
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThree-component Friedel–Crafts-type difunctionalization of ynamides with (hetero)arenes and iodine(III) electrophile†
Jun
Kikuchi
 *a,
Toya
Nagata
a,
Shingo
Ito
b and
Naohiko
Yoshikai
*a
*a,
Toya
Nagata
a,
Shingo
Ito
b and
Naohiko
Yoshikai
*a
aGraduate School of Pharmaceutical Sciences, Tohoku University, Sendai 980-8578, Japan. E-mail: jun.kikuchi.e8@tohoku.ac.jp; naohiko.yoshikai.c5@tohoku.ac.jp
bSchool of Chemistry, Chemical Engineering and Biotechnology, Nanyang Technological University, Singapore 637371, Singapore
First published on 9th April 2024
Abstract
The three-component Friedel–Crafts type functionalization of an electron-rich arene using an electrophile-activated alkyne can offer a strategy for rapidly constructing densely functionalized vinylarenes in a regio- and stereoselective fashion. An analogous process utilizing ynamides, a distinct class of electron-rich alkynes, has remained elusive despite its considerable potential for the synthesis of highly substituted and stereodefined enamides. This transformation is particularly challenging with respect to the stereochemical control, as the pivotal intermediate, the electrophile-bound keteniminium ion, can inherently undergo nucleophilic attack from either side of the cumulated double bonds. Here, we report the development of a three-component difunctionalization of ynamides with a cationic iodine(III) electrophile (benziodoxole triflate) and electron-rich (hetero)arenes that consistently displays trans-selectivity irrespective of the steric profiles of the ynamide and the arene. The reaction proceeds quickly under mild conditions across diverse arenes and heteroarenes, producing α-aryl-β-iodanyl enamides in a regio- and stereocontrolled manner. The robust trans-selectivity of this iodo(III)arylation process is attributed to the reactivity of a highly distorted keteniminium species generated from the ynamide and the iodine(III) electrophile. The iodanyl moiety in these reaction products is amenable to a variety of transition metal-mediated coupling, thereby facilitating access to a wide array of densely functionalized enamides.
Introduction
The Friedel–Crafts reaction represents one of the quintessential methods in synthetic organic chemistry, offering a robust avenue for the functionalization of electron-rich aromatic compounds.1 Within a prototypical mechanistic framework, a carbocation generated by the protonation of an unsaturated reactant, such as an alkene or alkyne, undergoes an electrophilic attack on the arene substrate. An intriguing variant emerges when a heteroatom electrophile such as a halenium ion substitutes the proton as the activating agent, paving the way for an analogous Friedel–Crafts-type reaction involving the formation of a three-membered cyclic cationic species (e.g., halonium ion). Despite the apparent simplicity of this mechanistic scenario, the practical execution of this intermolecular three-component assembly, enabling the vicinal difunctionalization of unsaturated bonds with the heteroatom electrophile alongside an arene nucleophile, has been met with sporadic success (Scheme 1A). Barluenga and coworkers reported iodoarylation of simple terminal and internal alkynes using cationic iodine(I) electrophile (IPy2·BF4) in combination with excess HBF4.2 Later, Kitamura and coworkers disclosed iodoarylation utilizing the combination of molecular iodine and PhI(OBz)2, where hypoiodide (IOBz) generated from these reagents served as the reactive electrophile.3 Chu and Ellman recently developed a reaction system employing N-iodosuccinimide (NIS) and 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) solvent to achieve iodoarylation of alkynes under mild conditions.4 The Ellman group also demonstrated (PhSO2)2NSCF3 as a viable sulfur-based electrophile for analogous trifluoromethylthioarylation. The trans-selectivity of this difunctionalization, guaranteed by the arene nucleophile's backside attack on the iodirenium ion, renders this three-component conjugation highly attractive in the context of stereocontrolled synthesis of multisubstituted olefins. This is particularly pertinent given the utility of the carbon–iodine bond in subsequent synthetic elaborations. Consequently, further development of the electrophile-mediated arylative difunctionalization encompassing broader acetylenic compounds with carbo- and heterocyclic arenes is strongly desired.5 | ||
| Scheme 1 Arylative difunctionalization of alkynes via electrophilic activation. | ||
Ynamides, characterized by their electron-rich and polarized C–C triple bond, have become valuable building blocks for synthesizing nitrogenous compounds like enamides and N-heterocycles.6 Their unique electronic nature makes them behave distinctly from common alkynes toward electrophilic activation (Scheme 1B-1). Thus, an electrophile undergoes addition onto the distal acetylenic carbon, resulting in a keteniminium ion. Unlike electrophile-activated simple alkynes, the keteniminium species is susceptible to nucleophilic attack from either side of the cumulated double bonds, presenting a significant challenge for stereochemical control within the reaction. Addressing this challenge in the context of arylative difunctionalization, Sahoo et al. demonstrated an elegant solution through Pd-catalyzed cis-diarylation (Scheme 1B-2),7 arylalkenylation,8 and arylmethylation,9 wherein a keteniminium intermediate, generated from the ynamide and an arylpalladium(II) species, undergoes intramolecular migration of the aryl group. Yamaguchi and Studer exploited analogous strategies for intramolecular aryl transfer, achieving cis-arylboration of ynamides with electrophilic dichloroarylboranes.10 Despite these remarkable achievements employing functionalized aryl donors, the intermolecular arylative difunctionalization of ynamides using readily available unfunctionalized arenes and heteroarenes, paralleling the aforementioned iodoarylation, has remained elusive.11,12
Herein, we report a three-component Friedel–Crafts-type arylative trans-difunctionalization of ynamides using unfunctionalized (hetero)arenes and benziodoxole triflate (BXT), an iodine(III) electrophile (Scheme 1C). The reaction engages various electron-rich arenes and heteroarenes to produce α-(hetero)aryl-β-iodanyl enamides in good yields in a regio- and stereoselective fashion. DFT calculations suggested that the approach of the arene nucleophile is directed by a significantly distorted keteniminium intermediate formed between the ynamide and the bulky iodine(III) electrophile, leading to the consistently trans-selective difunctionalization process irrespective of the steric properties of the arene nucleophile and the ynamide substituent. The iodanyl group in the products serves as a versatile handle for further synthetic elaborations to prepare structurally diverse multisubstituted enamides. Given the use of unfunctionalized (hetero)arenes as arylating agents and the versatility of the iodanyl group, this reaction would complement the aforementioned arylative difunctionalization reactions of ynamides. Furthermore, in the context of hypervalent iodine chemistry, the present multisubstituted β-iodanyl enamides feature distinct structural patterns that are complementary to those accessible via hydroamidation of ethynylbenziodoxoles (EBXs)13 or iodo(III)amidation of internal alkynes.14
Results and discussion
As a part of our studies on iodo(III)functionalization reactions of alkynes with iodine(III) electrophile and nucleophiles for the synthesis of vinylbenziodoxoles (VBXs),14–17 we recently identified ynamides as excellent substrates for the trans-iodo(III)etherification using benziodoxole triflate (BXT) and alcohols, affording β-alkoxy-β-amido VBXs.18 Meanwhile, we also demonstrated the capacity of BXT as a prominent electrophile for the C–H λ3-iodanation of electron-rich (hetero)arenes.19 Given these backgrounds, we posited that the iodine(III) electrophile could engage electron-rich (hetero)arenes as nucleophiles for the iodo(III)functionalization of ynamides, but only when the competing C–H λ3-iodanation could be effectively suppressed. With this potential pitfall in mind, we took the oxazolidinone-containing ynamide 1a and 1-methoxy-2-methylbenzene (2a) as model substrates to explore the feasibility of the desired iodo(III)arylation (Table 1). The reaction between 1a (1.2 equiv.), 2a (1 equiv.), and BXT (1.2 equiv.) was found to proceed smoothly in HFIP (0.2 M) at 0 °C, affording the C4-alkenylated product 3aa in 58% yield as a single regio- and stereoisomer (entry 1). However, this reaction was accompanied by a substantial amount (27%) of the aromatic C–H iodanation product 4a. To our delight, the chemoselectivity for 3aa could be significantly improved by modifying the reaction stoichiometry and the concentration (entries 2–6). The reaction using 1a as the limiting agent and 2 equiv. each of 2a and BXT at a lower concentration (0.05 M) was completed within 10 min, affording 3aa in 92% isolated yield along with a much-diminished amount (7%) of 4a (entry 6). Importantly, HFIP proved crucial for the present reaction. The reaction in analogous fluorinated alcohol, TFE, failed to produce the three-component product 3aa but afforded a large quantity (89%) of 4a (entry 7). Meanwhile, neither 3aa nor 4a was obtained in other solvents such as MeCN and CH2Cl2, where the formation of several unidentified products arising from 1a and 2a was observed (entries 8 and 9). As is commonly conceived for HFIP-facilitated Friedel–Crafts reactions,20 we speculate that HFIP stabilizes cationic intermediates such as the iodine(III)-bound keteniminium ion and the ensuing arenium ion (see the following mechanistic discussion).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Solvent | x | y | z | Conc. [M] | 3aa [%] | 4a [%] |
| a The reaction was performed on a 0.1 mmol scale. b Determined by 1H NMR using 1,1,2,2-tetrachloroethane as an internal standard. c Isolated yield. | |||||||
| 1 | HFIP | 1.2 | 1.0 | 1.2 | 0.20 | 58 | 27 |
| 2 | HFIP | 1.2 | 1.0 | 1.2 | 0.10 | 66 | 23 |
| 3 | HFIP | 1.2 | 1.0 | 1.2 | 0.05 | 74 | 23 |
| 4 | HFIP | 1.0 | 1.2 | 1.2 | 0.05 | 80 | 19 |
| 5 | HFIP | 1.0 | 1.5 | 1.5 | 0.05 | 82 | 17 |
| 6 | HFIP | 1.0 | 2.0 | 2.0 | 0.05 | 92c | 7c |
| 7 | TFE | 1.0 | 2.0 | 2.0 | 0.05 | 0 | 89 |
| 8 | MeCN | 1.0 | 2.0 | 2.0 | 0.05 | 0 | 0 |
| 9 | CH2Cl2 | 1.0 | 2.0 | 2.0 | 0.05 | 0 | 0 |

Having identified the optimized conditions, we explored the scope of the three-component coupling (Table 2). Various alkoxy-substituted arenes smoothly participated in the reaction with 2a at the electron-rich and less hindered position to afford the enamide products 3aa–3af in moderate to good yields. 1-Bromo-3,5-dimethoxybenzene was exclusively alkenylated at the 2-position (see 3ac), whereas anisole afforded a mixture of para- and ortho-alkenylated products in a ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (see 3ad). The reaction using 1a could be performed on a 1 mmol scale to give the product 3aa in a respectable yield of 80%. Mesitylene, m-xylene, and toluene also afforded the desired products 3ag–3ai. While sluggish, the reaction of toluene took place exclusively at the para-position. Tetrahydronaphthalene and naphthalene both smoothly underwent the three-component alkenylation. The former was functionalized predominantly at the C6 position (C6:C5 = 11:1), while the latter preferentially reacted at the C1 position (C1:C2 = 5:1). Besides 2a, the iodo(III)arylation using 1a tolerated oxazolidinone-based ynamides bearing benzyl, isopropyl, cyclohexyl, methoxymethyl, and cyanopropyl groups, affording the respective products 3ba–3fa in moderate to high yields. Meanwhile, a phenyl-substituted ynamide failed to participate in the present reaction. The oxazolidinone moiety in the ynamide could be replaced by a pyrrolidinone moiety or a benzoimidazolone moiety (see 3ga and 3ha). Unfortunately, ynamides containing acyclic carboxamide (NMeAc) and sulfonamide (NMeTs) failed to undergo the desired iodo(III)arylation under the present conditions.
:1 (see 3ad). The reaction using 1a could be performed on a 1 mmol scale to give the product 3aa in a respectable yield of 80%. Mesitylene, m-xylene, and toluene also afforded the desired products 3ag–3ai. While sluggish, the reaction of toluene took place exclusively at the para-position. Tetrahydronaphthalene and naphthalene both smoothly underwent the three-component alkenylation. The former was functionalized predominantly at the C6 position (C6:C5 = 11:1), while the latter preferentially reacted at the C1 position (C1:C2 = 5:1). Besides 2a, the iodo(III)arylation using 1a tolerated oxazolidinone-based ynamides bearing benzyl, isopropyl, cyclohexyl, methoxymethyl, and cyanopropyl groups, affording the respective products 3ba–3fa in moderate to high yields. Meanwhile, a phenyl-substituted ynamide failed to participate in the present reaction. The oxazolidinone moiety in the ynamide could be replaced by a pyrrolidinone moiety or a benzoimidazolone moiety (see 3ga and 3ha). Unfortunately, ynamides containing acyclic carboxamide (NMeAc) and sulfonamide (NMeTs) failed to undergo the desired iodo(III)arylation under the present conditions.
| a Unless otherwise noted, the reaction was performed on a 0.1 mmol scale under the conditions in Table 1, entry 6. The symbol IIII in each product refers to the benziodoxole moiety. b The reaction was performed using 3 equiv. (0.3 mmol) of toluene, 0.1 mmol of 2a, and 0.1 mmol of BXT. c The reaction of a heteroaromatic substrate was performed using 1.2 equiv. (0.12 mmol) of the heteroarene, 0.1 mmol of ynamide, and 0.12 mmol of BXT. |
|---|

|
The present three-component coupling proved feasible with electron-rich heteroarenes. Using a near equimolar mixture of 2-methylthiophene (1.2 equiv.), ynamide 2a (1 equiv.), and BXT (1.2 equiv.) under diluted conditions (0.05 M), the alkenylation took place smoothly with high regioselectivity at the C5 position over the C4 position (C5:C4 = 16:1), furnishing the desired enamide product 3al in 82% yield (see Table S1†). Notably, unlike the case of the arene 1a, we did not observe the direct C–H iodanation of the thiophene ring as a competing side reaction. 2-Bromo- and 2-iodo thiophenes also took part in the C5-selective reaction to afford the corresponding products 3am and 3an, respectively. Analogous 2-substituted furan and pyrrole derivatives were also alkenylated at the C5 position, albeit in moderate yields (see 3ao–3aq). Interestingly, 1-tosyl-2-methylindole underwent regioselective alkenylation at the C6 position rather than the expected C3 position to give the product 3ar in 56% yield, which represents a rare selectivity case of the Friedel–Crafts reaction of C3-free indole derivatives.21 Besides 2a, different ynamides such as 2b, 2f, and 2h smoothly underwent the reaction with 2-methylthiophene to give the desired products 3bl, 3fl, and 3hl with high regioselectivity. Finally, an olefin such as 1,1-diphenylethene proved to serve as a reactant in the present three-component coupling, affording the dienamide derivative 3as in a moderate yield. Meanwhile, other olefins such as cyclohexene and α-methylstyrene failed to participate in the reaction.
For the known electrophile-mediated Friedel–Crafts alkenylation (Scheme 1b),2–4 the intermediacy of a three-membered cationic species (e.g., iodirenium and thiirenium) is invoked to account for the trans-selectivity, where the arene nucleophile can only approach from the opposite side of the heteroatom electrophile. On the other hand, acid- and electrophile-mediated functionalization of ynamides is generally assumed to proceed via a keteniminium species having an N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(α)C(β) array.6 In this case, the nucleophile can potentially approach from either side of the α-carbon, where the favorable reaction pathway would heavily depend on the relative steric size of the two substituents on the β-carbon (cf.Scheme 1B-1). Thus, while cis-addition is common in proton-mediated nucleophile addition to ynamides, high stereoselectivity would not be a priori guaranteed for electrophile-mediated variants. With these considerations in mind, we performed DFT calculations to gain insight into the trans-selectivity of the present reaction (Fig. 1a). Starting from 1a, model ynamide 2a′, and BXT (ΔG = 0.0 kcal mol−1), the latter two form a highly distorted keteniminium species CP0 (ΔG = 1.2 kcal mol−1) upon dissociation of the triflate anion from the iodine center. This species can engage 1a to give rise to complexes CP1t or CP1c, where 1a is loosely positioned around the opposite or same side of the iodanyl group at the expense of free energy of ca. 4–5 kcal mol−1. CP1t proceeds to the transition state of trans-iodo(III)arylation, TS1t (ΔG = 12.6 kcal mol−1), which leads to a Wheland-type intermediate Int1t (ΔG = −0.5 kcal mol−1). Unlike the typical energy landscape of Friedel–Crafts reaction, Int1t is on a similar energy level as the starting materials and readily undergoes deprotonative rearomatization viaTS2 (ΔG = 3.1 kcal mol−1) with the aid of the internal triflate, furnishing the thermodynamically stable enamide product PD (ΔG = −15.9 kcal mol−1). The alternative cis-iodo(III)arylation pathway was found to be feasible, albeit with much higher activation energy (by >5 kcal mol−1) viaTS1c (ΔG = 17.8 kcal mol−1). Note that the competing C–H iodanation of 1a with BXT to give 4a (cf.Table 1) was calculated to proceed with an activation energy of 19.6 kcal mol−1 (see the ESI†). While quantitative comparison of the three-component and two-component reactions is nontrivial, the calculated activation free energies appear consistent with the preference for the three-component reaction, especially when using the arene as the limiting substrate (cf.Table 1).
C(α)C(β) array.6 In this case, the nucleophile can potentially approach from either side of the α-carbon, where the favorable reaction pathway would heavily depend on the relative steric size of the two substituents on the β-carbon (cf.Scheme 1B-1). Thus, while cis-addition is common in proton-mediated nucleophile addition to ynamides, high stereoselectivity would not be a priori guaranteed for electrophile-mediated variants. With these considerations in mind, we performed DFT calculations to gain insight into the trans-selectivity of the present reaction (Fig. 1a). Starting from 1a, model ynamide 2a′, and BXT (ΔG = 0.0 kcal mol−1), the latter two form a highly distorted keteniminium species CP0 (ΔG = 1.2 kcal mol−1) upon dissociation of the triflate anion from the iodine center. This species can engage 1a to give rise to complexes CP1t or CP1c, where 1a is loosely positioned around the opposite or same side of the iodanyl group at the expense of free energy of ca. 4–5 kcal mol−1. CP1t proceeds to the transition state of trans-iodo(III)arylation, TS1t (ΔG = 12.6 kcal mol−1), which leads to a Wheland-type intermediate Int1t (ΔG = −0.5 kcal mol−1). Unlike the typical energy landscape of Friedel–Crafts reaction, Int1t is on a similar energy level as the starting materials and readily undergoes deprotonative rearomatization viaTS2 (ΔG = 3.1 kcal mol−1) with the aid of the internal triflate, furnishing the thermodynamically stable enamide product PD (ΔG = −15.9 kcal mol−1). The alternative cis-iodo(III)arylation pathway was found to be feasible, albeit with much higher activation energy (by >5 kcal mol−1) viaTS1c (ΔG = 17.8 kcal mol−1). Note that the competing C–H iodanation of 1a with BXT to give 4a (cf.Table 1) was calculated to proceed with an activation energy of 19.6 kcal mol−1 (see the ESI†). While quantitative comparison of the three-component and two-component reactions is nontrivial, the calculated activation free energies appear consistent with the preference for the three-component reaction, especially when using the arene as the limiting substrate (cf.Table 1).
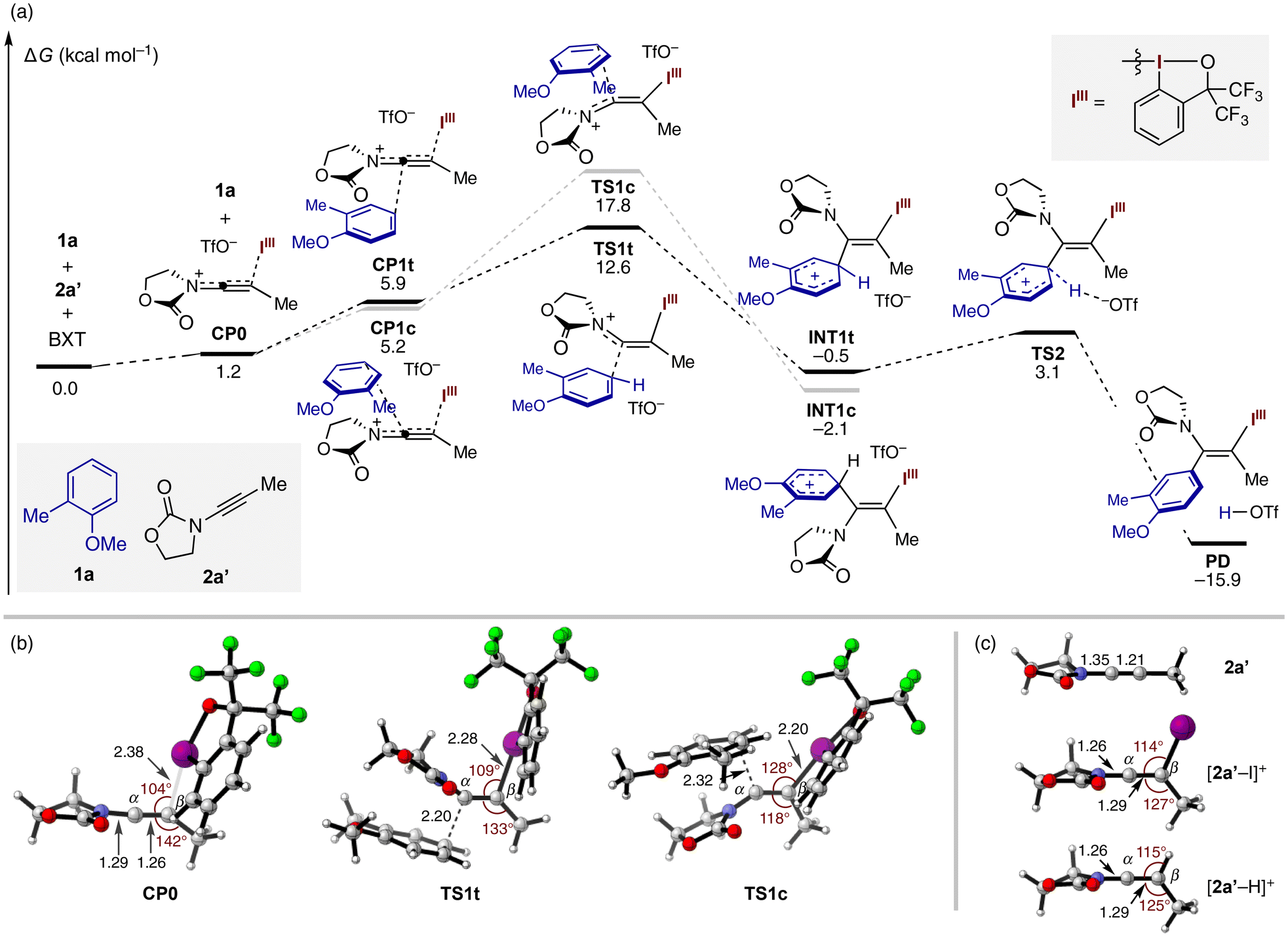 | ||
| Fig. 1 (a) The energy profile and (b) the structures of the key intermediate and transition states (triflate is omitted for clarity) of three-component coupling between 1-methoxy-2-methylbenzene (1a), ynamide 2a′, and BXT (M06-2X(CPCM, HFIP)/6-311+G(2df,2p)-SDD(for I)//M06-2X/6-31+G(d,p)-SDD(for I)). (c) Optimized structures of ynamide 2a′ and keteniminium ions generated from 2a′ and I+ or H+. Distances are in Å. | ||
A close look at the intermediate CP0 reveals its significant structural deviation from typical keteniminium geometry, with the β-carbon being far from an ideal trigonal geometry (Fig. 1b). Thus, the iodine center is significantly tilted toward the α-carbon (∠C(α)–C(β)–I = 104°), while the C(α)–C(β)–C(Me) angle (142°) is exceedingly wider than 120°. This angle distortion, along with the much longer C(β)–I length (2.38 Å) compared to the final product PD (2.18 Å), is suggestive of incomplete charge transfer from the ynamide to the BX cation in CP0. A quantum theory of atoms in molecules (QTAIM) analysis of the bond critical point between I and C(β) in CP0 and PD also underlined a significantly weaker nature of the C(β)–I bonding in CP0 (see Table S2†). The unique structural feature of CP0 can be manifested by its comparison with keteniminium ions [2a′–I]+ and [2a′–H]+ generated upon attaching I+ and H+, respectively, to 2a′ (Fig. 1c). Thus, the distinction between these normal keteniminiums and CP0 is reflected in their more trigonal-like β-carbons, shorter C(α)–N bonds, and longer C(α)–C(β) bonds, with which CP0 may be formally regarded as “half-keteniminium” featuring partial C(α)–N double bond, partial C(α)–C(β) triple bond, and partial C(β)–I bond. The transition from CP0 to the trans-iodo(III)arylation TS (TS1t) requires a relatively small degree of deformation around the β-carbon (∠C(α)–C(β)–I = 109°, ∠C(α)–C(β)–C(Me) = 133°). By contrast, to accommodate 1a on the same side of the bulky iodanyl group while avoiding steric clash, the cis-iodo(III)arylation TS (TS1c) has to go through a significant structural deformation (∠C(α)–C(β)–I = 128°, ∠C(α)–C(β)–C(Me) = 118°). These structural features account for the robust trans-selectivity observed across diverse aromatic and heteroaromatic nucleophiles.
The present three-component coupling product is amenable to a variety of transition metal-mediated conversion of the C–I(III) bond (Scheme 2). Suzuki–Miyaura and Sonogashira couplings of 3aa afforded the arylated and alkynylated enamides 5 and 6, respectively, in good yields with retention of the stereochemistry. Miyaura borylation of 3aa furnished the β-borylated enamide 7,22 albeit in a modest yield. Exposure of 3aa to stoichiometric CuBr or CuCN resulted in efficient conversion of the iodane moiety to Br or CN group, producing the β-bromo- and β-cyano enamides 8 and 9 in excellent yields. Although sluggish, Ullmann-type C–S coupling on 3aa also proceeded to give the β-arylthio enamide 10. These transformations collectively demonstrate the versatility of the β-iodanyl enamides13,14 as precursors to densely functionalized enamides, many of which would not be readily accessible by existing approaches.
 | ||
| Scheme 2 Product transformations. See the ESI† for the detailed reaction conditions for each transformation. | ||
Conclusions
In summary, we have developed a three-component Friedel–Crafts-type arylative difunctionalization of ynamides mediated by iodine(III) electrophile. The reaction encompasses a broad spectrum of electron-rich arenes and heteroarenes as competent nucleophiles, thus providing access to structurally diverse α-aryl-β-iodanyl enamides (or β-(hetero)aryl-β-amido VBXs) in a regio- and stereocontrolled manner. The uniform trans-selectivity of the reaction is ascribed to the nature of the bulky BX cation to form a distorted and incomplete keteniminium species with ynamide, which imposes distinct steric hindrances to two possible nucleophilic approaches. It is also worthwhile noting that the enamide products exhibit remarkable stereochemical integrity, undeterred by the generation of strongly acidic HOTf during the reaction, which could ostensibly cause post-reaction isomerization. The versatility of the BX group as a leaving group in the follow-up chemistry allows for converting the present products into structurally diverse, densely functionalized enamides in a stereoretentive fashion. The extension of the present three-component Friedel–Crafts chemistry to other classes of alkynes and unsaturated compounds and further exploration of iodo(III)functionalization of ynamides are underway.Author contributions
J. K. and N. Y. conceived the project. T. N. performed and analyzed experiments with the assistance of J. K., S. I., and N. Y., J. K. performed the DFT calculations. N. Y. wrote the manuscript with the assistance of J. K. and S. I.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI (Grant No. 20K23375 (N. Y.) and 19K15552 (J. K.)), The Uehara Memorial Foundation (N. Y.), Research Support Project for Life Science and Drug Discovery (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED (Grant No. JP23ama121040 (N. Y.)), and the Agency for Science, Technology and Research (A*STAR) AME IRG grant (A2083c0056 (S. I.)). We thank Central Glass Co., Ltd. for the generous donation of HFIP and 1,1,1,3,3,3-hexafluoro-2-phenylpropan-2-ol (HFAB).References
- G. A. Olah, R. Khrishnamurti and G. K. S. Prakash, Friedel–Crafts Alkylations, Compr. Org. Synth., 1991, 3, 293–335 CrossRef.
- J. Barluenga, M. A. Rodriguez, J. M. Gonzalez and P. J. Campos, Iodo-Carbofunctionalization of Alkynes with Aromatic Rings and IPy2BF4, Tetrahedron Lett., 1990, 31, 4207–4210 CrossRef CAS.
- (a) M. A. Rahman and T. Kitamura, Regio- and stereoselective iodoarylation of arylacetylenes using molecular iodine promoted by hypervalent iodine, Tetrahedron Lett., 2009, 50, 4759–4761 CrossRef CAS; (b) M. A. Rahman and T. Kitamura, Iodoarylation of Arylalkynes with Molecular Iodine in the Presence of Hypervalent Iodine Reagents, Molecules, 2009, 14, 3132–3141 CrossRef CAS PubMed.
- D. Chu and J. A. Ellman, Three-Component Friedel-Crafts Transformations: Synthesis of Alkyl and Alkenyl Trifluoromethyl Sulfides and Alkenyl Iodides, Org. Lett., 2022, 24, 2921–2925 CrossRef CAS PubMed.
- B. Maji, A. Bhattacharya and S. Hazra, Halogen-Induced Friedel-Crafts Alkenylation Reactions with Haloalkynes: Direct Access to gem-1,1-Dihaloalkenes, ChemistrySelect, 2017, 2, 10375–10378 CrossRef CAS.
- (a) G. Evano, A. Coste and K. Jouvin, Ynamides: Versatile Tools in Organic Synthesis, Angew. Chem., Int. Ed., 2010, 49, 2840–2859 CrossRef CAS PubMed; (b) C. Madelaine, V. Valerio and N. Maulide, Revisiting Keteniminium Salts: More than the Nitrogen Analogs of Ketenes, Chem. – Asian J., 2011, 6, 2224–2239 CrossRef CAS PubMed; (c) G. Evano, N. Blanchard, G. Compain, A. Coste, C. S. Demmer, W. Gati, C. Guissart, J. Heimburger, N. Henry, K. Jouvin, G. Karthikeyan, A. Laouiti, M. Lecomte, A. Martin-Mingot, B. Métayer, B. Michelet, A. Nitelet, C. Theunissen, S. Thibaudeau, J. Wang, M. Zarca and C. Zhang, A Journey in the Chemistry of Ynamides: From Synthesis to Applications, Chem. Lett., 2016, 45, 574–585 CrossRef CAS; (d) Y.-B. Chen, P.-C. Qian and L.-W. Ye, Brønsted acid-mediated reactions of ynamides, Chem. Soc. Rev., 2020, 49, 8897–8909 RSC; (e) S. Dutta, R. K. Mallick and A. K. Sahoo, Regioselective Difunctionalization and Annulation of Ynamide, Angew. Chem., Int. Ed., 2023, 62, e202300816 CrossRef CAS PubMed.
- S. Dutta, S. Yang, R. Vanjari, R. K. Mallick, V. Gandon and A. K. Sahoo, Keteniminium-Driven Umpolung Difunctionalization of Ynamides, Angew. Chem., Int. Ed., 2020, 59, 10785–10790 CrossRef CAS PubMed.
- R. Vanjari, S. Dutta, S. Yang, V. Gandon and A. K. Sahoo, Palladium-Catalyzed Regioselective Arylalkenylation of Ynamides, Org. Lett., 2022, 24, 1524–1529 CrossRef CAS PubMed.
- S. Dutta and A. K. Sahoo, Three Component syn-1,2-Arylmethylation of Internal Alkynes, Angew. Chem., Int. Ed., 2023, 62, e202300610 CrossRef CAS PubMed.
- C. You, M. Sakai, C. G. Daniliuc, K. Bergander, S. Yamaguchi and A. Studer, Regio- and Stereoselective 1,2-Carboboration of Ynamides with Aryldichloroboranes, Angew. Chem., Int. Ed., 2021, 60, 21697–21701 CrossRef CAS PubMed.
- For examples of Friedel–Crafts-type hydroarylation using specific types of (hetero)arenes, see: (a) Y.-W. Liu, R.-J. Ma, Q.-E. Wang, C.-M. Si and B.-G. Wei, Er(OTf)3-catalyzed approach to 3-alkenylindoles through regioselective addition of ynamides and indoles, Tetrahedron, 2020, 76, 131649 CrossRef CAS; (b) Y. Xiao, L. Tang, T.-T. Xu and J.-J. Feng, Boron Lewis Acid Catalyzed Intermolecular trans-Hydroarylation of Ynamides with Hydroxyarenes, Org. Lett., 2022, 24, 2619–2624 CrossRef CAS PubMed.
- For intramolecular iodoarylation leading to amide-functionalized phenanthrene skeletons, see: T. Okitsu, M. Itoh, A. Inui, E. Muta, R. Nakamoto, Y. Adachi, A. Wada and M. Hatano, Rapid Synthesis of Polycyclic Aromatic Compounds by Iodocyclization of Ynamides, Asian J. Org. Chem., 2022, 11, e202200275 CrossRef CAS.
- (a) P. Caramenti, N. Declas, R. Tessier, M. D. Wodrich and J. Waser, Stereoselective synthesis of alkyl-, aryl-, vinyl- and alkynyl-substituted Z-enamides and enol ethers, Chem. Sci., 2019, 10, 3223–3230 RSC; (b) D. Shimbo, A. Shibata, M. Yudasaka, T. Maruyama, N. Tada, B. Uno and A. Itoh, Synthesis of cis-β-Amidevinyl Benziodoxolones from the Ethynyl Benziodoxolone-Chloroform Complex and Sulfonamides, Org. Lett., 2019, 21, 9769–9773 CrossRef CAS PubMed; (c) D. Shimbo, T. Maruyama, N. Tada and A. Itoh, N-Alkenylation of hydroxamic acid derivatives with ethynyl benziodoxolone to synthesize cis-enamides through vinyl benziodoxolones, Org. Biomol. Chem., 2021, 19, 2442–2447 RSC.
- J. Chai, W. Ding, C. Wang, S. Ito, J. Wu and N. Yoshikai, Ritter-type iodo(III)amidation of unactivated alkynes for the stereoselective synthesis of multisubstituted enamides, Chem. Sci., 2021, 12, 15128–15133 RSC.
- (a) W. Ding, J. Chai, C. Wang, J. L. Wu and N. Yoshikai, Stereoselective Access to Highly Substituted Vinyl Ethers via trans-Difunctionalization of Alkynes with Alcohols and Iodine(III) Electrophile, J. Am. Chem. Soc., 2020, 142, 8619–8624 CrossRef CAS PubMed; (b) B. Wu, J. Wu and N. Yoshikai, Benziodoxole Triflate as a Versatile Reagent for Iodo(III)cyclization of Alkynes, Chem. – Asian J., 2017, 12, 3123–3127 CrossRef CAS PubMed; (c) J. Chai, W. Ding, J. Wu and N. Yoshikai, Fluorobenziodoxole-BF3 Reagent for Iodo(III)etherification of Alkynes in Ethereal Solvent, Chem. – Asian J., 2020, 15, 2166–2169 CrossRef CAS PubMed; (d) C. Wang, P. S. L. Tan, W. Ding, S. Ito and N. Yoshikai, Regio- and Stereoselective Synthesis of Enol Carboxylate, Phosphate, and Sulfonate Esters via Iodo(III)functionalization of Alkynes, Org. Lett., 2022, 24, 430–434 CrossRef CAS PubMed; (e) R. A. Laskar, W. Ding and N. Yoshikai, Iodo(III)-Meyer-Schuster Rearrangement of Propargylic Alcohols Promoted by Benziodoxole Triflate, Org. Lett., 2021, 23, 1113–1117 CrossRef CAS PubMed.
- For reviews on VBXs and related reagents, see: (a) N. Declas, G. Pisella and J. Waser, Vinylbenziodoxol(on)es: Synthetic Methods and Applications, Helv. Chim. Acta, 2020, 103, e2000191 CrossRef CAS; (b) I. A. Mironova, D. M. Noskov, A. Yoshimura, M. S. Yusubov and V. V. Zhdankin, Aryl-, Akynyl-, and Alkenylbenziodoxoles: Synthesis and Synthetic Applications, Molecules, 2023, 28, 2136 CrossRef CAS PubMed; (c) D. P. Hari, P. Caramenti and J. Waser, Cyclic Hypervalent Iodine Reagents: Enabling Tools for Bond Disconnection via Reactivity Umpolung, Acc. Chem. Res., 2018, 51, 3212–3225 CrossRef CAS PubMed; (d) A. Yoshimura, A. Saito and V. V. Zhdankin, Recent Progress in Synthetic Applications of Cyclic Hypervalent Iodine(III) Reagents, Adv. Synth. Catal., 2023, 365, 2653–2675 CrossRef CAS.
- For selected recent papers on the synthetic utility of VBXs, see: (a) N. Declas, J. R. J. Maynard, L. Menin, N. Gasilova, S. Götze, J. L. Sprague, P. Stallforth, S. Matile and J. Waser, Tyrosine bioconjugation with hypervalent iodine, Chem. Sci., 2022, 13, 12808–12817 RSC; (b) E. M. Di Tommaso, P.-O. Norrby and B. Olofsson, Explaining Regiodivergent Vinylations with Vinylbenziodoxolones, Angew. Chem., Int. Ed., 2022, 61, e202206347 CrossRef CAS PubMed; (c) T. M. Milzarek and J. Waser, Synthesis of Trifluoromethylated Alkenes: Hypervalent Iodine Meets High-Valent Copper, Angew. Chem., Int. Ed., 2023, 62, e202306128 CrossRef CAS PubMed; (d) K. B. Pal, E. M. Di Tommaso, A. K. Inge and B. Olofsson, Stereospecific Photoredox-Catalyzed Vinylations to Functionalized Alkenes and C-Glycosides, Angew. Chem., Int. Ed., 2023, 62, e202301368 CrossRef PubMed.
- J. Kikuchi, K. Maesaki, S. Sasaki, W. Wang, S. Ito and N. Yoshikai, Stereoselective Synthesis of β-Alkoxy-β-amido Vinylbenziodoxoles via Iodo(III)etherification of Ynamides, Org. Lett., 2022, 24, 6914–6918 CrossRef CAS PubMed.
- W. Ding, C. Wang, J. R. Tan, C. C. Ho, F. León, F. García and N. Yoshikai, Site-selective aromatic C–H λ3-iodanation with a cyclic iodine(III) electrophile in solution and solid phases, Chem. Sci., 2020, 11, 7356–7361 RSC.
- (a) H. F. Motiwala, A. M. Armaly, J. G. Cacioppo, T. C. Coombs, K. R. K. Koehn, V. M. Norwood IV and J. Aubé, HFIP in Organic Synthesis, Chem. Rev., 2022, 122, 12544–12747 CrossRef CAS PubMed; (b) V. Pozhydaiev, M. Power, V. Gandon, J. Moran and D. Lebœuf, Exploiting hexafluoroisopropanol (HFIP) in Lewis and Brønsted acid-catalyzed reactions, Chem. Commun., 2020, 56, 11548–11564 RSC.
- Y. Ling, D. An, Y. Zhou and W. Rao, Ga(OTf)3-Catalyzed Temperature-Controlled Regioselective Friedel-Crafts Alkylation of Trifluoromethylated 3-Indolylmethanols with 2-Substituted Indoles: Divergent Synthesis of Trifluoromethylated Unsymmetrical 3,3′- and 3,6′-Bis(indolyl)methanes, Org. Lett., 2019, 21, 3396–3401 CrossRef CAS PubMed.
- S. Liu, X. Zeng and B. Xu, Ligandless Copper-Catalyzed Carboborylation of Heteroatom-Substituted Alkynes, Adv. Synth. Catal., 2018, 360, 3249–3253 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qo00489b |
| This journal is © the Partner Organisations 2024 |