
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel/ligand loading-controlled divergent and selective coupling between redox-active methylenecyclopropanes and ArBr†
Ben
Mao
a,
Xiao-Yu
Zhang
a,
Xiu-Fen
Bi
c,
Xiao-Yin
Yang
*c,
Min
Shi
 *ab and
Yin
Wei
*b
*ab and
Yin
Wei
*b
aKey Laboratory for Advanced Materials and Institute of Fine Chemicals, School of Chemistry & Molecular Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China. E-mail: mshi@mail.sioc.ac.cn
bState Key Laboratory of Organometallic Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: weiyin@sioc.ac.cn
cHuafon Chemical Co. Ltd., 1688 Kaifaqu Road, Ruian Economic Development Zone, Zhejiang 325200, China. E-mail: yang.xiaoyin@huafeng.com
First published on 28th August 2024
Abstract
Divergent and selective synthesis has been widely achieved in transition-metal-catalyzed reactions through a ligand property tuning strategy. However, ligand-loading-controlled divergent synthesis has rarely been reported. Due to changes in ligand loading, different metal complexes should be formed and exhibit diverse catalytic properties. Herein, we disclose a Ni/photoredox-catalyzed divergent and selective coupling between redox-active methylenecyclopropanes and aryl bromides through ligand loading adjustment, providing ranges of alkyne derivatives and dibenzylethylene derivatives. Two different catalytic cycles are proposed to demonstrate the generation of two sets of products, where homopropargyl radicals and nickelacyclobutane species should be crucial intermediates in the corresponding catalytic cycle, respectively.
Introduction
A ligand-controlled divergent synthetic strategy has been widely explored in the field of transition-metal catalysis over the past few decades.1 By tuning the electronic properties and steric hindrance of ligands, the metal–ligand complex as the ligated catalyst can exhibit different catalytic activities and site selectivities.2 Hence, broader transformations of the same starting materials can be achieved under almost identical conditions, offering facile access to diverse synthetic applications. Nevertheless, this strategy requires chemists to invest more effort in the process of ligand screening and rational ligand design. Typically, the choice of ligands plays a crucial role in transition-metal-catalyzed reactions, but the ligand loading often does not receive careful attention in many reported protocols. In most cases, the dosage ratios of the ligand to metal are in the range of 1 to 2, and the specific ratios are not strictly controlled, only ensuring that the ligand is more than the metal to form the desired complex. Simple changes of ligand loading may lead to different metal–ligand complexes forming in the reaction system, providing metal centers with different catalytic activities and site selectivities. In particular, when the ligand loading is less than the metal loading, the free metal and ligand-coordinated metal in the system may show different properties, leading to some unexpected transformations of the substrates. Alternatively, when the ligand is far in excess, the multiple coordination of the ligand with the metal would also result in a catalytic center different from the single-coordinated metal–ligand complex. Compared with the differentiation of ligand selection, this scheme can also provide a potential path for divergent synthesis in transition-metal catalytic transformations (Scheme 1a). | ||
| Scheme 1 Project background and this work. | ||
In divergent and selective synthetic protocols, different catalytic transformations can be realized by changing the reaction conditions,3 which in turn requires the starting materials to have multiple reactivities to accomplish the corresponding conversions. Hence, chemists usually prefer substrates with multiple reactive sites since the rational design and selection of substrate structures are essential for achieving divergent synthesis.4 Methylenecyclopropanes (MCPs), a class of three-membered ring substrates with a strained exocyclic double bond, have been comprehensively explored as versatile C-3 building blocks since the 1970s.5 The highly strained structure with multiple active sites makes MCPs excellent candidates in the field of transition-metal-catalyzed carbon–carbon activation.6 Among them, ligand-controlled divergent transformations of MCPs have also been explored. For example, in 2023, Ge's group reported a cobalt-catalyzed ring-opening reaction of MCPs to obtain two sets of skipped diboronates through a ligand-tuning strategy (Scheme 1b).7
Our group has long been investigating applicable conversions of MCPs through careful substrate design and mild condition selection.8 In 2021, we successfully demonstrated that redox-active MCPs could be applied as alkyne building blocks through a facile radical rearrangement process (Scheme 1c).9 Under mild photocatalytic conditions, the MCP substrate tethered with N-(acyloxy)phthalimide (NHPI ester), a redox-active ester used as an efficient radical precursor,10 could afford a homopropargyl radical adding to the alkene in the protocol. To broaden the applications of these valuable building blocks, we envisioned that such substrates may be applied to nickel-catalyzed coupling reactions to develop potential transformations. Herein, we wish to report a ligand loading-controlled photoredox/nickel catalyzed divergent and selective coupling between redox-active MCPs and ArBr, delivering a series of alkyne derivatives and dibenzylethylene derivatives (Scheme 1d). Notably, the alkyne product was afforded when the loading of the bidentate ligand exceeded the nickel catalyst, while the dibenzylethylene product was obtained when the nickel catalyst was excessive.
Results and discussion
We commenced our studies by utilizing NHPI ester 1a (1.0 equiv.) with ArBr 2a (1.0 equiv.) as the model substrates. In the presence of Ir[dF(CF3)ppy]2(dtbbpy)PF6 (2 mol%), NiBr2·DME (10 mol%), 4,4′-di-tert-butyl-2,2′-bipyridine (L1, 15 mol%), Hantzsch ester (HEH, 2.0 equiv.), and Li2CO3 (4.0 equiv.) in NMP as the solvent under 30 W blue LED irradiation, the desired alkyne product 3aa could be obtained in 83% yield (Table 1, entry 1) (see ESI Tables S1–S9† for more details). Apart from ligand L1, other substituted 2,2′-bipyridine ligands were also screened in the reaction but did not offer better results (Table 1, entries 2–4). Notably, substituted 2,2′-bipyridine ligands containing electron-deficient groups (CF3, CO2Me) (Table 1, entry 3) were ineffective in this nickel-catalyzed coupling reaction. The polydentate ligand L6 showed no reactivity (Table 1, entry 5), while ligand 1,10-phenanthroline (L7) provided the alkyne product in 43% yield (Table 1, entry 6). Reducing the loading of the nickel source and ligand resulted in a slight decrease in the yield of 3aa (Table 1, entry 7). Increasing the concentration of the system and reducing the amount of base (Table 1, entry 9) would lower the yield of 3aa (see ESI Table S5† for more details). Control experiments revealed that HEH, light irradiation, nickel source, ligand and photocatalyst were indispensable for the generation of the alkyne product (Table 1, entry 10). It should be noted that in the process of nickel catalyst and ligand loading optimization, the excessive addition of a nickel salt over the ligand led to the disappearance of alkyne product 3aa (Table 1, entry 8) surprisingly, but gave a different coupling product 4aa (see ESI Table S9† for more details).|
|
||
|---|---|---|
| Entrya | Deviation | 3aa, yieldb (%) |
| a Reactions were performed on a 0.1 mmol scale. b Yields were determined by analyzing 1H NMR spectroscopic data using 1,3,5-trimethoxybenzene as an internal standard. c Isolated yield on a 0.2 mmol scale. | ||
| 1 | None | 83 (75)c |
| 2 | L2 | 50 |
| 3 | L3 or L4 | n.d. |
| 4 | L5 | 57 |
| 5 | L6 | n.d. |
| 6 | L7 | 43 |
| 7 | NiBr2·DME (5 mol%), dtbbpy (10 mol%) | 73 |
| 8 | NiBr2·DME (10 mol%), dtbbpy (5 mol%) | n.d. |
| 9 | NMP (0.1 M), Li2CO3 (2.0 equiv.) | 57 |
| 10 | w/o HEH, irradiation, nickel source, ligand or photocatalyst | n.d. |

|
||

Next, condition screening for the double aryl group coupling product 4aa was also conducted (Table 2). To better compare the differences in catalyst loading for producing two distinct products, we still utilized NHPI ester 1a (1.0 equiv.) with ArBr 2a (1.0 equiv.) as the model substrates under approximate reaction conditions. Product 4aa could be obtained in 81% yield (Table 2, entry 1) only by changing the loading of NiBr2·DME (15 mol%) and L1 (7.5 mol%) based on the optimized conditions for 3aa. Photocatalysts such as 4CzIPN, Ir(dtbbpy)(ppy)2PF6, and Ir[dF(Me)ppy]2(dtbbpy)PF6 were all competent in the catalytic process, albeit with moderate yields ranging from 51% to 69% for product 4aa (Table 2, entries 2–4). Lowering the Ni L−1 loading resulted in a significant decrease in the yield of 4aa to 50% (Table 2, entry 5), while increasing the Ni L−1 loading provided a similar yield (Table 2, entry 6). Analogous to our previous report,11 extra nickel salts seem to be indispensable for obtaining product 4aa (see ESI Table S9† for more details). When a commercially available NiBr2·dtbbpy complex was used as the sole catalyst, the double aryl group coupling product could also be delivered in 24% yield, probably due to the presence of dissociated nickel salts (Table 2, entry 7). NiCl2·DME was also an effective nickel source in this catalytic system (Table 2, entry 8), while Ni(acac)2 showed no catalytic activity in the catalytic process (Table 2, entry 9). Similarly, the use of ligands containing electron-deficient groups and polydentate ligands failed to yield the final product 4aa (Table 2, entry 10). The other screened ligands did not afford better results (Table 2, entries 11–13). A significant decrease was observed upon lowering the amount of HEH to one equivalent (Table 2, entry 14). The use of another aprotic polar solvent DMAc failed to provide 4aa in a comparable yield to that of NMP (Table 2, entry 15). In addition, such a dual coupling process was quite sensitive to the concentration of the reaction solution. The increase in concentration led to poor reactivity, giving the final product 4aa with a yield of 33% (Table 2, entry 16). Likewise, HEH, light irradiation, nickel source, ligand and photocatalyst were all indispensable for generating 4aa (Table 2, entry 17). We also analyzed the internal mixtures of these two reactions (see ESI page S32† for more details).
|
|
||
|---|---|---|
| Entrya | Deviation | 4aa, yieldb (%) |
| a Reactions were performed on a 0.1 mmol scale. b Yields were determined by analyzing 1H NMR spectroscopic data using 1,3,5-trimethoxybenzene as an internal standard and calculated on a 0.05 mmol. c Isolated yield on a 0.3 mmol scale. | ||
| 1 | None | 81 (72)c |
| 2 | 4CzlPN as the photocatalyst | 51 |
| 3 | Ir(dtbbpy)(ppy)2PF6 as the photocatalyst | 69 |
| 4 | Ir[dF(Me)ppy]2(dtbbpy)PF6 as the photocatalyst | 58 |
| 5 | NiBr2·DME (10 mol%), dtbbpy (5 mol%) | 50 |
| 6 | NiBr2·DME (20 mol%), dtbbpy (10 mol%) | 78 |
| 7 | NiBr2·dtbbpy (10 mol%) | 24 |
| 8 | NiCl2·DME instead of NiBr2·DME | 68 |
| 9 | Ni(acac)2 instead of NiBr2·DME | n.d. |
| 10 | L3, L4 or L6 instead of L1 | n.d. |
| 11 | L2 instead of L1 | 75 |
| 12 | L5 instead of L1 | 58 |
| 13 | L7 instead of L1 | 35 |
| 14 | 1.0 equiv. of HEH | 56 |
| 15 | DMAc instead of NMP | 32 |
| 16 | NMP (0.1 M) | 33 |
| 17 | w/o HEH, irradiation, nickel source, ligand or photocatalyst | n.d |

With the optimized conditions in hand, we then investigated the generality of such a nickel/ligand loading-controlled coupling protocol for producing distinct products 3 and 4. First, a series of NHPI esters bearing methylenecyclopropanes were coupled with ArBr (2a or 2b) under two slightly different conditions (Scheme 2). When the substituent R was an ethyl group (1b) or a propyl group (1c), two modes of coupling reactions could proceed smoothly to afford alkyne products (3bb, 3ca) and double aryl group coupling products (4bb, 4ca) in similar yields to those of substrate 1a. For substrate 1d where the substituent R was a hydrogen atom, the terminal alkyne coupling product 3db was obtained in 70% yield and the 1,1-dibenzylethylene product 4db was obtained in 65% yield. The cyclopropane group introduced in substrate 1e was retained in the generation of products 3eb and 4eb. When we introduced a terminal alkyne group in the NHPI ester substrate, the corresponding alkyne product 3fb was obtained in 36% yield, while only a trace amount of 4fb was generated (see ESI page S33† for more details). Replacing the terminal alkyne group with an internal alkyne group could increase the yield of alkyne product 3gb to 52%, though the double aryl group coupling product 4gb was still produced in a trace amount. The allyl group (1h) and methyl-substituted allyl group (1i) were well tolerated in this nickel-catalyzed coupling process, affording the corresponding products 3ha and 3ib in 60% and 65% yields, as well as 4ha and 4ib in 47% and 68% yields, respectively. An array of benzyl groups bearing diverse substituents introduced in the substrates (1j–1o) were tested under both sets of reaction conditions, affording the alkyne products (3jb–3ob) in yields ranging from 57 to 72% and tribenzylethylene products (4jb–4ob) in yields ranging from 59 to 68%. Notably, substrates containing the fused aromatic ring (1p) and aromatic heterocycle (1q) could also undergo the transformation processes smoothly to give the corresponding products 3pb and 3qb in 67% and 52% yields, as well as 4pb and 4qb in 62% and 40% yields, respectively.
 | ||
| Scheme 2 Scope of NHPI ester substrates. Reactions were performed on a 0.2 mmol scale for product 3 and on a 0.3 mmol scale for product 4. Isolated yields (yields of product 4 were calculated on a 0.15 mmol scale). See ESI page S12† for more experimental details. | ||
Next, we investigated the scope of the electrophilic coupling partners of aryl bromides under the two sets of reaction conditions (Scheme 3). Aryl bromides bearing electron-withdrawing groups at the para-position underwent the two modes of the coupling process smoothly yielding the alkyne products (3ab–3af) and 1,1-dibenzylethylene products (4ab–4ae) in moderate yields ranging from 56% to 82%. It should be noted that the cyano group was not well tolerated in the double aryl group coupling process, probably owing to coordination with the extra nickel salts resulting in the deactivation of the critical catalytic nickel species. For electron-neutral aryl bromides such as 4-bromobiphenyl, the corresponding coupling products (3ag/4ag) were obtained smoothly in 70% and 74% yields, respectively. Unfortunately, electron-rich aryl bromide namely 4-bromoanisole was not a competent coupling partner in this nickel-catalyzed coupling protocol. Aryl bromides containing meta-substituted groups (OMe, Cl) and ortho-substituted groups (F) were also tested, yielding the corresponding products 3ai–3ak and 4ai–4ak in moderate to good yields ranging from 50% to 77%. Boronic acid pinacol ester was also well tolerated under the two sets of mild conditions, affording the desired products (3ah/4ah) in 54% and 47% yields, respectively. Moreover, diverse polycyclic aryl bromides underwent the coupling processes smoothly to afford the final alkyne products (3al–3ar) and polysubstituted olefinic products (4al–4aq) in yields ranging from 50 to 78%. Notably, product 4ar could not be obtained successfully under the standard conditions B, probably due to the coordination between the nickel species, oxygen atom, and fluorine atom, which restricted the catalytic ability of the nickel catalysts (see ESI page S33† for more details). Furthermore, bromopyridine could not complete the double aryl group coupling process owing to the coordination interactions with the nickel species; however, the corresponding alkyne product 3as could be furnished in 81% yield.
 | ||
| Scheme 3 Scope of aryl bromides. Reactions were performed on a 0.2 mmol scale for product 3 and on a 0.3 mmol scale for product 4. Isolated yields (yields of product 4 were calculated on a 0.15 mmol scale). See ESI page S12† for more experimental details. | ||
Furthermore, to investigate the synthetic applicability of the nickel-catalyzed protocol, scale-up experiments of 1a and synthetic transformations of alkynes were also conducted (see ESI page S36† for more details).
Possible reaction pathways
Our group has recently disclosed that the NHPI ester substrates bearing methylenecyclopropanes could undergo the ring-opening reactions smoothly to deliver the homopropargyl radicals under photoreductive conditions.9 Specifically, radical anionic intermediate I formed from substrate 1 (NHPI ester) through a SET (single electron transfer) process could undergo homolytic fragmentation, releasing the phthalimide anion and CO2. The transiently formed ethylenic radical II underwent a thermodynamically favorable radical rearrangement process to afford homopropargyl radical III. In this protocol, such a C(sp3) hybridized fragment should engage in the nickel-catalyzed C(sp3)–C(sp2) cross-coupling process to give the final alkyne product 3 (Scheme 4, Path A).12 In addition, the double aryl group coupling product was also obtained in this photoredox/nickel catalyzed protocol. We initially speculated that the ethylenic radical II may undergo a HAT (hydrogen atom transfer) process or a SET process to provide the methylenecyclopropane intermediate IV. Then intermediate IV could undergo a Ni-catalyzed coupling process with ArBr via selective distal bond cleavage to afford product 4 (Scheme 4, Path B), a known process recently demonstrated by our group.11 However, considering that the radical rearrangement process is quite thermodynamically favored, Path B could not explain why the alkyne products 3 were not generated during the production of 4. Herein, we provide a tentative hypothesis to explain how the addition of extra nickel salts led to the production of two different products (Scheme 4, Path C). Due to the lack of coordination with bidentate ligands, the additional “free” nickel salts with electron deficiency and less steric hindrance were more inclined to undergo the oxidative addition process with the electron-rich methylenecyclopropane structure in the substrate (cyclopropane insertion). As a result, the nickelacyclobutene intermediate13 would inhibit the radical rearrangement in the homolytic fragmentation process, bypassing the generation of homopropargyl radical III. The following HAT process (or the SET process) and the coupling process delivered 4 as a single product. It should be noted that the sequence of fragmentation (P1), HAT or SET (P2) and the Ni-catalyzed coupling process (P3) was difficult to figure out in the generation of product 4 (Scheme 4, Path C). The nickel-catalyzed coupling process may take place before the reductive fragmentation process, or these three processes may occur simultaneously. | ||
| Scheme 4 Possible reaction pathways for the generation of products. | ||
Mechanistic investigations
To investigate the mechanism of NHPI ester activation, we first conducted UV/vis absorption studies of the redox-active substrate 1a and HEH. Consistent with the previous reports,14 a bathochromic shift (Scheme 5a, red band) was observed for a mixture of NHPI ester 1a and HEH, indicating that the formation of an electron donor–acceptor (EDA) complex was probably involved in this nickel-catalyzed protocol. Hence, the EDA-complex could provide a potential pathway for the single electron reduction of the NHPI ester substrate. Next, fluorescence quenching studies of the photocatalyst with HEH, substrate 1a and NiBr2·DME were conducted (Scheme 5b). The excited state of Ir[dF(CF3)ppy]2(dtbbpy)PF6 was more strongly quenched by HEH, implying that a reductive quenching cycle15 was involved in the reaction process and a reducing Ir(II) center (Ered1/2 = −1.37 V vs. SCE in MeCN)16 was generated. Though substrate 1a exhibited no quenching effect on the excited photocatalyst, we suggested that the NHPI ester 1a (Ered1/2 = −1.11 V vs. SCE in DMAc, see ESI Fig. S8† for more details) could also receive an electron from the reducing Ir(II) center.9 The quantum yields were measured as Φ = 0.15 and Φ = 0.04 (see ESI page S19† for more details) in the generation of alkyne product 3aa and dibenzylethylene product 4aa, respectively, suggesting that a radical chain process was possibly not involved in the two sets of reactions. | ||
| Scheme 5 UV/vis absorption studies and fluorescence quenching studies. | ||
A series of control experiments were also conducted to gain more insights into the two sets of reaction mechanisms. We first investigated the transformations of the NHPI ester substrate under the photocatalytic conditions. Rearrangement product 10 could be isolated in 82% yield from substrate 1n as a single product (Scheme 6a, above). This result suggests that such a rearrangement product was formed from the homopropargyl radical 1n-IV. In particular, substrate 1n underwent a SET process to form the radical anionic intermediate 1n-I, which could release the phthalimide anion and CO2 along with a rearrangement process to give intermediate 1n-IV through intermediate 1n-II or 1n-III. Though the classical MCP substrate 1j′ could also deliver the corresponding product 4jb in 68% yield under conditions B (Scheme 6c),11 we did not observe the cyclopropane-maintained product 11, which could be generated from the possible vinyl radical 1n-III through a HAT (or SET) process. In addition, considering that the nickel species may promote the hydrogenation of vinyl radical 1n-III, we added extra NiBr2·DME (10 mol%) into the reaction (Scheme 6a, below). However, we still did not observe any cyclopropane-maintained product 11. The rearrangement product 10 was not detected owing to the destruction of the MCP moiety by the nickel species (see ESI page S14† for more details). These results implied that the possible intermediate IV proposed in Scheme 4 (Path B) was possibly not involved in the reaction process of generating product 4 and path B was unlikely. Next, 2,2,6,6-tetramethylpiperidinooxy (TEMPO) used as the radical scavenger was added to the two sets of reactions, both reactions were inhibited completely and the corresponding products were not detected (Scheme 6b). These two experiments suggest that radical reaction pathways were possibly involved in the two sets of coupling reactions. Deuterium labeling studies using d3-HEH and D2O were conducted, and product d1-4aa was obtained with 30% and 47% deuterium incorporation, respectively (Scheme 6d). These results indicate that the olefinic hydrogen atom in the double aryl group coupling product 4aa may originate from HEH or a trace amount of water in the reaction system through SET and HAT processes. A crossover experiment was carried out using substrates 1a, 2a, and 2b under the conditions B. Apart from the production of the corresponding products 4aa and 4ab, a mixed coupling product 4aab was also isolated in 36% yield, presumably indicating that the two aryl group coupling process involved two discrete steps (Scheme 6e).
 | ||
| Scheme 6 Control experiments. | ||
When a catalytic amount of (dtbbpy)Ni(p-CF3Ar)Br (10 mol%) was applied in the reaction, 3aa could be smoothly obtained in 63% yield while 4aa was not detected, indicating that the Ni complex (dtbbpy)NiArBr was a possible catalytically active species in the generation of product 3 but not responsible for the cleavage of the cyclopropane ring in the NHPI ester substrate (Scheme 6f, eqn (I)). Stoichiometric experiments with (dtbbpy)Ni(p-CF3Ar)Br as a possible Ni(II) intermediate were next conducted (Scheme 6f, eqn (II)), where alkyne product 3aa could be obtained in 24% yield in the presence of the photocatalyst (x = 2) and HEH (y = 2). In addition, when the sacrificial reductant HEH was omitted (y = 0), no coupling product (3aa or 4aa) was detected. However, in the absence of a photocatalyst (x = 0), product 3aa could be generated in 12% yield. Such stoichiometric control experiments revealed that activating the NHPI ester mediated by the formation of an EDA complex was a feasible pathway.14a Notably, product 4aa was not detected in these stoichiometric experiments, highlighting the indispensable role of “free” nickel salts in generating the dibenzylethylene product 4. When extra NiBr2·DME was added, 4aa was obtained in 14% yield, while 3aa was not detected (Scheme 6f, eqn (III)). All these results suggest that (i) (dtbbpy)NiArBr should be a catalytically active species in the nickel catalytic cycle for the generation of product 3; (ii) the “free” nickel species, rather than the ligand-coordinated nickel species, should play the role of activating the cyclopropane ring in the NHPI ester substrate for the generation of product 4; and (iii) the SET activation of the NHPI ester substrate via the EDA complex was also feasible in this protocol.
On the basis of the above mechanistic studies and previous reports, we propose a preliminary plausible mechanism for this nickel/ligand loading controlled divergent synthetic protocol (Scheme 7). Initially, the excited photocatalyst Ir(III) (*Ered1/2 = +1.21 V vs. SCE in MeCN)16 was quenched by the sacrificial reductant HEH (Eox1/2 = +0.89 V vs. SCE in DMF),17 delivering a strongly reducing center Ir(II) (Ered1/2 = −1.37 V vs. SCE in MeCN),16 which is a competent reducing center for the reduction of the NHPI ester substrate and nickel species. With regard to the single-electron reduction of the NHPI ester substrate 1, two pathways should be responsible for the generation of a radical anionic intermediate (I).14a On the one hand, substrate 1 could be directly reduced by the reducing Ir(II) center to afford the radical anionic intermediate I; on the other hand, an EDA complex formed by the electron-deficient NHPI ester and the electron-rich HEH would also result in the single electron reduction of substrate 1 to generate intermediate I. Subsequently, when there were no “free” nickel salts present in the reaction system, the entity intermediate I could undergo radical fragmentation and a thermodynamically favored radical rearrangement process smoothly to give the homopropargyl radical III. According to the reported detailed computational studies,18 such a C(sp3) radical fragment may go through two reaction pathways: (i) captured by the LnNiIIArBr (LnNi: nickel species with bidentate ligand coordination) species B, which was formed from the oxidative addition of the aryl halides to Ni0 species A, to form the high-valent NiIII species C; (ii) combined with the initial Ni0Ln species A to form the NiI intermediate E. In the first reaction pathway, the NiIII species C underwent a reductive elimination process to yield the final C(sp2)–C(sp3) coupling product 3 and the corresponding NiI species D. The Ni0Ln species A could be regenerated by the reduction of the NiI species D (Eredp(NiILn/Ni0Ln) = −1.17 V vs. SCE in THF)19 through a SET event with the Ir(II) reducing center. In the second reaction pathway, subsequent oxidative addition of ArBr to the NiI intermediate E would occur, affording the same NiIII species C entering into the catalytic cycle (Scheme 7, bottom left).
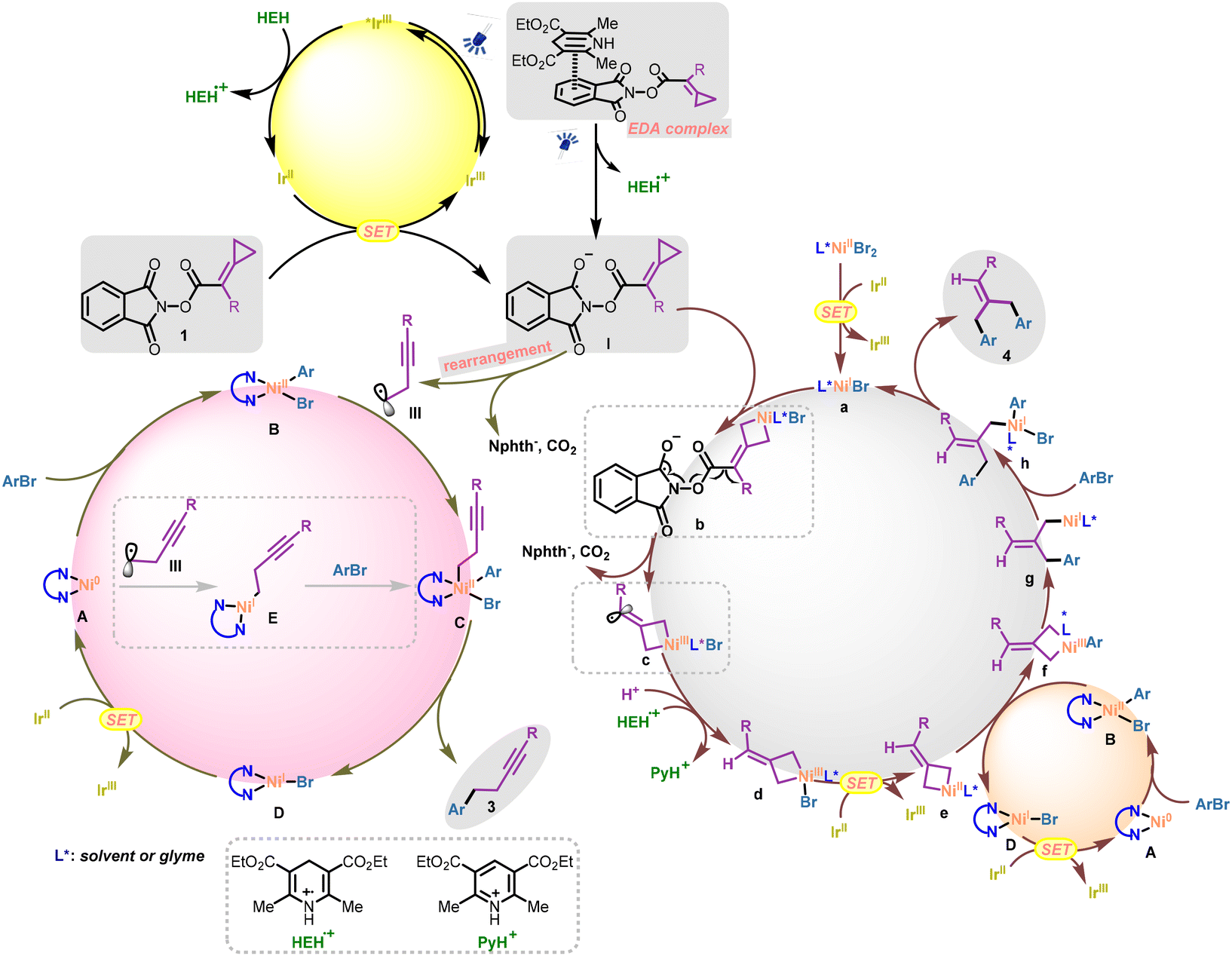 | ||
| Scheme 7 Proposed reaction mechanism. | ||
In contrast, when the nickel salts were excessive in the reactions, the cyclopropane ring in the radical anionic intermediate I could not be maintained for the following radical rearrangement process. In this scenario (Scheme 7, bottom right), the electron-deficient nickel center of L*NiIBr a (“free” nickel, L*: solvent or glyme) with less steric hindrance, speculatively reduced by the Ir(II) center (Ered1/2(NiII/NiI) = −1.70 V vs. Fc/Fc+ in DMF),20 could insert into the cyclopropane ring forming the nickelacyclobutane species b that inhibited the radical rearrangement. In this case, intermediate b possibly undergoes the radical fragmentation process to release the phthalimide anion and CO2, affording the vinyl radical intermediate c. After the SET and HAT processes, the free MCP intermediate d with nickel insertion was generated. The following transformations of such nickel(III)acyclobutane d were previously investigated by our group.11 In particular, the NiIII species d could be reduced to NiII species e by Ir(II) through a SET event. Meanwhile, oxidative addition of ArBr to the electron-rich Ni0Ln species A provided the LnNiIIArBr complex B. An aryl group transfer (disproportionation) event was considered to take place between nickelacyclobutane species e and LnNiIIArBr B,21 affording the corresponding aryl-nickelacyclobutane species f and LnNiIBr species D. The LnNiIBr species D could be reduced by the reducing Ir(II) center to regenerate the Ni0Ln species. Furthermore, the first reductive elimination from the aryl-nickelacyclobutane species f delivered the NiI intermediate g, which could undergo oxidative addition with ArBr or potentially another aryl ligand transfer process to deliver intermediate h. Intermediate h then underwent a second reductive elimination, yielding the final product 4 and regenerating the L*NiIBr species a. Notably, the aryl group transfer event occurring between nickel(III)acyclobutane d and LnNiIIArBr B may also be feasible21a (see ESI page S35† for more details).
Conclusions
In summary, we have developed a nickel/ligand loading-controlled divergent synthesis protocol, offering an array of alkyne derivatives and dibenzylethylene derivatives in moderate to good yields under mild conditions along with a broad substrate scope. Selective cleavage of carbon–carbon bonds on redox-active MCP substrates can be achieved efficiently by adjusting the loading of the nickel catalyst and bidentate ligand. Specifically, alkyne products can be obtained when the nickel-to-ligand ratio is 2 to 3, while dibenzylethylene products can be obtained when the ratio is 2 to 1 under almost identical conditions. Temporary mechanistic studies demonstrate that the generation of alkyne products should involve homopropargyl radicals stemming from the rearrangement of the MCP structure in the substrates, while the generation of dibenzylethylene products should involve nickelacyclobutane intermediates. Further investigations into this interesting loading-controlled catalytic mode and mechanistic studies are ongoing in our group. We hope such a ligand loading-controlled scheme could raise attention to ligand loading effects in transition-metal catalysis and bring more unexpected but applicable transformations in synthetic chemistry.Author contributions
B. Mao contributed to the experimental work. B. Mao, X.-Y. Zhang, X.-F. Bi, X.-Y. Yang, M. Shi and Y. Wei contributed to conceptualizations and writing of the paper.Data availability
Experimental and computational data have been made available as the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Key R & D Program of China (2023YFA1506700), the National Natural Science Foundation of China (21372250, 21121062, 21302203, 21772037, 21772226, 21861132014, 91956115 and 22171078) and the Fundamental Research Funds for the Central Universities 222201717003.References
- (a) Y. Ping, X. Li, Q. Pan and W. Kong, Ni-Catalyzed Divergent Synthesis of 2-Benzazepine Derivatives via Tunable Cyclization and 1,4-Acyl Transfer Triggered by Amide N-C Bond Cleavage, Angew. Chem., Int. Ed., 2022, 61, e202201574 CrossRef CAS PubMed; (b) Y. Ke, W. Li, W. Liu and W. Kong, Ni-catalyzed ligand-controlled divergent and selective synthesis, Sci. China: Chem., 2023, 66, 2951–2976 CrossRef CAS; (c) Q. Pan, Y. Ping and W. Kong, Nickel-Catalyzed Ligand-Controlled Selective Reductive Cyclization/Cross-Couplings, Acc. Chem. Res., 2023, 56, 515–535 CrossRef CAS PubMed; (d) Y. Wang, J. Feng, E.-Q. Li, Z. Jia and T.-P. Loh, Recent advances in ligand-enabled palladium-catalyzed divergent synthesis, Org. Biomol. Chem., 2024, 22, 37–54 RSC; (e) I. P. Beletskaya, C. Nájera and M. Yus, Chemodivergent reactions, Chem. Soc. Rev., 2020, 49, 7101–7166 RSC; (f) T. Long, C. Zhu, L. Li, L. Shao, S. Zhu, M. Rueping and L. Chu, Ligand-controlled stereodivergent alkenylation of alkynes to access functionalized trans- and cis-1,3-dienes, Nat. Commun., 2023, 14, 55 CrossRef CAS PubMed.
- (a) A. M. Allgeier and C. A. Mirkin, Ligand Design for Electrochemically Controlling Stoichiometric and Catalytic Reactivity of Transition Metals, Angew. Chem., Int. Ed., 1998, 37, 894–908 CrossRef; (b) R. Martin and S. L. Buchwald, Palladium-Catalyzed Suzuki−Miyaura Cross-Coupling Reactions Employing Dialkylbiaryl Phosphine Ligands, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS PubMed; (c) C. C. Chintawar, A. K. Yadav, A. Kumar, S. P. Sancheti and N. T. Patil, Divergent Gold Catalysis: Unlocking Molecular Diversity through Catalyst Control, Chem. Rev., 2021, 121, 8478–8558 CrossRef CAS PubMed; (d) Y. Himeda, Conversion of CO2 into Formate by Homogeneously Catalyzed Hydrogenation in Water: Tuning Catalytic Activity and Water Solubility through the Acid–Base Equilibrium of the Ligand, Eur. J. Inorg. Chem., 2007, 2007, 3927–3941 CrossRef; (e) M. D. Bairagya, R. J. Bujol and N. Elgrishi, Fighting Deactivation: Classical and Emerging Strategies for Efficient Stabilization of Molecular Electrocatalysts, Chem. – Eur. J., 2020, 26, 3991–4000 CrossRef PubMed; (f) T. Piou and T. Rovis, Electronic and Steric Tuning of a Prototypical Piano Stool Complex: Rh(III) Catalysis for C–H Functionalization, Acc. Chem. Res., 2018, 51, 170–180 CrossRef CAS PubMed.
- (a) Y. Sakakibara and K. Murakami, Switchable Divergent Synthesis Using Photocatalysis, ACS Catal., 2022, 12, 1857–1878 CrossRef CAS; (b) L. Li, Z. Chen, X. Zhang and Y. Jia, Divergent Strategy in Natural Product Total Synthesis, Chem. Rev., 2018, 118, 3752–3832 CrossRef CAS PubMed; (c) G. Zhan, W. Du and Y.-C. Chen, Switchable divergent asymmetric synthesis via organocatalysis, Chem. Soc. Rev., 2017, 46, 1675–1692 RSC.
- (a) Y.-C. Lee, K. Kumar and H. Waldmann, Ligand-Directed Divergent Synthesis of Carbo- and Heterocyclic Ring Systems, Angew. Chem., Int. Ed., 2018, 57, 5212–5226 CrossRef CAS PubMed; (b) Y.-C. Lee, S. Patil, C. Golz, C. Strohmann, S. Ziegler, K. Kumar and H. Waldmann, A ligand-directed divergent catalytic approach to establish structural and functional scaffold diversity, Nat. Commun., 2017, 8, 14043 CrossRef PubMed; (c) Y. Shi, T. Huang, T. Wang, J. Chen, X. Liu, Z. Wu, X. Huang, Y. Zheng, Z. Yang and Y. Wu, Divergent Construction of Diverse Scaffolds through Catalyst-Controlled C−H Activation Cascades of Quinazolinones and Cyclopropenones, Chem. – Eur. J., 2021, 27, 13346–13351 CrossRef CAS PubMed; (d) T. N. Nguyen and J. A. May, Branched Amine Synthesis via Aziridine or Azetidine Opening with Organotrifluoroborates by Cooperative Brønsted/Lewis Acid Catalysis: An Acid-Dependent Divergent Mechanism, Org. Lett., 2018, 20, 3618–3621 CrossRef CAS PubMed; (e) G. Chen and B. Xu, Divergent Synthesis of Sulfonyl Quinolines, Formyl Indoles, and Quinolones from Ethynyl Benzoxazinanones via AuI Catalysis, AuI-ArI Co-Catalysis, and Silver Catalysis, ACS Catal., 2022, 12, 7134–7141 CrossRef CAS.
- (a) P. Binger, Cyclodimerization of Methylenecyclopropane on a Ni(0) Catalyst, Angew. Chem., Int. Ed. Engl., 1972, 11, 309–310 CrossRef CAS; (b) L. Yu, M. Liu, F. Chen and Q. Xu, Heterocycles from methylenecyclopropanes, Org. Biomol. Chem., 2015, 13, 8379–8392 RSC; (c) L.-X. Shao and M. Shi, Lewis and Bronsted Acid Mediated Ring-Opening Reactions of Methylenecyclopropanes and Further Transformation of the Ring-Opened Products, Curr. Org. Chem., 2007, 11, 1135–1153 CrossRef CAS; (d) H. Pellissier, Recent developments in the reactivity of methylene- and alkylidenecyclopropane derivatives, Tetrahedron, 2010, 66, 8341–8375 CrossRef CAS; (e) H. Cao, F. Chen, C. Su and L. Yu, Construction of Carbocycles from Methylenecyclopropanes, Adv. Synth. Catal., 2020, 362, 438–461 CrossRef CAS; (f) H.-Z. Wei, M. Shi and Y. Wei, Visible-light-induced reactions of methylenecyclopropanes (MCPs), Chem. Commun., 2023, 59, 2726–2738 RSC.
- (a) A. Masarwa and I. Marek, Selectivity in Metal-Catalyzed Carbon-Carbon Bond Cleavage of Alkylidenecyclopropanes, Chem. – Eur. J., 2010, 16, 9712–9721 CrossRef CAS PubMed; (b) G. Fumagalli, S. Stanton and J. F. Bower, Recent Methodologies That Exploit C-C Single-Bond Cleavage of Strained Ring Systems by Transition Metal Complexes, Chem. Rev., 2017, 117, 9404–9432 CrossRef CAS PubMed; (c) I. Nakamura and Y. Yamamoto, Transition Metal-Catalyzed Reactions of Methylenecyclopropanes, Adv. Synth. Catal., 2002, 344, 111–129 CrossRef CAS; (d) P.-h. Chen, B. A. Billett, T. Tsukamoto and G. Dong, “Cut and Sew” Transformations via Transition-Metal-Catalyzed Carbon–Carbon Bond Activation, ACS Catal., 2017, 7, 1340–1360 CrossRef CAS PubMed; (e) D. Li, W. Zang, M. J. Bird, C. J. T. Hyland and M. Shi, Gold-Catalyzed Conversion of Highly Strained Compounds, Chem. Rev., 2021, 121, 8685–8755 CrossRef CAS PubMed.
- B. B. Tan, M. Hu and S. Ge, Cobalt-Catalyzed Regiodivergent Ring-Opening Dihydroboration of Arylidenecyclopropanes to Access Skipped Diboronates, Angew. Chem., Int. Ed., 2023, 62, e202307176 CrossRef CAS PubMed.
- (a) M. Shi, J.-M. Lu, Y. Wei and L.-X. Shao, Rapid Generation of Molecular Complexity in the Lewis or Brønsted Acid-Mediated Reactions of Methylenecyclopropanes, Acc. Chem. Res., 2012, 45, 641–652 CrossRef CAS PubMed; (b) L.-Z. Yu, Q. Xu, X.-Y. Tang and M. Shi, Iron- or Copper-Catalyzed Trifluoromethylation of Acrylamide-Tethered Alkylidenecyclopropanes: Facile Synthesis of CF3-Containing Polycyclic Benzazepine Derivatives, ACS Catal., 2016, 6, 526–531 CrossRef CAS; (c) M. Chen, Y. Wei and M. Shi, Cascade cyclization reactions of alkylidenecyclopropanes for the construction of polycyclic lactams and lactones by visible light photoredox catalysis, Org. Chem. Front., 2020, 7, 374–379 RSC; (d) X.-Y. Zhang, X.-Y. Wu, B. Zhang, Y. Wei and M. Shi, Silyl Radical-Mediated Carbocyclization of Acrylamide-/Vinyl Sulfonamide-Attached Alkylidenecyclopropanes via Photoredox Catalysis with a Catalytic Amount of Silane Reagent, ACS Catal., 2021, 11, 4372–4380 CrossRef CAS; (e) B. Mao, X.-Y. Zhang, Y. Wei and M. Shi, Visible-light-mediated intramolecular radical cyclization of α-brominated amide-tethered alkylidenecyclopropanes, Chem. Commun., 2022, 58, 3653–3656 RSC.
- X.-Y. Zhang, C. Ning, B. Mao, Y. Wei and M. Shi, A visible-light mediated ring opening reaction of alkylidenecyclopropanes for the generation of homopropargyl radicals, Chem. Sci., 2021, 12, 9088–9095 RSC.
- (a) S. Murarka, N-(Acyloxy)phthalimides as Redox-Active Esters in Cross-Coupling Reactions, Adv. Synth. Catal., 2018, 360, 1735–1753 CrossRef CAS; (b) P. Niu, J. Li, Y. Zhang and C. Huo, One-Electron Reduction of Redox-Active Esters to Generate Carbon-Centered Radicals, Eur. J. Org. Chem., 2020, 5801–5814 CrossRef CAS; (c) S. K. Parida, T. Mandal, S. Das, S. K. Hota, S. De Sarkar and S. Murarka, Single Electron Transfer-Induced Redox Processes Involving N-(Acyloxy)phthalimides, ACS Catal., 2021, 11, 1640–1683 CrossRef CAS.
- B. Mao, M. Shi and Y. Wei, Ni/Photoredox-catalyzed coupling of aryl bromides and methylenecyclopropanes via selective distal bond cleavage, Org. Chem. Front., 2024 10.1039/D4QO01072H.
- (a) J. C. Tellis, C. B. Kelly, D. N. Primer, M. Jouffroy, N. R. Patel and G. A. Molander, Single-Electron Transmetalation via Photoredox/Nickel Dual Catalysis: Unlocking a New Paradigm for sp3−sp2 Cross-Coupling, Acc. Chem. Res., 2016, 49, 1429–1439 CrossRef CAS PubMed; (b) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, The merger of transition metal and photocatalysis, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- M. L. G. Sansores-Paredes, P. M. Pérez-García and M.-E. Moret, Nickelacyclobutanes: Versatile Reactivity and Role as Catalytic Intermediates, Eur. J. Inorg. Chem., 2023, 26, e202300192 CrossRef CAS.
- (a) L. M. Kammer, S. O. Badir, R. M. Hu and G. A. Molander, Photoactive electron donor–acceptor complex platform for Ni-mediated C(sp3)–C(sp2) bond formation, Chem. Sci., 2021, 12, 5450–5457 RSC; (b) F. Cong, X. Y. Lv, C. S. Day and R. Martin, Dual Catalytic Strategy for Forging sp2−sp3 and sp3−sp3 Architectures via β-Scission of Aliphatic Alcohol Derivatives, J. Am. Chem. Soc., 2020, 142, 20594–20599 CrossRef CAS PubMed.
- (a) X. Xi, Y. Luo, W. Li, M. Xu, H. Zhao, Y. Chen, S. Zheng, X. Qi and W. Yuan, From Esters to Ketones via a Photoredox-Assisted Reductive Acyl Cross-Coupling Strategy, Angew. Chem., Int. Ed., 2022, 61, e202114731 CrossRef CAS PubMed; (b) L. J. Rono, H. G. Yayla, D. Y. Wang, M. F. Armstrong and R. R. Knowles, Enantioselective Photoredox Catalysis Enabled by Proton-Coupled Electron Transfer: Development of an Asymmetric Aza-Pinacol Cyclization, J. Am. Chem. Soc., 2013, 135, 17735–17738 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- (a) G. Park, S. Y. Yi, J. Jung, E. J. Cho and Y. You, Mechanism and Applications of the Photoredox Catalytic Coupling of Benzyl Bromides, Chem. – Eur. J., 2016, 22, 17790–17799 CrossRef CAS PubMed; (b) K. N. Lee, Z. Lei and M.-Y. Ngai, β-Selective Reductive Coupling of Alkenylpyridines with Aldehydes and Imines via Synergistic Lewis Acid/Photoredox Catalysis, J. Am. Chem. Soc., 2017, 139, 5003–5006 CrossRef CAS PubMed.
- O. Gutierrez, J. C. Tellis, D. N. Primer, G. A. Molander and M. C. Kozlowski, Nickel-Catalyzed Cross-Coupling of Photoredox-Generated Radicals: Uncovering a General Manifold for Stereoconvergence in Nickel-Catalyzed Cross-Couplings, J. Am. Chem. Soc., 2015, 137, 4896–4899 CrossRef CAS PubMed.
- T. J. Steiman, J. Liu, A. Mengiste and A. G. Doyle, Synthesis of β-Phenethylamines via Ni/Photoredox Cross-Electrophile Coupling of Aliphatic Aziridines and Aryl Iodides, J. Am. Chem. Soc., 2020, 142, 7598–7605 CrossRef CAS PubMed.
- Q.-Y. Meng, S. Wang and B. König, Carboxylation of Aromatic and Aliphatic Bromides and Triflates with CO2 by Dual Visible-Light-Nickel Catalysis, Angew. Chem., Int. Ed., 2017, 56, 13426–13430 CrossRef CAS PubMed.
- (a) K. Osakada and T. Yamamoto, Transmetallation of alkynyl and aryl complexes of Group 10 transition metals, Coord. Chem. Rev., 2000, 198, 379–399 CrossRef CAS; (b) G. S. Kumar, C. Zhu, R. Kancherla, P. S. Shinde and M. Rueping, Metal Cations from Sacrificial Anodes Act as a Lewis Acid Co-Catalyst in Electrochemical Cross-Coupling of Aryl Bromides and Aziridines, ACS Catal., 2023, 13, 8813–8820 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of new compounds. See DOI: https://doi.org/10.1039/d4qo01346h |
| This journal is © the Partner Organisations 2024 |