
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Acyl radicals generated from aldehydes with NHPI as electrocatalyst: aldehydes and alcohols as carbon-centered radical precursors†
Rodrigo G.
Enríquez
 a,
Juan S.
Dato-Santiago
a,
Roberto
del Río-Rodríguez
a,
José
Alemán
*abc and
Jose A.
Fernández-Salas
*ab
a,
Juan S.
Dato-Santiago
a,
Roberto
del Río-Rodríguez
a,
José
Alemán
*abc and
Jose A.
Fernández-Salas
*ab
aOrganic Chemistry Department, Módulo 2, Universidad Autónoma de Madrid, 28049 Madrid, Spain. E-mail: j.fernandez@uam.es
bInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, Madrid, Spain
cCenter for Innovation in Advanced Chemistry (ORFEO-CINQA), Universidad Autónoma de Madrid, 28049 Madrid, Spain
First published on 17th July 2024
Abstract
In this work, we describe how N-hydroxyphthalimide (NHPI) promotes hydrogen atom transfer from aldehydes under electrocatalytic chemical conditions. This involves the generation of phthalimide-N-oxyl (PINO), which abstracts an acyl hydrogen to produce acyl radicals from aldehydes. These acyl radicals then react with oximes or another PINO radical to form redox-active esters (RAEs). Additionally, we detail a Giese-type reaction with electron-deficient alkenes via electrochemical reduction of the generated RAE derivatives. This process produces sp3-carbon centered radicals from aldehydes. We also extend this method to aliphatic alcohols, involving a multi-step electrochemical process to form carbon-centered radicals from readily available alcohols.
Introduction
Nowadays, there is increasing demand from society for the development of new synthetic strategies that are more sustainable, environmentally friendly and exhibit high atom economy.1 In this context, the selective functionalization of C–H bonds has attracted enormous attention from both the chemical industry and academia as it has clearly been recognized as one of the prime challenges in modern synthetic chemistry.2 Inspired by visible light driven HAT processes,2i,3 electrochemistry, which has experienced a renaissance in the past decade,4 has also emerged as a new means for the in situ generation of HAT abstractors from various redox mediators with the ability to activate C–H bonds.5 From this point of view, various systems have been studied in the past few years. However, it should be highlighted that there are only a few methods that describe direct C–H functionalization using a HAT mediator under electrochemical conditions.5,6 These methods are even fewer if we consider those that use the generated carbon-centered radical for the formation of new C–C bonds promoted by substoichiometric amounts of the HAT promoter since the field is dominated by the oxidation of alcohols, and oxygenation, halogenation and azidation reactions.7Acyl radicals are an interesting class of carbon-centered radicals due to their valuable synthetic applications towards the construction of carbonyl compounds.8 Although these radicals can be accessed by abstraction of carbonylic hydrogen,9 less straightforward approaches have also traditionally been used when the generation of acyl radicals was performed under electrochemical conditions, such as the cleavage of prefunctionalised RCO-X or carbonylation reactions.8,10 Despite the great efforts that have been made towards the generation of acyl radicals under electrochemical conditions, the direct electro-generation of acyl radicals from aldehydes remains challenging and its widespread generation and application are yet to be achieved.11 Therefore, the development of an electrochemically driven process that would directly abstract hydrogen from aldehydes to generate acyl radicals would be highly desirable. In this context, redox-active esters (RAEs) are very useful radical precursors in organic synthesis,12 and their syntheses are carried out either by esterifying carboxylic acids with N-hydroxyphthalimide13 or by forming acyl radicals with stoichiometric reagents,14 with both methods requiring superstoichiometric amounts of reagents (Scheme 1A). Although important advancements have been made towards the synthesis of RAEs from aldehydes through cross dehydrogenative type strategies,14 the need for the use of harsh reaction conditions (high temperatures),14b,e,d metal promoters14c,f and stoichiometric amounts of oxidants14a,c,e,d may still hamper their broad applicability. Therefore, the synthesis of redox active esters from aldehydes by using a hydrogen atom transfer mediator, with the potential to synergistically act as a reagent under electrochemical conditions, would be an ideal process that leads to the formation of H2 as the sole byproduct.15,16
 | ||
| Scheme 1 Previous work (A) and this work (B). | ||
Herein, we describe the use of N-hydroxyphthalimide (NHPI) as a hydrogen atom transfer mediator to generate acyl radicals (Scheme 1B). These versatile carbon-centered radicals have been exploited for the formation of C–C bonds and the synthesis of RAEs when the electrochemically generated N-oxy radical is additionally used to trap the acyl radical. Moreover, when performed in the presence of a deactivated alkene, the in situ generated RAE can be electrochemically reduced to generate a sp3-carbon-centered radical with the ability to perform a Giese-type reaction. This one-pot transformation opens the opportunity for considering aldehydes as alkyl radical precursors.
Results and discussion
Optimization of the model reaction
We began the study by choosing octanal (1a) as the model precursor and oxime ether 2a as the ideal substrate for trapping the acyl radical due to its known versatility17 (Table 1, entry 1). By using an electrochemical system with graphite as the working electrode and nickel foam as the counter electrode under 1 mA constant current for 12 h in an undivided cell with KCl as the electrolyte in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of MeCN and H2O, compound 3a was isolated in good yield (61%, entry 1). With the aim of developing a synthetically useful electrochemical system for generating acyl radicals, various parameters such as solvents, electrolytes, electrode materials and electrochemical parameters have been studied and summarized, as shown in Table 1. Firstly, the addition of an alternative electrolyte was studied. KCl showed the best results (entries 2 and 3 and the ESI†). We then tested the reaction at a higher intensity and a shorter reaction time, and the desired product was obtained with a significantly lower efficiency (entry 4). Different solvent systems (entries 5–7) were then evaluated. Thus, when MeCN or H2O was used as the sole solvent, the performance of the electrochemical system was substantially worsened (entries 5 and 6). A mixture of MeCN:H2O in 3:1 proportion was then identified as optimal, as a different ratio of the solvent system was shown to lead to worse results (entry 7). Electrode material selection had a significant impact on the reaction. Counter electrodes other than nickel foam, such as those based on graphite, platinum or zinc, did not satisfactorily lead to the desired α-dicarbonylic product, with all affording the desired product in less than 10% yield (entries 9 and 10). We then tried to reduce the loading of NHPI (entry 11). Although the reaction proceeded remarkably well with only 20 mol% of the electrocatalyst (53%), 50 mol% of it was established as optimal in order to allow and facilitate the general progress of the studied reaction. Finally, the reaction did not take place in the absence of the electrochemical mediator or electrical current (entries 12 and 13).
:1 mixture of MeCN and H2O, compound 3a was isolated in good yield (61%, entry 1). With the aim of developing a synthetically useful electrochemical system for generating acyl radicals, various parameters such as solvents, electrolytes, electrode materials and electrochemical parameters have been studied and summarized, as shown in Table 1. Firstly, the addition of an alternative electrolyte was studied. KCl showed the best results (entries 2 and 3 and the ESI†). We then tested the reaction at a higher intensity and a shorter reaction time, and the desired product was obtained with a significantly lower efficiency (entry 4). Different solvent systems (entries 5–7) were then evaluated. Thus, when MeCN or H2O was used as the sole solvent, the performance of the electrochemical system was substantially worsened (entries 5 and 6). A mixture of MeCN:H2O in 3:1 proportion was then identified as optimal, as a different ratio of the solvent system was shown to lead to worse results (entry 7). Electrode material selection had a significant impact on the reaction. Counter electrodes other than nickel foam, such as those based on graphite, platinum or zinc, did not satisfactorily lead to the desired α-dicarbonylic product, with all affording the desired product in less than 10% yield (entries 9 and 10). We then tried to reduce the loading of NHPI (entry 11). Although the reaction proceeded remarkably well with only 20 mol% of the electrocatalyst (53%), 50 mol% of it was established as optimal in order to allow and facilitate the general progress of the studied reaction. Finally, the reaction did not take place in the absence of the electrochemical mediator or electrical current (entries 12 and 13).
|
|
||
|---|---|---|
| Deviation from optimized conditions | Yieldb (%) | |
|
a Standard reaction conditions: 0.1 mmol of 2a and 0.4 mmol of octanal in 3 mL MeCN:H2O 3:1 with KCl (0.1 mmol), under an air atmosphere, were set at constant current of 1 mA for 12 h at room temperature.
b Yields determined by 1H-NMR using 1,3,5-trimethoxybenzene as the internal standard. Isolated yields in brackets.
|
||
| 1 | No deviation | 72 (61) |
| 2 | TBABF4 | 69 |
| 3 | LiClO4 | 43 |
| 4 | 2.5 mA 3 h vs. 1 mA 12 h | 28 |
| 5 | H2O vs. MeCN/H2O 3:1 |
6 |
| 6 | MeCN vs. MeCN/H2O 3:1 |
34 |
| 7 | MeCN/H2O 1:1 vs. MeCN/H2O 3:1 |
10 |
| 8 | GRC (−) vs. Nifoam (−) | 7 |
| 9 | Zn (−) vs. Nifoam (−) | 3 |
| 10 | Pt (−) vs. Nifoam (−) | 8 |
| 11 | NHPI 0.2 equiv. vs. 0.5 equiv. | 53 |
| 12 | No NHPI | 0 |
| 13 | No current | 0 |
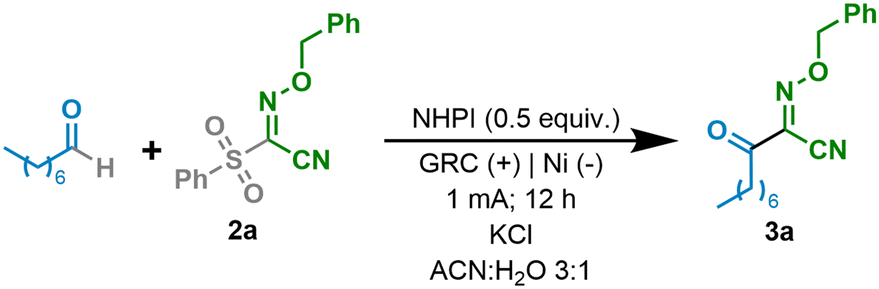
Substrate scope
Once the reaction conditions had been optimized, a variety of aldehydes were tested under the electrochemical reaction conditions in an undivided cell with the aim of studying the influence of the nature of the aldehyde on its electrochemical behaviour (Table 2). At first, various linear aldehydes were studied under the optimized reaction conditions. Thus, the carbon–carbon bond forming event through a HAT process took place efficiently and the desired products were obtained in good yields (3a–e). It should be highlighted that aromatic bromides (3c), which could get access to more versatile products upon further derivatizations, esters (3d) and free alcohols (3e) were found to be compatible with the electrochemical reaction conditions. More sterically hindered aldehydes decorated with an α-disubstituted carbon chain were also subjected to the electrochemical reaction. Thus, cyclopropyl and adamantyl decorated aldehydes led to the final product in synthetically useful yields (3f and 3g, respectively). A more sterically congested tert-butyl derivative was observed to react less efficiently and afforded the desired product in a low yield (3h). Aromatic aldehydes although consumed during the reaction did not lead to the desired product under the optimized reaction conditions. Benzylic or secondary and primary decorated amine aliphatic aldehydes were also incompatible with the present optimized reaction conditions. The electronics of the aromatic ring of the oxime partner were also considered. While an electron-poor phenyl ring led to a significantly lower yield (3i), its electron-rich analogue did not afford the desired product.|
a Reaction conditions: 1 (0.4 mmol) and 2 (0.1 mmol), NHPI (0.05 mmol) and KCI (0.1 mmol) in MeCN:H2O (3:1, 3 mL), r.t., under air, undivided cell (GRC (+), Nifoam (−)) at constant current (1.0 mA) for 12 h. Isolated yields.
|
|---|
|
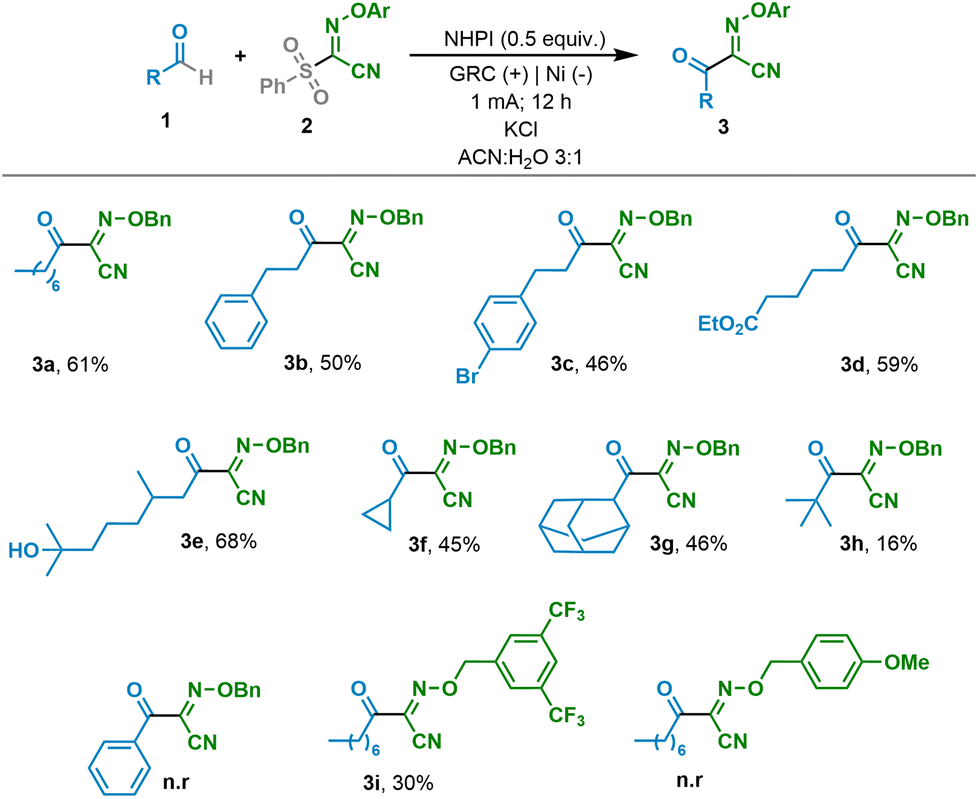
As mentioned above, with the aim of further exploiting the electrocatalytically generated acyl radicals, we envisioned the possibility of generating a redox active ester by exploiting the N-oxyl radical generated from NHPI not only to catalytically generate the acyl radical but also to quench it to form the carbon heteroatom bond, giving rise to the redox active ester. Therefore, after optimizing the reaction conditions (see the ESI†), we were able to obtain the desired RAE by simply coupling the aldehydes and NHPI (Table 3). It should be highlighted that primary, secondary and tertiary decorated aliphatic aldehydes were compatible and led to the desired RAEs in synthetically useful yields (4a–4e). In addition, benzaldehyde was efficiently coupled with the electrochemically generated PINO to yield the corresponding ester (4f).
|
a Reaction conditions: 1 (0.4 mmol), NHPI (0.05 mmol) and KCI (0.1 mmol) in MeCN:H2O (3:1, 3 mL), r.t., under air, undivided cell (GRC (+), Nifoam (−)) at constant current (1.0 mA) for 12 h. Isolated yields.
|
|---|
|
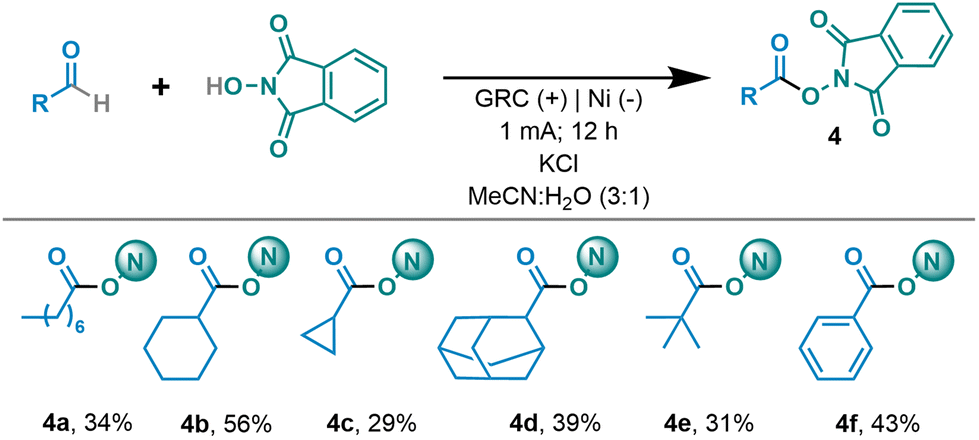
Given the encouraging results achieved and the synthetic potential of the developed method and considering the well-known ability of redox active esters to be electrochemically reduced,13 we speculated that under our optimized reaction conditions, the generated RAE might be in situ reduced, generating the corresponding alkyl radical upon decarboxylation. Therefore, considering that currently the primary application of redox active esters is to serve as precursors to generate sp3-carbon-centered radicals, we envisioned the possibility of extending the applicability of the method for generating the above-mentioned radical species directly from aldehydes. Thus, we studied the reaction of aldehydes in the presence of NHPI, which served as the catalyst and reagent, and vinyl sulfone, which served as the Giese acceptor (Scheme 2). To our delight, aldehydes as precursors of secondary and tertiary alkyl radicals performed efficiently, leading to the Giese adduct in moderate to good yields in the presence of vinyl sulfone and the phenyl ester (6a–6d).
 | ||
| Scheme 2 Giese reaction using aldehydes as radical precursors. Reaction conditions: 1 (0.3 mmol) and 5 (0.1 mmol), NHPI (0.2 mmol) and NH4PF6 (0.1 mmol) in MeCN:H2O (3:1, 3 mL), r.t., under air, undivided cell (GRC (+), Nifoam (−)) at constant current (10 mA) for 2 h. Isolated yields. b4 h. | ||
In addition, to expand this method, we envisioned the possibility of taking it one step further. Our approach would again take advantage of the versatility of the N-hydroxy radical derivatives generated electrochemically.5a,16a We propose a double oxidation process to generate the oxoammonium intermediate (shown to be feasible for similar substrates, Eox < 1.0 V vs. NHE).5a Once the active species is generated, an aliphatic alcohol derivative will form an adduct at the nitrogen atom of the oxoammonium species, followed by intramolecular hydride transfer to regenerate NHPI and the organic aldehyde. The aldehyde in situ generated would then trigger the acyl radical process, leading to the formation of the RAE and its subsequent reduction/decarboxylation to give rise to the sp3 carbon-centered radical. This overall process enables alcohols to serve as precursors of aliphatic carbon-centered radical species (Scheme 3). Therefore, after optimizing the reaction conditions (see the ESI†), we were able to obtain the desired RAE from a primary alcohol and NHPI (Scheme 3A). We then investigated alcohols as radical precursors in Giese-type reactions. Alcohols proved to be efficient precursors for tertiary, secondary and primary alkyl radicals, leading to the Giese adduct in synthetically useful yields (6b and 6e–g). It should be highlighted that primary alkyl radicals can be generated and they performed efficiently, in contrast to known alternatives.
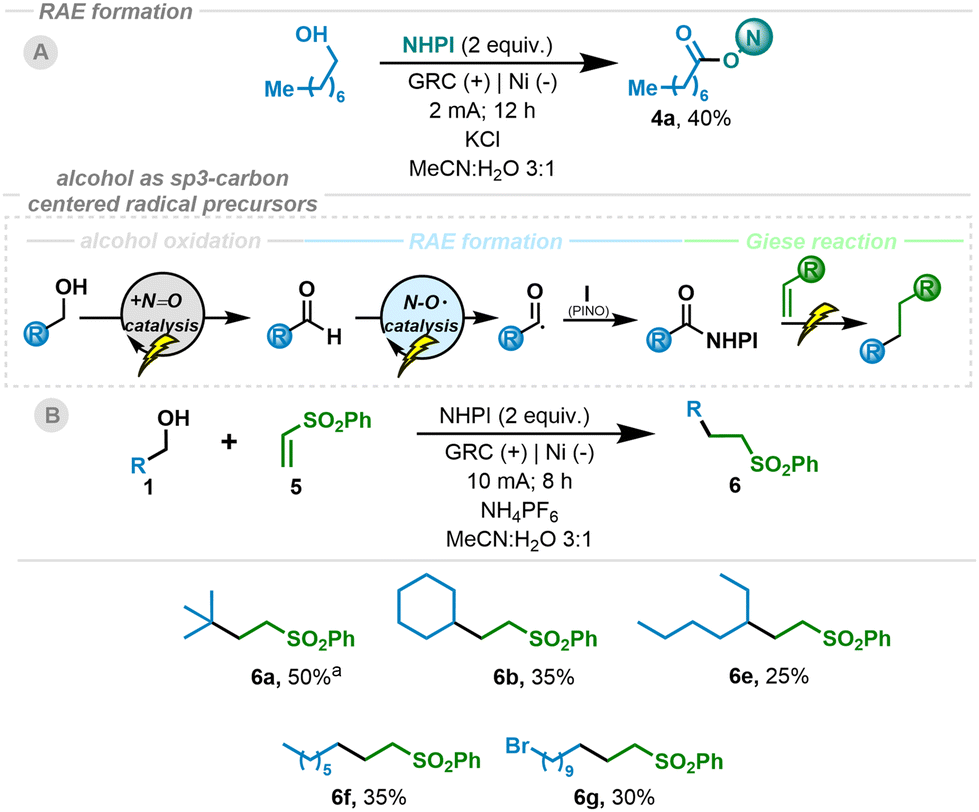 | ||
| Scheme 3 Giese reaction using alcohols as radical precursors. Reaction conditions: 1 (0.4 mmol) and 2 (0.1 mmol), NHPI (0.6 mmol) and NH4PF6 (0.1 mmol) in MeCH:H2O (3:1, 3 mL), r.t., under air, undivided cell (GRC (+), Nifoam (−)) at constant current (10 mA) for 8 h. Isolated yields. bUsing LiCIO4 as electrolyte. | ||
A mechanistic proposal of the electrochemical process is depicted in Scheme 4. Based on the literature,5a,16a the alkoxy radical I formed through the electrochemical oxidation of NHPI performed the hydrogen atom transfer event from the aldehyde, forming the corresponding acyl radical and regenerating the N-oxyl derivative. The radical formed can react either with oxime 2 to form compound 3 or be trapped by another alkoxy radical, giving rise to the RAE, as shown in the optimized reaction conditions in Schemes 2 and 3.
 | ||
| Scheme 4 Proposed reaction mechanism. | ||
The formation of compounds 3 and 4 provide mechanistic evidence for the formation of the acyl radical; trapping the acyl radical using the oxime or a radical scavenger such as PINO (I), respectively, would directly lead to these compounds. Once the RAE derivative is formed, it undergoes a well-documented electrochemical reduction, giving rise to the carbon-centered radical upon decarboxylation.13 In addition, to further support the source of the sp3-carbon-centered radical, we carried out a test reaction (see the ESI†) that showed that under the optimized reaction conditions, RAE 4b led to the desired Giese-type product 6b with similar efficiency compared to the reaction involving the aldehyde or the alcohol. Then, a conjugate addition of the radical to the Giese acceptor would take place.18 The resulting radical species III might then be reduced at the cathode and upon protonation, would lead to the Giese type product 6 from the aliphatic aldehyde.
Conclusions
In conclusion, we describe an electrochemical method for generating acyl radicals from aldehydes using NHPI as the promoter. The reaction system has demonstrated its robustness and generality as various aldehydes can be coupled successfully with oximes within this electrochemical system. The N-hydroxy radical generated upon anodic oxidation plays a multifunctional role in the present methodology, as it acts as substoichiometric promoter of the acyl radical and as a reagent towards the synthesis of redox active esters. Therefore, various RAEs have been prepared by this cross-dehydrogenative type electrochemical methodology using aldehydes as platforms and NHPI as the reagent, avoiding the use of coupling reagents. Moreover, aldehydes and even alcohols have been demonstrated to be valuable sp3-carbon-centered radical precursors in Giese type reactions through the electroreduction of RAEs in situ generated from them.Author contributions
R. G. E., J. S. D. S. and R. R. R. carried out the optimization and studied the scope of the reaction. J. A. and J. A. F. S. conceived the project and prepared the manuscript, which was edited by all the other authors.Data availability
All data supporting the findings of this study, including experimental details and spectroscopic characterization data for all compounds, are available within the article and the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the Spanish Government (PID2021-122299NB-I00, TED2021-130470B-I00, TED2021-129999B-C32), ‘Comunidad de Madrid’, European Structural Funds (S2018/NMT-4367), proyectos sinérgicos I + D (Y2020/NMT6469) and Comunidad Autónoma de Madrid (SI1/PJI/2019-00237). J. A. F.-S. thanks the Spanish Government for a Ramón y Cajal contract.References
- (a) N. S. Lewis, Toward Cost-Effective Solar Energy Use, Science, 2007, 315, 798–801 CrossRef CAS PubMed; (b) D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Key green chemistry research areas—a perspective from pharmaceutical manufacturers, Green Chem., 2007, 9, 411–420 RSC; (c) A. Albini and M. Fagnoni, Green chemistry and photochemistry were born at the same time, Green Chem., 2004, 6, 1–6 RSC.
- (a) A. S. Goldman and K. I. Goldberg, Organometallic C-H Bond Activation: An Introduction, ACS Symp. Ser., 2004, 885, 1–43 CrossRef CAS; (b) R. H. Crabtree, Introduction to Selective Functionalization of C−H Bonds, Chem. Rev., 2010, 110, 575–575 CrossRef CAS; (c) H. M. L. Davies;, J. DuBois and J.-Q. Yu, C–H Functionalization in organic synthesis, Chem. Soc. Rev., 2011, 40, 1855–1856 RSC; (d) M. P. Doyle and K. I. Goldberg, C–H Functionalization, Acc. Chem. Res., 2012, 45, 777–777 CrossRef CAS PubMed; (e) G. Rouquet and N. Chatani, Catalytic Functionalization of C(sp2)-H and C(sp3)-H Bonds by Using Bidentate Directing Groups, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS; (f) J. F. Hartwig, Evolution of C–H Bond Functionalization from Methane to Methodology, J. Am. Chem. Soc., 2016, 138, 2–24 CrossRef CAS; (g) M. D. Kärkäs, Electrochemical strategies for C–H functionalization and C–N bond formation, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC; (h) L. Chang, S. Wang, Q. An, L. Liu, H. Wang, Y. Li, K. Fengb and Z. Zuo, Resurgence and advancement of photochemical hydrogen atom transfer processes in selective alkane functionalizations, Chem. Sci., 2023, 14, 6841–6859 RSC; (i) P. Bellotti, H.-M. Huang, T. Faber and F. Glorius, Photocatalytic Late-Stage C–H Functionalization, Chem. Rev., 2023, 123, 4237–4352 CrossRef CAS PubMed.
- (a) L. Capaldo, D. Ravelli and M. Fagnoni, Direct Photocatalyzed Hydrogen Atom Transfer (HAT) for Aliphatic C–H Bonds Elaboration, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS; (b) L. Chang, S. Wang, Q. An, L. Liu, H. Wang, Y. Li, K. Feng and Z. Zuo, Resurgence and advancement of photochemical hydrogen atom transfer processes in selective alkane functionalizations, Chem. Sci., 2023, 14, 6841–6859 RSC.
- For recent reviews on organic electrochemistry, see: (a) T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Powering the Future: How Can Electrochemistry Make a Difference in Organic Synthesis?, Chem, 2020, 6, 1–13 CrossRef; (b) M. Yan, Y. Kawamata and P. S. Baran, Synthetic Organic Electrochemical Methods Since 2000: On theVerge of a Renaissance, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (c) D. Pollok and S. R. Waldvogel, Electro-organic synthesis – a 21st century technique, Chem. Sci., 2020, 11, 12386–12400 RSC; (d) S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Modern Electrochemical Aspects for the Synthesis of Value-Added Organic Products, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef; (e) N. E. S. Tay, D. Lehnherr and T. Rovis, Photons or Electrons? A Critical Comparison of Electrochemistry andPhotoredox Catalysis for Organic Synthesis, Chem. Rev., 2022, 122, 2487–2649 CrossRef CAS PubMed; (f) M. Klein and S. R. Waldvogel, Counter Electrode Reactions—Important Stumbling Blocks on theWay to a Working Electro-organic Synthesis, Angew. Chem., Int. Ed., 2022, 61, e202204140 CrossRef CAS; (g) L. Zeng, J. X. Wang, D. X. Wang, H. Yi and A. W. Lei, Comprehensive Comparisons between Directing and Alternating Current Electrolysis in Organic Synthesis, Angew. Chem., Int. Ed., 2023, 62, e202309620 CrossRef CAS; (h) W. Zhang, W. Guan, J. I. Martinez Alvarado, L. F. T. Novaes and S. Lin, Deep Electroreductive Chemistry: Harnessing Carbon- and Silicon-Based Reactive Intermediates in Organic Synthesis, ACS Catal., 2023, 13, 8038–8048 Search PubMed; (i) P. Villo, A. Shatskiy, M. D. Kärkäs and H. Lundberg, Electrosynthetic C−O Bond Activation in Alcohols and Alcohol Derivatives, Angew. Chem., Int. Ed., 2023, 62, e202211952 CrossRef CAS PubMed; (j) Y. Wang, S. Dana, H. Long, Y. Xu, Y. Li, N. Kaplaneris and L. Ackermann, Electrochemical Late-Stage Functionalization, Chem. Rev., 2023, 123, 11269–11335 CrossRef CAS and the references cited therein.
- (a) J. E. Nutting, M. Rafiee and S. S. Stahl, Tetramethylpiperidine N-Oxyl (TEMPO), Phthalimide N-Oxyl (PINO), and Related N-Oxyl Species: Electrochemical Properties and Their Use in Electrocatalytic Reactions, Chem. Rev., 2018, 118, 4834–4885 CrossRef CAS PubMed; (b) D. Leifert and A. Studer, Organic Synthesis Using Nitroxides, Chem. Rev., 2023, 123, 10302–10380 CrossRef CAS PubMed; (c) E. J. Horn, B. R. Rosen, Y. Chen, J. Tang, K. Chen, M. D. Eastgate and P. S. Baran, Scalable and sustainable electrochemical allylic C–H oxidation, Nature, 2016, 533, 77–81 CrossRef CAS PubMed; (d) S. Zhang and M. Findlater, Electrochemically Driven Hydrogen Atom Transfer Catalysis: A Tool for C(sp3)/Si–H Functionalization and Hydrofunctionalization of Alkenes, ACS Catal., 2023, 13, 8731–8751 CrossRef CAS; (e) C. Yang, L. A. Farmer, E. C. McFee, R. K. Jha and D. A. Pratt, Attenuating N-Oxyl Decomposition for Improved Hydrogen Atom Transfer Catalysts, Angew. Chem., Int. Ed., 2024, e202315917 CAS; (f) C. Yang, L. A. Farmer, D. A. Pratt, S. Maldonado and C. R. J. Stephenson, Revisiting the Reactivity of the Dismissed Hydrogen Atom Transfer Catalyst Succinimide-N-oxyl, J. Am. Chem. Soc., 2024, 146, 12511–12518 CrossRef CAS PubMed.
- For selected examples, see: (a) P. Xu, P.-Y. Chen and H.-C. Xu, Scalable Photoelectrochemical Dehydrogenative Cross-Coupling of Heteroarenes with Aliphatic C−H Bonds, Angew. Chem., Int. Ed., 2020, 59, 14275–14280 CrossRef CAS PubMed; (b) H. Li, J. Tong, Y. Zhu, C. Jiang, P. Liu and P. Sun, Electrochemical Minisci reaction via HAT-driven α-C(sp3)–H functionalization of alcohols, Green Chem., 2022, 24, 8406–8411 RSC; (c) L. Niu, C. Jiang, Y. Liang, D. Liu, F. Bu, R. Shi, H. Chen, A. D. Chowdhury and A. Lei, Manganese-Catalyzed Oxidative Azidation of C(sp3)–H Bonds under Electrophotocatalytic Conditions, J. Am. Chem. Soc., 2020, 142, 17693–17702 CrossRef CAS PubMed; (d) L. Rapisarda, A. Fermi, P. Ceroni, R. Giovanelli, G. Bertuzzi and M. Bandini, Electrochemical C(sp3)–H functionalization of ethers via hydrogen-atom transfer by means of cathodic reduction, Chem. Commun., 2023, 59, 2664–2667 RSC.
- J. Sim, B. Ryou, M. Choi, C. Lee and C.-M. Park, Electrochemical C(sp3)–H Functionalization of γ-Lactams Based on Hydrogen Atom Transfer, Org. Lett., 2022, 24, 4264–4269 CrossRef CAS PubMed.
- H. Zhang, S. Liang, D. Wei, K. Xu and C. Zeng, Electrocatalytic Generation of Acyl Radicals and Their Applications, Eur. J. Org. Chem., 2022, e202200794 CrossRef CAS.
- For a review about acyl radicals under visible light conditions, see: (a) A. Banerjee, Z. Lei and M.-Y. Ngai, Acyl Radical Chemistry via Visible-Light Photoredox Catalysis, Synthesis, 2019, 51, 303–333 CrossRef CAS PubMed For selected examples, see: (b) M. Lux and M. Klussmann, Additions of Aldehyde-Derived Radicals and Nucleophilic N-Alkylindoles to Styrenes by Photoredox Catalysis, Org. Lett., 2020, 22, 3697–3701 CrossRef CAS PubMed; (c) H. Y. Wang, T. Li, D. Y. Hu, X. G. Tong, L. Y. Zheng and C. F. Xia, Acylation of Arenes with Aldehydes through Dual C–H Activations by Merging Photocatalysis and Palladium Catalysis, Org. Lett., 2021, 23, 3772–3776 CrossRef CAS PubMed , and references cited therein.
- For selected articles, see: (a) X.-L. Lai, X.-M. Shu, J. Song and H.-C. Xu, Electrophotocatalytic Decarboxylative C−H Functionalization of Heteroarenes, Angew. Chem., Int. Ed., 2020, 59, 10626–10632 CrossRef CAS; (b) H. Ding, K. Xu and C. Zeng, Nickel-catalyzed electrochemical Minisci acylation of aromatic N-heterocycles with α-keto acids via ligand-to-metal electron transfer pathway, J. Catal., 2020, 381, 38–43 CrossRef CAS; (c) T. Feng, S. Y. Wang, Y. Liu, S. Z. Liu and Y. A. Qiu, Electrochemical Desaturative β-Acylation of Cyclic N-Aryl Amines, Angew. Chem., Int. Ed., 2022, 61, e202115178 CrossRef CAS PubMed; (d) Y. M. Li, S. Liang, D. H. Wang, K. Xu and C. Zeng, Electrochemical Decarboxylative Minisci-Type Acylation of Quinoxalines under Catalyst- and External-Oxidant-Free Conditions, Synthesis, 2023, 55, 3026–3032 CrossRef CAS; (e) H. N. Zhang, T. Wang, K. Xu and C. Zeng, N-Hydroxyphthalimide-Mediated Electrochemical Denitrogenation of Aroylhydrazides to Generate Acyl Radicals and Their Applications in the Syntheses of Fluorenones, J. Org. Chem., 2021, 86, 16171–16176 CrossRef CAS; (f) J. Xu, F. L. Lu, L. H. Sun, M. N. Huang, J. W. Jiang, K. Wang, D. D. Ouyang, L. J. Lu and A. W. Lei, Electrochemical reductive cross-coupling of acyl chlorides and sulfinic acids towards the synthesis of thioesters, Green Chem., 2022, 24, 7350–7354 RSC; (g) X. S. Luo and P. Wang, Ynonylation of Acyl Radicals by Electroinduced Homolysis of 4-Acyl-1,4-dihydropyridines, Org. Lett., 2021, 23, 4960–4964 CrossRef CAS PubMed.
- For articles on photoelectrochemistry, see: (a) H. He, Q. H. Wan, Z. W. Hou, Q. Zhou and L. Wang, Organoelectrophotocatalytic Generation of Acyl Radicals from Formamides and Aldehydes: Access to Acylated 3-CF3-2-Oxindoles, Org. Lett., 2023, 25, 7014–7019 CrossRef CAS; (b) H. He, C.-M. Pan, Z.-W. Hou, M. Sun and L. Wang, Organocatalyzed Photoelectrochemistry for the Generation of Acyl and Phosphoryl Radicals through Hydrogen Atom-Transfer Process, J. Org. Chem., 2024, 89, 7531–7540 CrossRef CAS.
- For a review, see: P. Niu, J. Li, Y. Zhang and C. Huo, One-Electron Reduction of Redox-Active Esters to Generate Carbon-Centered Radicals, Eur. J. Org. Chem., 2020, 5801–5814 CrossRef CAS.
- For selected reviews, see: (a) S. Murarka, N-(Acyloxy)phthalimides as Redox-Active Esters in Cross-Coupling Reactions, Adv. Synth. Catal., 2018, 360, 1735–1753 CrossRef CAS; (b) S. K. Parida, T. Mandal, S. Das, S. K. Hota, S. Sarkar and S. Murarka, Single Electron Transfer-Induced Redox Processes Involving N-(Acyloxy)phthalimides, ACS Catal., 2021, 11, 1640–1683 CrossRef CAS.
- (a) P.-F. Dai, Y.-P. Wang, J.-P. Qu and Y.-B. Kang, tert-Butyl Nitrite as a Twofold Hydrogen Abstractor for Dehydrogenative Coupling of Aldehydes with N-Hydroxyimides, Org. Lett., 2021, 23, 9360–9364 CrossRef CAS PubMed; (b) X. Xu, P. Li, Y. Huang, C. Tong, Y. Yan and Y. Xie, Atmospheric oxidative catalyst-free cross-dehydrogenative coupling of aldehydes with N-hydroxyimides, Tetrahedron Lett., 2017, 58, 1742–1746 CrossRef CAS; (c) Z. Guo, X. Jiang, C. Jin, J. Zhou, B. Sun and W. Su, Copper-Catalyzed Highly Efficient Esterification of Aldehydes with N-Hydroxyphthalimide via Cross-Dehydrogenative Coupling in Water at Room Temperature, Synlett, 2017, 28, 1321–1326 CrossRef CAS; (d) X. Xu, J. Sun, Y. Lin, J. Cheng, P. Li, X. Jiang, R. Bai and Y. Xie, Iron-Nitrate-Catalyzed Oxidative Esterification of Aldehydes and Alcohols with N-Hydroxyphthalimide: Efficient Synthesis of N-Hydroxyimide Esters, Eur. J. Org. Chem., 2017, 7160–7166 CrossRef CAS; (e) Y. Lv, K. Sun, W. Pu, S. Mao, G. Li, J. Niu, Q. Chena and T. Wang, Metal-free intermolecular C–O cross-coupling reactions: synthesis of N-hydroxyimide esters, RSC Adv., 2016, 6, 93486–93490 RSC; (f) M. Dinda, C. Bose, T. Ghosha and S. Maity, Cross dehydrogenative coupling (CDC) of aldehydes with N-hydroxyimides by visible light photoredox catalysis, RSC Adv., 2015, 5, 44928–44932 RSC.
- T. Tian, Z. Li and C.-J. Li, Cross-dehydrogenative coupling: a sustainable reaction for C–C bond formations, Green Chem., 2021, 23, 6789–6862 RSC.
- For recent reviews, see: (a) M. Caruso, M. Petroselli and M. Cametti, Design and Synthesis of Multipurpose Derivatives for N-Hydroxyimide and NHPI-based Catalysis Applications, ChemistrySelect, 2021, 6, 12975–12980 CrossRef CAS; (b) M. Bhardwaj, P. Grover, B. Rasool and D. Mukherjee, Recent Advances in N-Hyrdoxypthalimide: As a Free Radical Initiator and its Applications, Asian J. Org. Chem., 2022, 11, e202200442 CrossRef CAS. For selected examples, see: (c) C. Jiang, Y. Liao, H. Li, S. Zhang, P. Liu and P. Sun, Electrochemical Silylation of Electron-Deficient Heterocycles Using N-Hydroxyphthalimide as HAT Catalyst, Adv. Synth. Catal., 2023, 365, 1205–1210 CrossRef CAS; (d) J. Ke, W. Liu, X. Zhu, X. Tan and C. He, Electrochemical Radical Silyl-Oxygenation of Activated Alkenes, Angew. Chem., Int. Ed., 2021, 60, 8744–8749 CrossRef CAS PubMed.
- For selected articles using oximes as radical acceptors, see: (a) S. Sumino, T. Fukuyama, M. Sasano, I. Ryu, A. Jacquet, F. Robert and Y. Landais, Vicinal difunctionalization of alkenes by four-component radical cascade reaction of xanthogenates, alkenes, CO, and sulfonyl oxime ethers, Beilstein J. Org. Chem., 2019, 15, 1822–1828 CrossRef CAS PubMed; (b) X. Wang, M. Yu, H. Song, Y. Liu and Q. Wang, Radical Transformation of Aliphatic C−H Bonds to Oxime Ethers via Hydrogen Atom Transfer, Org. Lett., 2021, 23, 8353–8358 CrossRef CAS; (c) W.-H. Bao and X. Wu, Visible-Light-Driven Photocatalyst-Free Deoxygenative Radical Transformation of Alcohols to Oxime Ethers, J. Org. Chem., 2023, 88, 3975–3980 CrossRef CAS PubMed; (d) J. Dey, S. Paul, M. Bhakat and J. Guin, Photocatalytic Incorporation of an Oxime Ether Functional Group at Inert C(sp3)–H Bonds via HAT, Org. Lett., 2022, 24, 8047–8051 CrossRef CAS PubMed.
- (a) D. M. Kitcatt, S. Nicolle and A.-L. Lee, Direct decarboxylative Giese reactions, Chem. Soc. Rev., 2022, 51, 1415–1453 RSC; (b) C. R. Azpilcueta-Nicolas and J.-P. Lumb, Mechanisms for radical reactions initiating from N-hydroxyphthalimide esters, Beilstein J. Org. Chem., 2024, 20, 346–378 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qo01067a |
| This journal is © the Partner Organisations 2024 |