
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly stereoselective synthesis of α-glycosylated carboxylic acids by phenanthroline catalysis†
Nur-E.
Alom
,
Neha
Rani
,
H. Bernhard
Schlegel
 * and
Hien M.
Nguyen
*
* and
Hien M.
Nguyen
*
Department of Chemistry, Wayne State University, Detroit, Michigan 48202, USA. E-mail: hbs@chem.wayne.edu; hmnguyen@wayne.edu
First published on 20th August 2024
Abstract
Carbohydrate molecules with an α-glycosylated carboxylic acid motif provide access to biologically relevant chemical space but are difficult to synthesize with high selectivity. To address this challenge, we report a mild and operationally simple protocol to synthesize a wide range of functionally and structurally diverse α-glycosylated carboxylic acids in good yields with high diastereoselectivity. Although there is no apparent correlation between reaction conversion and pKa of carboxylic acids, we found that carboxylic acids with a pKa of 4–5 provide high selectivity while those of a pKa of 2.5 or lower do not. Our strategy utilizes readily available 2,9-dibutyl-1,10-phenanthroline as an effective nucleophilic catalyst to displace a bromide leaving group from an activated sugar electrophile in a nucleophilic substitution reaction, forming phenanthrolinium intermediates. The attack of the carboxylic acid takes place from the α-face of the more reactive intermediate, resulting in the formation of α-glycosylated carboxylic acid. Previous calculations suggested that the hydroxyl group participates in the hydrogen bond interaction with the basic C2-oxygen of a sugar moiety and serves as a nucleophile to attack the C1-anomeric center. In contrast, our computational studies reveal that the carbonyl oxygen of the carboxylic acid serves as a nucleophile, with the carboxylic acid-OH forming a hydrogen bond with the basic C2-oxygen of the sugar moiety. This strong hydrogen bond (1.65 Å) interaction increases the nucleophilicity of the carbonyl oxygen of carboxylic acid and plays a critical role in the selectivity-determining step. In contrast, when alcohol acts as a nucleophile, this scenario is not possible since the –OH group of the alcohol interacts with the C2-oxygen and attacks the C1-anomeric carbon of the sugar moiety. This is also reflected in alcohol-OH's weak hydrogen bond (1.95 Å) interaction with the C2-oxygen. The O(C2)–HO (carboxylic acid) angle was measured to be 171° while the O(C2)–HO (alcohol) angle at 122° deviates from linearity, resulting in weak hydrogen bonding.
Introduction
Carboxylic acids are widely used in various applications for human health.1 They are found in amino acids2 and fatty acids,3 contributing to the development of cell membranes, controlling nutrient utilization, and regulating metabolism within the human body. Additionally, they are used in the production of polymers,4 biopolymers,5 and pharmaceutical drugs.6 In addition, α- and β-glycosylated carboxylic acids (also referred to as glycosyl esters), whose monosaccharide and oligosaccharide are incorporated with carboxylic acid moieties at the C1-anomeric carbon, are found in natural products7 and pharmaceutical drugs (Fig. 1a).8 Glycosyl esters have also been reported as effective electrophilic donors due to their operational stability and ease of activation by transition metals (Fig. 1b).9–14 Therefore, robust and stereoselective protocols for the synthesis of α- or β-glycosyl esters are crucial, especially α-glycosyl esters that are attractive biomolecules and privileged donors in stereoselective glycosylations.9,10,14 However, access to their synthesis remains challenging,9,11,12,15 as a mixture of α- and β-glycosyl esters is often produced. In some cases, the newly-formed α-glycosyl esters need to be separated from their β-counterparts before their use as glycosyl electrophilic donors in the stereospecific glycosylation reactions (Fig. 1b).9,10 | ||
| Fig. 1 (a) Representative biologically relevant glycosyl ester molecules. (b) Glycosyl esters as electrophilic donors. | ||
Glycosylated carboxylic acid is typically formed through a nucleophilic substitution reaction, in which a carboxylic acid or carboxylate anion displaces a leaving group from an activated carbohydrate electrophile (Fig. 2a).16 This C–O bond-forming reaction creates a new stereocenter at the C1-carbon, wherein the α-1,2-cis- and/or β-1,2-trans-glycosyl ester (viewed in respect to C2 of carbohydrate moiety) could be formed.16 However, achieving highly stereoselective formation of glycosyl esters can be challenging and unpredictable due to the variable nature of sugar electrophilic partners.17 Various factors, such as protecting groups, specific reagents, temperature, and solvents, significantly influence the stereochemical outcome of the glycosylation products.18 Glycosylation strategies that utilize a Lewis basic protecting group at the C2 position of glycosyl donors afford β-1,2-trans-glycosyl esters (also referred to as β-glycosyl esters).19–21 However, this approach cannot be used for the diastereoselective construction of α-1,2-cis-glycosyl esters (also referred to as α-glycosyl esters) because of the absence of the C2-Lewis basic protecting groups. Given the importance of α- and β-glycosyl esters, several alternative strategies have been developed to construct these motifs.22–28
 | ||
| Fig. 2 (a) General glycosylation methodologies for synthesizing α- and β-glycosyl esters. (b) Reaction design and development. | ||
Despite recent advancements, the stereoselective synthesis of α-glycosyl esters bearing aliphatic and aromatic carboxylic acids remains limited.23–25 Aiming to expand the availability of glycosylation methods for creating biomedically relevant sugar libraries for tool development and research applications, small-molecule catalysts have emerged as a promising strategy to broaden the chemical space of glycosylation reactions. Our group29–33 and others34 discovered that small molecule catalysts can promote the selective addition of alcohols to glycosyl donors, producing α-O-glycosides. While there are reports on the selective coupling of aliphatic carboxylic acids,22 reactions with aromatic carboxylic acids remained underdeveloped, likely due to the lower reactivity of aromatic carboxylic acids compared to aliphatic carboxylic acids.26
Our group recently discovered that readily available phenanthrolines can effectively catalyze the stereoselective addition of alcohol nucleophiles to glycosyl bromide donors.29–33 The extension of this strategy to carboxylic acids remains unknown due to the reactivity and acidity difference between alcohol and carboxylic acid. To achieve the stereoselective synthesis of α-glycosylated carboxylic acids, the catalyst and reaction design must overcome the low reactivity of carboxylic acids to ensure efficient reaction and high α-selectivity. Small-molecule catalysts used for glycosylation must withstand acidic conditions without being too basic to deprotonate carboxylic acid. Herein, we describe a new glycosylation methodology for the diastereoselective construction of α-glycosylated carboxylic acids based on the readily available 2,9-dibutyl-1,10-phenanthroline catalyst to facilitate the coupling of aliphatic and aromatic carboxylic acids with glycosyl bromide donors (Fig. 2b). DFT computations have revealed that hydrogen bond interactions between the carboxylic acid-OH and the C2-oxygen of a sugar moiety enable α-selectivity and enhance the nucleophilicity of the carbonyl oxygen to attack the C1-anomeric carbon. We have demonstrated the utility of this new protocol by efficiently constructing a variety of α-glycosyl esters containing biologically relevant carboxylic acids in good yields with excellent selectivity.
Results and discussion
The use of readily available phenanthroline catalysts provides a range of options for finding the optimal catalyst. In a model reaction, the coupling of 2-naphthoic acid 2a with glycosyl bromide 1 was investigated (Table 1a). Initially, our previously reported 4,7-piperidine-1,10-phenanthroline (NPhen) was used as the catalyst,29–31 which resulted in poor yield (33%) and low α-selectivity (α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β = 3:1) of glycosyl ester 3a. It was speculated that the 4,7-piperidine substituents might be basic enough to deprotonate carboxylic acid, inhibiting catalyst activity. This led us to consider unsubstituted 1,10-phenanthroline (Phen), which provided an improvement in both yield (33% → 50%) and selectivity (α:β = 3:1 → 5:1). A further enhancement (α:β = 5:1 → 8:1) was observed with the 4,7-diphenyl-1,10-phennathroline (BPhen) catalyst.32 Other 4,7-substituted phenanthroline catalysts (C1–C4) resulted in reduced yield and/or selectivity. Evaluating 2,9 substituents on the phenanthroline framework confirmed their ability to modulate reaction reactivity and selectivity. For instance, 2,9-disubstituted alkyl catalysts (C6 and C7) significantly increased the α-selectivity of 3a (α:β = 14:1–15:1). However, increasing the steric bulk of the 2,9-disubstituted substituent (C8) diminished both the yield and selectivity of product 3a. The 9-monosubstituted (C5) and the 5,6-di-ketone (C9) catalysts decreased both yield and α-selectively. The 6,6′-dibutyl-2,2′-bipyridine (C10) catalyst exhibited lower α-selectivity and conversion than the phenanthroline C6 catalyst (Table 1a).
:β = 3:1) of glycosyl ester 3a. It was speculated that the 4,7-piperidine substituents might be basic enough to deprotonate carboxylic acid, inhibiting catalyst activity. This led us to consider unsubstituted 1,10-phenanthroline (Phen), which provided an improvement in both yield (33% → 50%) and selectivity (α:β = 3:1 → 5:1). A further enhancement (α:β = 5:1 → 8:1) was observed with the 4,7-diphenyl-1,10-phennathroline (BPhen) catalyst.32 Other 4,7-substituted phenanthroline catalysts (C1–C4) resulted in reduced yield and/or selectivity. Evaluating 2,9 substituents on the phenanthroline framework confirmed their ability to modulate reaction reactivity and selectivity. For instance, 2,9-disubstituted alkyl catalysts (C6 and C7) significantly increased the α-selectivity of 3a (α:β = 14:1–15:1). However, increasing the steric bulk of the 2,9-disubstituted substituent (C8) diminished both the yield and selectivity of product 3a. The 9-monosubstituted (C5) and the 5,6-di-ketone (C9) catalysts decreased both yield and α-selectively. The 6,6′-dibutyl-2,2′-bipyridine (C10) catalyst exhibited lower α-selectivity and conversion than the phenanthroline C6 catalyst (Table 1a).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Catalyst loading (mol%) | Additive | Solvent | Temp. (°C) | Yield (%) |
α:β ratio |
| a The reaction was carried out with glycosyl bromide 1 (0.2 mmol), 2a (0.1 mmol), and catalyst (7.5 mol%) with respect to donor 1. b The reaction was carried out using donor 1 (0.2 mmol), 2a (0.1 mmol), C6 or C7 (7.5–10 mol%) with respect to donor 1. The crude product's yield and α/β or ratio were determined by 1H NMR using 1,3-dinitrobenzene as an internal standard. | |||||||
| 1 | C6 | 7.5 | DTBMP | MTBE/DCE (5:1) |
50 | 67 | 14:1 |
| 2 | C7 | 7.5 | DTBMP | MTBE/DCE (5:1) |
50 | 58 | 15:1 |
| 3 | C7 | 10 | DTBMP | MTBE/DCE (5:1) |
50 | 65 | 19:1 |
| 4 | C6 | 10 | DTBMP | MTBE/DCE (5:1) |
50 | 85 | 19:1 |
| 5 | C6 | 10 | DTBMP | MTBE/DCE (5:1) |
25 | 14 | 10:1 |
| 6 | C6 | 10 | DTBMP | MTBE | 50 | 54 | 13:1 |
| 7 | C6 | 10 | DTBMP | DCE | 50 | 57 | 5:1 |
| 8 | C6 | 10 | None | MTBE/DCE (5:1) |
50 | 12 | 8:1 |
| 9 | None | None | DTBMP | MTBE/DCE (5:1) |
50 | 28 | 5:1 |
| 10 | None | None | None | MTBE/DCE (5:1) |
50 | Trace | ND |
| 11 | AgOTf | 200 | None | MTBE/DCE (5:1) |
50 | Trace | ND |

Given that C6 and C7 are the most effective phenanthroline catalysts, we assessed the impact of reaction parameters (Table 1b). The increase in catalyst loading to 10 mol% further improved the yield and α-selectivity of 3a (entries 3 and 4). Notably, C6 outperformed C7 (85% vs. 65%). As a result, we employed C6 to optimize reaction conditions. It was observed that reaction rate and selectivity are influenced by temperature. At 25 °C, the reaction proceeded slowly, drastically reducing yield and α-selectivity (entry 5). This outcome is likely due to the low reactivity of carboxylic acid 2a and the slow equilibration of the less reactive phenanthroline intermediate to the more reactive isomer at 25 °C.29,33 Solvent evaluations indicated that a 5:1 mixture of MTBE and DCE solvent (entry 4) gave rise to higher α-anomeric selectivity. Conversely, using MTBE (entry 6) or DCE (entry 7) individually led to decreased α-selectivity. It was found that the presence of C6 and acid scavenger 2,6-di-tert-butylmethyl pyridine (DTBMP) is essential to achieve both high product yield and α-selectivity (entries 8–10). For comparison, we conducted the reaction with silver triflate known to activate glycosyl bromide,35 and product 3a did not form (entry 11). Collectively, the optimal reaction conditions, employing 10 mol% of 2,9-dibutyl-1,10-phenanthroline C6 in MTBE/DCE (5:1) at 50 °C for 18 h, yielded 3a in 85% NMR yield with α:β = 19:1 (Table 1b, entry 4).
Next, a series of benzoic acids were examined to assess their relative reactivity and selectivity. It was observed that the pKa of carboxylic acids has a significant impact on both product yield and α-selectivity based on a subset of benzoic acids listed in Table 2. Specifically, electron-donating benzoic acids with pKa values of 3.84–4.22 resulted in high α-selectivity (α:β = 11:1–20:1) and good yield (59–88%) for products 3a–3g (Table 2). Conversely, electron-withdrawing benzoic acids with pKa values of 1.48–3.65 produced 3h–3m in lower α-selectivity (Table 2). Benzoic acids with pKa values of 2.9–3.7 produced 3h–3j in moderate to good diastereocontrol (α:β = 5:1–9:1), while carboxylic acids with pKa values of 2.5 or lower showed unsatisfactory selectivity (3k–3m). The decrease in reactivity of catalyst C6 as the pKa of the benzoic acid decreases can be attributed to the likely protonation of its pyridine nitrogen atoms (see Fig. S4†), leading to a shift in the reaction closer to the SN1 end. Notably, reactions with nitro-substituted benzoic acids 2i and 2k were sluggish, and the α-glycosylated products 3i and 3k were obtained in lower yields (∼32%) relative to other benzoic acids (47–88%).
|
a The reaction was carried out with glycosyl electrophilic bromide 1 (0.2 mmol), 2a–2m (0.1 mmol), C6 catalyst (10 mol%) with respect to glycosyl bromide 1, DTBMP (2 equiv.) at 50 °C in 5:1 MTBE/DCE for 18 h. Isolated yield of the purified product was calculated. The α/β ratio was determined by 1H NMR of the crude product.
|
|---|

|
Next, we investigated a series of aromatic, heteroaromatic, and unsaturated carboxylic acids with a pKa ∼4 (Table 3) to establish the scope and limitation. The coupling of three benzoic acids with both electron-donating and electron-withdrawing groups yielded α-glycosyl esters 4–6 in good yield (58–89%) and selectivity (α:β = 15:1–18:1). Heterocyclic-2-carboxylic acids, commonly found in various pharmaceuticals,36 also produced 7–10 in 66–79% yield with α:β = 15:1–21:1. Indole-2-carboxylic acid 11 exhibited slightly lower selectivity (α:β = 9:1). Cinnamic acid proved to be an excellent nucleophile, producing 12 in 73% yield with excellent selectivity (α:β = 20:1). A number of aliphatic carboxylic acids, such as levulinic acid and palmitic acid, reacted to produce 13–15 with α:β = 10:1–20:1.37 Glycosylated ibuprofen 16 was readily synthesized in 60% yield with α:β = 20:1. Reaction with hindered cyclohexane-, pyran- and piperidine-4-, and 1-adamantane-carboxylic acid resulted in 17–20 in 74–92% yield with α:β = 17:1–20:1. Glycosyl esters 21–23 were synthesized from indomethacin, deoxycholic acid, and oleanolic acid, respectively, in 63–87% yield with high diastereocontrol (α:β = 17:1–20:1). We also attempted glycosylation of amino acids. Products 24–27 were formed with varying selectivity (α:β = 8:1–20:1). We observed that Fmoc-protected proline, with a pKa value similar to that of a typical carboxylic acid (pKa ∼4–5),38 exhibited more α-selectivity compared to other Fmoc-protected cysteine and tryptophan amino acids with a pKa value of around 3.3.

Altering sugar structures with distinct stereochemistry or protecting groups located away from the C1-coupling site can significantly affect the reactivity and selectivity of the reaction.39 We investigated whether the developed protocol could override substrate effects. Initially, we examined the coupling of four aliphatic and aromatic carboxylic acids with benzyl-protected L-fucose electrophile (see Table 4a). We successfully obtained coupling products 28–31 in 62–69% yield with excellent selectivity (α:β = 16:1–20:1). Encouraged by these results, we further assessed whether C6 catalyst could selectively promote the formation of α-glycosyl ester resulting from the coupling of the heterocyclic carboxylic acid with D-galactosyl bromide bearing an axial C(4) group.40 Galactosylated carboxylic acids 32 and 33 were obtained with good α-selectivity (α:β = 8:1), indicating that this catalysis protocol is partially α-selective of D-galactose substrate. In contrast, using phase-transfer glycosylation conditions (K2CO3 and TBABr)41 afforded 32 in 69% yield with poor selectivity (α:β = 1:2), favoring the expected β-isomer. The absence of the C6-methylhydroxyl renders D-xylose and L-arabinose substrates more likely to proceed via SN1 pathways.42 Nevertheless, the use of a C6 catalyst partially overrides the inherent bias of the substrate, resulting in arabinosyl esters 34 and 35 in 78% and 67% yield, respectively, with synthetically useful β-selectivity (α:β ∼ 8:1). A similar selectivity trend was also observed with D-xylose, affording 36 with moderate stereocontrol (α:β = 6:1).
|
a The reaction was carried out using glycosyl bromide (0.2 mmol), carboxylic acid (0.1 mmol), C6 (10 mol%) with respect to glycosyl bromide, DTBMP (2 equiv.) at 50 °C in 5:1 MTBE/DCE for 18 h. Isolated yield was calculated. 1H NMR of the crude product determined the α/β ratio. (a) Coupling of donors with carboxylic acids. (b) Stereoselective synthesis of α-glycosyl ester donors.
b Glycosyl bromide (0.3 mmol), carboxylic acid (0.1 mmol), and C6 (7 mol%) with respect to glycosyl bromide.
|
|---|

|
Next, we evaluated the scope and limitations of the established protocol with the more electron-withdrawing 2-azido-D-glucosyl bromide (Table 4a).43 As expected, the reaction with the less reactive 2-azido-D-glucose substrate was slow, resulting in 37 in 33% yield albeit with high selectivity (α:β = 15:1). The cis-1,2-O-glycosylation of D-mannosyl bromide is strongly disfavored, both sterically and electronically.44 Consequently, the glycosylation of D-mannosyl bromide with carboxylic acids predominantly yielded 38 and 39 as the 1,2-trans-α-products (Table 4a). In our study of phenanthroline-catalyzed glycosylation of alcohol nucleophiles, we observed that the type of furanosyl bromide donors significantly affect the selectivity of the furanosyl product.32 This has prompted us to explore whether this trend also applies to carboxylic acid nucleophiles. Our findings with products 40–42 (Table 4a) indicate that regardless of their stereochemical nature, we consistently obtained moderate α-selectivity for furanosyl substrates. We also evaluated the potential extension of the developed protocol to the unprotected amide. Our findings revealed that no reaction was observed. Additionally, it was noted that this protocol is not well-suited for a regioselective reaction when the acceptor bears two unprotected carboxylic acids.
The question remains whether the phenanthroline catalysis system could be used for the highly α-selective synthesis of glycosyl ester donors. To demonstrate this potential, we examined the coupling of several carboxylic acids with glucosyl bromide 1 (Table 4b). Both glycosyl ester products 43 and 44 were previously synthesized as a mixture of α- and β-isomers.11,15 In our approach, the C6 catalyst proved effective in providing highly α-selective products 43 (α:β > 20:1) and 44 (α:β = 15:1). It is challenging to obtain α-glycosyl ester 45 in high yield and selectivity as it can undergo 6-endo-dig cyclization to form six-membered ring lactone.12 Nevertheless, product 45 was formed with excellent diastereocontrol (α:β = 20:1) despite a low yield (33%). Finally, glycosylated carboxylic acid 46, the key intermediate of α-glycosyl ester donor bearing an amide-directing group (see Fig. 1b), was obtained in 75% yield with α:β > 20:1.9
Next, we initiated our mechanistic studies by examining the reaction of glucosyl bromide 1 with phenanthroline C6 (Fig. 3a). Although the pyranosyl phenanthrolinium ion intermediate 1a could not be detected by 1H NMR, it was confirmed by ESI mass spectrometry (Fig. 3b). This finding implies that the glycosyl phenanthrolinium ion intermediate 1a is likely to revert to the initial bromide donor 1.29,30 However, when carboxylic acid 2a was added, product 3a formed in similar yield and α-selectivity to that obtained under optimized conditions (see Table 1b, entry 4). We also subjected α- and β-glycosyl ester 3a to the optimized conditions and found no change in the anomeric ratio (Fig. S3†), indicating that the high α-selectivity is not the result of anomerization.
 | ||
| Fig. 3 (a) Detection of phenanthrolinium ion intermediate. (b) Control experiments. | ||
In our NMR experiment, we observed that 2,6-di-tert-butylmethyl pyridine (DTBMP) is not a sufficiently strong base to deprotonate 2-naptholic acid 2a in deuterated chloroform forming carboxylate (Fig. S4†). To further confirm if carboxylate could directly attack the reacting β-phenanthrolinium ion intermediate,31 sodium carboxylate 47 was subjected to the reaction with glucosyl bromide 1 in the presence and the absence of C6 catalyst (Fig. 3b1). We found poor yield and α-selectivity for product 3a in both cases, likely due to the solubility of sodium carboxylate 47. Control experiments were carried out to address this solubility issue in the presence of a strong base, penta-methylpiperidine (PMP). It is hypothesized that PMP, with pKa value of 11.5, could effectively deprotonate carboxylic acid in situ. Accordingly, 3a was obtained with excellent yield (88–93%) despite low selectivity (α:β = 3:1) in the presence or absence of C6 (Fig. 3b2). These results suggest that the reaction of carboxylic acid with glycosyl bromide does not occur via a direct SN2 pathway.
Next, we conducted a comparison experiment involving the reaction of aromatic carboxylic acid 2a with glycosyl bromide 1 using benzo[h]quinoline (C11, Fig. 3b3). It was observed that the C11 catalyst, in contrast to the phenanthroline C6 catalyst, exhibited lower reactivity and α-selectivity, resulting in 3a in a yield of only 36% and moderate selectivity (α:β = 5:1). This finding confirms the significance of the second nitrogen atom of phenanthroline in influencing the reactivity and selectivity of the reaction. Previous NMR evidence has indicated the existence of a hydrogen bonding interaction between the second nitrogen of phenanthroline and the C1-anomeric proton. This hydrogen bond interaction stabilizes the reacting β-phenanthrolinium ion complex, thereby enhancing the attack of carboxylic acid on its α-face.
It has been reported that the hydrogen bonding interaction between the alcohol-OH and C2-oxygen of a carbohydrate unit plays a crucial role in modulating reaction selectivity.45 To elucidate this interaction, a control experiment was performed using 2-deoxy-glucosyl bromide 45 (Fig. 3c) to probe the nature of the interaction of carboxylic acid-OH with the Lewis basic C2-oxygen of a carbohydrate moiety. The 2-deoxy substrate was unstable under our reaction conditions, leading to significant decomposition. Consequently, the desired α-2-deoxy glycosyl ester 46 was obtained in only 12% yield (Fig. 3d), albeit with good selectivity (α:β = 10:1). As a result, drawing a conclusive inference from the study with 2-deoxy donor substrate 45 is impeded due to the potential decomposition of the β-product at a faster rate than the α-product.
To determine the hydrogen bonding interaction between the carboxylic acid-OH and the C2-ether oxygen of the sugar substrate, we need to evaluate the chemical shift of the sugar C2-carbon. This will help us determine if it has a more pronounced shift than the adjacent carbons of other ether linkages. However, we cannot specifically measure the chemical shift of the sugar C2-carbon using 13C NMR because the hydrogen bonding interaction takes place in the transition state. Therefore, we examined the correlation between the 13C NMR chemical shift for the carbonyl carbon of carboxylic acid 2b to determine potential hydrogen bond interactions between benzoic acid-OH and the basic oxygen atoms of glucoside 50. We selected benzoic acid 2b as a model nucleophile. We carried out 1H NMR titrations in CDCl3 at 25 °C to determine the complexation-induced changes in the chemical shift of the carbonyl carbon C7 of 2b.46,47 The 13C NMR chemical shifts of carbon C7 were significantly shifted upfield (Δδ = 0.044–0.7717 ppm) with increased sugar 50 concentration, indicating increased hydrogen bond interactions between benzoic acid 2b and glucoside 50. This suggests that shielding effects are likely due to the redistribution of electron density upon the formation of new intermolecular hydrogen bonds of benzoic acid-OH with the basic oxygens of glucoside 50.46
Carboxylic acids form dimers in concentrated solution (>1 mM) in alkanes, held together by two cooperative hydrogen bonding interactions.48,49 In its dimeric form, the carboxylic acid's hydrogen-bond donor site cannot be involved in a hydrogen bond with the hydrogen-bond acceptor molecule. Since our glycosylation reaction was conducted at 0.5 M concentration (see Table 1), competition between the dimerization process of benzoic acid and hydrogen bond interactions of benzoic acid with the basic oxygen acceptors of sugar may play a critical role in determining reactivity and selectivity. As a result, the association constant, KA, for hydrogen-bonded complexes of benzoic acid 2b and perfluorobenzoic acid 2m with glucoside 50 was measured using UV-vis absorption titrations (Fig. 4b). The KA value was calculated based on the absorbance of the benzoic acid·glucoside complex at 228, 230, and 232 nm and the absorbance of perfluorobenzoic acid·glucoside complex at 226, 228, and 230 nm. The titration data were then fitted to a 1:1 binding isotherm with a linear correction to account for the absorbance of carboxylic acid at higher glucoside concentrations. The KA value for the interaction of benzoic acid 2a with 50 was 263 M−1, while the KA value for perfluorobenzoic acid 2m was 387 M−1. The data suggest that 2a and 2m are sufficiently exposed to form hydrogen bonds with 50, and 2m forms a stronger hydrogen bond with 50 than 2b. The KA value was determined based on the absorbance of the benzoic acid·glucoside complex at 228, 230, and 232 nm, as well as the absorbance of perfluorobenzoic acid·glucoside complex at 226, 228, and 230 nm. The titration data were then fitted to a 1:1 binding isotherm with a linear correction to account for the absorbance of carboxylic acid at higher glucoside concentrations. The KA value for the interaction of benzoic acid 2a with 50 was found to be 263 M−1, while the KA value for perfluorobenzoic acid 2m was 387 M−1. These results indicate that 2a and 2m have sufficient exposure to form hydrogen bonds with 50, with 2m showing a stronger hydrogen bond formation with 50 compared to 2b.
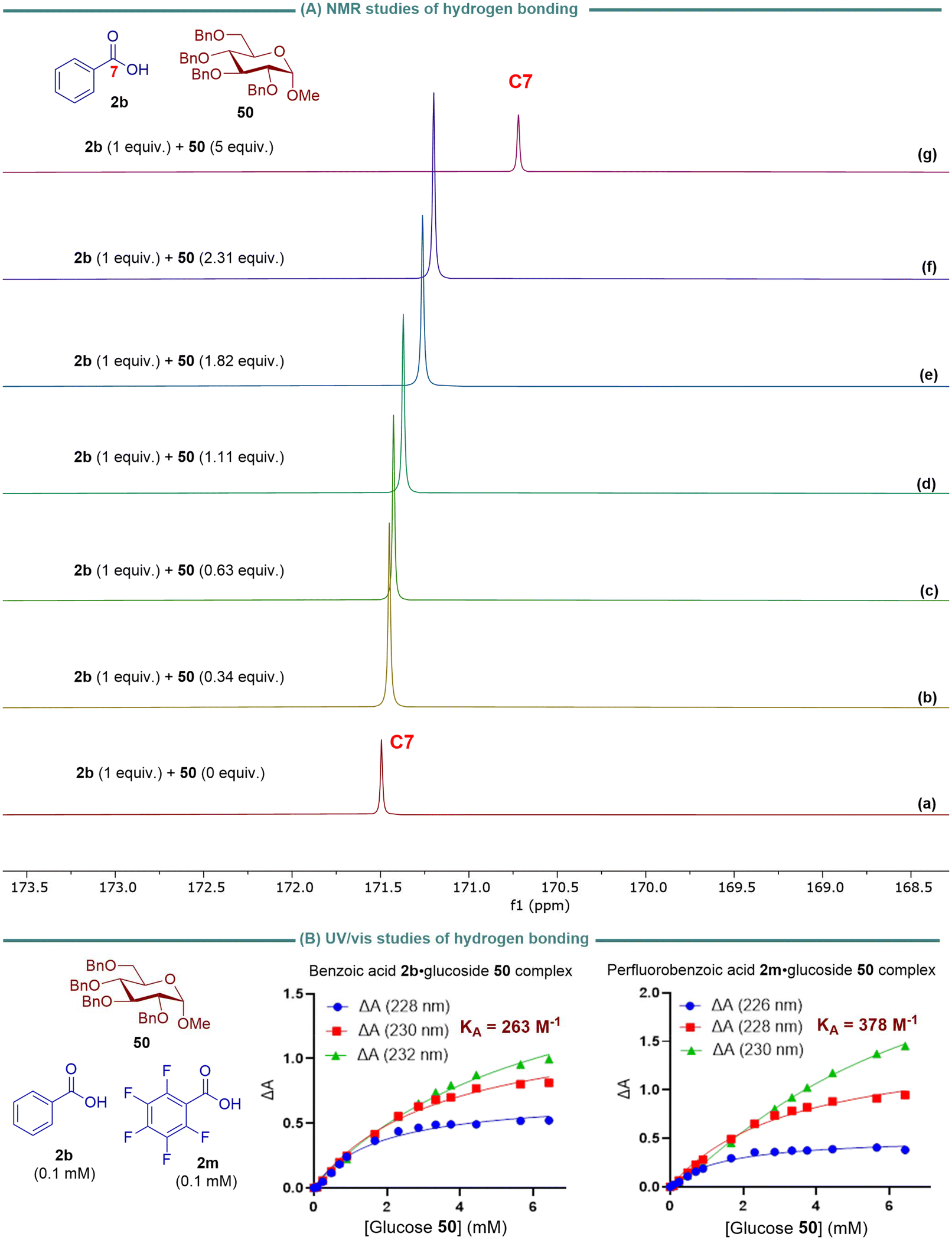 | ||
| Fig. 4 (A) NMR studies of interactions between benzoic acid OH and oxygen atoms of 50 in CDCl3 at 20 mM. (B) Binding isotherms for the formation of complexes of carboxylic acids 2b and 2m with glucoside 50 – fit of the experimental shifts of the UV-vis signals of 2a and 2m upon titration with 50 in dichloromethane. | ||
Next, we conducted density functional theory (DFT) calculations to examine the hydrogen bond interaction between carboxylic acid-OH and the glycosyl C2-oxygen. We focus on analyzing the rate-limiting transition states for the formation of both α- and β-glycosyl ester products (Fig. 5). To simplify the calculations, we used tetramethyl-protected glucosyl bromide and acetic acid as the model coupling partners, with phenanthroline (Phen) as the catalyst. The computed free energy profile and the optimized structures of key intermediates and transition states are shown in Fig. 5. The Phen catalyst displaces the bromide leaving group of the glycosyl donor, forming the β-phenanthrolinium ion intermediate Int1, where phenanthroline is bound to the C1-carbon of a sugar moiety. The subsequent attack of acetic acid on the intermediate Int1, via transition state TS-alpha, leads to the formation of the major product, α-glycosyl ester, with a barrier of 16.90 kcal mol−1 (Fig. 5). On the other hand, if phenanthroline is bound to the C1-carbon on the α-face, the phenanthrolinium ion intermediate Int1′ is 3.02 kcal mol−1 higher in energy than Int1. When carboxylic acid attacks from the β-face of the higher energy Int1′, the minor product, β-glycosyl ester, is formed via transition state TS-beta with a barrier of 19.79 kcal mol−1. Overall, TS-alpha is 2.88 kcal mol−1 lower in energy than TS-beta, thus strongly favoring the formation of α-glycosyl ester over β-glycosyl ester. This computational result is consistent with our experimental findings. Inspection of TS-beta and TS-alpha transition states revealed the CH3 group of carboxylic acid had an unfavorable interaction with the catalyst's phenyl ring in TS2-beta, leading to the minor β-product, while this CH3 group pointed away from the phenyl rings in TS-alpha leading to the major α-product.
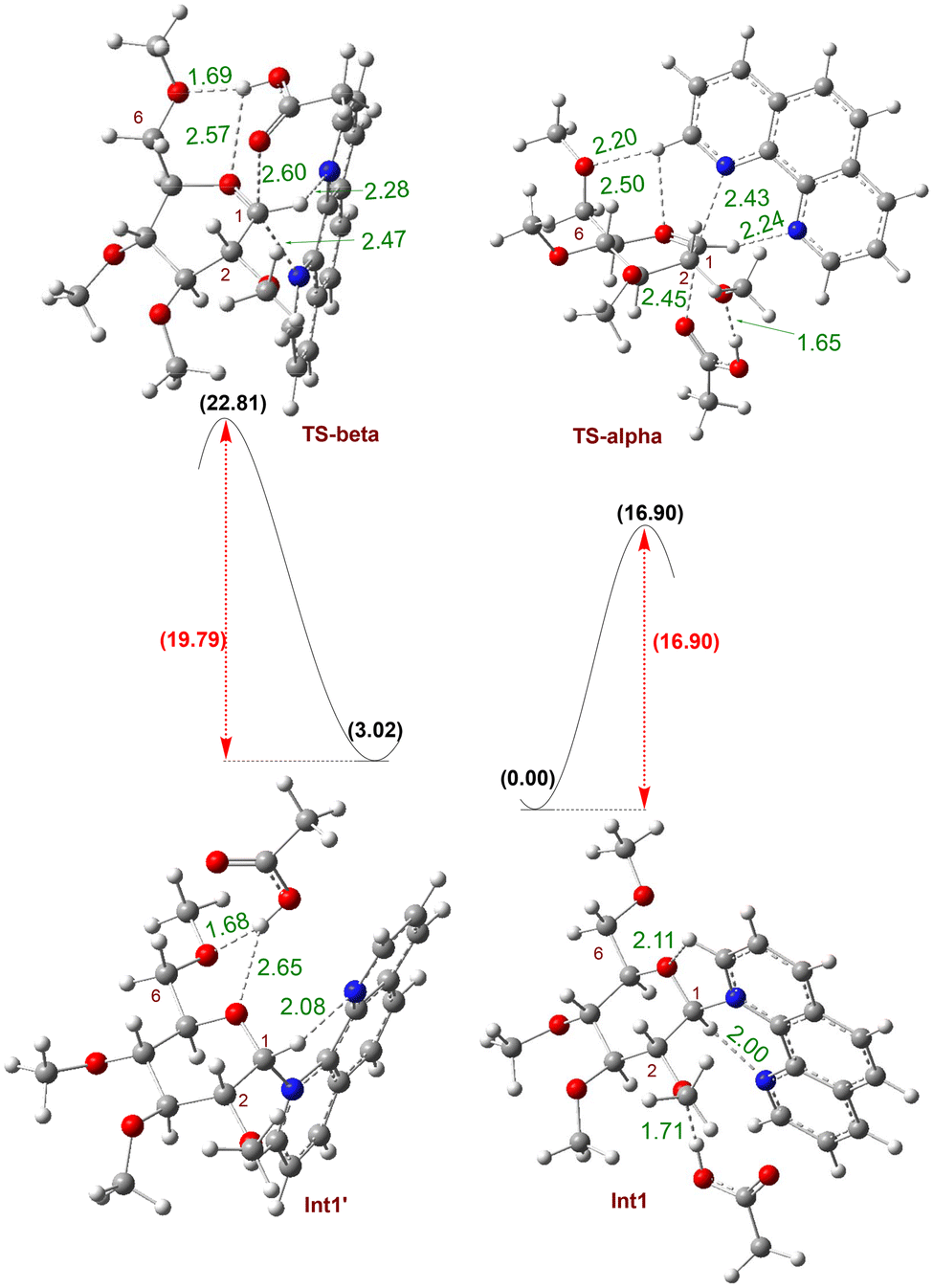 | ||
| Fig. 5 Kinetic barriers for the α- and β-glycosyl ester products from glucosyl bromide. The free energy changes (ΔG at 50 °C) are in kcal mol−1 and are computed with the Gaussian 16 program50 and at M06-2X/def2-TZVPP //M06-2X/def2-SVP level of theory51 using diethyl ether with SMD implicit solvation.52 Hydrogen bonding interactions are reported in Å. | ||
In examining the impact of hydrogen bonding on enhancing the nucleophilicity of carboxylic acid, conducted a comparative analysis of the optimized geometries of transition states for the formation of α-glycosyl ester and α-O-glycosides products from glucosyl bromide with carboxylic acid and alcohol as nucleophiles, respectively (Fig. 6a). Previous DFT calculations have focused on the hydrogen bonding interaction between the C2-oxygen of sugar electrophile and the hydroxyl group (–OH) nucleophile.45a Our DFT calculations primarily centered on the disparity between alcohol and carboxylic acid. An α-glycosyl ester is produced upon nucleophilic attack from the α-face of the reacting β-phenanthrolinium ion intermediate. The calculations underscored the pivotal role of hydrogen bond interaction between the hydroxyl group (–OH) of carboxylic acid and the C2-oxygen of a carbohydrate moiety, influencing the nucleophilicity of the incoming carboxylic acid. The strong hydrogen bond (1.65 Å) between carboxylic acid-OH and the basic O atom at the C2-carbon of sugar increases the nucleophilicity of the carbonyl oxygen (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of carboxylic acid. In contrast, when alcohol acts as a nucleophile, this scenario is not possible since the –OH group of the alcohol interacts with the C2-oxygen and attacks the C1-anomeric carbon of the sugar moiety. This is further evident in alcohol-OH's weak hydrogen bond (1.95 Å) interaction with the C2-oxygen. The weak hydrogen bond interaction is further supported by the fact that hydrogen bonds between two electronegative atoms and hydrogen prefer a linear arrangement, maximizing orbital overlap for a stronger hydrogen bond interaction. As illustrated in Fig. 6a, the O(C2)–HO (carboxylic acid) angle was calculated to be 171°. Conversely, the O(C2)–HO (alcohol) angle at 122° deviates from linearity, resulting in weak hydrogen bonding when alcohol approaches the C2 carbon of the sugar moiety.
O) of carboxylic acid. In contrast, when alcohol acts as a nucleophile, this scenario is not possible since the –OH group of the alcohol interacts with the C2-oxygen and attacks the C1-anomeric carbon of the sugar moiety. This is further evident in alcohol-OH's weak hydrogen bond (1.95 Å) interaction with the C2-oxygen. The weak hydrogen bond interaction is further supported by the fact that hydrogen bonds between two electronegative atoms and hydrogen prefer a linear arrangement, maximizing orbital overlap for a stronger hydrogen bond interaction. As illustrated in Fig. 6a, the O(C2)–HO (carboxylic acid) angle was calculated to be 171°. Conversely, the O(C2)–HO (alcohol) angle at 122° deviates from linearity, resulting in weak hydrogen bonding when alcohol approaches the C2 carbon of the sugar moiety.
 | ||
| Fig. 6 (a) Comparison between the optimized transition states leading α-glycosyl ester products using carboxylic acid and alcohol nucleophiles. Hydrogen bonding interactions are reported in Å. (b) Proposed reaction mechanism. | ||
Based on both experimental and computational results, we propose a plausible mechanism for the phenanthroline C6-catalyzed stereoselective formation of α-glycosyl ester (Fig. 6b). Upon displacement of a bromide leaving group from glycosyl bromide donor with the C6 catalyst, the major reacting 4C1 chair-liked β-phenanthrolinium ion intermediate is likely to be formed in the reaction.31 To achieve high conversion and selectivity, we propose the formation of hydrogen bonding interaction between carboxylic acid OH and the basic C2-oxygen of the carbohydrate moiety (O–H⋯O),45 forming the hydrogen-bond phenanthrolinium complex. This consequently increases the nucleophilicity of carboxylic acid and directs its attack from the α-face of the β-phenanthrolinium ion intermediate. As previously observed in related phenanthroline-catalyzed glycosylation of alcohol acceptors with glycosyl bromide donors,31 there was a hydrogen bonding interaction between the C1-anomeric proton and the second nitrogen of phenanthroline. This could rule out the possibility that the carboxylic acid-OH forms a hydrogen bond with the secondary nitrogen of phenanthroline before attacking the C1-site. Subsequent attack of carboxylic acid from the axial face of the reacting 4C1 chair-like equatorial complex affords α-glycosyl ester as the major product. The minor β-glycosyl ester product will likely be derived from a minor B2,5 boat-liked α-phenanthrolinium ion.31 The two phenanthrolinium ion conformers, a 4C1 chair-liked β-conformer and a B2,5 boat-like α-conformer, have been detected previously in a ratio of 2:1 (β:α) using variable temperature NMR experiments.31
Conclusions
A newly developed glycosylation methodology utilizes a precisely tailored phenanthroline catalyst to produce challenging α-glycosylated carboxylic acids selectively. This readily available catalyst facilitates the displacement of the bromide leaving group from glycosyl bromide donors, forming the reactive β-phenanthrolinium ion intermediate. Various alkyl and aromatic carboxylic acids and amino acid residues selectively attack the α-face of this intermediate, resulting in the formation of α-glycosyl esters with high selectivity and good yields. Both theoretical calculations and experimental data demonstrate the crucial role of hydrogen bonding between the carboxylic acid-OH and the basic C2-oxygen of the sugar moiety of the phenanthrolinium ion intermediate in the stereoselectivity-determining step of glycosylation. In addition, this hydrogen bond interaction enhances the nucleophilicity of the carboxylic acid, allowing it to attack the C1-anomeric carbon of the sugar moiety of the reactive intermediate. It has been observed that carboxylic acids with pKa values between 4 and 5 produce α-glycosyl esters with high selectivity, whereas those with pKa values of 2.5 or lower do not yield satisfactory results. This developed glycosylation protocol is mild, operationally simple, tolerant of various functional groups, and broadly applicable for the synthesis of a range of α-glycosylated carboxylic acids.Author contributions
H.M.N. conceived the idea. N.E.A. designed the experiments and performed all of the experiments. N.R. performed the DFT calculations. H.B.S. helped with the analysis of computational data. H.M.N. supervised the research and co-wrote the manuscript.Data availability
All relevant experimental and computational data and characterization details are provided in the ESI.†The data supporting this article have been included in the ESI,† including experimental procedures, spectral characterization of all the new products, detection of the phenanthrolinium intermediate, experiment for isomerization of α- and β-glycosyl ester, effect of carboxylic acid's pKa, NMR titration experiment for hydrogen bonding, UV-vis for hydrogen bonding constant, computational data, and the 1H, 13C, and 19F NMR spectra of all the new products (PDF).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
H.M.N. acknowledges financial support from Carl Johnson and A. Paul Schaap Endowed Chair and NIH (R35GM149213). H.B.S. acknowledges financial support from NSF (CHE1856437). The Wayne State University Lumigen Center received support from NIH (S10OD028488 for NMR and R01GM098285 for Mass Spectrometry). We would also like to express our gratitude to the Wayne State University Grid for providing computing resources.References
- B. Godlewska-Zylkiewicz, R. Swislocka, M. Kalinowska, A. Golonko, G. Swiderski, Z. Arciszewska, E. Nalewajko-Sieliwoniuk, M. Naumowicz and W. Lewandowski, Biologically Active Compounds of Plants: Structure-Related Antioxidant, Microbiological and Cytotoxic Activity of Selected Carboxylic Acids, Materials, 2020, 13, 4454–4490 CrossRef PubMed.
- R. G. Almquist, W. R. Chao and C. Jenningswhite, Synthesis and Biological Activity of Carboxylic-Acid Replacement Analogs of the Potent Angiotensin Converting Enzyme Inhibitor 5(S)-Benzamido-4-Oxo-6-Phenylhexanoyl-L-Proline, J. Med. Chem., 1985, 28, 1067–1071 CrossRef PubMed.
- S. Offermanns, Free Fatty Acid (FFA) and Hydroxy Carboxylic Acid (HCA) Receptors, Annu. Rev. Pharmacol. Toxicol., 2014, 54, 407–434 CrossRef PubMed.
- D. Dionisi, M. Majone, V. Papa and M. Beccari, Biodegradable Polymers from Organic Acids By Using Activated Sludge Enriched by Aerobic Periodic Feeding, Biotechnol. Bioeng., 2004, 85, 569–579 CrossRef PubMed.
- F. Ayadi, S. Mamzed, C. Portella and P. Dole, Synthesis of Bis(Pyrrolidone-4-Carboxylic Acid)- Based Polyamides Derived From Renewable Itaconic Acid-Application as a Compatibilizer in Biopolymer Blends, Polym. J., 2013, 45, 766–774 CrossRef CAS.
- C. Ballatore, D. M. Huryn and A. B. Smith, Carboxylic Acid (Bio)Isosteres in Drug Design, ChemMedChem, 2013, 8, 385–395 CrossRef CAS PubMed.
- (a) B. Yu, J. S. Sun and X. Y. Yang, Assembly of Naturally Occurring Glycosides, Evolved Tactics, and Glycosylation Methods, Acc. Chem. Res., 2012, 45, 1227–1236 CrossRef CAS PubMed; (b) S. D. Burke, J. E. Cobb and K. Takeuchi, Total Synthesis of (+)-Phyllanthocin - Introduction of Intramolecular Hydroformylation for Complex Molecule Functionalization, J. Org. Chem., 1990, 55, 2138–2151 CrossRef CAS; (c) A. B. Smith and M. Fukui, Total Synthesis of (+)-Phyllanthocin, J. Am. Chem. Soc., 1987, 109, 1269–1272 CrossRef CAS; (d) A. B. Smith, M. Fukui, H. A. Vaccaro and J. R. Empfield, Phyllanthoside-Phyllanthostatin Synthetic Studies .7. Total Synthesis of (+)-Phyllanthocin and (+)-Phyllanthocindiol, J. Am. Chem. Soc., 1991, 113, 2071–2092 CrossRef; (e) K. C. Nicolaou, J. A. Pfefferkorn, A. J. Roecker, G. Q. Cao, S. Barluenga and H. J. Mitchell, Natural product-like combinatorial libraries based on privileged structures. 1. General principles and solid-phase synthesis of benzopyrans, J. Am. Chem. Soc., 2000, 122, 9939–9953 CrossRef; (f) K. S. Feldman and M. D. Lawlor, Ellagitannin chemistry. The first total synthesis of a dimeric ellagitannin, coriariin A., J. Am. Chem. Soc., 2000, 122, 7396–7397 CrossRef; (g) S. Quideau, Ellagitannin chemistry, Chem. Rev., 1996, 96, 475–503 CrossRef PubMed.
- (a) P. R. Bradshaw, T. J. Athersuch, A. V. Stachulski and I. D. Wilson, Acyl glucuronide reactivity in perspective, Drug Discovery Today, 2020, 25, 1639–1650 CrossRef CAS; (b) Q. L. He, I. Q. L. Minn, Q. L. Wang, P. Xu, S. A. Head, E. Datan, B. Yu, M. G. Pomper and J. O. Liu, Targeted Delivery and Sustained Antitumor Activity of Triptolide through Glucose Conjugation, Angew. Chem., Int. Ed., 2016, 55, 12035–12039 CrossRef CAS; (c) E. Datan, I. Minn, P. Xu;, Q. L. He, H. H. Ahn, B. Yu, M. G. Pomper and J. O. Liu, A Glucose-Triptolide Conjugate Selectively Targets Cancer Cells under Hypoxia, iScience, 2020, 23, 101582 CrossRef; (d) L. Tong, Q. F. Zhao, E. Datan, G. Q. Lin, I. Minn, M. G. Pomper, B. A. Yu, D. Romo, Q. L. He and J. O. Liu, Triptolide: reflections on two decades of research and prospects for the future, Nat. Prod. Rep., 2021, 38, 843–860 RSC; (e) M. Bekhit, H.-Y. Huang, S. McKardy and W. Gorski, Infection screening in biofluids with glucose Strips, Anal. Chem., 2020, 92, 3860–3866 CrossRef CAS.
- X. Ma, Y. L. Zhang, X. J. Zhu, Y. L. Wei and L. M. Zhang, Directed SN2 Glycosylation Employing an Amide-Functionalized 1-Naphthoate Platform Featuring a Selectivity- Safeguarding Mechanism, J. Am. Chem. Soc., 2023, 145, 11921–11926 CrossRef CAS PubMed.
- X. Ma, Z. Zheng, Y. Fu, X. Zhu, P. Liu and L. A. Zhang, A “Traceless” Directing Group Enables Catalytic SN2 Glycosylation toward 1,2-cis-Glycopyranosides, J. Am. Chem. Soc., 2021, 143, 11908–11913 CrossRef.
- H. Y. Wang, C. J. Simmons, S. A. Blaszczyk, P. G. Balzer, R. S. Luo, X. Y. Duan and W. P. Tang, Isoquinoline-1-Carboxylate as a Traceless Leaving Group for Chelation-Assisted Glycosylation under Mild and Neutral Reaction Conditions, Angew. Chem., Int. Ed., 2017, 56, 15698–15702 CrossRef.
- Y. Tang, J. Li, Y. Zhu, Y. Li and B. Yu, Mechanistic Insights into the Gold(I)-Catalyzed Activation of Glycosyl ortho-Alkynylbenzoates for Glycosidation, J. Am. Chem. Soc., 2013, 135, 18396–18405 CrossRef PubMed.
- (a) P. Li, H. He, Y. Zhang, R. Yang, L. Xu, Z. Chen, Y. Huang, L. Bao and G. Xiao, Glycosyl ortho-(1-phenylvinyl)benzoates versatile glycosyl donors for highly efficient synthesis of both o-glycosides and nucleosides, Nat. Commun., 2020, 11, 405 CrossRef PubMed; (b) J. P. Chen, Y. Tang and B. A. Yu, A Mild Glycosylation Protocol with Glycosyl 1-Methylimidazole-2-carboxylates as Donors, Eur. J. Org. Chem., 2021, 4333–4344 CrossRef CAS; (c) B. Yu, Gold(I)-Catalyzed Glycosylation with Glycosyl o-Alkynylbenzoates as Donors, Acc. Chem. Res., 2018, 51, 507–516 CrossRef CAS PubMed; (d) B. B. Zhan, K. S. Xie, Q. Zhu, W. P. Zhou, D. P. Zhu and B. Yu, A Cu(OTf)2-C atalyzed Glycosylation with Glycosyl ortho-N-Phthalimidoylpropynyl Benzoates as Donors, Org. Lett., 2023, 25, 3841–3846 CrossRef CAS.
- (a) X. Ma, Y. L. Zhang, X. J. Zhu and L. M. Zhang, An SN2-Type Strategy toward 1,2-cis- Furanosides, CCS Chem., 2022, 4, 3677–3685 CrossRef CAS; (b) X. Dong, L. Chen, Z. T. Zheng, X. Ma, Z. G. Luo and L. M. Zhang, Silver-catalyzed stereoselective formation of glycosides using glycosyl ynenoates as donors, Chem. Commun., 2018, 54, 8626–8629 RSC.
- Y. Li, Y. Yang and B. Yu, An efficient glycosylation protocol with glycosyl ortho- alkynylbenzoates as donors under the catalysis of Ph3PAuOTf, Tetrahedron Lett., 2008, 49, 3604–3608 CrossRef.
- X. M. Zhu and R. R. Schmidt, New Principles for Glycoside-Bond Formation, Angew. Chem., Int. Ed., 2009, 48, 1900–1934 CrossRef.
- W. L. Leng, H. Yao, J. X. He and X. W. Liu, Venturing beyond Donor-Controlled Glycosylation: New Perspectives toward Anomeric Selectivity, Acc. Chem. Res., 2018, 51, 628–639 CrossRef.
- P. R. Andreana and D. Crich, Guidelines for O-Glycoside Formation from First Principles, ACS Cent. Sci., 2021, 7, 1454–1462 CrossRef PubMed.
- K. Morimoto, K. Yanase, T. Kajimoto and Y. Kita, Metal-Free Synthesis of Acyl Glycosides and Application to Oligosaccharide Synthesis, Org. Lett., 2022, 24, 9028–9032 CrossRef CAS PubMed.
- J. Weber, S. Krauter, T. Schwarz, C. Hametner and H. Mikula, (2-Benzyloxyphenyl)acetyl (BnPAc): A Participating Relay Protecting Group for Diastereoselective Glycosylation and the Synthesis of 1,2-trans Glycosyl Esters, Synlett, 2018, 29, 2265–2268 CrossRef CAS.
- (a) S. Feng and C. Li, Stereospecific, High-Yielding, and Green Synthesis of beta-Glycosyl Esters, J. Agric. Food Chem., 2015, 63, 5732–5739 CrossRef CAS PubMed; (b) S. Galland, N. Mora, M. Abert-Vian, N. Rakotomanomana and O. Dangles, Chemical synthesis of hydroxycinnamic acid glucosides and evaluation of their ability to stabilize natural Colors via anthocyanin copigmentation, J. Agric. Food Chem., 2007, 55, 7573–7579 CrossRef CAS PubMed; (c) T. Y. Yang, F. Zhu and M. A. Walczak, Stereoselective oxidative glycosylation of anomeric nucleophiles with alcohols and carboxylic acids, Nat. Commun., 2018, 9, 3650 CrossRef PubMed.
- Z. Liu, D. Liu, D. Zhu and B. Yu, Stereoselective Synthesis of & beta;-glycosyl Esters via 1- Hydroxybenzotriazole Mediated Acylation of Glycosyl Hemiacetals, Org. Lett., 2023, 25, 5372–5377 CrossRef PubMed.
- (a) H. Y. Wang, C. J. Simmons, Y. Zhang, A. M. Smits, P. G. Balzer, S. Wang and W. Tang, Chiral Catalyst-Directed Dynamic Kinetic Diastereoselective Acylation of Anomeric Hydroxyl Groups and a Controlled Reduction of the Glycosyl Ester Products, Org. Lett., 2017, 19, 508–511 CrossRef; (b) H. Y. Wang, K. Yang, D. Yin, C. Liu, D. A. Glazier and W. Tang, Chiral Catalyst- Directed Dynamic Kinetic Diastereoselective Acylation of Lactols for De Novo Synthesis of Chiral Catalyst-Directed Dynamic Kinetic Diastereoselective Acylation of Lactols for De Novo Synthesis of Carbohydrate, Org. Lett., 2015, 17, 5272–5275 CrossRef.
- C. G. Zhao, F. Li and J. Wang, N-Heterocyclic Carbene Catalyzed Dynamic Kinetic Resolution of Pyranones.Wang, Angew. Chem., Int. Ed., 2016, 55, 1820–1824 CrossRef.
- C. G. Zhao and J. Wang, Divergent Synthesis of Dihydropyranone Stereoisomers via N- Heterocyclic Carbene Catalysis, Adv. Synth. Catal., 2019, 361, 1668–1672 CrossRef CAS.
- N. Majumdar, Carboxylic Acids as Building Blocks in Catalytic Asymmetric Reactions, ACS Catal., 2022, 12, 8291–8324 CrossRef CAS.
- V. B. Birman and X. M. Li, Benzotetramisole: A Remarkably Enantioselective Acyl Transfer Catalyst, Org. Lett., 2006, 8, 1351–1354 CrossRef CAS PubMed.
- H. Zuo, C. Zhang, Y. Zhang and D. W. Niu, Base Promoted Glycosylation Allows Protecting Group-Free and Stereoselective Promoted Glycosylation Allows Protecting Group-Free and Stereoselective O-Glycosylationof CarboxylicAcids, Angew. Chem., Int. Ed., 2023, 62, e202309887 CrossRef CAS.
- F. Yu, J. Li, P. M. DeMent, Y. J. Tu, H. B. Schlegel and H. M. Nguyen, Phenanthroline-Catalyzed Stereoretentive Glycosylations, Nguyen, Angew. Chem., Int. Ed., 2019, 58, 6957–6961 CrossRef CAS.
- P. M. DeMent, C. L. Liu, J. Wakpal, R. N. Schaugaard, H. B. Schlegel and H. M. Nguyen, Phenanthroline-Catalyzed Stereoselective Formation of alpha-1,2-cis 2-Deoxy-2-Fluoro Glycosides, ACS Catal., 2021, 11, 2108–2120 CrossRef.
- J. Y. Li and H. M. Nguyen, A Mechanistic Probe into 1,2-cis Glycoside Formation Catalyzed by Phenanthroline and Further Expansion of Scope, Adv. Synth. Catal., 2021, 363, 4054–4066 CrossRef.
- H. F. Xu, R. N. Schaugaard, J. Y. Li, H. B. Schlegel and H. M. Nguyen, Stereoselective 1,2-cis Furanosylations Catalyzed by Phenanthroline, J. Am. Chem. Soc., 2022, 144, 7441–7456 CrossRef PubMed.
- J. Y. Li and H. M. Nguyen, Phenanthroline Catalysis in Stereoselective 1,2-cis Glycosylations, Acc. Chem. Res., 2022, 55, 3738–3751 CrossRef PubMed.
- (a) Y. Q. Geng, A. Kumar, H. M. Faidallah, H. A. Albar, I. A. Mhkalid and R. R. Schmidt, Cooperative catalysis in glycosidation reactions with O-glycosyl trichloroacetimidates as glycosyl donors, Angew. Chem., Int. Ed., 2013, 52, 10089–10092 CrossRef CAS PubMed; (b) T. Kimura, M. Sekine, D. Takahashi and K. Toshima, Chiral Brønsted acid mediated glycosylation with recognition of alcohol chirality, Angew. Chem., Int. Ed., 2013, 52, 12131–12134 CrossRef CAS; (c) Y. Park, K. C. Harper, N. Kuhl, E. E. Kwan, R. Y. Liu and E. N. Jacobsen, Macrocyclic bis-thioureas catalyze stereospecific glycosylation reactions, Science, 2017, 355, 162–166 CrossRef CAS PubMed; (d) S. M. Levi, Q. Li, A. R. Rotheli and E. N. Jacobsen, Catalytic activation of glycosyl phosphates for stereoselective coupling reactions, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 35–39 CrossRef CAS; (e) A. B. Mayfield, J. B. Metternich, A. H. Trotta and E. N. Jacobsen, Stereospecific Furanosylations Catalyzed by Bis- thiourea Hydrogen-Bond Donors, J. Am. Chem. Soc., 2020, 142, 4061–4069 CrossRef CAS; (f) Q. Li, S. M. Levi and E. N. Jacobsen, Highly Selective beta-Mannosylations and beta-Rhamnosylations Catalyzed by Bis-thiourea, J. Am. Chem. Soc., 2020, 142, 11865–11872 CrossRef; (g) Q. Li, S. M. Levi, C. C. Wagen, A. E. Wendlandt and E. N. Jacobsen, Site-selective, stereocontrolled glycosylation of minimally protected sugars, Nature, 2022, 608, 74–79 CrossRef; (h) M. M. Nielsen, T. Holmstrom and C. M. Pedersen, Stereoselective O-Glycosylations by Pyrylium Salt Organocatalysis, Angew. Chem., Int. Ed., 2022, 61, e202115394 CrossRef.
- Y. Singh and A. V. Demchenko, Defining the Scope of the Acid-Catalyzed Glycosidation of Glycosyl Bromides, Chem. – Eur. J., 2020, 26, 1042–1051 CrossRef PubMed.
- B. Yu and X. Y. Yang, Why are heterocycles so special in medicinal chemistry?, Chem. Biol. Drug Des., 2022, 100, 763–764 CrossRef CAS.
- S. An, Q. Q. Wang, W. J. Zhu, Q. K. Sun, G. He and G. Chen, Palladium-Catalyzed - and -Glycosylation with Glycosyl Chlorides, CCS Chem., 2020, 2, 1821–1829 Search PubMed.
- M. J. Harms, C. A. Castañeda, J. L. Schlessman, G. R. Sue, D. G. Isom, B. R. Cannon and B. García-Moreno, Values of Acidic and Basic Residues Buried at the Same Internal Location in a Protein Are Governed by Different Factors, J. Mol. Biol., 2009, 389, 34–47 CrossRef CAS.
- (a) U. Ellervik and G. Magnusson, A high-yielding chemical synthesis of sialyl Lewis x tetrasaccharide and Lewis x trisaccharide; Examples of regio- and stereodifferentiated glycosylations, J. Org. Chem., 1998, 63, 9314–9322 CrossRef; (b) D. Crich and V. Dudkin, Why are the hydroxy groups of partially protected N-acetylglucosamine derivatives such poor glycosyl acceptors, and what can be done about it? A comparative study of the reactivity of N- acetyl-, N-phthalimido-, and 2-azido-2-deoxy-glucosamine derivatives in glycosylation. 2- picolinyl ethers as reactivity-enhancing replacements for benzyl ethers, J. Am. Chem. Soc., 2001, 123, 6819–6825 CrossRef; (c) B. Hagen, S. Ali, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codee, Mapping the Reactivity and Selectivity of 2-Azidofucosyl Donors for the Assembly of N-Acetylfucosamine-Containing Bacterial Oligosaccharides, J. Org. Chem., 2017, 82, 848–868 CrossRef; (d) S. van der Vorm, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codee, Stereoselectivity of Conformationally Restricted Glucosazide Donors, J. Org. Chem., 2017, 82, 4793–4811 CrossRef; (e) S. van der Vorm, T. Hansen, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codee, The influence of acceptor nucleophilicity on the glycosylation reaction mechanism, Chem. Sci., 2017, 8, 1867–1875 RSC; (f) S. van der Vorm, J. M. A. van Hengst, M. Bakker, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codee, Mapping the Relationship between Glycosyl Acceptor Reactivity and Glycosylation Stereoselectivity, Angew. Chem., Int. Ed., 2018, 57, 8240–8244 CrossRef CAS PubMed.
- S. Chatterjee, S. Moon, F. Hentschel, K. Gilmore and P. H. Seeberger, An Empirical Understanding of the Glycosylation Reaction, J. Am. Chem. Soc., 2018, 140, 11942–11953 CrossRef CAS PubMed.
- K. Gao, Y. Qin, S. Liu, L. Wang, R. Xing, H. Yu, X. Chen and P. Li, A Review of the Preparation, Derivatization, and Functions of Glucosamine and N-Acetyl-Glucosamine from Chitin, Carbohydr. Polym. Technol. Appl., 2023, 5, 10029642 Search PubMed.
- P. O. Adero, H. Amarasekara, P. Wen, L. Bohé and D. Crich, The Experimental Evidence in Support of Glycosylation Mechanisms at the SN1−SN2 Interface, Chem. Rev., 2018, 118, 8242–8284 CrossRef CAS PubMed.
- E. T. Sletten, Y.-T. Tu, H. B. Schlegel, H. M. Nguyen and H. M. Are, Brønsted Acids the True Promoter of Metal Triflate Catalyzed Glycosylations? A Mechanistic Probe into 1,2-cis- Aminoglycoside Formation by Nickel Triflate, ACS Catal., 2019, 9, 2110–2123 CrossRef CAS PubMed.
- S. S. Nigudkar and A. V. Demchenko, Stereocontrolled 1,2-cis Glycosylation as the Driving Force of Progress in Synthetic Carbohydrate Chemistry, Chem. Sci., 2015, 6, 2687–2704 RSC.
- (a) C. Zhang, H. Zuo, G. Y. Lee, Y. Zou, Q. D. Dang, K. N. Houk and D. Niu, Halogen-Bond- Assisted Radical Activation of Glycosyl Donors Enables Mild and Stereoconvergent 1,2-cis- Glycosylation, Nat. Chem., 2022, 14, 686–694 CrossRef; (b) J. P. Yasomanee and A. V. Demchenko, Effect of Remote Picolinyl and Picoloyl Substituents on the Stereoselectivity of Chemical Glycosylation, J. Am. Chem. Soc., 2012, 49, 20097–20102 CrossRef PubMed; (c) W. Ma, J. L. Kirchhoff, C. Strohmann, B. Grabe and C. C. J. Loh, Cooperative Bifurcated Chalcogen Bonding and Hydrogen Bonding as Stereocontrolling Elements for Selective Strain-Release Septanosylation, J. Am. Chem. Soc., 2023, 145, 26611–26622 CrossRef PubMed.
- P. Novak, D. Vikictopic, Z. Meic, S. Sekusak and A. Sabljic, Investigation of Hydrogen-Bond Structure in Benzoic-Acid Solutions, J. Mol. Struct., 1995, 356, 131–141 CrossRef CAS.
- Y. Hiraga, S. Chaki and S. Niwayama, 13C NMR Spectroscopic Studies of the Behaviors of Carbonyl Compounds in Various Solutions, Tetrahedron Lett., 2017, 58, 4677–4681 CrossRef CAS.
- M. H. Abraham, P. P. Duce, R. A. Schulz, J. J. Morris, P. J. Taylor and D. G. Barratt, Hydrogen- Bonding .1. Equilibrium-Constants and Enthalpies of Complexation for Monomeric Carboxylic- Acids with N-Methylpyrrolidinone in 1,1,1-Trichloroethane, J. Chem. Soc., Faraday Trans. 1, 1986, 82, 3501–3514 RSC.
- V. Amenta, J. L. Cook, C. A. Hunter, C. M. R. Low, H. M. Sun and J. G. Vinter, Interplay of Self-Association and Solvation in Polar Liquids, J. Am. Chem. Soc., 2013, 135, 12091–12100 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, et al., Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- (a) Y. Wang, P. Verma, X. Jin, D. G. Truhlar and X. He, Revised M06 Density Functional for Main-Group and Transition-Metal Chemistry, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 10257–10262 CrossRef CAS; (b) Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main Group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed; (c) A. Schäfer, C. Huber and R. Ahlrichs, Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef; (d) F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qo00710g |
| This journal is © the Partner Organisations 2024 |