
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegioselective pyridazine synthesis from tetrazines and alkynyl sulfides†
Chika
Yamamoto
a,
Koyo
Numata
a,
Minori
Suzuki
ab and
Suguru
Yoshida
 *a
*a
aDepartment of Biological Science and Technology, Faculty of Advanced Engineering, Tokyo University of Science, 6-3-1 Niijuku, Katsushika-ku, Tokyo 125-8585, Japan. E-mail: s-yoshida@rs.tus.ac.jp
bLaboratory of Chemical Bioscience, Institute of Biomaterials and Bioengineering, Tokyo Medical and Dental University (TMDU), 2-3-10 Kanda-Surugadai, Chiyoda-ku, Tokyo 101-0062, Japan
First published on 29th August 2024
Abstract
A regioselective synthesis of trisubstituted pyridazines from tetrazines and alkynyl sulfides is described. Various pyridazines were selectively prepared by the inverse-electron-demand Diels–Alder reaction and subsequent denitrogenation. Good transformability of sulfur substituents allowed us to synthesize a range of pyridazines without regioisomers.
Introduction
Regioselective Diels–Alder reactions are fundamental ways to synthesize diverse molecules having 6-membered rings, serving in a broad range of disciplines including natural product synthesis, pharmaceutical sciences, and materials chemistry.1 For example, electron-rich Danishefsky–Kitahara diene 1 reacts with electron-poor dienophiles, such as methacrolein (2), to afford cyclohexanones with high regioselectivity (Fig. 1A).2 This is achieved through the significant interaction between the HOMO of dienes and the LUMO of dienophiles, aligning the carbon atoms with the highest coefficients. Herein, we describe a unique regioselective synthesis of pyridazines through the inverse-electron-demand Diels–Alder (IEDDA) reactions of tetrazines and alkynyl sulfides. | ||
| Fig. 1 (A) Conventional regioselective Diels–Alder reaction. (B) Pioneering studies. (C) Our previous study. (D) This work. | ||
Tetrazines play pivotal roles as reactive dienes in IEDDA reactions due to their remarkable electron-deficient nature.3–6 Indeed, a few distinctive transformations were found in pioneering studies on pyridazine synthesis through the IEDDA reactions of tetrazines with alkynes followed by denitrogenative aromatization.6 In 1970, Sauer found that 3-phenyl-1,2,4,5-tetrazine reacts with ynamine 5 without activators to provide pyridazine 6 in excellent yield with interesting regioselectivity (Fig. 1B, upper).6a Also, regioselective pyridazine synthesis was achieved from tetrazine 4a and phenylacetylene (7a) by Meresz (Fig. 1B, lower).6b However, it is not easy to synthesize multisubstituted pyridazines from tetrazines owing to the immature IEDDA chemistry of tetrazines with alkynes from synthetic and theoretical aspects.
Recently, we revisited the tetrazine reactions with alkynes,7 in which efficient conditions using 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP)8 as a solvent and detailed scope of accessible disubstituted pyridazines were disclosed (Fig. 1C, upper). Indeed, regioselective pyridazine formation was achieved by treatment of tetrazine 4a with aromatic alkyne 7a in HFIP (Fig. 1C, upper). Unfortunately, we found that it is challenging to accomplish the regioselective reaction of tetrazine 4a with aliphatic alkyne 7b (Fig. 1C, lower). In addition, the DFT calculation of the reaction between tetrazines and alkynes served in the prediction of regioselectivities. It is worth noting that regioselective IEDDA reactions took place although substantial differences were not observed in the frontier molecular orbitals of 3-phenyl-1,2,4,5-tetrazine (4a) and phenylacetylene (7a), suggesting that significant secondary orbital interaction between two benzene rings led to the regioselectivity.
With the success of our previous study in mind, we directed our attention to the reactions of tetrazines with heteroatom-substituted alkynes (Fig. 1D). Inspired by the pioneering study of Sauer (Fig. 1B, upper),6a we hypothesized that heteroatom substituents can control the regioselectivity and serve in the synthesis of a wide variety of multisubstituted pyridazines by further transformations. Pyridazines are of great importance as bioactive compounds.9,10 Therefore, facile synthesis of multisubstituted pyridazines from simple modules including heteroatom-substituted alkynes would contribute to various research fields such as pharmaceutical sciences and agrochemistry. Good accessibility of stable heteroatom-substituted alkynes,11 compared to reactive and unstable ynamines,12 should be a clear benefit for pyridazine synthesis. In this study, after screening a broad range of heteroatom-substituted alkynes in the tetrazine reaction, we have developed a facile method to prepare trisubstituted pyridazines regioselectively from tetrazines and alkynyl sulfides through uncommon interactions between tetrazine substituents and sulfanyl groups.
Results and discussion
Diels–Alder reaction and subsequent denitrogenation efficiently took place to afford trisubstituted pyridazines 11 and/or 12 bearing heteroatom substituents from tetrazine 4a with various heteroatom-substituted alkynes 10 (Fig. 2). After examining the reactions in HFIP at 40 °C, we changed the reaction conditions in several cases. For example, aromatic alkynes having N, S, and P substituents showed good reactivities with tetrazine 4a (Fig. 2A and B). When ynamide 10a was treated with tetrazine 4a in p-xylene at 150 °C, we achieved the synthesis of aminopyridazines 11a and 12a in moderate yields, where 4-amino-substituted pyridazine 12a was obtained as a major product. Desired pyridazines 11a and 12a were not obtained when the reaction was performed in HFIP at 40 °C. Treatment of alkynyl sulfide 10b with tetrazine 4a in toluene at 110 °C or in HFIP at 40 °C provided pyridazines 11b and 12b in high yields, in which the formation of 5-sulfanylpyridazine 11b was slightly favored. Pyridazine 11c bearing a phosphinyl group was prepared selectively from alkynyl phosphine 10c without forming 4-phosphinyl-substituted pyridazine 12c, although desired pyridazine 11c was not obtained when the reaction was conducted in HFIP at 40 °C. Notably, efficient pyridazine synthesis was accomplished by using a variety of heteroatom-substituted alkynes 10a–10c which showed low reactivities and thus were easy to purify with silica-gel column chromatography, although the pioneering study was conducted using significantly reactive and easily hydrolyzable ynamine 5. Considering that 3,4-diphenyl-1,2,4,5-pyridazine (8a) was obtained selectively by treatment of tetrazine 4a with phenylacetylene (7a) by the significant interaction between two phenyl groups, competitive effects of the amino or sulfanyl group would lead to the formation of 4-heteroatom-substituted pyridazines 12a and 12b.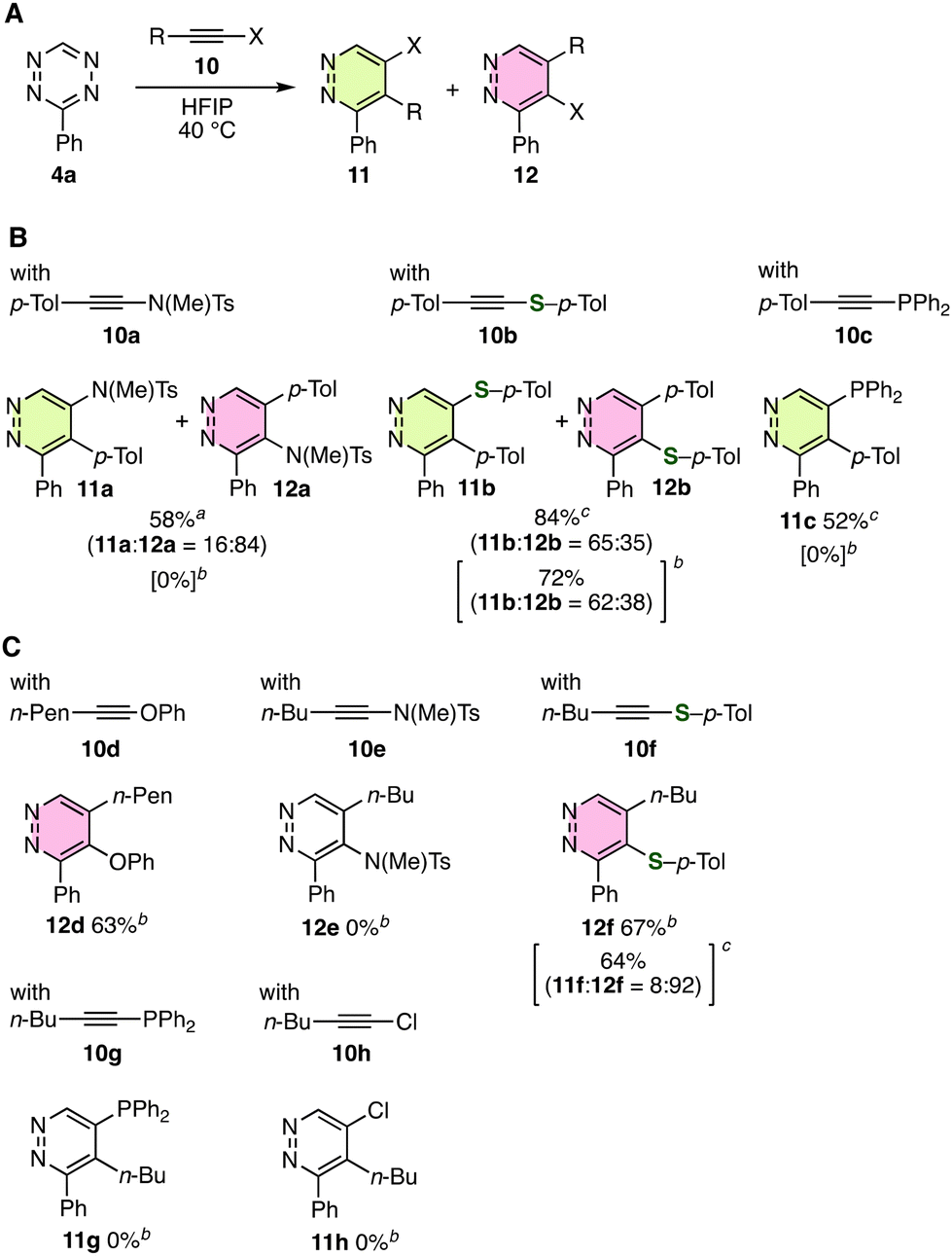 | ||
Fig. 2 Synthesis of pyridazines from tetrazine 4a using various heteroatom-substituted alkynes. (A) General scheme. (B) Products when using aromatic alkynes. (C) Products when using aliphatic alkynes. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) The reaction was conducted in p-xylene at 150 °C. bThe reaction was conducted in HFIP at 40 °C. cThe reaction was conducted in toluene at 110 °C. See the ESI† for details. The reaction was conducted in p-xylene at 150 °C. bThe reaction was conducted in HFIP at 40 °C. cThe reaction was conducted in toluene at 110 °C. See the ESI† for details. | ||
We then examined pyridazine synthesis from tetrazine 4a and heteroatom-substituted aliphatic alkynes 10d–10h (Fig. 2A and C). As a result, the selective preparation of pyridazines 12d and 12f was achieved when using alkynyl ether 10d and sulfide 10f, respectively. Particularly, it is noteworthy that 4-sulfanylpyridazine 12f was selectively synthesized by treatment of tetrazine 4a with alkynyl sulfide 10f in HFIP at 40 °C due to the good transformability of sulfanyl groups. The selectivity was slightly decreased when the reaction was performed in toluene at 110 °C. In contrast, pyridazines 11e/12e, 11g/12g, or 11h/12h were not obtained by the reactions of 4a with alkyne 10e, 10g, or 10h, where the formation of complex mixtures of products was observed. These results clearly showed that alkynyl sulfides efficiently reacted with tetrazine 4a and the unique selectivity was controlled by the sulfanyl group.
A wide range of pyridazyl sulfides 12 were successfully synthesized from 3-phenyl-1,2,4,5-tetrazine (4a) and various alkynyl sulfides (Fig. 3A and B). For example, 4-ethylthio-substituted pyridazine 12i was selectively obtained in good yield by treatment of tetrazine 4a with ethyl-4-tolylethynyl sulfide (10i) in HFIP at 40 °C, where 5-sulfanylpyridazine 11i was not observed. We accomplished the selective synthesis of pyridazines 12j and 12k in high yields from the corresponding alkynyl sulfides leaving sulfanyl, hydroxy, and ester moieties untouched. Of note, ethyl-3-hydroxy-3-methyl-1-butynyl sulfide also reacted with tetrazine 4a to afford pyridazine 12l having a bulky tertiary alcohol moiety. Pyridazines 12m and 12n with n-dodecyl and 4-chlorophenyl groups were efficiently prepared from tetrazine 4a and the corresponding alkynyl sulfides. Also, we synthesized a mixture of pyridazines 11o and 12o from tetrazine 4a and ethyl 4-tolylethynyl sulfide (10o).
 | ||
| Fig. 3 Synthesis of pyridazines from tetrazines and alkynyl sulfides. (A) General scheme. (B) Products from 4a with various alkynyl sulfides. (C) Products from ethyl 1-hexyn-1-yl sulfide (10i) with various tetrazines. See the ESI† for details. aThe reaction was conducted in toluene at 110 °C instead of HFIP at 40 °C. | ||
Various tetrazines participated in the selective pyridazine synthesis with alkynyl sulfide 10i (Fig. 3A and C). Indeed, pyridazines 12p and 12q were successfully synthesized from tetrazines having electron-rich aromatic rings without forming 5-sulfanyl-substituted pyridazines. Reactions of tetrazines having electron-deficient aryl groups with alkynyl sulfide 10i took place smoothly to furnish pyridazines 12r–12t without affecting fluoro, chloro, and bromo groups. It is worth noting that the selective pyridazine formation was also realized when using 3-benzyl- or 3-(2-(methoxycarbonyl)ethyl)-1,2,4,5-tetrazine to provide 12u or 12v, respectively, in which 5-sulfanylpyridazines 11 were not obtained. Unfortunately, we failed in the preparation of tetrasubstituted pyridazine 12w from 3,6-bis(2-pyridyl)-1,2,4,5-tetrazine and sulfide 10i.
To our surprise, the regioselectivity was completely switched only by changing the sulfur substituents from sulfanyl to sulfinyl or sulfonyl groups (Fig. 4A and B).13 For instance, 5-sulfinyl-substituted pyridazine 11x was synthesized selectively from tetrazine 4a with ethyl 1-hexyn-1-yl sulfoxide (10p). Reactions of alkynyl sulfoxides with tetrazine 4a proceeded smoothly to afford 5-sulfinyl-substituted pyridazines 11y–11aa without the formation of regioisomers 12. We also accomplished the selective synthesis of 5-sulfonyl-substituted pyridazines 11ab and 11ac from the corresponding alkynyl sulfones, where 4-sulfonylpyridazines 12 were not observed.
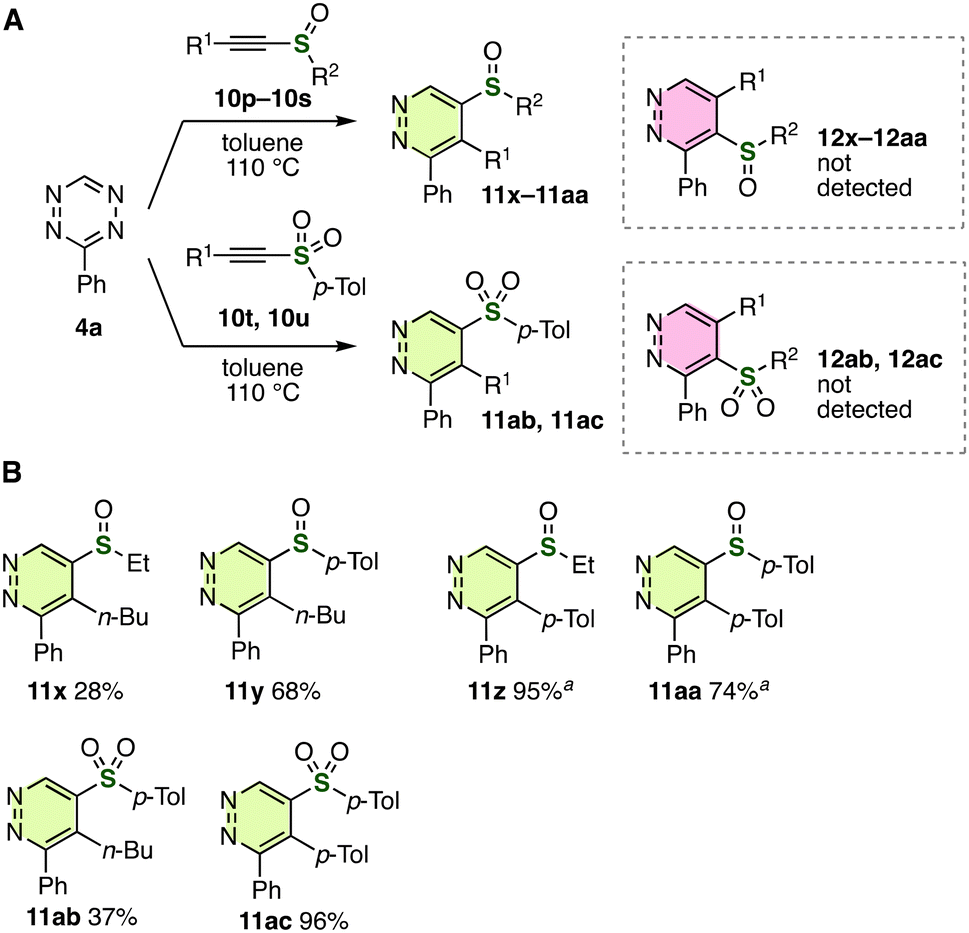 | ||
| Fig. 4 Synthesis of pyridazines from tetrazine 4a and alkynyl sulfoxides or sulfones. (A) General scheme. (B) Products. See the ESI† for details. aThe reaction was conducted in toluene at 110 °C instead of HFIP at 40 °C. | ||
Transformations of 4-sulfanylpyridazine 12i indicated that a wide variety of pyridazines can be synthesized through the selective pyridazine formations from tetrazines and alkynyl sulfides (Fig. 5A). Sulfoxide 12x was prepared by the oxidation of sulfide 12i in the presence of a molybdenum catalyst in moderate yield (Fig. 5A, upper).14 Since the synthesis of 4-sulfinyl-substituted pyridazine 12x was difficult by the reaction of 4a with alkynyl sulfoxide 10p, this result demonstrates the advantage of this protocol via the reaction of tetrazines with alkynyl sulfides and subsequent S-oxidation. We then succeeded in the reduction of sulfide 12i furnishing 5-butyl-3-phenylpyridazine (13) under palladium-catalyzed conditions (Fig. 5A, lower).15 Since it is not easy to prepare pyridazine 13 from tetrazine 4a and 1-hexyne due to the poor selectivity in the case of aliphatic alkyne (Fig. 1C, lower), it is worth noting that the selective synthesis of disubstituted pyridazines having alkyl substituents was realized through pyridazine formation followed by reduction. The reduction of sulfoxide 11aa was realized under conditions using titanium(IV) chloride and triphenylphosphine to afford sulfide 11b (Fig. 5B).16 A regioisomeric mixture of 11b and 12b was obtained when tetrazine 4a was treated with alkynyl sulfide 10b (Fig. 2C). In contrast, the selective pyridazyl sulfoxide formation from 4a and p-tolyl-p-tolylethynyl sulfoxide and subsequent S-reduction resulted in the preparation of pyridazine 11b without 4-sulfanyl-substituted pyridazine 12b. These results clearly show that selective pyridazine formation from tetrazines and sulfur-substituted alkynes followed by further transformations allowed us to synthesize diverse multisubstituted pyridazines without isomers.
 | ||
| Fig. 5 (A) Transformations of pyridazine 12i. (B) Reduction of sulfoxide 11aa. See the ESI† for details. | ||
To gain insight into the origin of the unique selectivity for the tetrazine reaction with alkynyl sulfides, we analyzed the reaction mechanism by theoretical calculations based on the DFT method (ωB97X-D/6-311+G(d,p))5n (Fig. 6). These results obviously support the reaction pathways forming pyridazines 11ad and 12advia the IEDDA reaction between tetrazine 4a and alkynyl sulfide 10i and subsequent removal of nitrogen, in which activation energies for the denitrogenation were significantly smaller than those for the IEDDA reactions (Fig. 6A and B). The theoretical pathway in the gas phase for 4-sulfanylpyridazine 12i requires 2.4 kcal mol−1 lower activation energy than that for the isomer 11i, showing good agreement between the DFT calculations and experimental results. The difference of activation energies was slightly increased when the calculation was conducted in polar solvents. These results are in good agreement with the experimental result that 4-sulfanylpyridazine 12f was obtained as a major isomer not only in HFIP at 40 °C but also in toluene at 110 °C.
 | ||
| Fig. 6 Calculated plausible reaction pathways for the synthesis of pyridazines 12ad (A) and 11ad (B) by a DFT method (ωB97X-D/6-311+G(d,p)). Respective energy differences are shown. aCalculated energy differences in polar solvents. See the ESI† for details. | ||
The detailed computational analysis provided a clue for the favored transition state structure TS1 forming 4-sulfanylpyridazine 12ad compared to TS3 for isomer 11ad (Fig. 7 and 8). The frontier molecular orbital (FMO) theory analysis of tetrazine 4a and alkynyl sulfide 10v indicates the energy gap between LUMO+1 of tetrazine 4a and HOMO of alkynyl sulfide 10v is significantly lower than that between HOMO−1 of 4a and LUMO of 10v (Fig. 7A–C), supporting that the pyridazine formation proceeds via the IEDDA reaction, while the regioselective pyridazine formation is not explained by the orbital analysis. In the transition state structure TS1 for major isomer 12ad, the n–π* interaction between the sulfur atom of alkynyl sulfide 10v and the phenyl group of tetrazine 4a was suggested by the positive charge at the sulfur atom due to the electron donation to the electron-deficient benzene ring (Fig. 8A–C), LUMO of the transition state (Fig. 8E), and shorter distances of S–C2-2 and S–C2-3 than the sum of van der Waals radii (Fig. 8C) in comparison to TS3 for isomer 11ad (Fig. 8D and F). These results suggested that the significant n–π* interaction of the sulfur with the tetrazine substituent realized the selective synthesis of 4-sulfanyl-substituted pyridazines.
 | ||
| Fig. 7 Calculated structures by a DFT method (ωB97X-D/6-311+G(d,p)). See the ESI† for details. (A) Optimized structure of 4a. (B) Optimized structure of 10v. (C) FMO analysis between 4a and 10v with structures and orbital levels of LUMO+1, LUMO, HOMO, and HOMO−1 of 4a and LUMO and HOMO of 10v. | ||
 | ||
| Fig. 8 Charge distribution analysis of 4a (A), 10v (B), TS1 (C), and TS3 (D). Natural atomic charges were calculated by a DFT method (ωB97X-D/6-311+G(d,p)). The calculated charges are shown as red numbers. Atomic distances are shown as black numbers. See the ESI† for details. LUMOs of TS1 (E) and TS3 (F). | ||
Conclusions
In summary, we found that a wide variety of pyridazines could be synthesized from 3-substituted 1,2,4,5-tetrazines and alkynyl sulfides. Unique regioselectivities were controlled by the interaction between sulfanyl groups and tetrazine substituents which was supported by theoretical calculations. Further studies such as applications to construct a vast pyridazine library for drug discovery, regiocontrolled IEDDA reactions using alkynyl sulfides with various electron-deficient dienes, and detailed theoretical studies of IEDDA reactions involving energy decomposition analysis based on the distortion/interaction model17,18 are ongoing in our laboratory.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Central Glass Co. Ltd for providing Tf2O. This work was supported by JSPS KAKENHI Grant Number JP22H02086 (S. Y.); the Uehara Memorial Foundation (S. Y.); Tokuyama Science Foundation (S. Y.); the Ube Foundation (S. Y.); and Inamori Research Grants (S. Y.).References
- F. Fringuelli and A. Taticchi, The Diels-Alder Reaction: Selected Practical Methods, John Wiley & Sons, West Sussex, 2002 Search PubMed.
- S. Danishefsky and T. Kitahara, Useful diene for the Diels-Alder reaction, J. Am. Chem. Soc., 1974, 96, 7807 Search PubMed.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Inverse electron demand Diels–Alder reactions in chemical biology, Chem. Soc. Rev., 2017, 46, 4895 Search PubMed.
- (a) A. C. Knall and C. Slugovc, Inverse electron demand Diels–Alder (iEDDA)-initiated conjugation: a (high) potential click chemistry scheme, Chem. Soc. Rev., 2013, 42, 5131 Search PubMed; (b) K. Kang, J. Park and E. Kim, Tetrazine ligation for chemical proteomics, Proteome Sci., 2016, 15, 15 Search PubMed; (c) S. Mayer and K. Lang, Tetrazines in Inverse-Electron-Demand Diels–Alder Cycloadditions and Their Use in Biology, Synthesis, 2017, 49, 830 Search PubMed; (d) V. Rigolot, C. Biot and C. Lion, To View Your Biomolecule, Click inside the Cell, Angew. Chem., Int. Ed., 2021, 60, 23084 Search PubMed.
- (a) S. B. Engelsma, L. I. Willems, C. E. van Paaschen, S. I. van Kasteren, G. A. van der Marel, H. S. Overkleeft and D. V. Filippov, Acylazetine as a Dienophile in Bioorthogonal Inverse Electron-Demand Diels–Alder Ligation, Org. Lett., 2014, 16, 2744 Search PubMed; (b) J. Henise, B. R. Hearn, G. W. Ashley and D. V. Santi, Biodegradable Tetra-PEG Hydrogels as Carriers for a Releasable Drug Delivery System, Bioconjugate Chem., 2015, 26, 270 Search PubMed; (c) S. Eising, F. Lelivelt and K. M. Bonger, Vinylboronic Acids as Fast Reacting, Synthetically Accessible, and Stable Bioorthogonal Reactants in the Carboni–Lindsey Reaction, Angew. Chem., Int., Ed., 2016, 55, 12243 Search PubMed; (d) A.-C. Knall, M. Hollauf, R. Saf and C. Slugovc, A trifunctional linker suitable for conducting three orthogonal click chemistries in one pot, Org. Biomol. Chem., 2016, 14, 10576 Search PubMed; (e) K. Liu, B. Enns, B. Evans, N. Wang, X. Shang, W. Sittiwong, P. H. Dussault and J. Guo, A genetically encoded cyclobutene probe for labelling of live cells, Chem. Commun., 2017, 53, 10604 Search PubMed; (f) X. Shang, X. Song, C. Faller, R. Lai, H. Li, R. Cerny, W. Niu and J. Guo, Fluorogenic protein labeling using a genetically encoded unstrained alkene, Chem. Sci., 2017, 8, 1141 Search PubMed; (g) H. Lebraud, D. J. Wright, C. N. Johnson and T. D. Heightman, Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras, ACS Cent. Sci., 2016, 2, 927 Search PubMed; (h) Y. Qu, F.-X. Sauvage, G. Clavier, F. Miomandre and P. Audebert, Metal-Free Synthetic Approach to 3-Monosubstituted Unsymmetrical 1,2,4,5-Tetrazines Useful for Bioorthogonal Reactions, Angew. Chem., Int. Ed., 2018, 57, 12057 Search PubMed; (i) S. Eising, A. H. J. Engwerda, X. Riedijk, F. M. Bickelhaupt and K. M. Bonger, Highly Stable and Selective Tetrazines for the Coordination-Assisted Bioorthogonal Ligation with Vinylboronic Acids, Bioconjugate Chem., 2018, 29, 3054 Search PubMed; (j) B. J. Umlauf, K. A. Mix, V. A. Grosskopf, R. T. Raines and E. V. Shusta, Site-Specific Antibody Functionalization Using Tetrazine–Styrene Cycloaddition, Bioconjugate Chem., 2019, 29, 1605 Search PubMed; (k) M. Gupta, M. Singha, D. B. Rasale, Z. Zhou, S. Bhandari, S. Beasley, J. Sakr, S. M. Parker and R. C. Spitale, Mutually Orthogonal Bioconjugation of Vinyl Nucleosides for RNA Metabolic Labeling, Org. Lett., 2021, 23, 7183 Search PubMed; (l) A. Turlik, K. N. Houk and D. Svatunek, Origin of Increased Reactivity in Rhenium-Mediated Cycloadditions of Tetrazines, J. Org. Chem., 2021, 86, 13129 Search PubMed; (m) U. M. Battisti, R. García-Vázquez, D. Svatunek, B. Herrmann, A. Löffler, H. Mikula and M. M. Herth, Synergistic Experimental and Computational Investigation of the Bioorthogonal Reactivity of Substituted Aryltetrazines, Bioconjugate Chem., 2022, 33, 608 Search PubMed; (n) D. Svatunek, M. Wilkovitsch, L. Hartmann, K. N. Houk and H. Mikula, Uncovering the Key Role of Distortion in Bioorthogonal Tetrazine Tools That Defy the Reactivity/Stability Trade-Off, J. Am. Chem. Soc., 2022, 144, 8171 Search PubMed.
- (a) A. Steigel and J. Sauer, (4 + 2)-cycloadditionen 6-gliedriger heterocyclen mit inaminen, Tetrahedron Lett., 1970, 11, 3357 Search PubMed; (b) O. Meresz and P. A. Foster-Verner, Synthesis of 3-monosubstituted s-tetrazines and their reactions with monosubstituted acetylenes, J. Chem. Soc., Chem. Commun., 1972, 950 Search PubMed; (c) L. Birkofer and E. Hënsel, Silyl-Derivate von Pyridazin, Chem. Ber., 1981, 114, 3154 Search PubMed; (d) J. Sauer and D. K. Heldmann, Synthesis of 3,5-disubstituted pyridazines by regioselective [4 + 2] cycloadditions with ethynyltributyltin and subsequent replacement of the organotin substituent, Tetrahedron, 1998, 54, 4297 Search PubMed; (e) J. Sauer, D. K. Heldmann, J. Hetzenegger, H. Sichert and J. Schuster, 1,2,4,5-Tetrazine: Synthesis and Reactivity in [4 + 2] Cycloadditions, Eur. J. Org. Chem., 1998, 2885 Search PubMed.
- C. Yamamoto, M. Suzuki and S. Yoshida, Pyridazine Synthesis from 1,2,4,5-Tetrazines and Alkynes in 1,1,1,3,3,3-Hexafluoro-2-propanol through the Inverse Electron Demand Diels–Alder Reaction, Bull. Chem. Soc. Jpn., 2022, 95, 1741 Search PubMed.
- (a) J. Ichikawa, S. Miyazaki, M. Fujiwara and T. Minami, Fluorine-Directed Nazarov Cyclizations: A Controlled Synthesis of Cross-Conjugated 2-Cyclopenten-1-ones, J. Org. Chem., 1995, 60, 2320 Search PubMed; (b) J.-P. Bégué, D. Bonnet-Delpon and B. Crousse, Fluorinated Alcohols: A New Medium for Selective and Clean Reaction, Synlett, 2004, 18 Search PubMed; (c) T. Dohi, N. Yamaoka and Y. Kita, Fluoroalcohols: versatile solvents in hypervalent iodine chemistry and syntheses of diaryliodonium(III) salts, Tetrahedron, 2010, 66, 5775 Search PubMed; (d) Y.-F. Yang, P. Yu and K. N. Houk, Computational Exploration of Concerted and Zwitterionic Mechanisms of Diels–Alder Reactions between 1,2,3-Triazines and Enamines and Acceleration by Hydrogen-Bonding Solvents, J. Am. Chem. Soc., 2017, 139, 18213 Search PubMed; (e) H. F. Motiwala, A. M. Armaly, J. G. Cacioppo, T. C. Coombs, K. R. K. Koehn, V. M. Norwood IV and J. Aubé, HFIP in Organic Synthesis, Chem. Rev., 2022, 122, 12544 Search PubMed; (f) P. Ma, D. Svatunek, Z. Zhu, D. L. Boger, X.-H. Duan and K. N. Houk, Computational Studies of Reactions of 1,2,4,5-Tetrazines with Enamines in MeOH and HFIP, J. Am. Chem. Soc., 2024, 146, 18706 Search PubMed.
- Z.-X. He, Y.-P. Gong, X. Zhang, L.-Y. Ma and W. Zhao, Pyridazine as a privileged structure: An updated review on anticancer activity of pyridazine containing bioactive molecules, Eur. J. Med. Chem., 2021, 209, 112946 Search PubMed.
- (a) S. Elmeligie, E. M. Ahmed, S. M. Abuel-Maaty, S. A.-B. Zaitone and D. S. Mikhail, Design and Synthesis of Pyridazine Containing Compounds with Promising Anticancer Activity, Chem. Pharm. Bull., 2017, 65, 236 Search PubMed; (b) C. Hamze, J. Brossier, K. Karaghiosoff, E. Godineau and P. Knochel, Selective and Stepwise Functionalization of the Pyridazine Scaffold by Using Thio-Substituted Pyridazine Building Blocks, Chem. – Eur. J., 2023, 29, e202302156 Search PubMed.
- (a) K. A. DeKorver, H. Li, A. G. Lohse, R. Hayashi, Z. Lu, Y. Zhang and R. P. Hsung, Ynamides: A Modern Functional Group for the New Millennium, Chem. Rev., 2010, 110, 5064 Search PubMed; (b) D. Campeau and F. Gagosz, Heteroatom-substituted alkynes as three-atom components in (3 + 2) cycloadditions, Cell Rep. Phys. Sci., 2023, 4, 101212 Search PubMed.
- H. G. Viehe, Synthesis and Reactions of the Alkynylamines, Angew. Chem., Int. Ed. Engl., 1967, 6, 767 Search PubMed.
- Sulfanyl-, sulfinyl, and sulfonyl-substituted dienophiles reacted with Danishefsky–Kitahara diene (1) to afford cycloadducts with same regioselectivities. See: M. Iwao and T. Kuraishi, Utilization of Sulfide, Sulfoxide, and Sulfone Groups as Regiochemical Control Elements in the Diels–Alder Reaction of Naphthoquinones, Bull. Chem. Soc. Jpn., 1987, 60, 4051 Search PubMed.
- A. Bayat, M. Shakourian-Fard and M. M. Hashemi, Selective oxidation of sulfides to sulfoxides by a molybdate-based catalyst using 30% hydrogen peroxide, Catal. Commun., 2014, 52, 16 Search PubMed.
- T. Matsumura, T. Niwa and M. Nakada, Pd-catalyzed reductive cleavage of alkyl aryl sulfides with triethylsilane that is accelerated by trialkylsilyl chloride, Tetrahedron Lett., 2012, 53, 4313 Search PubMed.
- S. Kikuchi, H. Konishi and Y. Hashimoto, The Deoxygenation of Sulfoxide Mediated by the PhsP/Lewis Acid Combination and the Application to the Kinetic Resolution of Racemic Phosphines Using Optically Active Sulfoxide, Tetrahedron, 2005, 61, 3587 Search PubMed.
- D. H. Ess and K. N. Houk, Theory of 1,3-Dipolar Cycloadditions: Distortion/Interaction and Frontier Molecular Orbital Models, J. Am. Chem. Soc., 2008, 130, 10187 Search PubMed.
- (a) F. M. Bickelhaupt and K. N. Houk, Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model, Angew. Chem., Int. Ed., 2017, 56, 10070 Search PubMed; (b) P. Vermeeren, T. A. Hamlin and F. M. Bickelhaupt, Origin of asynchronicity in Diels–Alder reactions, Phys. Chem. Chem. Phys., 2021, 23, 20095 Search PubMed; (c) N. Houszka, H. Mikula and D. Svatunek, Substituent Effects in Bioorthogonal Diels–Alder Reactions of 1,2,4,5-Tetrazines, Chem. – Eur. J., 2023, 29, e202300345 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, and characterization for new compounds including NMR spectra. See DOI: https://doi.org/10.1039/d4qo01286k |
| This journal is © the Partner Organisations 2024 |