pollutant could be fully dechlorinated by interconversion with simple alkenes. The authors designed this reaction with the following possible mechanism. Initially, the single-electron reduction of the dihalide at the cathode produces the X− and releases the alkene with simultaneous reduction of the carbon-centered radical and subsequent loss of X−. Subsequently, oxidation of X− at the anode generates X+, which then undergoes electrophilic addition to the alkene to form the desired vicinal products.
 |
| | Scheme 4 Electrochemical vicinal dibromination and dichlorination of alkenes.18 | |
Apart from the consecutive paired electrolysis, Hilt and co-workers developed a linear paired electrolysis for the dibromination of alkenes during the same period (Scheme 5).19 The protocol shows a good substrate scope and affords the desired dibromination products in good to excellent yields with perfect current efficiency (up to 200%). Moreover, the functional group compatibility test indicates that several sensitive groups such as alkyne, ester, halides, and ethers are tolerated under the electrochemical conditions. Furthermore, the diiodination of cyclohexene is compatible with the linear paired electrolysis. However, the analogous dichlorination of cyclohexene failed due to the higher oxidation potential of the chloride anion. The plausible mechanism for the dibromination of alkenes outlined in Scheme 5 was proposed by the authors. At the cathode, the reduction of oxygen in the presence of a proton generates hydrogen peroxide, which then oxidizes the Br− to bromine. Bromination of the alkene then yields the final dibrominated product. Meanwhile, the Br− is oxidized at the anode to also afford bromine, making this reaction perform with perfect current efficiency.
 |
| | Scheme 5 Electrochemical vicinal dibromination of alkenes.19 | |
As one of the earliest organic reactions reported in 1877, dichlorination of alkenes typically delivers trans-dichlorinated products. Until 2015, the Denmark group disclosed the first elegant example of cis-dichlorination of alkenes, in which diselenide was employed as a precatalyst, benzyltriethylammonium chloride (BnEt3NCl) as a chlorine source, and the N-fluoropyridinium salt as an oxidant.20 Inspired by this work and to obviate the use of stochiometric amounts of oxidant, the Hilt group described an electrochemical version for the cis-dichlorination of alkenes (Scheme 6).21 This strategy is carried out with 50 mol% phenylselenyl chloride (PhSeCl) as a substoichiometric mediator and TBACl as the electrolyte and partially source of chloride in DMF. Under their optimized conditions, a variety of cyclic and acyclic produced cis-dichlorides with high diastereoselectivities. Typically, aliphatic alkene substrates worked well, while styrene derivatives afforded only moderate yields. In combination with control experiments and cyclic voltammetric (CV) studies, the authors proposed a possible mechanism for this cis-dichlorination of alkenes. First, phenylselenyl chloride serves two roles. On the one hand, it reacts with alkene to generate the phenylselenyl chloride-alkene adduct I. On the other hand, it is oxidized at the anode and combines with the Cl− to give PhSeCl3, which then reacts with phenylselenyl chloride-alkene adduct I to produce intermediate II. Finally, nucleophilic substitution by the Cl− leads to the cis-dichlorinated product.
 |
| | Scheme 6 Electrochemical vicinal cis-dichlorination of alkenes.21 | |
In 2019, the Lei group used HBr as a bromide source to realize the dibromination of alkenes and alkynes under electrochemical oxidation conditions (Scheme 7).22 The reaction tolerated a wide variety of aromatic alkenes and aliphatic alkenes and provided the dibrominated products in moderate to high yields. Moreover, this method was not limited to terminal alkenes, internal alkenes were also compatible and produced exclusive trans-dibromides. Notably, the reaction was easily scaled up to 200 mmol and afforded the pure product in 86% yield (50.9 g), which is inaccessible using the orthodox method. It is worth noting that this protocol can be applied to the dibromination of alkynes and furnished E-isomers as sole isomerism. Control experiments of the reaction of alkene with molecular Br2 also afforded the same dibromination product, indicating that the Br2 intermediate is involved.
 |
| | Scheme 7 Electrochemical vicinal dibromination of alkenes and alkynes.22 | |
3. Electrochemical intermolecular halofunctionalization of alkenes and alkynes
The heterodifunctionalization of alkenes and alkynes, leading to the concurrent formation of compounds with two useful functionalities, is a step-economy and sustainable strategy.23 Among them, halofunctionalization of alkenes and alkynes for the synthesis of compounds with halide and another functional group in a vicinal position is synthetically attractive since the C–X bond could easily participate in the array of transformations. This section focuses on summarizing the electrochemical halofunctionalization of alkenes and alkynes.
3.1 The simultaneous formation of C–X and C–C bonds
After accomplishing the Mn-catalyzed electrochemical dichlorination of alkenes,15 the Lin group achieved an anodically coupled electrolysis for the chlorotrifluoromethylation of alkenes (Scheme 8).24 The success of this proof-of-principle reaction is due to the redox potentials of CF3SO2˙/CF3SO2− (Ep/2 = 800 mV) and [MnII]–Cl/[MnIII]–Cl (E = 780 mV) are matched well, thereby fulfilling these two open-shell species generated simultaneously at the anode under mild conditions. Compared to the previously described photochemical method,25 this protocol showcases a wide substrate scope. For example, styrene derivatives bearing oxidatively sensitive functional groups and tetrasubstituted alkenes could be well tolerated. However, electron-rich styrenes failed due to the competing arene trifluoromethylation. By simply using KBr instead of MgCl2 as a bromide source, the reaction could be applied for bromotrifluoromethylation. Moreover, alkynes could also be chlorotrifluoromethylated using this electrocatalytic method. After a year, the same group accomplished the chloroalkylation of alkenes using malononitriles or cyanoacetates and NaCl as a chloride donor via the similar Mn-catalyzed anodically coupled electrolysis.26
 |
| | Scheme 8 Electrochemical chlorotrifluoromethylation/chloroalkylation of alkenes and alkynes.24,26 | |
In the same year, the Xu group disclosed an electrochemical method for the fluoroalkynylation of alkenes using Et4NF·3HF as a fluoride source and alkynyltrifluoroborate salts as the alkynylation source (Scheme 9).27 The alkene is firstly oxidized to the corresponding radical cation, which is then trapped by the alkynyltrifluoroborate to generate a carbon-centered radical intermediate. This intermediate further undergoes single-electron oxidation to give rise to the cation species, which is attacked by nucleophile F− to afford the final difunctionalized product.
 |
| | Scheme 9 Electrochemical fluoroalkynylation of alkenes.27 | |
3.2 The simultaneous formation of C–X and C–N3 bonds
The electrochemical azidoiodination of alkenes was first reported by the Zeng group in 2016 (Scheme 10).28 In this method, IN3, an explosive reagent that is conventionally used to react with alkenes to access vicinal iodoazides, could be generated by indirect electrolysis using NaI as an iodide source followed by reacting with NaN3. Driven by constant current electrolysis, the Markovnikov addition products were exclusively obtained. In the proposed mechanism, the in situ generated IN3 predominantly acts as an I+ source for the reaction with alkene to form an iodonium intermediate, which was trapped by azide to complete the reaction. Very recently, Ren, Meng, and co-workers reported an interesting electrode-switchable regiodivergent azidoiodination of alkenes (Scheme 10).29 When the reaction was conducted in an undivided cell with a graphite plate as the anode and a Pt plate as the cathode, Markovnikov α-azidoiodination products were obtained. Interestingly, when a Pt plate was used as the anode, the reaction afforded anti-Markovnikov β-azidoiodination products under otherwise identical conditions. Density functional theory (DFT) calculations revealed that the adsorption energy of I− on graphite and Pt plates is about −0.56 and −2.81 eV, respectively. Control experiments and cyclic voltammogram (CV) studies showed that I− underwent successive anodic oxidation processes to produce I2 at the graphite plate anode. In contrast, intermediate IO4− was generated at the Pt plate surface. Based on these findings, the authors proposed the following mechanism for this regiodivergent azidoiodination of alkenes (Scheme 10). When a graphite plate was used as the anode, the reaction proceeded via an iodonium ion mechanism to deliver Markovnikov addition products. When the Pt anode was employed, I− was oxidized to produce hypervalent IO4− due to the higher adsorption energy I− on the Pt surface. IO4− was able to oxidize TMSN3 to produce an azide radical, which underwent addition to the alkene, providing the corresponding α-carbon-centered radical. The resulting radical may undergo three processes. It could be further oxidized to the carbocation intermediate, which was attacked by N3− to yield an anti-Markovnikov product. Alternatively, it could also react with I˙ or I2 to deliver the final product, and the latter is more favorable.
 |
| | Scheme 10 Electrochemical azidoiodination of alkenes.28,29 | |
3.3 The simultaneous formation of C–X and C–P bonds
To further expand their strategy of Mn-catalyzed anodically coupled electrolysis, Lin and co-workers successfully developed the chlorophosphinoylation of alkenes in 2019 (Scheme 11).30 This electrocatalytic chlorophosphinoylation reaction tolerates a wide range of alkene derivatives with high efficiency and regioselectivity. In addition, alkynes were also compatible with this reaction and afforded the corresponding E-isomer as a single product. However, the attempt for bromophosphinoylation failed since only the dibromination product was detected.
 |
| | Scheme 11 Electrochemical chlorophosphinoylation of alkenes.30 | |
3.4 The simultaneous formation of C–X and C–O bonds
In 2019, the Li group reported a three-component electrochemical oxidative bromoesterification of alkenes,31 in which N-bromosuccinimide (NBS) as the Br source, TEMPO as the promoter with a constant current of 10 mA (Scheme 12, top). During the reaction, TEMPO+ could be generated by a single oxidation of TEMPO at the anode. Subsequent addition of NBS to the alkene yielded the bromonium ion intermediate with the assistance of TEMPO+. The bromoesterification product was ultimately formed through nucleophilic addition and deprotonation. Four years later, He and Guan used tetrabutylammonium iodide as both an electrolyte and an iodine source to realize iodoesterization under chemical oxidant-free conditions (Scheme 12, below).32 Furthermore, this method is also applicable to the chloroesterification and bromoesterification of alkenes using the appropriate halide source instead.
 |
| | Scheme 12 Electrochemical haloesterification of alkenes and alkynes.31,32 | |
In 2019, Hu, Fang, and co-workers developed an electrochemical oxidative method for the haloformyloxylation of alkenes using readily available NaCl or NaBr as the halide source and DMF as the formyloxylation reagent (Scheme 13).33 During the reaction, Cl− or Br− underwent a single-electron oxidation and formed the Cl˙ or Br˙. Then the resultant radical was added to the alkene and generated the corresponding benzyl radical, which was further oxidized to the benzyl carbocation. After nucleophilic attack and hydrolysis via an imine intermediate, the corresponding product was formed along with a hydrogen ion. In addition, the synthetic method could be used to synthesize trifluoromethylation-formyloxylation products by employing Langlois reagent (CF3SO2Na) as a trifluoromethyl radical source.
 |
| | Scheme 13 Electrochemical haloformyloxylation of alkenes.33 | |
The halide sources could also be acquired from organohalides. In 2020, the Li group reported a convergent electrochemical method for the alkoxybromination of alkenes (Scheme 14).34 Alkyl halides or aryl bromides could offer the halide anion either in the presence of MeOH and nBu4NOH or through reduction at the cathode. Meanwhile, the alkene was oxidized to the radical cation with the assistance of Cp2Fe. The combination of halide anion and radical cation delivered the alkyl radical, which was subsequently oxidized to the alkyl cation. Subsequent nucleophilic addition by alcohol generated the product. Besides, the halide anion could also be oxidized and provided the product via a halonium ion mechanism. A couple of years later, the Cai group also achieved the alkoxybromination of alkenes by employing dibromomethane (CH2Br2) as a bromide source under electrochemical oxidation conditions.35 Initially, single-electron oxidation of CH2Br2 at the anode generates bromine radical, which would undergo addition to an alkene, resulting in a carbon-centered radical. This radical would further undergo another SET and afford the corresponding carbocation intermediate. The addition of alcohol as a nucleophilic reagent and subsequent deprotonation leads to the formation of the product.
 |
| | Scheme 14 Electrochemical alkoxybromination of alkenes.34,35 | |
Inspired by previous Mn-catalyzed paired electrosynthesis,15,24 the Chen group reported a system for oxychlorination of styrenes (Scheme 15).36 Anodic oxidation of Ln−1MnIICl2 to Ln−1MnIIICl2 which could transfer chlorine radical to the alkene, affording the transient alkyl radical III. Single-electron reduction of oxygen produces the persistent superoxide ion O2˙−. The combination of these two species results in intermediate IV, which then decomposes to form compound V. Further oxidation by superoxide or electricity affords the chloroacetophenone product.
 |
| | Scheme 15 Electrochemical oxyclorination of alkenes.36 | |
The electrochemical oxydihalogenation of alkynes was soon reported by several groups (Scheme 16).37–40 In 2020, Wang, Zha, and co-workers disclosed the electrochemistry for the oxyhalogenation of alkynes using HCl or KBr as a halide source in a divided cell.37 The authors proposed that anodic oxidation of Cl− generates Cl2, which reacts with H2O to afford HClO. HClO then transforms into Cl2O which would add across the alkyne to finally provide the product. During the same period, the Huang group reported another electrochemical method to access the same α,α-dihaloketones using readily available organohalides such as CHCl3, CH2Cl2, ClCH2CH2Cl, and CH2Br2 as halogen sources in an undivided cell.38 Subsequently, the Lei group developed the electrochemical oxydibromination of arylalkynes using LiBr as both the halogen source and supporting electrolyte.39 Recently, the Chen group developed an efficient photoelectrochemical cerium-catalyzed method to synthesize α,α-dichloroketones via electricity enabling the ligand to metal charge transfer (LMCT) of Ce(IV) to provide the key Cl˙ intermediate.40
 |
| | Scheme 16 Electrochemical oxydihalogenation of alkynes.37–40 | |
The electrochemical hydroxybromination of alkenes was achieved by Terent'ev group in 2021 (Scheme 17).41 The choice of DMSO as a solvent to stabilize the anodically generated bromide cation to generate VI is crucial for the success of this method. This species then reacts with the alkene to produce the sulfonimsalt VII, which is attacked by water to furnish the desired product. The reaction proceeds via a cyclic bromonium cation mechanism, which is also a possible pathway, and the radical pathway cannot be ruled out completely. However, the observation that bromine drips from the electrodes during the reaction suggests that the radical route seems unlikely. The addition of AcOH as an additive is also very important as it could lower the concentration of hydroxide ions, thereby diminishing the by-production of epoxide and diol. Besides, by replacing the solvent with the mixture of CH3CN/ROH, the reaction could also provide the corresponding alkoxybromination product. The generation of bromide from HBr in an electrochemical flow reactor was demonstrated by Wirth in the same year.42 This methodology converts various alkenes into their corresponding dibromination and hydroxybromination products. In addition, the use of alcohols as nucleophiles and internal nucleophiles would also provide the desired alkoxybromination and cyclization products.
 |
| | Scheme 17 Electrochemical hydroxybromination of alkenes.41 | |
3.5 The simultaneous formation of C–X and C–S/Se bonds
The construction of the C–S bond is of great interest in organic synthesis because the resulting organosulfur compounds occur in many natural products and pharmaceuticals as well as versatile building blocks.43 Mechanistically different from Mn-catalyzed anodically coupled electrolysis for three-component difunctionalization of alkenes,15,24,26,32 the Lei group reported a Mn-catalyzed two-component chlorosulfonylation of alkenes/alkynes via convergent paired electrolysis (Scheme 18).44 As described, sulfonyl chloride, as a bifunctional reagent, is reduced to a sulfonyl radical and a chloride ion. Subsequently, the radical reacts with the alkene to furnish the alkyl radical intermediate. The resulting chloride anion is oxidized to the chlorine radical at the anode and subsequently combines with Mn(III)Cl2 to form Mn(III)Cl3, which acts as the atom-transfer reagent to pass the chlorine atom to the alkyl radical intermediate, and finally delivers the target product.
 |
| | Scheme 18 Electrochemical chlorosulfonylation of alkenes/alkynes.44 | |
Shortly thereafter, the groups of Ye45 and Xue46 independently developed an anodically oxidative electrolysis for fluorosulfonylation, starting from alkenes, sulfonylhydrazides, and triethylamine trihydrofluoride as the fluorination reagent (Scheme 19). They proposed the mechanism as follows: the anodically generated sulfonyl radical adds to the alkene to form a benzyl radical, which subsequently undergoes anodic oxidation to give the carbocation intermediate. Subsequent nucleophilic fluorination with NEt3·HF affords the final product. Given the important applications of fluorine- and sulfone-containing molecules and their continued interest in electrosynthesis, the Ye group reported a similar fluoroselenation reaction of olefins to access diverse β-fluoroselenides.47
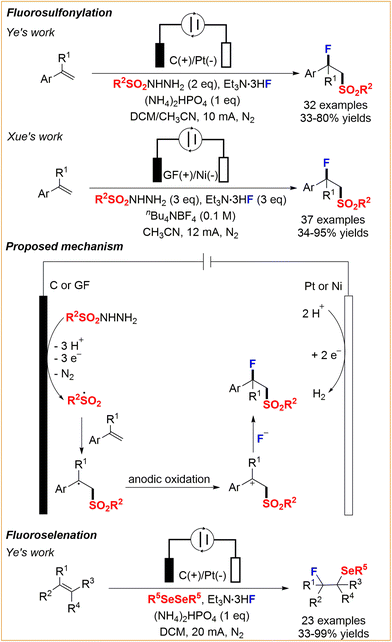 |
| | Scheme 19 Electrochemical fluorosulfonylation and fluoroselenation of alkenes.45,46,47 | |
One of the advantages of electrochemistry in organic chemistry is its promising capability to control reactivity by properly adjusting the applied potential. A good example of this was reported by Ye's group, in which the controllable fluorosulfenylation and flurosulfoxidation of alkenes was realized by applying an appropriate potential (Scheme 20).48 CV studies and sampling experiments support that a judicious choice of the applied potential is pivotal to controlling selectivity. By combining these results with other mechanistic experiments, the authors proposed the mechanism shown in Scheme 20. The thiyl radical would be obtained by a single-electron oxidation of thiophenol. Then the radical–radical combination affords disulfide, which could undergo either secondary oxidation or cathode reduction to release the thiyl radical. This radical then adds to the alkene, followed by anodic oxidation, delivering an episulfonium ion. Subsequent nucleophilic fluorination by NEt3·HF gives the desired fluorosulfenylation product. Alternatively, the disulfide loses an electron at the anode to generate a bis(arylthio)sulfonium ion which then reacts with the alkene and could also afford the episulfonium ion. When a higher cell potential was applied, fluorosulfides would be smoothly oxidized to fluorosulfoxides in the presence of CCl4.
 |
| | Scheme 20 Electrochemical fluorosulfonylation and flurosulfoxidation of alkenes.48 | |
The analogous chorosulfuration and chorosulfoxidation of alkenes using thiols and hydrochloride as a chloride source was soon reported by the Wang group (Scheme 21, top).49 The reaction generally produced the chorosulfenylated product within 10 hours under the optimized conditions. By Simply prolonging the reaction time to 22 hours, the desired sulfoxide products were obtained. During the same period, Ye et al. extended their previous work and reported the same reaction with the same starting materials and similar reaction conditions (Scheme 21, below).50 Importantly, diverse post-derivatizations of chlorosulfoxides by taking advantage of chlorine substituent as a practical handle highlight the synthetic potential.
 |
| | Scheme 21 Electrochemical chorosulfuration and chorosulfoxidation of alkenes.49,50 | |
Very recently, Waldvogel and Morandi documented another electrochemically enabled shuttle reaction for the bromothiolation of alkynes via a consecutive paired mechanism (Scheme 22).51 This protocol employed β-bromosulfides as a bifunctional reagent, enabling the construction of diverse bromothiolated products in high chemo-, regio-, and stereoselectivity. Furthermore, terminal alkenes were also applicable to this method and the anticipated chlorothiolated products could also be obtained using the corresponding β-chlorosulfide in place of β-bromosulfide. The diverse transformations of sulfide and bromide moieties in the product highlight its versatile synthetic values. CV analysis reveals that the aryl thiolate anion (ca. −0.2 V) is preferentially oxidized over the bromide anion (ca. 0.6 V), which is crucial for controlling the chemoselectivity. Therefore, the reduction of β-bromosulfides generates the aryl thiolate anion and the bromide anion along with 1,1-dichloroethene as a byproduct. The thiolate anion is then oxidized prior to the bromide anion and transformed to disulfide. Subsequently, the bromide anion is oxidized to the bromide radical or cation, which would react with disulfide to generate the reactive RSBr. The subsequent addition of alkyne provides the final products.
 |
| | Scheme 22 Electrochemical bromothiolation of alkynes.51 | |
4. Electrochemical intramolecular halocyclization
Electrochemically promoted difunctionalization of the unsaturated C–C bond via cascade intramolecular cyclization provides an efficient strategy for constructing functionalized cyclic and heterocyclic compounds. In this section, we summarize representative examples of the synthesis of halogenated cycles by intramolecular cyclization of enynes, alkenes, and alkynes under electrochemical conditions.
4.1 Intramolecular halocyclization of enynes
In 2018, the Lin group reported an electrochemical intramolecular cyclization of 1,6-enynes, leading to the synthesis of chlorotrifluoromethylated pyrrolidine derivatives (Scheme 23).52 Similar to their previous work, the reaction involves the parallel generation of two key intermediates, ˙CF3 and [MnIII]–Cl, at the anode, followed by addition to the 1,6-enyne substrate via trifluoromethylation, radical cyclization, and chlorination to afford the final product. Importantly, the introduction of 2,2′-bipyridine as a chelating ligand was key to guaranteeing the reaction with high stereoselectivity.
 |
| | Scheme 23 Electrochemical cyclization of 1,6-enynes toward pyrrolidines.52 | |
In 2020, Jiang, Tu, Hao, and co-workers developed an electrochemical method for the halosulfonylation of 1,6-enynes (Scheme 24).53 This radical-triggered three-component cascade cyclization enables to access halosulfonated 1-indanones in good yields with good stereoselectivities. Mechanistic studies showed that the reaction begins with the anodic oxidation of the iodide ion to its corresponding cation, which reacts with arylsulfonyl hydrazide to produce the arylsulfonyl iodide. This resulting intermediate then undergoes homolysis to generate an arylsulfonyl radical and an iodine radical. The former attacks the double bond of 1,6-enyne, followed by a 5-exo-dig cyclization to afford a vinyl radical. Due to the strong steric hindrance between the aryl and arylsulfonyl groups, the vinyl radical (Z)-VIII is the major stereoisomer, which is the reason for the good stereoselectivity. This vinyl radical can undergo two pathways to generate the final product either reacting with arylsulfonyl iodide or oxidizing to a vinyl cation and further reacting with iodide ion. Following the similar radical addition and cyclization mechanism, the same group reported an example of electrochemical iodosulfonylation of 1,5-enynes to access spiroindenes.54
 |
| | Scheme 24 Electrochemical cyclization of 1,6-enynes toward 1-indanones.53 | |
4.2 Intramolecular halocyclization of alkenes
Using the in situ electrochemical generation of hypervalent iodine as a key mediator in lieu of chemical oxidants, the Waldvogel group reported the synthesis of 5-fluoromethyl-2-oxazoline by fluorocyclization of N-allylcarboxamides under electrochemical conditions (Scheme 25).55 Soon after, the same group applied this strategy to the electrochemical fluorocyclization of N-propargylamides, providing the corresponding 5-fluoromethyl-2-oxazoles.56 To avoid exposure risk and solve scalability issues, the Wirth group developed the same fluorocyclization reaction in flow,57 which shows high efficiency compared to the batch reaction. Afterward, the Lennox group described a method for the preparation of 3-fluorinated chromanes from allylic phenol esters also using the electrochemical generation of hypervalent iodine species.58
 |
| | Scheme 25 Electrochemical fluorocyclization of alkenes toward oxazolines.55 | |
In 2019, Zhang, Chen, and colleagues developed an efficient electrochemical generation of molecular bromine that could be further used to trigger the semi-pinacol rearrangement of allylic alcohols for the synthesize of β-halocarbonyls (Scheme 26).59 It is worth noting that the simulated seawaters can be used as a halogen source in this system for the synthesis of two key intermediates of natural products. In the last several years, electrooxidation of halide salts to their radical or cation intermediates followed by reaction with alkene and subsequent cyclization has been successfully adopted to construct various cyclic and heterocyclic compounds.60
 |
| | Scheme 26 Electrochemical halogenation/semi-pinacol rearrangement of allylic alcohols.59 | |
4.3 Intramolecular halocyclization of alkynes
Similar to alkenes, halocyclization of functionalized alkynes under electrochemical conditions has emerged as an efficient strategy to construct various heterocyclic and carbocyclic compounds. For example, the Chen and Xu group reported an electrochemical method for the synthesis of spiro[4,5]troenones from N-aryl alkynamides (Scheme 27).61 Several groups including us reported electrochemical annulation of o-alkynylanilines to construct C3-halogenated indoles, respectively (Scheme 27).62 The electrochemical oxidative halocyclization of o-alkynylbenzamides to access isobenzofuran-1-imines was demonstrated by the Anandhan group (Scheme 27).63 These transformations generally proceed through the electrochemical oxidation of halide salt to halide radical or halide cation, which reacts with alkyne and subsequent cyclization to simultaneously forge C–X and C–C/N/O bonds.
 |
| | Scheme 27 Selected examples of electrochemical intramolecular halocyclization of alkynes.61–63 | |
5. Conclusions
This review summarizes recent representative examples of electrosynthesis of organohalides by difunctionalization of alkenes and alkynes. These reactions generally enabled the synthesis of organohalides without the need for exogenous chemical oxidants under mild conditions. Reaction types include electrochemical dihalogenation, electrochemical intermolecular halofunctionalization, and electrochemical intramolecular halocyclization of alkenes and alkynes. The electrochemical dihalogenation reaction mainly relies on the generation of halonium ion as a key intermediate, resulting in vicinal dihalide products with anti-selectivity. Although an example of cis-dichlorination of alkenes has been developed,21 the strategy for cis-difluorination, cis-dibromination, and cis-diiodination is still highly desirable as they are more valuable and reactive compared with their chloride analogue. The electrochemical intermolecular halofunctionalization of alkenes and alkynes has succeed in forging C–X and C–C/N/O/P/S/Se bonds simultaneously. However, there is no electrochemical method for introducing C–X and C–B/Si bonds. Despite asymmetric dihalogenation and halofunctionalization of alkenes have been well-established,64 the enantioselective variants of such reactions using electrocatalysis remain unexploited. This direction can be fulfilled by combining with transition metal catalysis or enzyme catalysis. Furthermore, the application of these approaches in the synthesis of natural products and bioactive compounds would be another promising direction given the synthetic value of the resulting organohalides.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (22201133 and 32171724) and the Natural Science Foundation of Jiangsu Province (BK20210607) for their financial support of this work.
References
-
(a) P. Jeschke, The unique role of halogen substituents in the design of modern agrochemicals, Pest Manage. Sci., 2010, 66, 10–27 CrossRef CAS;
(b) R. Wilcken, M. O. Zimmermann, A. Lange, A. C. Joerger and F. M. Boeckler, Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology, J. Med. Chem., 2013, 56, 1363–1388 CrossRef CAS PubMed;
(c) W.-j. Chung and C. D. Vanderwal, Stereoselective Halogenation in Natural Product Synthesis, Angew. Chem., Int. Ed., 2016, 55, 4396–4434 CrossRef CAS;
(d) V. H. Woolner, R. M. A. Gordon, J. H. Miller, M. Lein, P. T. Northcote and R. A. Keyzers, Halogenated Meroditerpenoids froma South Pacific Collection of the Red Alga Callophycus serratus, J. Nat. Prod., 2018, 81, 2446–2454 CrossRef CAS PubMed.
-
(a) J. Terao and N. Kambe, Cross-Coupling Reaction of Alkyl Halides with Grignard Reagents Catalyzed by Ni, Pd, or Cu Complexes with π-Carbon Ligand(s), Acc. Chem. Res., 2008, 41, 1545–1554 CrossRef CAS;
(b) N. Kambe, T. Iwasaki and J. Terao, Pd-catalyzed cross coupling reactions of alkyl halides, Chem. Soc. Rev., 2011, 40, 4937–4947 RSC;
(c) I. Saikia, A. J. Borah and P. Phukan, Use of Bromine and Bromo-Organic Compounds in Organic Synthesis, Chem. Rev., 2016, 116, 6837–7042 CrossRef CAS PubMed;
(d) X.-M. Cai, S. Li, W.-J. Wang, Y. Lin, W. Zhong, Y. Yang, F. E. Kühn, Y. Li, Z. Zhao and B. Z. Tang, Natural Acceptor of Coumarin-Isomerized Red-Emissive BioAIEgen for Monitoring Cu2+ Concentration in Live Cells via FLIM, Adv. Sci., 2024, 11, 2307078 CrossRef CAS;
(e) W. Zhong, Y. Wu, Y. Lin, S. Li, J. Zhang and X.-M. Cai, Fabrication of efficient and red-emissive salicylaldehyde Schiff base isomers for multi-scenario information decryption, J. Mater. Chem. C, 2024, 12, 11394–11401 RSC.
-
(a) D. A. Petrone, J. Ye and M. Lautens, Modern Transition-Metal-Catalyzed Carbon–Halogen Bond Formation, Chem. Rev., 2016, 116, 8003–8104 CrossRef CAS;
(b) J. Barluenga, J. M. Alvarez-Gutierrez and A. Ballesteros, Direct ortho Iodination of β- and γ-Aryl Alkylamine Derivatives, Angew. Chem., Int. Ed., 2007, 46, 1281–1283 CrossRef CAS;
(c) C. J. Teskey, A. Y. W. Lui and M. F. Greaney, Ruthenium-Catalyzed Meta-Selective C−H Bromination, Angew. Chem., Int. Ed., 2015, 54, 11677–11680 CrossRef CAS;
(d) X.-Q. Chu, M.-L. Zhi, S.-X. Zhang, Y.-W. Wang, W. Rao, H. Xu and Z.-L. Shen, Palladium-catalyzed defluorinative alkynylation of polyfluoroalkyl ketones with alkynes for the synthesis of fluorinated fused furans, Org. Chem. Front., 2021, 8, 572–578 RSC.
-
(a) A. Atterberg and O. Widman, Neue Chlornaphtaline, Ber. Dtsch. Chem. Ges., 1877, 10, 1841–1844 CrossRef;
(b)
H. O. House, Modern Synthetic Reactions, ed. W. A. Benjamin, New York, 2nd edn, 1972, pp. 263–274 Search PubMed;
(c)
M. B. Smith, Advanced Organic Chemistry, ed. Wiley, New York, 7th edn, 2013, p. 982 Search PubMed.
-
(a) T. Arai, K. Horigane, T. K. R. Suzuki, R. Itoh and M. Yamanaka, Catalytic Asymmetric Iodoesterification of Simple Alkenes, Angew. Chem., Int. Ed., 2020, 59, 12680–12683 CrossRef CAS;
(b) M. Eissen and D. Lenoir, Electrophilic Bromination of Alkenes: Environmental, Health and Safety Aspects of New Alternative Methods, Chem. – Eur. J., 2008, 14, 9830–9841 CrossRef CAS;
(c) C. Nilewski, R. W. Geisser and E. M. Carreira, Total synthesis of a chlorosulpholipid cytotoxin associated with seafood poisoning, Nature, 2009, 457, 573–576 CrossRef CAS;
(d) K. C. Nicolaou, N. L. Simmons, Y. Ying, P. M. Heretsch and J. S. Chen, Enantioselective Dichlorination of Allylic Alcohols, J. Am. Chem. Soc., 2011, 133, 8134–8137 CrossRef CAS;
(e) J. Salazar and R. Dorta, Pentylpyridinium Tribromide: A Vapor Pressure Free Room Temperature Ionic Liquid Analogue of Bromine, Synlett, 2004, 1318–1320 CrossRef CAS;
(f) G. A. Olah, J. T. Welch, Y. D. Vankar, M. Nojima, I. Kerekes and J. A. Olah, Synthetic Methods and Reactions. 63. Pyridinium Poly(hydrogen fluoride) (30% pyridine-70% hydrogen fluoride): a Convenient Reagent for Organic Fluorination Reactions, J. Org. Chem., 1979, 44, 3872–3881 CrossRef CAS;
(g) X. Zeng, C. Gong, J. Zhang and J. Xie, A simple and highly diasteroselective approach for the vicinal dichlorination of functional olefins, RSC Adv., 2016, 6, 85182–85185 RSC.
-
(a) M. Finkelstein, S. A. Hart, S. D. Ross and L. Eberson, Complexes between N-bromosuccinimide and quaternary ammonium bromides and their role in the addition of bromine to olefins, J. Org. Chem., 1986, 51, 3548–3551 CrossRef CAS;
(b) L.-X. Shao and M. Shi,
N-Bromosuccinimide and Lithium Bromide: An Efficient Combination for the Dibromination of Carbon-Carbon Unsaturated Bonds, Synlett, 2006, 1269–1271 CrossRef CAS;
(c) D. X. Hu, G. M. Shibuya and N. Z. Burns, Catalytic Enantioselective Dibromination of Allylic Alcohols, J. Am. Chem. Soc., 2013, 135, 12960–12963 CrossRef CAS PubMed.
-
(a) P. F. Richardson and I. E. Markó, On the Permanganate-Mediated Dichlorination of Olefins, Synlett, 1991, 733–736 CrossRef CAS;
(b) K. D. Donnelly, W. E. Fristad, B. J. Gellerman, J. Peterson and B. Selle, Chlorination of Alkenes by Manganese(III) Chloride Species, Tetrahedron Lett., 1984, 25, 607–610 CrossRef CAS;
(c) S. V. Kohlhepp and T. Gulder, Hypervalent Iodine(III) Fluorinations of alkenes and diazo compounds: new opportunities in fluorination chemistry, Chem. Soc. Rev., 2016, 45, 6270–6288 RSC;
(d) G. K. Dewkar, S. V. Narina and A. Sudalai, NaIO4-Mediated Selective Oxidative Halogenation of Alkenes and Aromatics Using Alkali Metal Halides, Org. Lett., 2003, 5, 4501–4504 CrossRef CAS PubMed;
(e) J. Ren and R. Tong, Convenient in situ generation of various dichlorinating agents from oxone and chloride: diastereoselective dichlorination of allylic and homoallylic alcohol derivatives, Org. Biomol. Chem., 2013, 11, 4312–4315 RSC;
(f) S. Liu, Y. Zhang, K. Gong, X. Zeng, F. Xie, B. Xu and J. Han, Widely applicable (radio)dihalogenation of alkynes and alkenes using two different nucleophilic alkali metal halides, Org. Chem. Front., 2022, 9, 6826–6832 RSC.
-
(a) K. Ren, M. Wang and L. Wang, FeX3-Promoted Intermolecular Addition of Benzylic Alcohols to Aromatic Alkynes: A Mild and Efficient Strategy for the Synthesis of Alkenyl Halides, Eur. J. Org. Chem., 2010, 565–571 CrossRef CAS;
(b) Y. F. Zheng, J. Yu, G. B. Yan, X. Li and S. Luo, FeBr3-catalyzed dibromination of alkenes and alkynes, Chin. Chem. Lett., 2011, 22, 1195–1198 CrossRef CAS;
(c) D. X. Hu, G. M. Shibuya and N. Burns, Catalytic Enantioselective Dibromination of Allylic Alcohols, J. Am. Chem. Soc., 2013, 135, 12960–12963 CrossRef CAS PubMed;
(d) X. Li, S. He and Q. Song, Diethylzinc-Mediated Radical 1,2-Addition of Alkenes and Alkynes, Org. Lett., 2021, 23, 2994–2999 CrossRef CAS;
(e) X. Song, S. Meng, H. Zhang, Y. Jiang, A. S. C. Chanab and Y. Zou, Dibrominated addition and substitution of alkenes catalyzed by Mn2(CO)10, Chem. Commun., 2021, 57, 13385–13388 RSC.
-
(a) J. D. Griffin, C. L. Cavanaugh and D. A. Nicewicz, Reversing the Regioselectivity of Halofunctionalization Reactions through Cooperative Photoredox and Copper Catalysis, Angew. Chem., Int. Ed., 2017, 56, 2097–2100 CrossRef CAS;
(b) P. Lian, W. Long, J. Li, Y. Zheng and X. Wan, Visible-light-induced Vicinal Dichlorination of Alkenes through LMCT Excitation of CuCl2, Angew. Chem., Int. Ed., 2020, 59, 23603–23608 CrossRef CAS PubMed;
(c) H. Singh, S. I. N. H. Inaththappulige, R. K. Tak and R. Gir, α-Bromoacetate as a Mild and Safe Brominating Agent in the Light Driven Vicinal Dibromination of Unactivated Alkenes and Alkynes, Org. Lett., 2024, 26, 5478–5481 CrossRef CAS;
(d) J. N. Mo, W. L. Yu, J. Q. Chen, X. Q. Hu and P. F. Xu, Regiospecific Three-Component Aminofluorination of Olefins via Photoredox Catalysis., Org. Lett., 2018, 20, 4471–4474 CrossRef CAS PubMed;
(e) J. Fang, Z. K. Wang, S. W. Wu, W. G. Shen, G. Z. Ao and F. Liu, Photoredox-catalysed chloro-, bromo- and trifluoromethylthio-trifluoromethylation of unactivated alkenes with sodium triflinate, Chem. Commun., 2017, 53, 7638–7641 RSC;
(f) Y. Yu, A. Liu, J. He, C. Wang, H. Mei and J. Han, Visible-light-irradiated tandem sulfonylation/cyclization of indole tethered alkenes for the synthesis of tetrahydrocarbazoles, Chin. Chem. Lett., 2022, 33, 4886–4890 CrossRef CAS;
(g) H. Mei, A. Liu, J. He, Y. Yu and J. Han, Visible-Light-Irradiated Cascade Reaction of Indole-Tethered Alkenes to Access Tetracyclic Tetrahydro-γ-carbolines, Org. Lett., 2022, 24, 2630–2635 CrossRef CAS;
(h) A. A. Festa, O. A. Storozhenko, L. G. Voskressensky and E. V. Van der Eycken, Visible light-mediated halogenation of organic compounds, Chem. Soc. Rev., 2023, 52, 8678–8698 RSC.
-
(a) W. Z. Yu, F. Chen, Y. A. Cheng and Y.-Y. Yeung, Catalyst-Free and Metal-Free Electrophilic Bromoamidation of Unactivated Olefins Using the N-Bromosuccinimide/Sulfonamide Protocol, J. Org. Chem., 2015, 80, 2815–2821 CrossRef CAS PubMed;
(b) L.-H. Lu, S.-J. Zhou, M. Sun, J.-L. Chen, W. Xia, X. Yu, X. Xu and W.-M. He, Metal- and Solvent-Free Ultrasonic Multicomponent Synthesis of (Z)-β-Iodo Vinylthiocyanates, ACS Sustainable Chem. Eng., 2019, 7, 1574–1579 CrossRef CAS.
-
(a) L. Song, S. Luo and J.-P. Cheng, Visible-light promoted intermolecular halofunctionalization of alkenes with N-halogen saccharins, Org. Chem. Front., 2016, 3, 447–452 RSC;
(b) Z. Liu, H. Chen, Y. Lv, X. Tan, H. Shen, H.-Z. Yu and C. Li, Radical Carbofluorination of Unactivated Alkenes with Fluoride Ions, J. Am. Chem. Soc., 2018, 140, 6169–6175 CrossRef CAS;
(c) W. Deng, W. Feng, Y. Li and H. Bao, Merging Visible-Light Photocatalysis and Transition-Metal Catalysis in Three-Component Alkyl-Fluorination of Olefins with a Fluoride Ion, Org. Lett., 2018, 20, 4245–4249 CrossRef CAS;
(d) P. Guo, J.-F. Han, G.-C. Yuan, L. Chen, J.-B. Liao and K.-Y. Ye, Cobalt-Catalyzed Divergent Aminofluorination and Diamination of Styrenes with N-Fluorosulfonamides, Org. Lett., 2021, 23, 4067–4071 CrossRef CAS PubMed.
- For selected reviews, see:
(a) R. Francke and R. D. Little, Redox catalysis in organic electrosynthesis: basic principles and recent developments, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC;
(b) M. Yan, Y. Kawamata and P. S. Baran, Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed;
(c) Y. Jiang, K. Xu and C. Zeng, Use of Electrochemistry in the Synthesis of Heterocyclic Structures, Chem. Rev., 2018, 118, 4485–4540 CrossRef CAS PubMed;
(d) M. D. Kärkäs, Electrochemical strategies for C–H functionalization and C–N bond formation, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC;
(e) A. Wiebe, T. Gieshoff, S. Moehle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Electrifying Organic Synthesis, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed;
(f) S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and J. C. Kampf, Electrochemical Arylation Reaction, Chem. Rev., 2018, 118, 6706–6765 CrossRef CAS PubMed;
(g) J. C. Siu, N. Fu and S. Lin, Catalyzing Electrosynthesis: A Homogeneous Electrocatalytic Approach to Reaction Discovery, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS PubMed;
(h) K.-J. Jiao, Y.-K. Xing, Q.-L. Yang, H. Qiu and T.-S. Mei, Site-Selective C–H Functionalization via Synergistic Use of Electrochemistry and Transition Metal Catalysis, Acc. Chem. Res., 2020, 53, 300–310 CrossRef CAS;
(i) J. Liu, L. Lu, D. Wood and S. Lin, New Redox Strategies in Organic Synthesis by Means of Electrochemistry and Photochemistry, ACS Cent. Sci., 2020, 6, 1317–1340 CrossRef CAS;
(j) Y. Yuan, J. Yang and A. Lei, Recent advances in electrochemical oxidative cross-coupling with hydrogen evolution involving radicals, Chem. Soc. Rev., 2021, 50, 10058–10086 RSC;
(k) C. Ma, P. Fang, Z.-R. Liu, S.-S. Xu, K. Xu, X. Cheng, A. Lei, H.-C. Xu, C. Zeng and T.-S. Mei, Recent advances in organic electrosynthesis employing transition metal complexes as electrocatalysts, Sci. Bull., 2021, 66, 2412–2429 CrossRef CAS;
(l) L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Electrocatalysis as an enabling technology for organic synthesis, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC;
(m) X. Cheng, A. Lei, T.-S. Mei, H.-C. Xu, K. Xu and C. Zeng, Recent Applications of Homogeneous Catalysis in Electrochemical Organic Synthesis, CCS Chem., 2022, 4, 1120–1152 CrossRef CAS;
(n) C. A. Malapit, M. B. Prater, J. R. C.-Pardo, M. Li, T. D. Pham, T. P. McFadden, S. Blank and S. D. Minteer, Advances on the Merger of Electrochemistry and Transition Metal Catalysis for Organic Synthesis, Chem. Rev., 2022, 122, 3180–3218 CrossRef CAS PubMed;
(o) C. Yin, S. Tang, J. Mei, X. Hu and H. Zhang, Electrochemical synthesis and transformation of organoboron compounds, Org. Chem. Front., 2023, 10, 3361–3377 RSC.
-
(a) G. S. Sauer and S. Lin, An Electrocatalytic Approach to the Radical Difunctionalization of Alkenes, ACS Catal., 2018, 8, 5175–5187 CrossRef CAS;
(b) G. M. Martins, B. Shirinfar, T. Hardwick and N. Ahmed, A Green Approach: Vicinal Oxidative Electrochemical Alkene Difunctionalization, ChemElectroChem, 2019, 6, 1300–1315 CrossRef CAS;
(c) G. M. Martins, B. Shirinfar, T. Hardwick, A. Murtazae and N. Ahmed, Organic electrosynthesis: electrochemical alkyne functionalization, Catal. Sci. Technol., 2019, 9, 5868–5881 RSC;
(d) H. Mei, Z. Yin, J. Liu, H. Sun and J. Han, Recent Advances on the Electrochemical Difunctionalization of Alkenes/Alkynes, Chin. J. Chem., 2019, 37, 292–301 CrossRef CAS;
(e) M. Bortolami, R. Petrucci, D. Rocco, V. Scarano and I. Chiarotto, Alkynes as Building Blocks, Intermediates and Products in the Electrochemical Procedures Since 2000, ChemElectroChem, 2021, 8, 3604–3613 CrossRef CAS;
(f) D. A. Thadathil, A. Varghese and K. V. Radhakrishnan, The Renaissance of Electro-Organic Synthesis for the Difunctionalization of Alkenes and Alkynes: A Sustainable Approach, Asian J. Org. Chem., 2021, 10, 2820–2847 CrossRef CAS;
(g) X. Li, P. Tao, Y. Cheng, Q. Hu, W. Huang, Y. Li, Z. Luo and G. Huang, Recent Progress on the Electrochemical Difunctionalization of Alkenes/Alkynes, Chin. J. Org. Chem., 2022, 42, 4169–4201 CrossRef CAS;
(h) J.-H. Qin, Ni. Nan and J.-H. Li, Electrochemical Difunctionalization of Alkenes, Synthesis, 2023, 55, 2843–2859 CrossRef CAS;
(i) B. Dong, J. Shen and L.-G. Xie, Recent developments on 1,2-difunctionalization and hydrofunctionalization of unactivated alkenes and alkynes involving C–S bond formation, Org. Chem. Front., 2023, 10, 1322–1345 RSC.
- S. Parisotto, E. Azzi, A. Lanfranco, P. Renzi and A. Deagostino, Recent Progresses in the Preparation of Chlorinated Molecules: Electrocatalysis and Photoredox Catalysis in the Spotlight, Reactions, 2022, 3, 233–253 CrossRef CAS.
- N. Fu, G. S. Sauer and S. Lin, Electrocatalytic Radical Dichlorination of Alkenes with Nucleophilic Chlorine Sources, J. Am. Chem. Soc., 2017, 139, 15548–15553 CrossRef CAS PubMed.
-
(a) A. E. Feiring, S. Rozen and E. R. Wonchoba, Synthesis of arylperfluoroalkyl ethers by direct fluorination, J. Fluorine Chem., 1998, 89, 31–34 CrossRef CAS;
(b) W. Dmowski and T. Kozłowski, Electrochemical fluorination of methyl cinnamates in Et3N·3HF-MeCN, Electrochim. Acta, 1997, 42, 513–523 CrossRef CAS;
(c) J. H. H. Meurs and W. Eilenberg, Oxidative Fluorination in Amine-hf Mixtures, Tetrahedron, 1991, 47, 705–714 CrossRef CAS;
(d) G. S. Lal, Site-selective fluorination of organic compounds using 1-alkyl-4-fluoro-1,4-diazabicyclo[2.2.2]octane salts (selectfluor reagents), J. Org. Chem., 1993, 58, 2791–2796 CrossRef CAS;
(e) S. Hara, J. Nakahigashi, K. Ishi-i, M. Sawaguchi, H. Sakai, T. Fukuhara and N. Yoneda, Difluorination of Alkenes with Iodotoluene Difluoride, Synlett, 1998, 495–496 CrossRef CAS;
(f) I. G. Molnr and R. Gilmour, Catalytic Difluorination of Olefins, J. Am. Chem. Soc., 2016, 138, 5004–5007 CrossRef PubMed;
(g) S. M. Banik, J. W. Medley and E. N. Jacobsen, Catalytic, Diastereoselective 1,2-Difluorination of Alkenes, J. Am. Chem. Soc., 2016, 138, 5000–5003 CrossRef CAS PubMed.
- S. Doobary, A. T. Sedikides, H. P. Caldora and D. L. Poole, Electrochemical Vicinal Difluorination of Alkenes: Scalable and Amenable to Electron-Rich Substrates, Angew. Chem., Int. Ed., 2020, 59, 1155–1160 CrossRef CAS PubMed.
- X. Dong, J. L. Roecki, S. R. Waldvogel and B. Morandi, Merging shuttle reactions and paired electrolysis for reversible vicinal dihalogenations, Science, 2021, 371, 507–514 CrossRef CAS PubMed.
- J. Strehl, M. L. Abraham and G. Hilt, Linear Paired Electrolysis—Realising 200% Current Efficiency for Stoichiometric Transformations—The Electrochemical Bromination of Alkenes, Angew. Chem., Int. Ed., 2021, 60, 9996–10000 CrossRef CAS PubMed.
- A. J. Cresswell, S. T.-C. Eey and S. E. Denmark, Catalytic, stereospecific syn-dichlorination of alkenes, Nat. Chem., 2015, 7, 146–152 CrossRef CAS PubMed.
- J. Strehl, C. Fastie and G. Hilt, The Electrochemical cis-Chlorination of Alkenes, Chem. – Eur. J., 2021, 27, 17341–17345 CrossRef CAS PubMed.
- Y. Yuan, A. Yao, Y. Zheng, M. Gao, Z. Zhou, J. Qiao, J. Hu, B. Ye, J. Zhao, H. Wen and A. Lei, Electrochemical Oxidative Clean Halogenation Using HX/NaX with Hydrogen Evolution, iScience, 2019, 12, 293–303 CrossRef CAS PubMed.
-
(a) B. Fu, J. Escorihuela, J. Han, S. Fustero, P. Barrio, M. Sodeoka, S. Kawamura, A. Sorochinsky and V. A. Soloshonok, Recent Advances on the Halo- and Cyano-Trifluoromethylation of Alkenes and Alkynes, Molecules, 2021, 26, 7221 CrossRef CAS PubMed;
(b) X.-D. Song, X.-R. Li, Y.-W. Wang, X.-Q. Chu, W. Rao, H. Xu, G.-Z. Han and Z.-L. Shen, Indium-mediated difunctionalization of iodoalkyl-tethered unactivated alkenes via an intramolecular cyclization and an ensuing palladium-catalyzed cross-coupling reaction with aryl halides, Org. Chem. Front., 2020, 7, 2703–2709 RSC;
(c) T. Yang, X. Chen, W. Rao and M. J. Koh, Broadly Applicable Directed Catalytic Reductive Difunctionalization of Alkenyl Carbonyl Compounds, Chem, 2020, 6, 738–751 CrossRef CAS;
(d) Z. Dong, L. Song and L.-A. Chen, Enantioselective Ni-Catalyzed Three-Component Dicarbofunctionalization of Alkenes, ChemCatChem, 2023, 15, e202300803 CrossRef CAS.
- K. Ye, G. Pombar, N. Fu, G. S. Sauer, I. Keresztes and S. Lin, Anodically Coupled Electrolysis for the Heterodifunctionalization of Alkenes, J. Am. Chem. Soc., 2018, 140, 2438–2441 CrossRef CAS PubMed.
- S. H. Oh, Y. R. Malpani, N. Ha, Y.-S. Jung and S. B. Han, Vicinal Difunctionalization of Alkenes: Chlorotrifluoromethylation with CF3SO2Cl by Photoredox Catalysis, Org. Lett., 2014, 16, 1310–1313 CrossRef CAS PubMed.
- N. Fu, Y. Shen, A. R. Allen, L. Song, A. Ozaki and S. Lin, Mn-Catalyzed Electrochemical Chloroalkylation of Alkenes, ACS Catal., 2019, 9, 746–754 CrossRef CAS PubMed.
- P. Xiong, H. Long and H.-C. Xu, Electrochemical Fluoroalkynylation of Aryl Alkenes with Fluoride Ions and Alkynyltrifluoroborate Salts, Asian J. Org. Chem., 2019, 8, 658–660 CrossRef CAS.
- W.-Q. Yan, M.-Y. Lin, R. D. Little and C.-C. Zeng, Electrochemical regioselective azidoiodination of alkenes, Tetrahedron, 2017, 73, 764–770 CrossRef CAS.
- X.-L. Sun, C.-X. Xia, Y. Ren, Y.-J. Li, Z.-Q. Cao and L.-G. Meng, Electrode-switchable: exploring this new strategy to achieve regiodivergent azidoiodination of alkenes, Org. Chem. Front., 2024, 11, 2189–2194 RSC.
- L. Lu, N. Fu and S. Lin, Three-Component Chlorophosphinoylation of Alkenes via Anodically Coupled Electrolysis, Synlett, 2019, 30, 1199–1203 CrossRef CAS.
- C. Wan, R.-J. Song and J.-H. Li, Electrooxidative 1,2-Bromoesterification of Alkenes with Acids and N-Bromosuccinimide, Org. Lett., 2019, 21, 2800–2803 CrossRef CAS PubMed.
- Y.-F. Tan, Y.-N. Zhao, D. Yang, J.-F. Lv, Z. Guan and Y.-H. He, Electrochemical Synthesis of β-Iodoesters by 1,2-Iodoesterization of Unactivated Alkenes with Carboxylic Acids and Tetrabutylammonium Iodide, J. Org. Chem., 2023, 88, 5161–5171 CrossRef CAS PubMed.
- X. Sun, H.-X. Ma, T.-S. Mei, P. Fang and Y.-L. Hu, Electrochemical Radical Formyloxylation–Bromination, −Chlorination, and −Trifluoromethylation of Alkenes, Org. Lett., 2019, 21, 3167–3171 CrossRef CAS PubMed.
- T.-T. Zhang, M.-J. Luo, Y. Li, R.-J. Song and J.-H. Li, Electrochemical Alkoxyhalogenation of Alkenes with Organohalides as the Halide Sources via Dehalogenation, Org. Lett., 2020, 22, 7250–7254 CrossRef CAS PubMed.
- Z. Fu, F. Chen, G. Hao, X. Yi, J. Zeng and H. Cai, Electrochemical Difunctionalization of Alkenes towards the Synthesis of β-Bromoethers under Metal-Free Conditions, Synthesis, 2023, 55, 3040–3046 CrossRef CAS.
- S. Tian, X. Jia, L. Wang, B. Li, S. Liu, L. Ma, W. Gao, Y. Wei and J. Chen, The Mn-catalyzed paired electrochemical facile oxychlorination of styrenes via the oxygen reduction reaction, Chem. Commun., 2019, 55, 12104–12107 RSC.
- Z. Li, Q. Sun, P. Qian, K. Hu, Z. Zha and Z. Wang, Electrochemical synthesis of α,α-dihaloacetophenones from terminal alkyne derivatives, Chin. Chem. Lett., 2020, 31, 1855–1858 CrossRef CAS.
- X. Meng, Y. Zhang, J. Luo, F. Wang, X. Cao and S. Huang, Electrochemical Oxidative Oxydihalogenation of Alkynes for the Synthesis of α,α-Dihaloketones, Org. Lett., 2020, 22, 1169–1174 CrossRef CAS PubMed.
- D. Wang, Z. Wan, H. Zhang and A. Lei, Electrochemical Oxidative Functionalization of Arylalkynes: Access to α,α-Dibromo Aryl Ketones, Adv. Synth. Catal., 2021, 363, 1022–1027 CrossRef CAS.
- D. Shao, Y. Wu, S. Hu, W. Gao, Y. Du, X. Jia, S. Liu, M. Zhou and J. Chen, Decoupled Photoelectrochemical Cerium-Catalyzed Oxydichlorination of Alkynes: Slow Releasing of Chloride Ions and Chlorine Radicals, ACS Sustainable Chem. Eng., 2022, 10, 10294–10302 CrossRef CAS.
- O. V. Bityukov, V. A. Vil’, G. I. Nikishin and A. O. Terent'ev, Alkene, Bromide, and ROH – How To Achieve Selectivity? Electrochemical Synthesis of Bromohydrins and Their Ethers, Adv. Synth. Catal., 2021, 363, 3070–3078 CrossRef CAS.
- J. Seitz and T. Wirth, Electrochemical Bromofunctionalization of Alkenes in a Flow Reactor, Org. Biomol. Chem., 2021, 19, 6892–6896 RSC.
-
(a) N. Wang, P. Saidhareddy and X. Jiang, Construction of sulfur-containing moieties in the total synthesis of natural products, Nat. Prod. Rep., 2020, 37, 246–275 RSC;
(b) C. Zhao, K. P. Rakesh, L. Ravidar, W. Y. Fang and H. L. Qin, Pharmaceutical and medicinal significance of sulfur (SVI)-Containing motifs for drug discovery: A critical review, Eur. J. Med. Chem., 2019, 162, 679–734 CrossRef CAS PubMed;
(c) P. Pei, M. Zhao, D. Lin, Z. Dong, L. Song and L.-A. Chen, Manganese-Catalyzed Redox-Neutral Thiolation of Alkyl Halides with Thioformates, Angew. Chem., Int. Ed., 2023, 62, e202305510 CrossRef CAS PubMed.
- M. Yu, H. Wang, Y. Gao, F. Bu, H. Cong and A. Lei, Manganese-catalyzed chlorosulfonylation of terminal alkene and alkyne via convergent paired electrolysis, Cell Rep. Phys. Sci., 2021, 2, 100476 CrossRef CAS.
- Y.-M. Jiang, Y. Yu, S.-F. Wu, H. Yan, Y. Yuan and K.-Y. Ye, Electrochemical Fluorosulfonylation of Styrenes, Chem. Commun., 2021, 57, 11481–11484 RSC.
- B. Zhao, Z. Pan, A. Zhu, Y. Yue, M. Ma and F. Xue, Electrochemical fluorosulfonylation of alkenes to access vicinal fluorinated sulfones derivatives, Tetrahedron, 2022, 106–107, 132651 CrossRef CAS.
- S.-F. Wu, Y. Yu, Y. Yuan, Z. Li and K.-Y. Ye, Electrochemical Synthesis of β-Fluoroselenides, Eur. J. Org. Chem., 2022, e202201032 CrossRef CAS.
- Y. Yu, Y. Jiang, S. Wu, Z. Shi, J. Wu, Y. Yuan and K. Ye, Electrochemistry enabled selective vicinal fluorosulfenylation and fluorosulfoxidation of alkenes, Chin. Chem. Lett., 2022, 33, 2009–2014 CrossRef CAS.
- P. Zhou, K. Niu, H. Song, Y. Liu and Q. Wang, Difunctionalization of unactivated olefins via selective electrochemical chlorosulfuration or chlorosulfoxidation, Green Chem., 2022, 24, 5760–5763 RSC.
- Y. Yu, Y.-M. Jiang, X.-B. Zhu, Y.-Y. Lin, Y. Yuan and K.-Y. Ye, Electrochemical β-chlorosulfoxidation of alkenes, Org. Chem. Front., 2022, 9, 5586–5559 RSC.
- X. Dong, M. Klein, S. R. Waldvogel and B. Morandi, Controlling Selectivity in Shuttle Hetero-difunctionalization Reactions: Electrochemical Transfer Halo-thiolation of Alkynes, Angew. Chem., Int. Ed., 2023, 62, e202213630 CrossRef CAS PubMed.
- K.-Y. Ye, Z. Song, G. S. Sauer, J. H. Harenberg, N. Fu and S. Lin, Synthesis of Chlorotrifluoromethylated Pyrrolidines by Electrocatalytic Radical Ene-Yne Cyclization, Chem. – Eur. J., 2018, 24, 12274–12279 CrossRef CAS PubMed.
- T.-S. Zhang, W.-J. Hao, R. Wang, S.-C. Wang, S.-J. Tu and B. Jiang, Electrocatalytic three-component annulation-halosulfonylation of 1,6-Enynes toward 1-indanones using sodium halides as both halogen sources and electrolytes, Green Chem., 2020, 22, 4259–4269 RSC.
- H.-D. Zuo, W.-J. Hao, C.-F. Zhu, C. Guo, S.-J. Tu and B. Jiang, Electrochemical Annulation−Iodosulfonylation of 1,5-Enynecontaining para-Quinone Methides (p-QMs) to Access (E)-Spiroindenes, Org. Lett., 2020, 22, 4471–4477 CrossRef CAS PubMed.
- J. D. Haupt, M. Berger and S. R. Waldvogel, Electrochemical Fluorocyclization of N-Allylcarboxamides to 2-Oxazolines by Hypervalent Iodine Mediator, Org. Lett., 2019, 21, 242–245 CrossRef CAS PubMed.
- J. D. Herszman, M. Berger and S. R. Waldvogel, Fluorocyclization of N-Propargylamides to Oxazoles by Electrochemically Generated ArIF2, Org. Lett., 2019, 21, 7893–7896 CrossRef CAS PubMed.
- B. Winterson, T. Rennigholtz and T. Wirth, Flow electrochemistry: a safe tool for fluorine chemistry, Chem. Sci., 2021, 12, 9053–9059 RSC.
- S. Doobary, D. L. Poole and A. J. J. Lennox, Intramolecular Alkene Fluoroarylation of Phenolic Ethers Enabled by Electrochemically Generated Iodane, J. Org. Chem., 2021, 86, 16095–16103 CrossRef CAS PubMed.
- C. Chen, J.-C. Kang, C. Mao, J.-W. Dong, Y.-Y. Xie, T.-M. Ding, Y.-Q. Tu, Z.-M. Chen and S.-Y. Zhang, Electrochemical halogenation/semi-pinacol rearrangement of allylic alcohols using inorganic halide salt: an eco-friendly route to the synthesis of β-Halocarbonyls, Green Chem., 2019, 21, 4014–4019 RSC.
-
(a) D. Li, P. W. Seavill and J. D. Wilden, Application of Electrochemical Processes to Classical Iodocyclisation: Utility for Selectivity and Mechanistic Insight, ChemElectroChem, 2019, 6, 5829–5835 CrossRef CAS;
(b) A. P. Kale, P. Nikolaienko, K. Smirnova and M. Rueping, Intramolecular Electrochemical Oxybromination of Olefins for the Synthesis of Isoxazolines in Batch and Continuous Flow, Eur. J. Org. Chem., 2021, 24, 3496–3500 CrossRef;
(c) R. Kim, J. Ha, J. Woo and D. Y. Kim, Electrochemical oxidative bromolactonization of unsaturated carboxylic acids with sodium bromide: Synthesis of bromomethylated γ-lactones, Tetrahedron Lett., 2022, 88, 153567 CrossRef CAS;
(d) Z.-J. Shen, B. Huang, N. Ma, L. Yao, C. Yang, L. Guo and W. Xia, Transition Metal-Free Synthesis of Sulfonyl- and BromoSubstituted Indolo[2,1-α]isoquinoline Derivatives through Electrochemical Radical Cascade Cyclization, Adv. Synth. Catal., 2021, 363, 1944–1954 CrossRef CAS;
(e) J. He, H. Mei, J. Escorihuela and J. Han, Electrochemical Multicomponent Cascade Radical Process Enabling Synthesis of Iodomethyl Spiropyrrolidinyl-Oxindoles, Chin. J. Chem., 2024, 42, 1691–1698 CrossRef CAS.
- K. Yu, X. Kong, J. Yang, G. Li, B. Xu and Q. Chen, Electrochemical Oxidative Halogenation of N-Aryl Alkynamides for the Synthesis of Spiro[4.5]trienones, J. Org. Chem., 2021, 86, 917–928 CrossRef CAS PubMed.
-
(a) J. Zhang, S.-Q. Shi, W.-J. Hao, G.-Y. Dong, S.-J. Tu and B. Jiang, Tunable Electrocatalytic Annulations of o-Arylalkynylanilines: Green and Switchable Syntheses of Skeletally Diverse Indoles, J. Org. Chem., 2021, 86, 15886–15896 CrossRef CAS PubMed;
(b) H. Xu, X. Meng, Y. Zheng, J. Luo and S. Huang, Electrochemical Annulations of o-Alkynylanilines for Synthesis of 3-Iodoindoles, Chin. J. Org. Chem., 2021, 41, 4696–4703 CrossRef CAS;
(c) B. Huang, G. Chen, H. Zhang, X. Tang, J. Yuan, C. Lu and J. Wang, Divergent electrosynthesis of 3-iodoindoles and indoles from 2-ethynylanilines under ambient and aqueous conditions, Org. Chem. Front., 2023, 10, 3515–3521 RSC.
- M. B. Reddy, R. Peri, M. Bhagavathiachari and R. Anandhan, Electrochemical synthesis of isobenzofuran-1-imines using oxidative halocyclization of o-alkynylbenzamides, Org. Biomol. Chem., 2021, 19, 6792–6796 RSC.
- For selected reviews and examples, see:
(a) S. E. Denmark, W. E. Kuester and M. T. Burk, Catalytic, Asymmetric Halofunctionalization of Alkenes—A Critical Perspective, Angew. Chem., Int. Ed., 2012, 51, 10938–10953 CrossRef CAS;
(b) E. P. A. Talbot, T. de A. Fernandes, J. M. McKenna and F. D. Toste, Asymmetric Palladium-Catalyzed Directed Intermolecular Fluoroarylation of Styrenes, J. Am. Chem. Soc., 2014, 136, 4101–4104 CrossRef CAS;
(c) A. J. Cresswell, S. T.-C. Eey and S. E. Denmark, Catalytic, Stereoselective Dihalogenation of Alkenes: Challenges and Opportunities, Angew. Chem., Int. Ed., 2015, 54, 15642–15682 CrossRef CAS;
(d) M. L. Landry and N. Z. Burns, Catalytic Enantioselective Dihalogenation in Total Synthesis, Acc. Chem. Res., 2018, 51, 1260–1271 CrossRef CAS PubMed;
(e) T. Arai, K. Horigane, T. K. Suzuki, R. Itoh and M. Yamanaka, Catalytic Asymmetric Iodoesterification of Simple Alkenes, Angew. Chem., Int. Ed., 2020, 59, 12680–12683 CrossRef CAS.
|
| This journal is © the Partner Organisations 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *,
Wenguang
Lu
,
Chunxi
Chen
,
Yanju
Lu
*,
Wenguang
Lu
,
Chunxi
Chen
,
Yanju
Lu


























