
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpiro-centre substitution effects in the intramolecular spin–spin interactions of spirobiacridine diradicals†
Shinichi
Ogawa
,
Takuya
Kanetomo
 * and
Masaya
Enomoto
*
* and
Masaya
Enomoto
*
Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: kanetomo@rs.tus.ac.jp
First published on 2nd April 2024
Abstract
Spirobiacridine diradicals exhibit intramolecular ferromagnetic coupling owing to their unique cruciform structure (D2d molecular symmetry). In this study, novel spirodiradicals 2,2′,7,7′-tetra(tert-butyl)-9,9′(10H,10′H)-spirobisilaacridine-10,10′-dioxyl (1-Si) and a Ge-centred derivative (1-Ge) were synthesized. These compounds exhibited a ground triplet spin state (S = 1), which was confirmed by spectroscopic and magnetic studies. Interestingly, the exchange coupling constant of 1-Ge (+19.3(2) K) was larger than that of 1-Si (+14.2(3) K), although the distance between intramolecular radical centres was increased owing to its larger bonding lengths. This result implies that σ*(Si–Cα)–π* hyperconjugation has a negative effect on spin–spin coupling.
Introduction
Organic radicals have many applications in spin trapping,1 spin labelling,2 dynamic nuclear polarization (DNP),3 organic catalysts,4 photochromic materials5 and molecular magnetic materials.6 In molecules containing more than two unpaired spins, spin–spin interaction is determined by two factors: (i) spin polarization and (ii) molecular symmetry. The former exploits the periodicity of the π-conjugation system7 and has been extensively studied in this field,8 whereas the latter utilizes the degeneracy of orbitals caused by symmetry.9 However, in this approach it is difficult to suppress Jahn–Teller distortions,10 resulting in slower progress compared to the spin-polarization approach.The compound 9,9′(10H,10′H)spirobiacridine-10,10′-dioxyl (SBDO-C) has two orthogonal π systems, resulting in D2d symmetry.11 Each dihydroacridine moiety possesses one unpaired electron, and singly occupied molecular orbitals (SOMOs) are symmetric to the bisecting plane, resulting in the ground triplet state (S = 1). In fact, the spin state of this spirobiacridine diradical is dictated by its tetra(tert-butyl) derivative, 2,2′,7,7′-tetra(tert-butyl)-9,9′(10H,10′H)spirobiacridine-10,10′-dioxyl (1-C).12 While the structural analysis of 1-C recognises the distortion in these acridine moieties, deviating from a strict D2d symmetry, magnetic studies confirm the ground triplet state (2J/kB = +23(1) K in 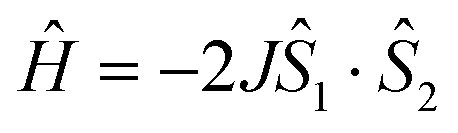 ). Spirobiacridine diradicals, which can manifest the triplet state despite structural distortion, represent an ideal class of substances for exploring molecular designs based on molecular symmetry.13
). Spirobiacridine diradicals, which can manifest the triplet state despite structural distortion, represent an ideal class of substances for exploring molecular designs based on molecular symmetry.13
A spiro centre plays a crucial role in the development of spiro compounds. Group 14 elements such as C, Si and Ge have been intensively studied.14–16 In particular, silicon compounds exhibit a significant reduction in lowest unoccupied molecular orbital (LUMO) energy levels in π-conjugated systems, which is attributed to a robust σ*(Si–C) and π* hyperconjugation effect.17 The effect of spiro-centre elements on intramolecular spin–spin interaction through a spiro-skeleton has not been sufficiently investigated.18 In this study, we synthesized Si and Ge substitutes of 1-C, namely 2,2′,7,7′-tetra(tert-butyl)-9,9′(10H,10′H)spirobisilaacridine-10,10′-dioxyl (1-Si) and 2,2′,7,7′-tetra(tert-butyl)-9,9′(10H,10′H)spirobigermaacridine-10,10′-dioxyl (1-Ge). Based on the theoretical and experimental results, both compounds exhibited ground triplet states. Interestingly, the intramolecular exchange coupling constant of 1-Ge was larger than that of 1-Si, despite the longer distance between the radical centres.
Results and discussion
Synthesis and characterization
Compounds 1-Si and 1-Ge were synthesized according to the procedure shown in Scheme 1. The starting material, bis(4-tert-butylphenyl)amine, was dibrominated at the 2,2′-position using N-bromosuccinimide (NBS), resulting in 2. The amino site of 2 was protected by a p-methoxybenzyl (PMB) group. The subsequent lithiation of 3 with n-butyllithium was followed by the addition of tetrachlorosilane/germane, resulting in the formation of the spiro compounds 4-Si and 4-Ge. This spiro annulation reaction was based on the method reported by Ito and co-workers.19 After the deprotection reaction with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), 1-Si and 1-Ge were synthesized via the oxidation of diamines 5-Si and 5-Ge, respectively, with m-chloroperbenzoic acid (m-CPBA). Dark red platelet polycrystals of 1-Si and 1-Ge were obtained through recrystallization from CH2Cl2 and n-hexane. Both compounds had a melting point above 200 °C, indicating their high thermal stability. The products were characterized using spectroscopic and X-ray crystallographic analyses. | ||
| Scheme 1 Synthetic routes for 1-Si and 1-Ge. | ||
Electron spin resonance (ESR) spectroscopy
The ESR spectra of 1-Si and 1-Ge, measured in toluene at RT, showed a five-line pattern (Figs. S1 and S2†), which is attributed to the presence of two nitrogen atoms (I = 1) derived from intramolecular exchange coupling. These spectra reproduce those of 1-C.12 The g values of 1-Si and 1-Ge were calculated to be 2.0064 and 2.0061, respectively. The hyperfine splitting constants aN were characterized as 0.46 and 0.47 mT, respectively.The frozen-solution ESR spectra of 1-Si and 1-Ge in degassed toluene at 100 K are shown in Figs. 1a and b, respectively. The experimental spectra exhibit the sum of zero-field splitting (ZFS) structures based on D2d (D ≠ 0 and E = 0) and D2 (D ≠ 0 and E ≠ 0) symmetries. The small central signal can be assigned to monoradicals as an impurity. These findings provide a good reproduction of the results for 1-C. In addition, a forbidden signal with |Δms| = 2 appeared at half field (Figs. S3 and S4†), indicating the presence of the triplet spin state (S = 1). ZFS parameters for 1-Si and 1-Ge were estimated using EasySpin software.20 For 1-Si, the experimental spectrum was reproduced by simulating D2/D2d/monoradical components with an intensity ratio of 89.6%/6.6%/3.8%. The simulation gave gxx = gyy = 2.0048, gzz = 2.0087, |D| = 7.37 × 10−3 mT and |E| = 7.67 × 10−4 mT for D2 symmetry; gxx = 2.0061, gyy = 2.0036, gzz = 2.0035 and |D| = 7.38 × 10−3 mT for D2d symmetry; and gxx = 2.0046, gyy = 2.0039 and gzz = 2.0147 for monoradical impurity. For 1-Ge, the experimental spectrum was reproduced by simulating three components with the intensity ratio of 91.2%, 5.6% and 3.2%. The ZFS parameters were gxx = 2.0046, gyy = 2.0048, gzz = 2.0081, |D| = 6.64 × 10−3 mT and |E| = 7.83 × 10−4 mT for the D2 symmetry; gxx = gyy = 2.0048, gzz = 2.0019 and |D| = 6.67 × 10−3 mT for D2d symmetry; and gxx = 2.0047, gyy = 2.0045 and gzz = 2.0082 for monoradical impurity. The experimental |D| and g values of D2 symmetry for 1-Si and 1-Ge led to dipole–dipole distances of 7.1 and 7.3 Å, respectively, from the point dipole approximation 2D = 3g2μB/r3. These values reproduced the crystallographically determined distances of 7.4 Å for 1-Si and 7.5 Å for 1-Ge (more details are given below). Compound 1-C had a dipole–dipole distance of 6.9 Å and an experimentally determined distance of 7.2 Å.12
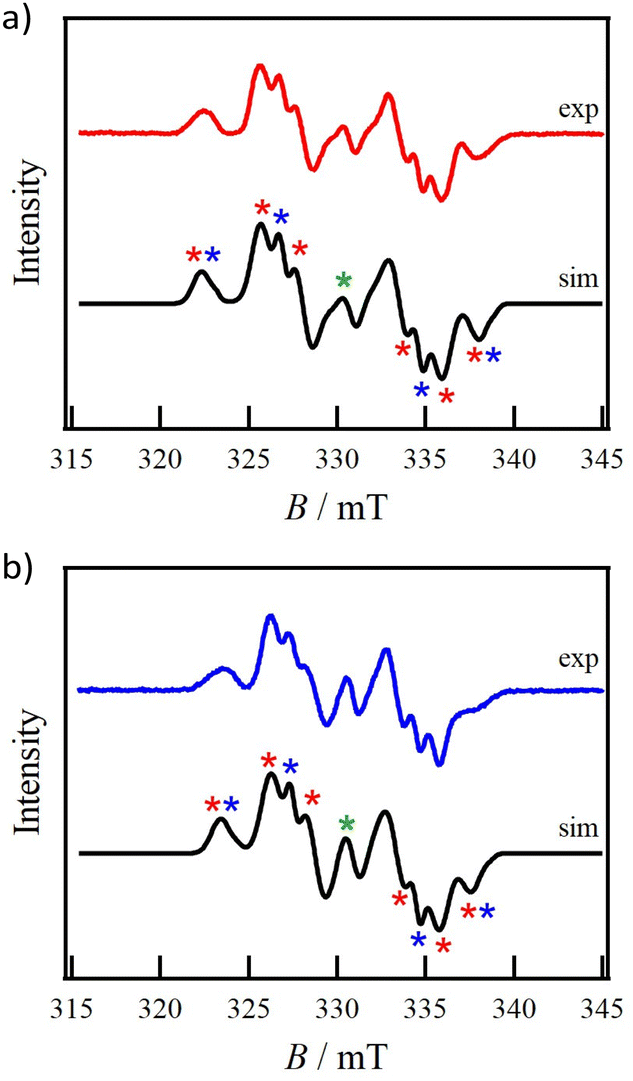 | ||
| Fig. 1 Frozen-solution ESR spectra for (a) 1-Si and (b) 1-Ge in degassed toluene solution at 100 K. Red and blue solid lines represent the experimental spectra. Black solid lines represent the simulated spectra. Red, blue and green markers correspond to D2, D2d and monoradical components. | ||
Crystal structures
Compound 1-Si crystallized in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group (Table S1†). The crystal structure and molecular packing diagram are shown in Fig. 2. Two crystallographically independent molecules were present in a unit cell. The two independent molecules were denoted as 1-Si-a (the Si1 atom) and 1-Si-b (the Si2 atom). The N1–O1, N2–O2, N3–O3 and N4–O4 bond lengths are 1.288(1), 1.289(1), 1.288(1) and 1.288(1) Å, respectively, which are close to the typical values for nitroxides.21 The bond lengths between Si and neighbouring carbon (Cα) atoms are 1.850(1)–1.866(1) Å around the Si1 atom and 1.850(1)–1.868(1) Å around the Si2 atom. These values reproduce those of typical Si–Csp2 bonds.14c,16b,22,23 The Si atom connects two dihydroacridines, forming a cruciform structure with dihedral angles of 80.33° and 82.02° between the two dihydroacridine moieties. In addition, the dihedral angles between the terminal phenyl rings are 28.78° and 6.42° for the N1 and N2 sides, respectively, in 1-Si-a and 5.17° and 38.67° for the N3 and N4 sides, respectively, in 1-Si-b. High-planarity skeletons (small dihedral angles of 6.42 and 5.17°) result from the intermolecular π–π contact between the dihydroacridines, including the N2 and N3 atoms, as shown in Fig. 2a. In this contact, the N2–O2 and N3–O3 sites are positioned above the C41–C46 and C21–C26 sites, respectively. The N2⋯C41 and N3⋯C21 distances are 3.558(1) and 3.546(1) Å, respectively, slightly exceeding the sum of the van der Waals (vdW) radii (C/N: 3.3 Å),24 indicating weak intermolecular contacts. Alternatively, low-planarity skeletons (large dihedral angles of 28.78 and 38.67°) appeared to be related to the intermolecular contacts around the tert-butyl groups (C33 and C53 atoms) and the dihydroacridine moieties (Fig. 2b). The intermolecular O1⋯O4 distance is 3.828(1) Å, exceeding the sum of the vdW radii (O/O: 3.04 Å).24 Nevertheless, this can still contribute to strong intermolecular antiferromagnetic coupling owing to the direct contacts between the radical centres.
space group (Table S1†). The crystal structure and molecular packing diagram are shown in Fig. 2. Two crystallographically independent molecules were present in a unit cell. The two independent molecules were denoted as 1-Si-a (the Si1 atom) and 1-Si-b (the Si2 atom). The N1–O1, N2–O2, N3–O3 and N4–O4 bond lengths are 1.288(1), 1.289(1), 1.288(1) and 1.288(1) Å, respectively, which are close to the typical values for nitroxides.21 The bond lengths between Si and neighbouring carbon (Cα) atoms are 1.850(1)–1.866(1) Å around the Si1 atom and 1.850(1)–1.868(1) Å around the Si2 atom. These values reproduce those of typical Si–Csp2 bonds.14c,16b,22,23 The Si atom connects two dihydroacridines, forming a cruciform structure with dihedral angles of 80.33° and 82.02° between the two dihydroacridine moieties. In addition, the dihedral angles between the terminal phenyl rings are 28.78° and 6.42° for the N1 and N2 sides, respectively, in 1-Si-a and 5.17° and 38.67° for the N3 and N4 sides, respectively, in 1-Si-b. High-planarity skeletons (small dihedral angles of 6.42 and 5.17°) result from the intermolecular π–π contact between the dihydroacridines, including the N2 and N3 atoms, as shown in Fig. 2a. In this contact, the N2–O2 and N3–O3 sites are positioned above the C41–C46 and C21–C26 sites, respectively. The N2⋯C41 and N3⋯C21 distances are 3.558(1) and 3.546(1) Å, respectively, slightly exceeding the sum of the van der Waals (vdW) radii (C/N: 3.3 Å),24 indicating weak intermolecular contacts. Alternatively, low-planarity skeletons (large dihedral angles of 28.78 and 38.67°) appeared to be related to the intermolecular contacts around the tert-butyl groups (C33 and C53 atoms) and the dihydroacridine moieties (Fig. 2b). The intermolecular O1⋯O4 distance is 3.828(1) Å, exceeding the sum of the vdW radii (O/O: 3.04 Å).24 Nevertheless, this can still contribute to strong intermolecular antiferromagnetic coupling owing to the direct contacts between the radical centres.
 | ||
| Fig. 2 Crystal structures of 1-Si. (a) Asymmetric units in the unit cell. (b) Intermolecular contacts around the O1 and O4 atoms. Symmetry codes: a = 1−x, 2−y, 1−z; b = x, 1+y, z; c = 1−x, 2−y, −z. Thermal ellipsoids for non-hydrogen atoms are drawn at the 50% probability level. The H atoms are omitted for clarity. The major configuration is shown with disordered tert-butyl groups. | ||
The compound 1-Ge crystallized in the monoclinic P21/n space group (Table S1†). The crystal structure and molecular packing diagram are shown in Fig. 3. The N1–O1 and N2–O2 bond lengths are 1.2868(15) and 1.2846(16) Å, respectively, which indicates the presence of a nitroxide group.21 Ge–Cα bond lengths are 1.9345(14)–1.9445(14) Å, which reproduce typical Ge–Csp2 bonds.16b,23,25 The Ge atom connects two dihydroacridines, forming a cruciform structure with a dihedral angle of 84.99°. In addition, the dihedral angles between the terminal phenyl rings are 40.20° and 4.61° for the N1 and N2 sides, respectively. The former is attributed to the contact with the neighbouring tert-butyl groups and 1-Si. The O1 atom is located at 5.653(2) Å from the nearest neighbouring O atom (O1*), suggesting weak intermolecular interaction. Alternatively, a high-planarity skeleton results from the intermolecular π–π contact between the dihydroacridine moieties, as shown in Fig. 3b. The N2–O2 site is positioned above the C21–C22 site, and the O2⋯C21 and N2⋯C22 distances are 3.340(2) and 3.396(2) Å, respectively. These values are close to the sum of the vdW radii (C/O, 3.2 Å; C/N, 3.3 Å),24 indicating strong interactions compared with 1-Si.
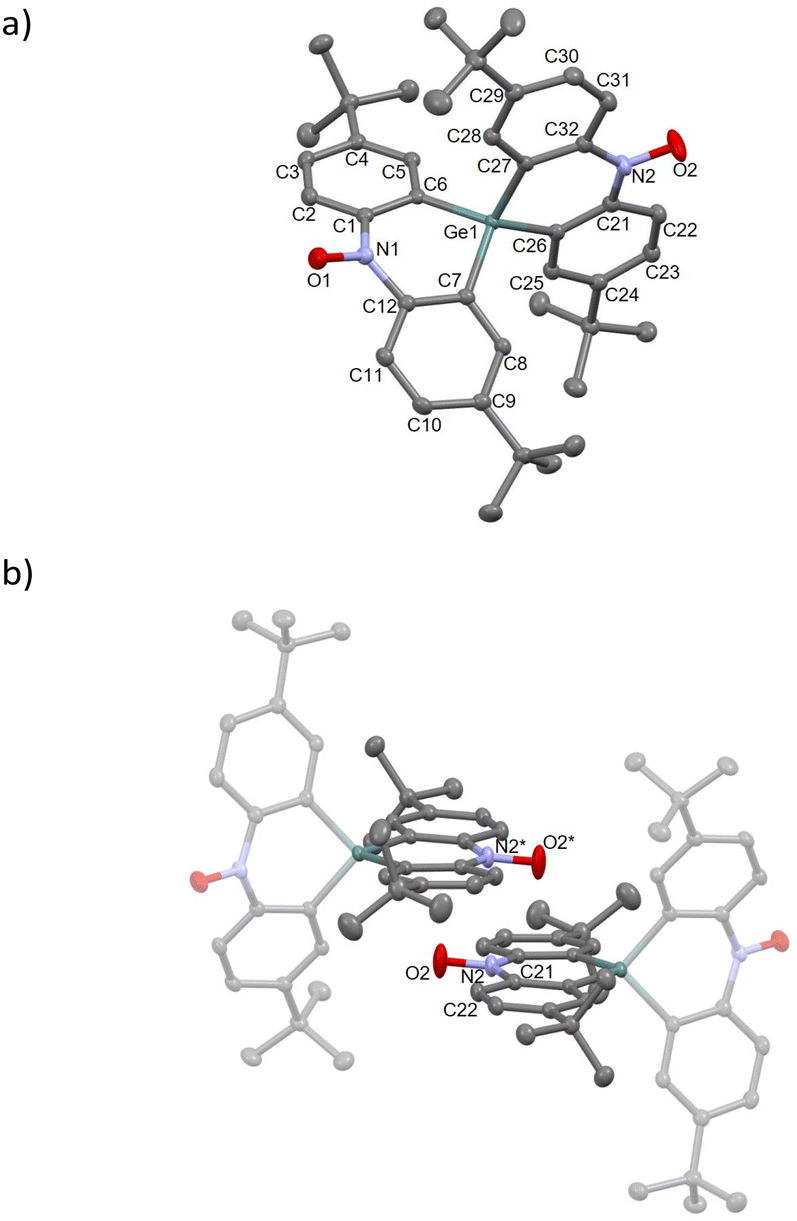 | ||
| Fig. 3 Crystal structures of 1-Ge. (a) Asymmetric units in the unit cell. (b) Intermolecular contacts around the O2 atom. Symmetry code: * = 2−x, 1−y, 1−z. Thermal ellipsoids for non-hydrogen atoms are drawn at the 50% probability level. The H atoms are omitted for clarity. | ||
Theoretical calculations
Density functional theory (DFT) calculations were performed for 1-Si (1-Si-a and 1-Si-b units) and 1-Ge during atomic coordination analysis based on the above crystallographic study. The calculated spin densities of the triplet (T) and broken-symmetry singlet (BS) states were mapped onto the molecular skeleton shown in Figs. S5–S7.† The triplet and singlet energy states are summarized in Tables S2–S4.† Exchange coupling constants 2J/kB were +12.93, +11.25 and +13.81 K, respectively, as estimated by using Yamaguchi's equation.26 Both 1-Si and 1-Ge, as well as 1-C (2J/kB = +30.26 K),12 exhibit the ground triplet state.The exchange coupling constant 2J/kB of +13.81 K for 1-Ge was larger than that of +12.93 and +11.25 K (a mean value of 12.09 K) for 1-Si. Despite the increase in the distance between the intramolecular radical centres (according to the ESR and structural studies), the observed enhancement in 2J/kB is an intriguing result. However, the MOs for 1-Si-a, 1-Si-b and 1-Ge are complicated owing to the distortion of the spiro structure and the dihydroacridine moieties. Therefore, we confirmed the magnitude of the interaction by the optimized structure for model compounds, SBDO-C and the Si and Ge derivatives SBDO-Si and SBDO-Ge (for details, see ESI, Figs. S8–S10 and Tables S8–S13†). These calculations gave 2J/kB values of +30.37, +18.42 and +21.06 K for SBDO-C, -Si and -Ge, respectively. The values for the Si and Ge derivatives were larger than the above-mentioned constants, implying the suppression of distortion for the dihydroacridine moieties.
To investigate the relationship between exchange couplings and intramolecular spin-centres distances, we calculated second model compounds (SBDO-C′ and -C′′), in which the spiro centres were replaced by a carbon atom in the optimised structural parameters of SBDO-Si and -Ge. The N–N distances for SBDO-C, -C′ and -C′′ were 5.93, 6.44 and 6.52 Å, respectively. The energy gaps between the bonding and antibonding orbitals resulting from spiro-conjugation (ΔEspiro), as shown in Fig. 4a, showed a narrowing trend (Table 1). The exchange coupling constants (2J/kB) for SBDO-C, -C′ and -C′′ were +30.37, +29.00 and +27.05 K, respectively. These values show a proportional change with respect to the distance between the radical centres. These findings imply that spirodiradicals not only rely on molecular symmetry, but also require space/bond interactions within each unit to enhance spin–spin interaction. In this case, the spiro-conjugation of the occupied MOs could play a role in the interaction of the two dihydroacridine radical units.
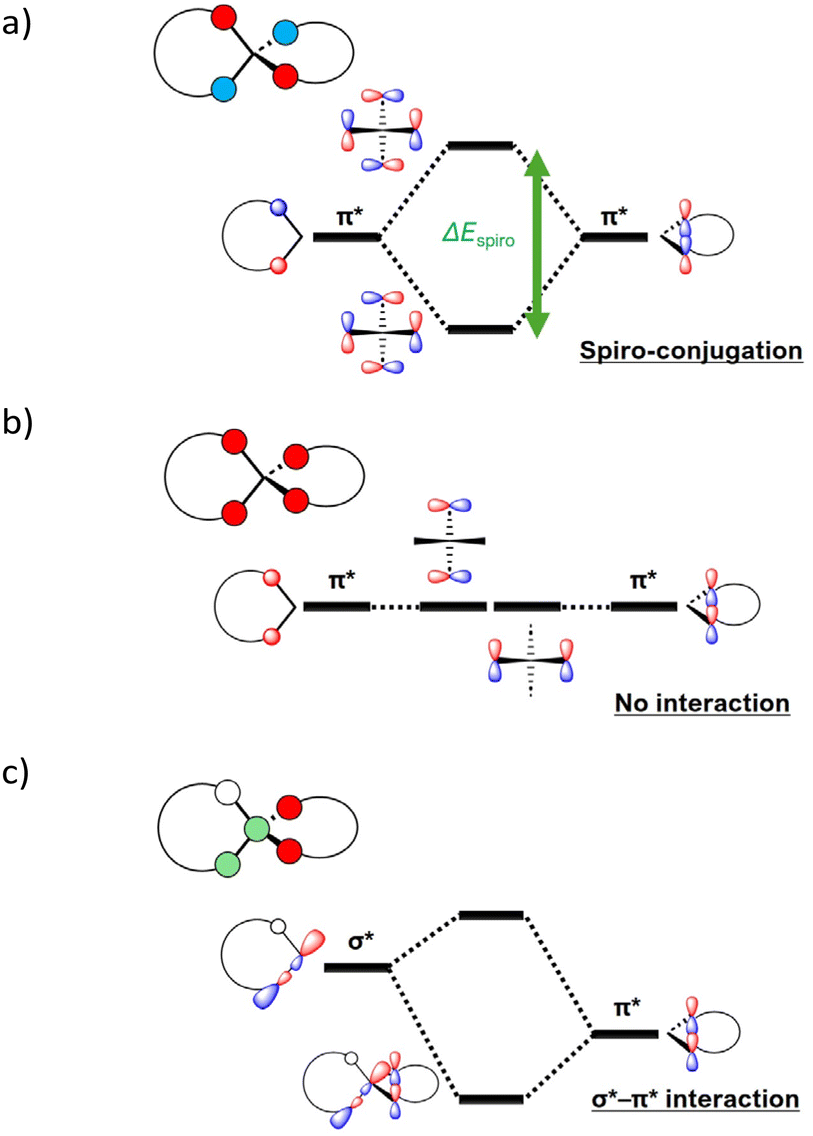 | ||
| Fig. 4 Interaction of two π* orbitals that are (a) antisymmetric with respect to two σd planes and (b) symmetric and antisymmetric with respect to two σd planes. (c) Interaction of the π* orbitals and one of the σ* bondings between the spiro centre and the neighbouring atom. | ||
| α spins | β spins | |||
|---|---|---|---|---|
| ΔEspiro | UMOs/eV | OMOs/eV | UMOs/eV | OMOs/eV |
| SBDO-C | 1.001 | 1.169 | 1.088 | 1.130 |
| SBDO-C′ | 0.438 | 0.429 | 0.461 | 0.423 |
| SBDO-C′′ | 0.380 | 0.358 | 0.401 | 0.354 |
| SBDO-Si | 0.646 | 0.564 | 0.719 | 0.546 |
| SBDO-Ge | 0.491 | 0.430 | 0.524 | 0.420 |
The difference in the interactions of SBDO-Si and -C′ (+18.42 K vs. +29.00 K) showed a more pronounced change compared to that of SBDO-Ge and -C′′ (+21.06 K vs. +27.05 K). For SBDO-C, -Si and -Ge, ΔEspiro values are summarized in Table 1. The tendency of the change in ΔEspiro values with the SBDO-C, -Si and -Ge agrees with the SBDO-C, -C′ and -C′′ cases, and thus the pronounced difference cannot be only explained by it. Therefore, we should consider the contribution of hyperconjugation between the molecular orbitals of the dihydroacridine moieties and the σ*(Si–Cα) orbital. Fig. 5 presents the energy diagrams of the α and β spins in the triplet state for SBDO-C, -Si and -Ge (for more details, see Tables S11–S13†). For SBDO-C, the highest occupied MO (HOMO) and the lowest unoccupied MO (LUMO) were assigned to the bonding and antibonding orbitals based on the spiro-conjugation in the OMO and UMO, respectively. However, for SBDO-Si and -Ge, the HOMOs were the antibonding orbitals from the spiro-conjugation, whereas the LUMOs were the π* orbital of the dihydroacridine units. The energy levels of this orbital for SBDO-Si and -Ge were lower than that of SBDO-C. Here, the LUMOs for SBDO-Si and -Ge have the same phase with lobes at two neighbouring carbon atoms to the spiro centre (Fig. 4b). This symmetry allows the σ*(X–Cα) orbital to interact with the π* orbital, namely σ*(X–Cα)–π* hyperconjugation, as shown in Fig. 4c. This facilitates electron transfer from the spiro centre to the dihydroacridine moieties. In fact, the calculated spin densities of the Si spiro centre of −0.030 was lower than those of the spiro centres for SBDO-C and -Ge, as shown in Table S14.† On the other hand, the O atom for SBDO-Si was +0.474, which was the largest for the three compounds. These results indicate that for SBDO-Si, radical spin is localized at the NO site, resulting in small exchange coupling.
 | ||
| Fig. 5 Selected MO energy-level diagram for (left column) α and (right column) β spins in the triplet state for (left) SBDO-C, (centre) -Si and (right) -Ge. The MO images are superposed with an isovalue of 0.03. The red and blue MOs represent SOMOs and the lowest degenerate π*orbital, respectively. | ||
For 1-Si, structural studies revealed two intermolecular contacts, namely indirect O2⋯O3 and direct O1⋯O4 interactions. Notably, to reduce computational costs, the dihydroacridine moiety, which is not involved in the contacts, and the tert-butyl groups were replaced by methyl groups. First, the indirect O2⋯O3 contact through C atoms was evaluated. The calculated spin densities of the T and BS states were mapped onto the molecular skeleton, as shown in Fig. S11.† The triplet and singlet energy states were ET = −2080.30652569 au with 〈S2〉T = 2.0024 and EBS = −2080.30652658 au with 〈S2〉BS = 0.2455. The intermolecular interaction 2J/kB was estimated to be −0.32 K, indicating weak antiferromagnetic coupling. Next, the direct O1⋯O4 contact was evaluated. The calculated spin densities were mapped onto the molecular skeleton, as shown in Fig. S12.† The triplet and singlet energy states were ET = −2080.31124125 au with 〈S2〉 T = 2.0005 and EBS = −2080.31133968 au with 〈S2〉 BS = 0.2159. The intermolecular interaction 2J/kB was estimated to be −34.81 K, indicating strong antiferromagnetic coupling.
For 1-Ge, an indirect O2⋯O2 contact was present, as shown in Fig. 3b. To reduce computational costs, the dihydroacridine moiety, which was not involved in the contacts, and the tert-butyl groups were replaced by methyl groups. The calculated spin densities of the T and BS states were mapped onto the molecular skeleton, as shown in Fig. S13.† The triplet and singlet energy states were ET = −5655.17316304 au with 〈S2〉T = 2.0006 and EBS = −5655.17317585 au with 〈S2〉BS = 0.2520. The intermolecular magnetic interaction 2J/kB was estimated to be −4.62 K, indicating antiferromagnetic coupling.
Magnetic properties
Magnetic susceptibilities were measured for the polycrystalline samples 1-Si and 1-Ge at 2–300 K (Fig. 6). First, for 1-Si (Fig. 6a), the χmT value at 300 K was 0.718 cm3 K mol−1, which was close to the value of 0.750 cm3 K mol−1 expected for a species having two magnetically isolated radical centres (S = 1/2; g = 2.0). Upon cooling, the χmT value was plateaued up to 120 K and then displayed a decreasing behaviour. At 12 K, there was a difference in the slope of the change in the χmT value (Fig. S14†). The experimental data for 1-Si were analysed using PHI software.27 The spin Hamiltonian is defined as the linear four S = 1/2 spin model: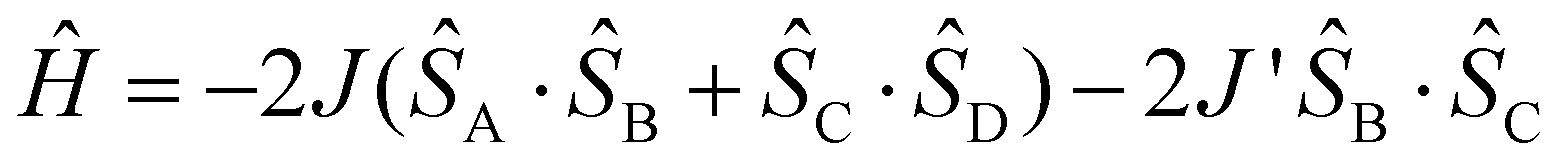 . This model assumes intramolecular coupling and the direct O1⋯O4 contact. The g value was fixed at 2.0064 from the ESR study. The best-fit curve was achieved with 2J/kB = +14.2(3) K, 2J′/kB = −41.2(2) K and TIP = −0.249(4) × 10−3 cm3 mol−1. These exchange coupling constants almost reproduced the DFT calculation results. The field dependence of the magnetization curve at 2 K is shown in the inset of Fig. 6a. The M value was saturated and reached 0.90 μB at 7 T. This value was lower than the expected M value of 2.0 μB (S = 1 and g = 2.0) and close to the expected 1.0 μB (S = 1/2 and g = 2.0). This result indicates strong intermolecular antiferromagnetic coupling (J′). Namely, in the dimer, described in the 4-spin model based on the O1⋯O4 contact, the spin state reflects the cancellation of two spins. Note the monomer exhibited the ground triplet state, as confirmed by the experimental (2J/kB = +14.2 K) and theoretical results.
. This model assumes intramolecular coupling and the direct O1⋯O4 contact. The g value was fixed at 2.0064 from the ESR study. The best-fit curve was achieved with 2J/kB = +14.2(3) K, 2J′/kB = −41.2(2) K and TIP = −0.249(4) × 10−3 cm3 mol−1. These exchange coupling constants almost reproduced the DFT calculation results. The field dependence of the magnetization curve at 2 K is shown in the inset of Fig. 6a. The M value was saturated and reached 0.90 μB at 7 T. This value was lower than the expected M value of 2.0 μB (S = 1 and g = 2.0) and close to the expected 1.0 μB (S = 1/2 and g = 2.0). This result indicates strong intermolecular antiferromagnetic coupling (J′). Namely, in the dimer, described in the 4-spin model based on the O1⋯O4 contact, the spin state reflects the cancellation of two spins. Note the monomer exhibited the ground triplet state, as confirmed by the experimental (2J/kB = +14.2 K) and theoretical results.
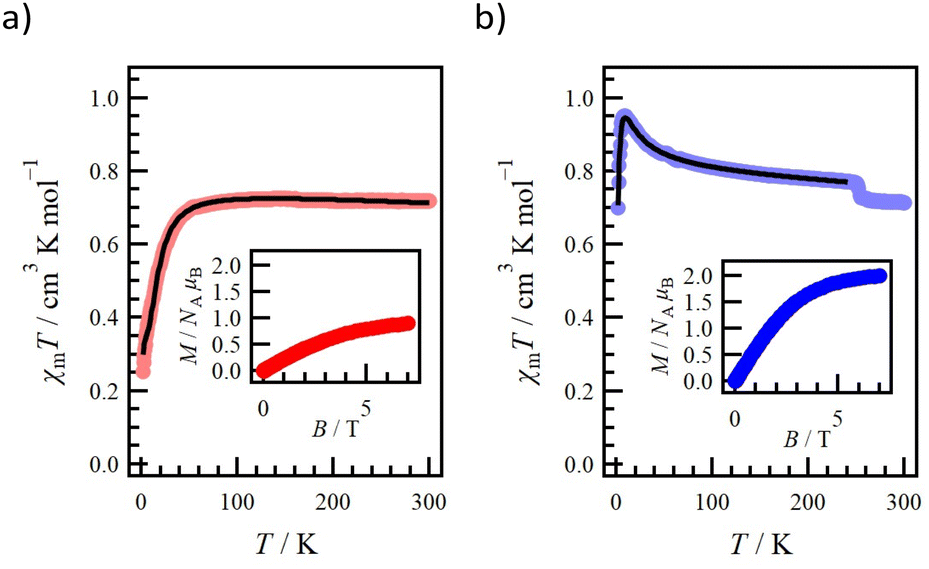 | ||
| Fig. 6 Temperature dependence of the product χmT measured at 5 kOe for (a) 1-Si and (b) 1-Ge. Solid lines represent the simulated curves. The inset shows the magnetization curve measured at 2.0 K. | ||
For 1-Ge (Fig. 6b), the χmT value at 300 K was 0.714 cm3 K mol−1, which was close to the expected value of 0.750 cm3 K mol−1. Upon cooling, the χmT value increased abruptly at 252 K, which was a reversible process according to the results of the cooling and second heating measurements (Fig. S15†). This unique behaviour has been further investigated and will be reported in another paper. Upon further cooling, the χmT value increased and reached 0.950 cm3 K mol−1 at 8.5 K, and then decreased to 0.699 cm3 K mol−1 at 2.0 K. The experimental data of 1-Ge were analysed using PHI software.27 The spin Hamiltonian is defined as the linear four S = 1/2 spin model:  . This model assumes intramolecular coupling and the indirect O2⋯O2 contact. The best-fit curve was achieved with 2J/kB = +19.3(2) K, 2J′/kB = −3.69(1) K, g = 2.059(1) and TIP = −0.323(7) × 10−3 cm3 mol−1. These exchange coupling constants almost reproduced the DFT calculation results. The field dependence of the magnetization curve at 2 K is shown in the inset of Fig. 6b. The M value was saturated and reached 2.00 μB at 7 T, which reproduces to be the expected M value (S = 1 and g = 2.0).
. This model assumes intramolecular coupling and the indirect O2⋯O2 contact. The best-fit curve was achieved with 2J/kB = +19.3(2) K, 2J′/kB = −3.69(1) K, g = 2.059(1) and TIP = −0.323(7) × 10−3 cm3 mol−1. These exchange coupling constants almost reproduced the DFT calculation results. The field dependence of the magnetization curve at 2 K is shown in the inset of Fig. 6b. The M value was saturated and reached 2.00 μB at 7 T, which reproduces to be the expected M value (S = 1 and g = 2.0).
Conclusions
Novel spirobiacridine diradicals 1-Si and 1-Ge were prepared. These exhibited the ground triplet state (S = 1), as supported by spectroscopic and magnetic measurements and theoretical calculations. The compound 1-Ge exhibited large ferromagnetic coupling compared with 1-Si. According to the theoretical calculations for the optimized compounds, the Si-centred derivative could reduce exchange coupling owing to the presence of σ*(Si–Cα)–π* hyperconjugation. Therefore, molecular design based on molecular symmetry should consider not only the suppression of Jahn–Teller distortion but also the effective exchange path between the units; in the case of spirodiradicals, spiro-conjugation except for the SOMOs and the suppression of the hyperconjugation. The compound 1-Ge exhibited the reversible change in the magnetic behaviour. We plan to clarify this finding and report on it in a separate paper. The results of this study elucidate the effect of spiro-centre substitution on the interaction between two orthogonal π-conjugated systems. While σ*–π* conjugation had the negative effect in this study, exploring systems where hyperconjugation acts positively may lead to enhancements in spin–spin interaction. Such investigations are expected to advance structural organic chemistry as well as contribute to applications in organic magnets.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Part of this work was supported by “Advanced Research Infrastructure for Materials and Nanotechnology in Japan (ARIM)” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Grant Number A-22-UT-0080 and JXMXP1222UE5271.References
- (a) E. G. Janzen, Spin trapping, Acc. Chem. Res., 1971, 4, 31–40 CrossRef CAS; (b) M. J. Davies, Detection and characterisation of radicals using electron paramagnetic resonance (EPR) spin trapping and related methods, Methods, 2016, 109, 21–30 CrossRef CAS PubMed.
- (a) J. F. W. Keana, Newer aspects of the synthesis and chemistry of nitroxide spin labels, Chem. Rev., 1978, 78, 37–64 CrossRef CAS; (b) A. Bonucci, O. Ouari, B. Guigliarelli, V. Belle and E. Mileo, In-Cell EPR: Progress towards Structural Studies Inside Cells, ChemBioChem, 2020, 21, 451–460 CrossRef CAS PubMed.
- (a) O. Haze, B. Corzilius, A. A. Smith, R. G. Griffin and T. M. Swager, Water-Soluble Narrow-Line Radicals for Dynamic Nuclear Polarization, J. Am. Chem. Soc., 2012, 134, 14287–14290 CrossRef CAS PubMed; (b) D. Wisser, G. Karthikeyan, A. Lund, G. Casano, H. Karoui, M. Yulikov, G. Menzildjian, A. C. Pinon, A. Purea, F. Engelke, S. R. Chaudhari, D. Kubicki, A. J. Rossini, I. B. Moroz, D. Gajan, C. Copéret, G. Jeschke, M. Lelli, L. Emsley, A. Lesage and O. Ouari, BDPA-Nitroxide Biradicals Tailored for Efficient Dynamic Nuclear Polarization Enhanced Solid-State NMR at Magnetic Fields up to 21.1 T, J. Am. Chem. Soc., 2018, 140, 13340–13349 CrossRef CAS PubMed.
- (a) L. Li, R. Matsuda, I. Tanaka, H. Sato, P. Kanoo, H. J. Jeon, M. L. Foo, A. Wakamiya, Y. Murata and S. Kitagawa, A Crystalline Porous Coordination Polymer Decorated with Nitroxyl Radicals Catalyzes Aerobic Oxidation of Alcohols, J. Am. Chem. Soc., 2014, 136, 7543–7546 CrossRef CAS PubMed; (b) P. Qu, M. Kuepfert, S. Jockusch and M. Weck, Compartmentalized Nanoreactors for One-Pot Redox-Driven Transformations, ACS Catal., 2019, 9, 2701–2706 CrossRef CAS.
- (a) R. Dessauer, Photochemistry, History, and Commercial Applications of Hexaarylbiimidazoles, Elsevier, 2006 Search PubMed; (b) A. Tokunaga, L. M. Uriarte, K. Mutoh, E. Fron, J. Hofkens, M. Sliwa and J. Abe, Photochromic Reaction by Red Light via Triplet Fusion Upconversion, J. Am. Chem. Soc., 2019, 141, 17744–17753 CrossRef CAS PubMed.
- (a) I. Ratera and J. Veciana, Playing with organic radicals as building blocks for functional molecular materials, Chem. Soc. Rev., 2012, 41, 303–349 RSC; (b) N. M. Gallagher, A. Olankitwanit and A. Rajca, High-Spin Organic Molecules, J. Org. Chem., 2015, 80, 1291–1298 CrossRef CAS PubMed; (c) E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Two-dimensional sp2 carbon–conjugated covalent organic frameworks, Science, 2017, 357, 673–676 CrossRef CAS PubMed.
- H. M. McConnel, Ferromagnetism in Solid Free Radicals, J. Phys. Chem., 1963, 39, 1910 CrossRef.
- (a) E. V. Tretyakov, P. V. Petunin, S. I. Zhivetyeva, D. E. Gorbunov, N. P. Gritsan, M. V. Fedin, D. V. Stass, R. I. Samoilova, I. Y. Bagryanskaya, I. K. Shundrina, A. S. Bogomyakov, M. S. Kazantsev, P. S. Postnikov, M. E. Trusova and V. I. Ovcharenko, Platform for High-Spin Molecules: A Verdazyl-Nitronyl Nitroxide Triradical with Quartet Ground State, J. Am. Chem. Soc., 2021, 143, 8164–8176 CrossRef CAS PubMed; (b) H. Zhang, M. Pink, Y. Wang, S. Rajca and A. Rajca, High-Spin S = 3/2 Ground-State Aminyl Triradicals: Toward High-Spin Oligo-Aza Nanographenes, J. Am. Chem. Soc., 2022, 144, 19576–19591 CrossRef CAS PubMed.
- (a) M. Saunders, R. Berger, A. Jaffe, J. M. McBride, J. O'Neill, R. Breslow, J. M. Hoffmann, C. Perchonock, E. Wasserman, R. S. Hutton and V. J. Kuck, Unsubstituted cyclopentadienyl cation, a ground-state triplet, J. Am. Chem. Soc., 1973, 95, 3017–3018 CrossRef CAS; (b) H. Thomann, M. Bernardo and G. P. Miller, Observation of triplet-state electron spin resonance in oxidized fullerene C60, J. Am. Chem. Soc., 1992, 114, 6593–6594 CrossRef CAS; (c) W. T. Broden, R. Hoffmann, T. Stuyver and B. Chen, Dioxygen: What Makes This Triplet Diradical kinetically Persistent?, J. Am. Chem. Soc., 2017, 139, 9010–9018 CrossRef PubMed.
- (a) T. Sugimoto, Y. Misaki, T. Kajita, T. Nagatomi, Z. Yoshida and J. Yamauchi, Tris(thioxanthen-9-ylidene)cyclopropane, and Its Radical Cation and Dication, Angew. Chem., Int. Ed. Engl., 1988, 27, 1078–1080 CrossRef; (b) T. Kodama, M. Aoba, Y. Hirao, S. M. Rivero, J. Casado and T. Kubo, Molecular and Spin Structures of a Through-Space Conjugated Triradical System, Angew. Chem., Int. Ed., 2022, 61, e20220688 CrossRef PubMed.
- T. Ishida, M. Ooishi, N. Ishii, H. Mori and T. Nogami, Mono- and dinitroxide radicals from 9,9′(10H,10′H)-spirobiacridine: An approach to a D2d triplet biradical, Polyhedron, 2007, 26, 1793–1799 CrossRef CAS.
- T. Kanetomo, K. Ichihashi, M. Enomoto and T. Ishida, Ground Triplet Spirobiradical: 2,2′,7,7′-Tetra(tert-butyl)-9,9′(10H,10′H)-spirobiacridine-10,10′-dioxyl, Org. Lett., 2019, 21, 3909–3912 CrossRef CAS PubMed.
- (a) N. L. Frank, R. Clérac, J.-P. Sutter, N. Daro, O. Kahn, C. Coulon, M. T. Green, S. Golhen and L. Ouahab, Synthesis, Crystal Structure, Magnetic, and Electron Paramagnetic Resonance Properties of a Spiroconjugated Biradical. Evidence for Spiroconjugation Exchange Pathway, J. Am. Chem. Soc., 2000, 122, 2053–2061 CrossRef CAS; (b) A. Ito, M. Urabe and K. Tanaka, A Spiro-Fused Triarylaminium Radical Cation with a Triplet Ground State, Angew. Chem., Int. Ed., 2003, 42, 921–924 CrossRef CAS PubMed; (c) Z. Yue, J. Liu, M. Baumgarten and D. Wang, Spirobifluorene Mediating the Spin–Spin Coupling of Nitronyl Nitroxide Diradicals, J. Phys. Chem. A, 2023, 127, 1565–1575 CrossRef CAS PubMed.
- (a) T. Iwamoto, M. Tamura, C. Kabuto and M. Kira, A Stable Bicyclic Compound with Two Si=Si Double Bonds, Science, 2000, 290, 504–506 CrossRef CAS PubMed; (b) S. H. Lee, B.-B. Jang and Z. H. Kafari, Highly Fluorescent Solid-State Asymmetric Spirosilabifluorene Derivatives, J. Am. Chem. Soc., 2005, 127, 9071–9078 CrossRef CAS PubMed; (c) X.-Y. Liu, X. Tang, Y. Zhao, D. Zhao, J. Fan and L.-S. Liao, Spirobi[dibenzo[b,e,][1,4]azasiline]: a novel platform for host materials in highly efficient organic light-emitting diodes, J. Mater. Chem. C, 2018, 6, 1023–1030 RSC.
- (a) K. Ueda, M. Yamanoha, T. Sugimoto, H. Fujita, A. Ugawa, K. Yakushi and K. Kano, New Spiro Donor Molecules: Bis(tetrathiafulvalenyldithio)-methane and -germane, Chem. Lett., 1997, 26, 461–462 CrossRef; (b) Y. Guo, Z. Xia, J. Liu, J. Yu, S. Yao, W. Shi, K. Hu, S. Chen, Y. Wang, A. Li, M. Driess and W. Wang, A Tetra-amido-Protected Ge5-Spiropentadiene, J. Am. Chem. Soc., 2019, 141, 19252–19256 CrossRef CAS PubMed.
- (a) B. H. Boo, J. Park, H. G. Yeo, S. Y. Lee, C. J. Park and J. H. Kim, Infrared and Raman Spectroscopy of 9,9′-Spirobifluorene, Bis(2,2′-biphenylene)silane, and Bis(2,2′-biphenylene)germane. Vibrational Assignment by Depolarization Measurement and HF and Density Functional Theory Studies, J. Phys. Chem. A, 1998, 102, 1139–1145 CrossRef CAS; (b) A. G. Russell, N. S. Spencer, D. Philp, B. M. Kariuki and J. S. Snaith, The First Spirobifluorenes Containing Two Binaphthyl Moieties, Organometallics, 2003, 22, 5589–5592 CrossRef CAS; (c) S. Furukawa, K. Hayashi, K. Yamagishi and M. Saito, Synthesis and properties of spiro-type heterasumanenes containing group 14 elements as bridging atoms, Mater. Chem. Front., 2018, 2, 929–934 RSC.
- S. Yamaguchi, Y. Itami and K. Tamao, Group 14 Metalloles with Thienyl Groups on 2,5-Positions: Effects of Group 14 Elements on Their π-Electronic Structures, Organometallics, 1998, 17, 4910–4916 CrossRef CAS.
- (a) A. L. Shain, J. P. Ackerman and M. W. Teague, Intramolecular Triplet Exciton Transfer in Some Noncoplanar aromatic systems, Chem. Phys. Lett., 1969, 3, 550–551 CrossRef CAS; (b) A. L. Shain, Dynamics of Intramolecular Triplet Exciton Transfer Using Electron Spin Resonance, J. Chem. Phys., 1972, 56, 6201–6212 CrossRef CAS.
- A. Ito, M. Urabe and K. Tanaka, Design of high-spin organic molecules: toward magnetic parts in molecular electronics, Curr. Appl. Phys., 2003, 3, 149–153 CrossRef.
- S. Stoll and A. Schweiger, EasySpin, a comprehensive software package for spectral simulation and analysis in EPR, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- T. Kanetomo, S. Ono, Y. Fukushima, Y. Takenouchi and M. Enomoto, Galvinoxyl-inspired dinitronyl nitroxide: structural, magnetic, and theoretical studies, Org. Chem. Front., 2023, 10, 3193–3200 RSC.
- (a) A. Ito, K. Hata, K. Kawamoto, Y. Hirao, K. Tanaka, M. Shiro, K. Furukawa and T. Kato, para-Phenylene-Bridged Spirobi(triarylamine) Dimer with Four Perpendicularly Linked Redox-Active π Systems, Chem. – Eur. J., 2010, 16, 10866–10878 CrossRef CAS PubMed; (b) N. Terada, K. Uematsu, R. Higuchi, Y. Tokimaru, Y. Sato, K. Nakano and K. Nozaki, Synthesis and Properties of Spiro-double Sila[7]helicene: The LUMO Spiro-conjugation, Chem. – Eur. J., 2021, 27, 9342–9349 CrossRef CAS PubMed.
- K. Clabron, B. Kahr and W. Kaminsky, Calculations of optical properties of the tetraphenyl-X family of isomorphous crystals (X = C, Si, Ge, Sn, Pb), CrystEngComm, 2002, 4, 252–256 RSC.
- A. Bondi, van der Waals Volumes and Radii, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- A. Karipides and D. A. Haller, The Crystal Structure of Tetraphenylgermanium, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 2889–2892 CrossRef CAS.
- K. Yamaguchi, T. Kawakami, Y. Takano, Y. Kitagawa, Y. Yamashita and H. Fujita, Analytical and ab initio studies of effective exchange interactions, polyradical character, unpaired electron density, and information entropy in radical clusters (R)N: Allyl radical cluster (N=2–10) and hydrogen radical cluster (N=50), J. Quantum Chem., 2002, 90, 370–385 CrossRef CAS.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, PHI: A powerful new program for the analysis of anisotropic monomeric and exchange-coupled polynuclear d- and f-block complexes, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 23349212334922. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qo00419a |
| This journal is © the Partner Organisations 2024 |