
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoselective strain-release Ferrier rearrangement: the dual role of catalysts†
Huajun
Zhang‡
,
Aoxin
Guo‡
,
Han
Ding‡
 ,
Yuan
Xu
,
Yuhan
Zhang
,
Dan
Yang
and
Xue-Wei
Liu
*
,
Yuan
Xu
,
Yuhan
Zhang
,
Dan
Yang
and
Xue-Wei
Liu
*
School of Chemistry, Chemical Engineering and Biotechnology, 21 Nanyang Link, 637371 Singapore. E-mail: xuewei@ntu.edu.sg
First published on 25th June 2024
Abstract
Herein, we report an efficient, stereoselective synthesis of 2,3-unsaturated glycosides under mild conditions through a novel Ferrier rearrangement of reasonably designed glycal donors with C3-position ortho-2,2-dimethoxycarbonycyclopropylbenzoyl (CCBz), facilitated by user-friendly, eco- and environmental-friendly Cu(OTf)2 or Fe(OTf)3. The newly devised rearrangement tactic is highly α-stereoselective and applies to a broad scope of nucleophile acceptors, enabling the construction of 2,3-unsaturated O-, S-, N-, and C-glycosides, with exceptional yields and stereoselectivities. DFT calculations were conducted to elucidate the reaction mechanism, unveiling the dual role of the metal catalysts in activating the glycal donor through promoting ring-opening of the intramolecularly incorporated donor–acceptor cyclopropane (DAC) of CCBz and directing the following α-face-preferential nucleophilic attack of the incoming acceptor mediated by H-bond interactions.
Introduction
Carbohydrates feature essential roles in manifold physiological processes, including cellular respiration, cell–cell contact and adhesion, modulating transcription and sophisticated cascades, inflammation, and post-translational modifications.1 Among the diverse carbohydrate structures, 2,3-unsaturated glycosides stand out for their significant pharmacological properties and retrofittable potential in organic synthesis (Fig. 1A). The naturally occurring unsaturated sugar derivative blasticidin S, fortuitously discovered in the 1950s,2 prevents rice blast disease. Synthetic compounds containing 2,3-unsaturated sugars also show potent bioactivities. 7-O-(4,6-di-O-acetyl-hex-2-enea-D-erythro-pyranosyl) genistein, a genistein derivative, suppresses tumor growth.3,4 Stavudine, a synthetic L-nucleoside that has been approved as the fourth anti-HIV drug, inhibits the HIV reverse transcriptase with fewer adverse effects on bone marrow stem cells and mitochondrial DNA replication5 than the predecessor AZT. Meanwhile, the 2,3-olefinic bond in a pyranose ring is amenable to facile functionalization at C2- and C3-positions via a variety of well-established reactions such as asymmetric dihydroxylation, amino hydroxylation, hydrogenation, and epoxidation, underscoring the wide applications of this type of scaffold in synthesizing diverse functionalized sugar structures.6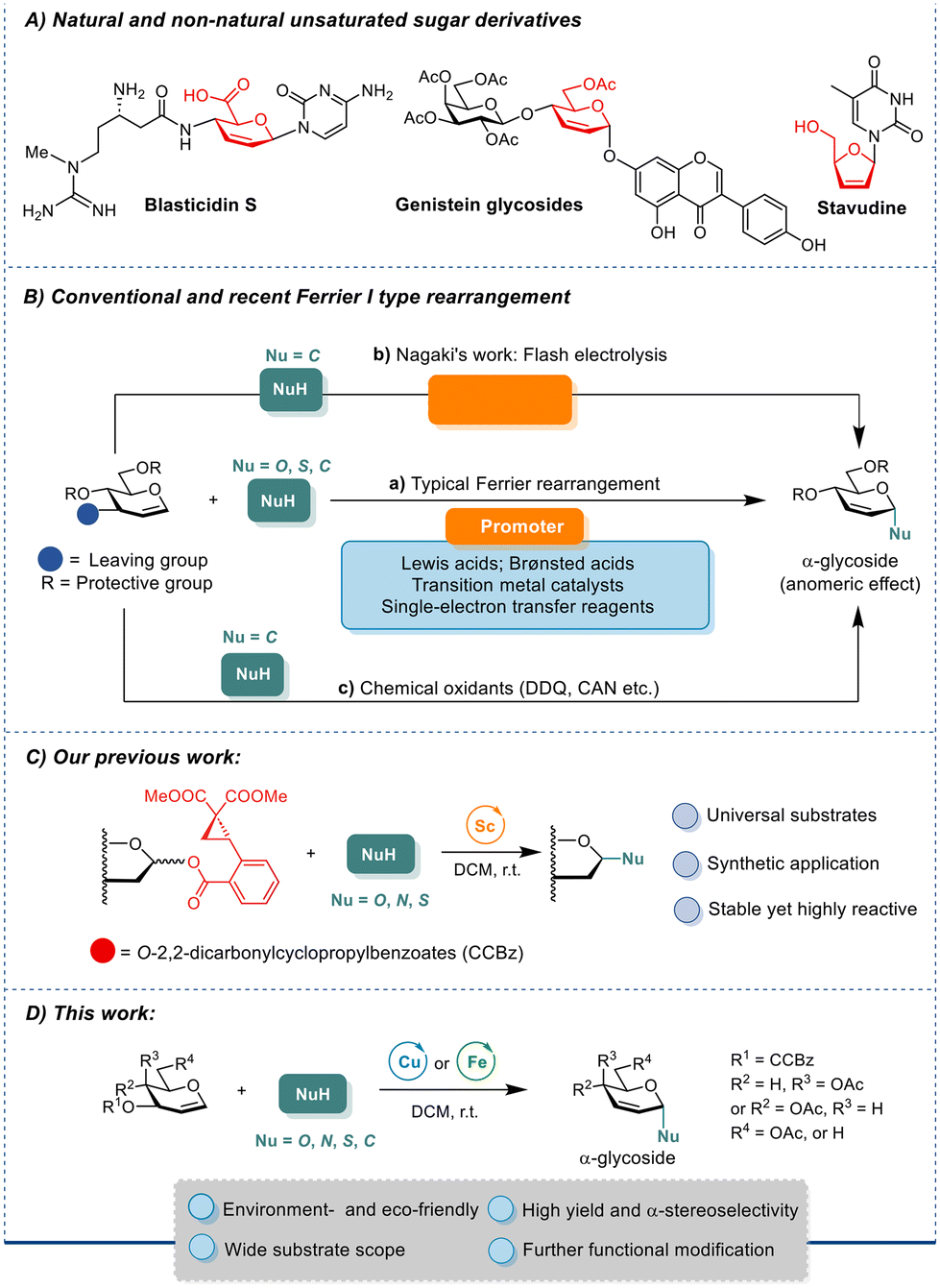 | ||
| Fig. 1 Significance of and synthetic approaches to 2,3-unsaturated glycosides and our strategy. | ||
Glycals have long been exploited as synthons for chemical derivatizations to various sugar adducts and derivatives. The Ferrier I rearrangement of glycals proves to be a facile and efficient approach for creating 2,3-unsaturated pyranose scaffolds. In a classical Ferrier I rearrangement, glycal with a good leaving group (LG) at the C3-position, such as acetate, carbonate, or trichloroacetimidate, is first activated, then an allylic rearrangement on the pyranose ring occurs, accompanied by the dissociation of the C3-LG and the ensuing attack of a nucleophile at the anomeric position, affording the 2,3-unsaturated glycoside (Fig. 1B, a).7 Many Ferrier rearrangement reactions under mild conditions with short reaction times consume stoichiometric amounts of Lewis acids as promoters, while other transformations are either promoted by Brønsted acids8 or transition metal catalysts.9,10 A Ferrier-type O-glycosylation through a radical pathway employing single-electron transfer reagents11 and chemical oxidants like 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) or ceric ammonium nitrate (CAN) has also been reported (Fig. 1B, c).12 Recently, Nagaki and coworkers reported flash electrochemical and superacid-promoted pathways leading to nitrogen-containing 2,3-unsaturated C-aryl glycosides and C-alkyl glycosides respectively via short-lived intermediates (Fig. 1B, b).13 Despite these advancements, current Ferrier reactions are often limited to narrow substrate scopes and poor-to-moderate α-selectivities during glycosidic linkage formation, which is influenced by multiple factors of the vinylogous anomeric effect (VAE)14 that is affected by the orientation of the C4-substituent, 1,3-diaxial interactions, the substituents at C5,15 as well as the solvent effect in most cases.16,17
Our group is long devoted to the development of novel glycosylation methodologies,18 and we have developed a novel ortho-2,2-dimethoxycarbonylcyclopropylbenzoate (CCBz)19 leaving group which can readily be activated by the Sc(OTf)3 recently (Fig. 1C). The ring-strain release of intramolecular incorporated donor–acceptor cyclopropanes (DACs) in CCBz during LG dissociation efficiently drives glycosylation reactions with various O-, S-, and N-nucleophiles.20 Drawing inspirations from the synthetic prowess of the CCBz LG, herein, we report the development of a novel Ferrier rearrangement for stereoselective construction of 2,3-unsaturated α-glycosides employing low-cost Cu(OTf)2 or Fe(OTf)3 catalysts under mild conditions by introducing CCBz to C3-position of glycal. Results from DFT simulations of the reaction pathways reveal that the α-stereoselectivity of the Cu(OTf)2-catalyzed Ferrier rearrangement arises from the face-preferential directed nucleophilic attack mediated by the non-covalent interactions between metal-bound triflate and the incoming alcohol, denoting a dual-tasked Cu(OTf)2 (Fig. 1D). Our approach provides facile, efficient, and stereoselective access to the synthetically valuable 2,3-unsaturated pyranose (pseudo-glycals) as chiral precursors for numerous further chemical transformations.
Results and discussion
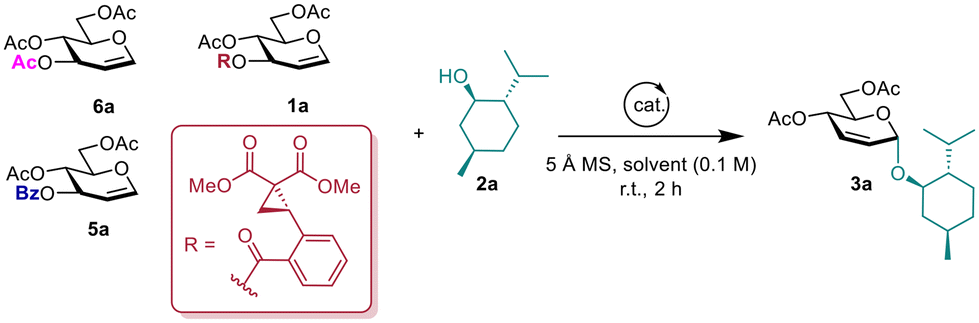
We commenced our study by evaluating the feasibility of the proposed reaction and optimizing reaction conditions, using L-menthol 2a as the model acceptor substrate. The results, detailed in Table 1, showed that employing glucal 1a as the donor had an impressive 98% conversion but unsatisfactory stereoselectivity using Sc(OTf)3 as the catalyst in dichloromethane (DCM, entry 1). Interestingly, significant improvements in stereoselectivities were observed employing Cu(OTf)2 or Fe(OTf)3, supplying the desired products in 98% yields with 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 α-selectivity (entries 2 and 3). Lowering the catalyst loading of Fe(OTf)3 in the reaction system resulted in a drastic decrease in yield, while the stereoselectivity was not affected (entry 4). In contrast to Fe(OTf)3, FeCl3 produced minimal product conversion, probably due to its hygroscopic and deliquescent nature (entry 6). The ferrous salts with lower valence (+2) iron ions were ineffective for catalyzing the reaction (entries 5–7). Similarly, non-chelatable Lewis acids and Brønsted acids did not facilitate the reaction (entries 8 and 9). A screening of reaction solvents revealed that 1,2-dichloroethane (DCE) was also an efficient solvent (entries 10 and 11). However, utilizing Et2O resulted in moderate yield and stereoselectivity (entry 12), and this reaction failed to proceed in toluene and acetonitrile (ACN) at room temperature (entries 13 and 14). Subjecting the contrast donor 3-O-benzoyl-4,6-di-O-acetyl-glucal 5a to the optimized conditions did not lead to the formation of the corresponding product. This result suggests that the catalyst's activation of the DAC site occurs through chelation with two carbonyl groups (entries 15 and 16).21 Lastly, when tri-O-acetyl-D-glucal (6a) as a donor to the optimized conditions did not obtain the desired product 3a in great yield and α-stereoselectivity (entries 17 and 18).
:1 α-selectivity (entries 2 and 3). Lowering the catalyst loading of Fe(OTf)3 in the reaction system resulted in a drastic decrease in yield, while the stereoselectivity was not affected (entry 4). In contrast to Fe(OTf)3, FeCl3 produced minimal product conversion, probably due to its hygroscopic and deliquescent nature (entry 6). The ferrous salts with lower valence (+2) iron ions were ineffective for catalyzing the reaction (entries 5–7). Similarly, non-chelatable Lewis acids and Brønsted acids did not facilitate the reaction (entries 8 and 9). A screening of reaction solvents revealed that 1,2-dichloroethane (DCE) was also an efficient solvent (entries 10 and 11). However, utilizing Et2O resulted in moderate yield and stereoselectivity (entry 12), and this reaction failed to proceed in toluene and acetonitrile (ACN) at room temperature (entries 13 and 14). Subjecting the contrast donor 3-O-benzoyl-4,6-di-O-acetyl-glucal 5a to the optimized conditions did not lead to the formation of the corresponding product. This result suggests that the catalyst's activation of the DAC site occurs through chelation with two carbonyl groups (entries 15 and 16).21 Lastly, when tri-O-acetyl-D-glucal (6a) as a donor to the optimized conditions did not obtain the desired product 3a in great yield and α-stereoselectivity (entries 17 and 18).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Donor | Catalyst | Solvent | Yield of 3a %b | α:βc |
|
a Unless otherwise noted, all reactions were carried out with 1a or 5a (0.1 mmol, 1.0 equiv.), 2a (1.2 equiv.), catalyst (0.1 equiv.) and 5 Å MS (50 mg) in corresponding solvent (1.0 mL, 0.1 M) under nitrogen atmosphere for 2 h.
b Isolated yields.
c The α:β ratios were determined by 1H NMR analysis.
d 0.05 equiv. of catalyst.
e DCM = dichloromethane.
f DCE = 1,2-dichloroethane.
g ACN = acetonitrile.
h N.R. = no reaction.
i N.D. = not detected.
j MS = molecular sieve.
|
|||||
| 1 | 1a | Sc(OTf)3 | DCM | 98 | 4:1 |
| 2 | 1a | Cu(OTf)2 | DCM | 98 | 10:1 |
| 3 | 1a | Fe(OTf)3 | DCM | 98 | 10:1 |
| 4d | 1a | Fe(OTf)3 | DCM | 53 | 10:1 |
| 5 | 1a | Fe(OTf)2 | DCMe | N.R.e | N.D.f |
| 6 | 1a | FeCl3 | DCM | 6 | — |
| 7 | 1a | FeCl2 | DCM | N.R. | N.D. |
| 8 | 1a | TMSOTf | DCM | N.R. | N.D. |
| 9 | 1a | TfOH | DCM | N.R. | N.D. |
| 10 | 1a | Cu(OTf)2 | DCEf | 98 | 10:1 |
| 11 | 1a | Fe(OTf)3 | DCE | 98 | 10:1 |
| 12 | 1a | Cu(OTf)2 | Et2O | 65 | 10:3 |
| 13 | 1a | Fe(OTf)3 | Toluene | 12 | 10:1 |
| 14 | 1a | Fe(OTf)3 | ACNg | N.R.h | N.D.i |
| 15 | 5a | Cu(OTf)2 | DCM | N.R. | N.D. |
| 16 | 5a | Fe(OTf)3 | DCM | N.R. | N.D. |
| 17 | 6a | Cu(OTf)2 | DCM | 74% | 3:1 |
| 18 | 6a | Fe(OTf)3 | DCM | 69% | 3:1 |
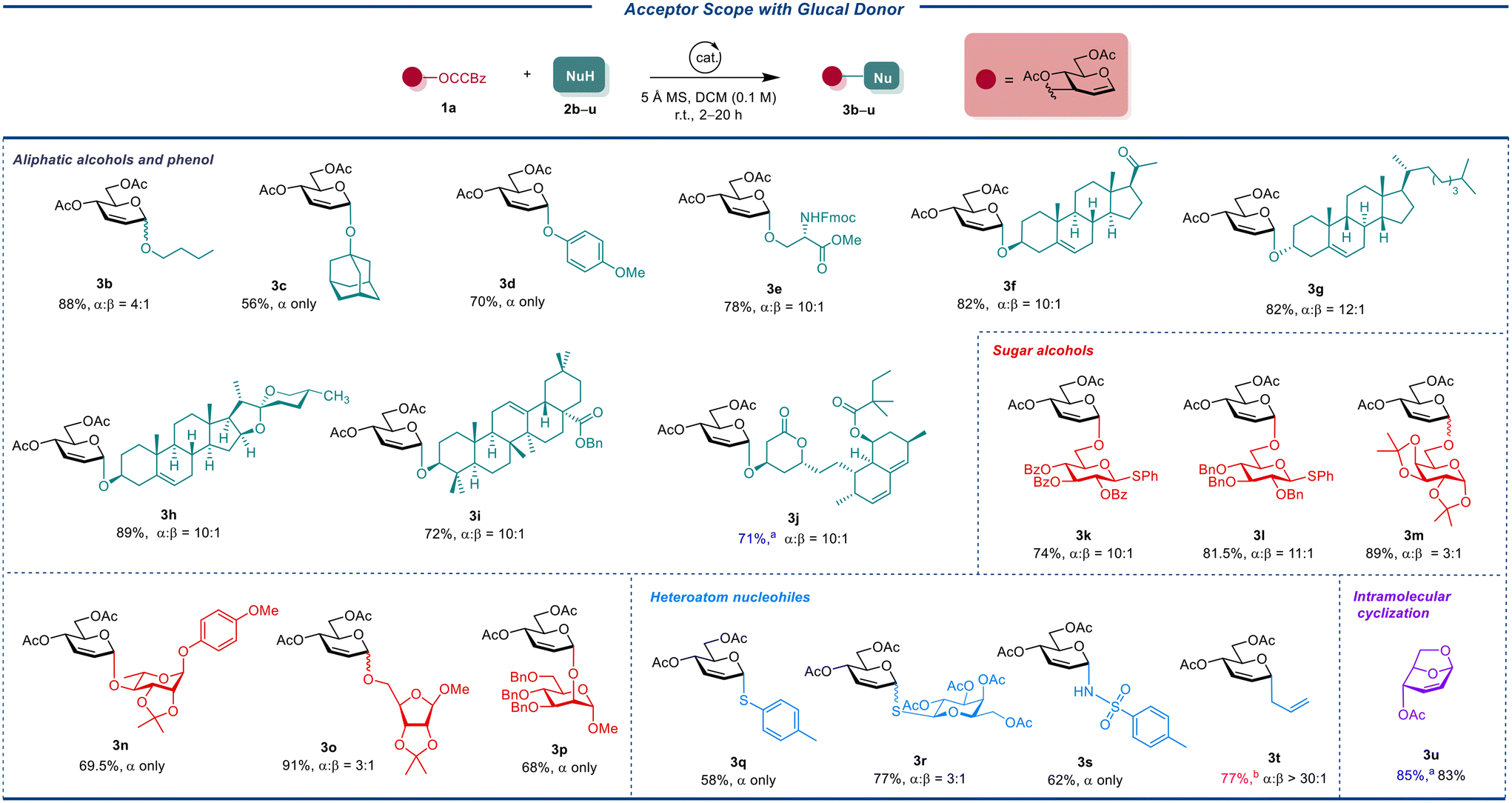
To explore the substrate scope of the Cu(OTf)2- or Fe(OTf)3-catalyzed Ferrier reaction, we subjected a library of structurally diverse acceptors (2b–2u) to the reaction with 1a under the optimized reaction conditions, as depicted in Table 2. This set of acceptors included aliphatic alcohols with primary, secondary, and tertiary hydroxyl groups, sugar alcohols, phenol, and heteroatom nucleophiles. The results were encouraging, yielding corresponding 2,3-unsaturated glucosides in 56% to 98% yields. Notably, the reaction results were predominantly α selectivities (α:β = 3:1 to α only, determined by 1H NMR analysis). Additionally, the strain-release glycosylation effectively promoted the reaction of 1a with thioglycoside acceptors, producing unsaturated disaccharide thioglycosides (3l and 3k). Interestingly, the typical aglycone transfer often seen with thioglycoside acceptors was not observed.22 Although reactions with 2m and 2o took 12 hours to complete, acid-sensitive acetonides were well tolerated, forming 3m and 3o products. Under our mild reaction conditions, we selectively constructed the α-O-glycosidic bond with the C2–OH of mannoside 2p and the C4–OH of rhamnoside 2n. Notably, only Cu(OTf)2 successfully promoted the Ferrier rearrangement of 2n, while Fe(OTf)3 resulted in a complex reaction mixture. A similar result was also observed when para-methoxyphenol 2d was subjected to reaction with 1a under the catalysis of Fe(OTf)3, possibly due to the oxidation of the para-methoxyphenyl group by the oxidizing ferric ion. We further applied this methodology to the syntheses of complex and bioactive natural products and drugs. The α-O-glycosides building block was successfully installed onto the C3–OH of cholesterol 2g, diosgenin 2h and triterpene oleanolate 2i, affording the respective glyconjugates 3g, 3h and 3i in good yields and stereoselectivities (72% to 89%, α:β = 10:1 to 12:1). Additionally, the hormone pregnenolone (2f) and the antihyperlipidemic drug simvastatin (2j) were also compatible with our method, although longer reaction times were required.
| Conditions: unless otherwise noted, all reactions were carried out with 1a (0.1 mmol, 1.0 equiv.), 2b–u (1.2 equiv.), Cu(OTf)2 (0.1 equiv.) and 5 Å MS (50 mg) in distilled DCM (1.0 mL, 0.1 M) under nitrogen atmosphere at the room temperature.a Fe(OTf)3 (0.1 equiv.) and 5 Å MS (50 mg) in distilled DCM (1.0 mL, 0.1 M) under nitrogen atmosphere at the room temperature.b Sc(OTf)3 (0.1 equiv.) and 5 Å MS (50 mg) in distilled DCM (1.0 mL, 0.1 M) under nitrogen atmosphere at the room temperature. MS = molecular sieve; DCM = dichloromethane. |
|---|
|
Heteroatom glycosidic linkages, i.e., which are glycosidic bonds where the anomeric oxygen in the hemiacetal sugars is replaced with a different atom, are widely found in a variety of important biological molecules, including glycoconjugates, glycopeptides,23 glycosylamines,24 glycolipids,25 as well as in DNA, RNA, and coenzyme A.26 To our delight, we confirmed that our Ferrier rearrangement applies to the synthesis of 2,3-unsaturated S-, N-, and C-glycosides, through glycosylation of the corresponding nucleophiles with 1a. Of note, in all successful glycosylation instances with heteroatom nucleophiles, there is no direct ring-opening of the DAC moiety. This suggests that intramolecular cyclization may be kinetically favored even in the presence of a heteroatom nucleophile. Following the hard–soft-acid–base (HSAB) principle, oxygen nucleophiles, being hard bases, preferentially add to the hard acid center at C1. Conversely, soft bases like sulfonamide and thiol moieties are inclined to add to the soft acid center at C3.27 Interestingly, we did not observe any C3-substituted byproducts. Under our conditions, 3s was produced with high yield and stereoselectivity, though the reaction of 1a with 2q and 2r yielded 3q and 3r with moderate yields and imperfect stereoselectivities, respectively. The relatively low yields for 3q and 3s were attributed to the unconsumed starting materials in lieu of the C3 attack of the acceptors. It is noteworthy that Fe(OTf)3 was rarely effective in forming C–S bonds. Neither Cu(OTf)2 nor Fe(OTf)3 was effective in creating unsaturated C-glycosides when allyltrimethylsilane was used as a nucleophile. Pleasingly, our initial catalyst, Sc(OTf)3, facilitated the formation of allyl C-glycosyl derivatives 3t with good yield (77%) and remarkable stereoselectivity (>30:1). Regarding the selectivity preference, according to Danishefsky's work,28 in our reaction, the preferred stereoselectivity in the construction of C-glycosides is not because of the function of the catalyst, but because of the own nature of the allyl nucleophilic reagent. This allylic rearrangement, leveraging ring-strain-release technology, is instrumental in synthesizing the key synthon of 1,6-anhydrosugar 3u with excellent yield (83%). According to the literature, 1,6-anhydrosugars like 3u can be transformed into polyfluorinated carbohydrates.29 These compounds, known for their challenging synthesis and impact on lipophilicity, have garnered significant research interest in recent years as chemical probes or potential therapeutic agents.30,31 Additionally, 1,6-anhydrosugar 3u can be used to synthesize biologically relevant polymers found in the cell wall of Mycobacterium tuberculosis,32 and it serves as a suitable intermediate for the total synthesis of bleomycin A2, and heparin-like oligosaccharides,33 as reported in previous studies.
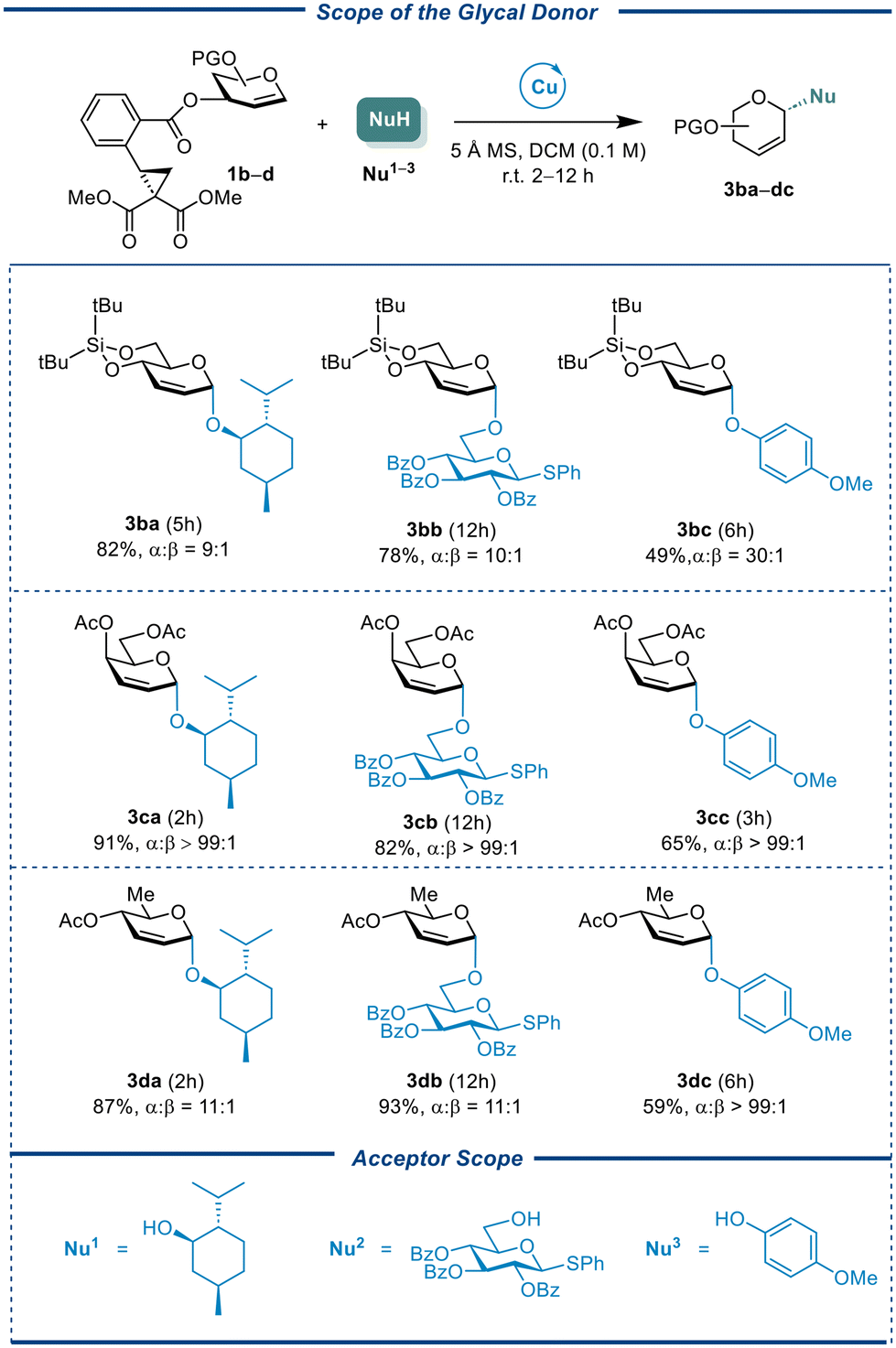
A series of differentially protected glycals were prepared including conformationally constrained donor CCBz 1b, galactal donor CCBz 1c, and L-rhamnal CCBz 1d. These donors were reacted with three types of acceptors, namely, L-menthol (Nu1) as an aliphatic nucleophile, phenyl 2,3,4-O-tri-benzoyl-1-thio-β-glucopyranoside as a sugar nucleophile (Nu2), para-methoxylphenol as a phenol nucleophile (Nu3). The results were showed in Table 3. Most cases had good-to-excellent yields and α-selectivities, leading to the formation of 2,3-unsaturated Ferrier-type products. While the 4,6-O-constrained glucal 1b afforded desired products in lower yields of 49%–82% with little change in stereoselectivity (α:β = 9:1 to 30:1). Like glucal CCBz 1a, galactal CCBz 1c could be coupled with Nu1, Nu2, and Nu3 to give the desired products in 65%–91% with nearly perfect α-stereocontrollability (α only). The three types of acceptors reacted with the L-rhamnal CCBz 1d to obtain the required compounds in 59%–93% and mostly α-selectivity (α:β = 11:1 to α only). It is worth noting that the aglycon transfer of thioglycoside acceptor was not observed for most glycal CCBz donors, highlighting this method's potential for synthesizing more valuable oligosaccharides.
| a Conditions: unless otherwise noted, all reported yields are isolated and purified products. For Ferrier rearrangement glycosylation reactions of Nu1, Nu2, and Nu3, 1.0 equiv. of the donor 1b–d, 1.2 equiv. of the acceptor Nu1–3, and 0.1 equiv. of Cu(OTf)2 was used. r.t. = room temperature; DCM = dichloromethane; Nu = nucleophile; Bz = benzoyl; Ac = acetyl; Ph = phenyl. |
|---|
|
Finally, galactal donor CCBz 1c was reacted with 6 (from dapagliflozin which is a drug treating type 2 diabetes) to produce a disaccharide compound 7 with perfect α-selectivity (Scheme 1, a). The rich chemistry of double bonds in this compound supplied a flexible handle for chemical modification to access structurally diverse sodium-glucose cotransporter-2 (SGLT 2) inhibitors.34 In addition, when deprotected donor 2u is subjected to Fe(OTf)3-catalyzed conditions, it can undergo synthesis to produce 1,6-anhydrosugar 3u in high yields on a gram-scale. Likewise, donor 1a can be effectively treated with diosgenin, a bioactive biomolecule known for its various medicinal properties such as hypolipidemic, hypoglycaemic, antioxidant, anti-inflammatory, and antiproliferative activities,35 to synthesize saponin compound 3h with a high yield and α-selectivity on a gram scale in using Cu(OTf)2 as a catalyst. The successful implementation of gram-scale reactions significantly enhances the competitiveness and appeal of the approach in pharmaceutical synthesis.
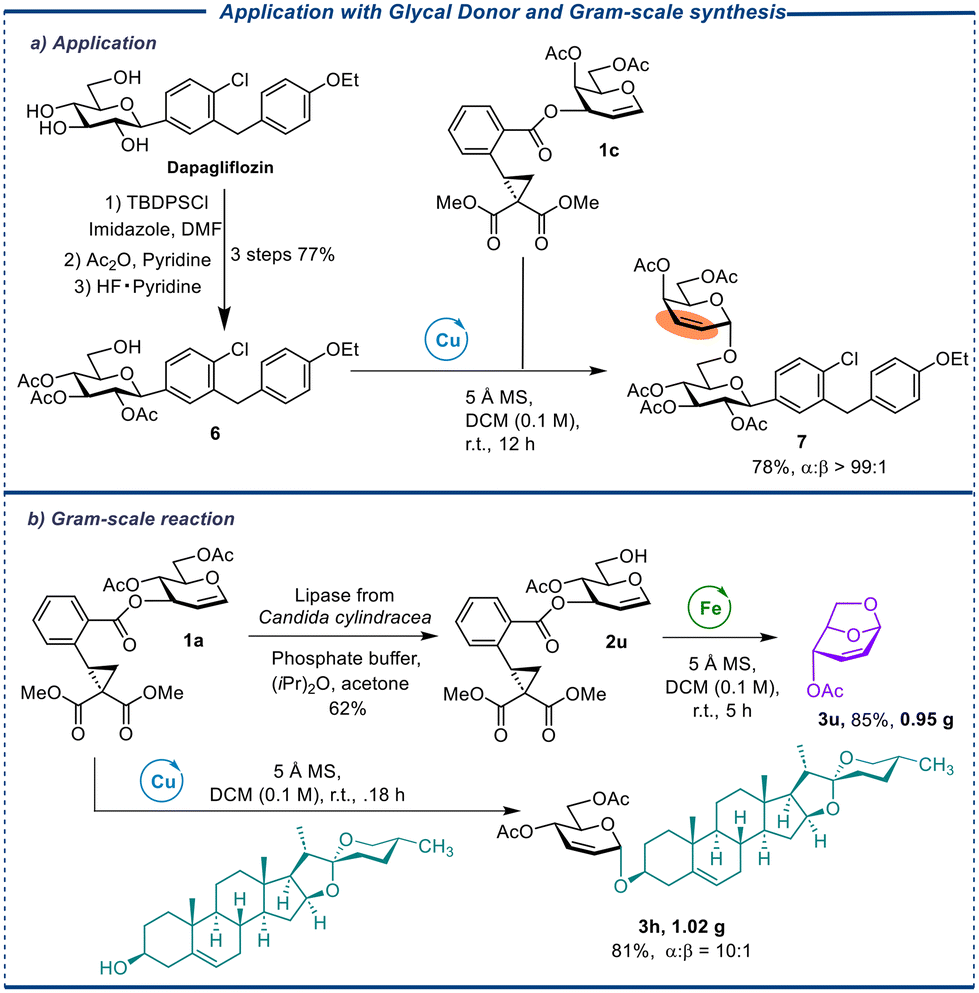 | ||
| Scheme 1 The application to modifiable dapagliflozin and gram-scale synthesis. Conditions: this reaction was carried out with 1c (0.1 mmol, 1.0 equiv.), 6 (1.2 equiv.), Cu(OTf)2 (0.1 equiv.); DCM = dichloromethane; DMF = dimethylformamide; Et = ethyl; Ac = acetyl; TBDPS = tert-butyldiphenylsilyl. | ||
To shed light on the stereoselectivity of the glycosylation reaction, we simulated employing model donor 1a and model acceptors methanol (MeOH), and p-methoxyphenol (p-OMePhOH) (see ESI†), and Cu(OTf)2, Sc(OTf)3 as the catalysts, we proposed that our Cu(OTf)2-catalyzed Ferrier rearrangement with α-selective glycosylation proceeds in three stages: Firstly, the copper(II)-promoted DAC ring of the CCBz opening resulting in the dissociation of C3-substituent and the migration of the 1,2-double bond, giving rise to an ion pair of the LG anion and glycosyl oxocarbenium. Then another copper(II) species coordinates to the 2,3-position of the glycosyl oxocarbenium IM4α, from either α- or β-face, bringing the coordinated alcohol acceptor to the proximity of the glycosyl oxocarbenium. Finally, a directed nucleophilic α-attack by the alcohol at the anomeric position forms the glycosidic bond (Scheme 2). We assumed that the metal center participating in coordination carries an accompanying triflate anion, which serves as a proton acceptor in the final O-glycosylation step. Structures of reagents, products, catalysts, proposed intermediates, and transition states were built with GaussView6.0 and pre-optimized at B3LYP/def2-SV(P) level of theory36 with Grimme's empirical dispersion D3(BJ),37 and the pre-optimized structures were subjected to geometry optimization and frequency analysis at MN15-L/def2-TZVP/SMD (solvent = DCM) level of theory.38–40 Accurate electronic energy is calculated from the optimized structures at MN15-L/ma-def2-TZVPP/SMD (solvent = DCM) level of theory.41 Gibbs free energies of the intermediates were electronic energy calculated as the sum of thermal corrections and the accurate. All calculations were performed with Gaussian 16 software.42
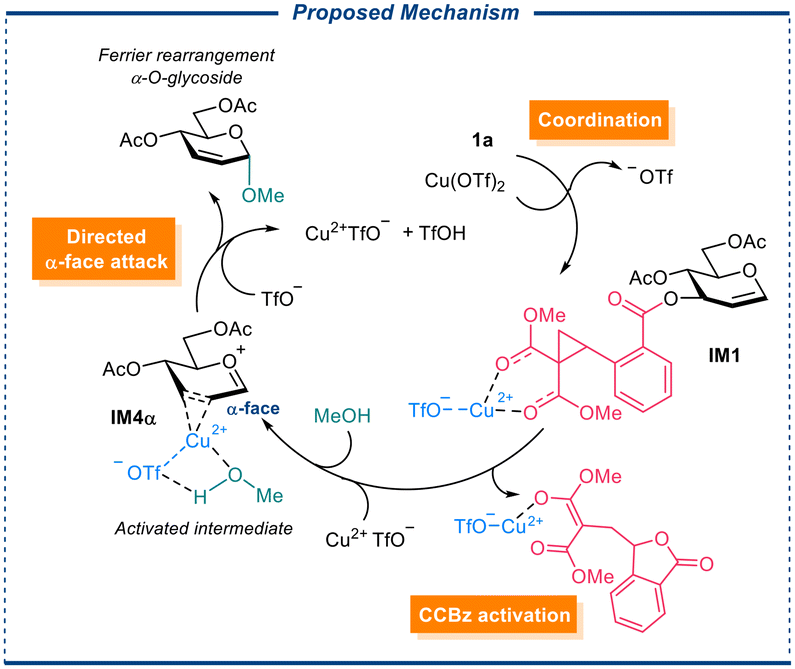 | ||
| Scheme 2 Proposed stepwise Ferrier rearrangement followed by directed α-face O-glycosylation (alcohol attack). | ||
Results indicate that in the most stable conformation of the model glucal 1a, the C3-position CCBz is positioned at the β-face. Coordination of the metal center to the carbonyl groups on CCBz affords IM1, with moderate stabilization of the system (ΔG = −5.5 kcal mol−1). Opening of the activated CCBz ring then occurs on IM1, and passing through TS1 (ΔG‡TS1 = +30.2 kcal mol−1) to give the C3-ring-opened intermediate IM2. Inspection of the structure of IM2 indicated that opening of the strained cyclopropane ring in IM1 did not directly lead to breakage of the C–O bond, and IM2 swiftly passes through a following transition state TS2 (ΔG‡TS2 = +3.3 kcal mol−1) to give the ion pair constituting the glycosyl oxocarbenium and the dissociated C3-position LG anion 4a, which remains coordinated to the Cu(OTf)2 and stay close to the glycosyl oxocarbenium at the β-face of the pyranose ring. We have also explored an alternative C3-activation route for CCBz ring opening and dissociation, starting from a stabler intermediate with the Cu(OTf)2 coordinated to both the CCBz carbonyls, C1 and C2 from the β-face, IM1coord, yet the reaction route from this IM1coord pass through transition states with higher energy barriers, probably due to internal strains (see ESI†), and is hence kinetically disfavored. Sc(OTf)3 the opening and dissociation of the C3-position CCBz following the same route with a similar free energy profile (see ESI†). Upon dissociation of the C3-substituent, the close contacting LG 4a may linger closely at the β-face of the pyranose ring and block the incoming nucleophiles, resulting in nucleophilic attack from exclusively α-face. Alternatively, if the LG 4a can freely move away from the glycosyl oxocarbenium, the calculated NBO charge of the atoms in the oxocarbenium indicated that C1 carries a significant positive charge whilst C2 and C3 do not (NBO charge C1 = +0.37, C2 = −0.38, C3 = +0.02), and a new incoming metal species bringing along with it the alcohol nucleophile (MeOH or p-OMePhOH) can presumably coordinate to C2 and C3 on the ring from either α- or β-face after the removal of LG 4a. DFT simulations of the following nucleophilic addition processes after glucal activation involving MeOH or p-OMePhOH acceptors produce similar results leading to the same conclusions, with the α-product from p-OMePhOH being more thermodynamically stable than the counterpart from MeOH (ΔG = +0.3 kcal mol−1 for β-glucoside from MeOH and +2.6 kcal mol−1 for β-glucoside from p-OMePhOH), which plausibly contribute to the enhanced α-selectivity of larger acceptors. (Structures from simulation employing MeOH are shown here for discussion, see ESI† for the optimized structures of intermediates and TSs along the reaction routes of p-OMePhOH). The optimized structure of IM4α and IM4β with copper(II) species coordinating from α- or β-face indicated that the metal center can indeed coordinate to C2 and C3 from α-face (Fig. 2c left), but not from β-face. Instead, in the β-face coordination scenario (IM4β), the copper(II) center drifts during geometry optimization and preferential coordinates to C3 and carbonyl oxygen at the 4-position of glucal, bringing the acceptor (MeOH or p-OMePhOH) away from the anomeric position, and disfavoring directed nucleophilic attack. Importantly, DFT simulations of putative intermediates of a scandium(III) species coordinating to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C π-ally system from either face resulted in optimized structures with the metal center dissociated from the CC system and precluded the formation of stable Sc(OTf)3 coordinated intermediates (see ESI†). Therefore, starting from the only stable copper(II) coordinated intermediate IM4α and passing through a transition state of directed nucleophilic attack TS4α (ΔG‡TS4α = +9.7 kcal mol−1) with moderate energy barrier gives rise to the α-glycosylation product intermediate IM5. It is important to note that in TS4α, the direction is not directly mediated by the coordination of MeOH to the α-face metal center, but rather the H-bond-like attractive non-covalent interaction between the α-face MeOH and metal bound triflate at the same face (Fig. 2c right). Therefore, DFT results suggest that the preferential α-face metal-triflate mediated direction results in the observed α-stereoselectivity in the glycosylation, which is absent when Sc(OTf)3 with a poorer capability to coordinate to the π-allyl-system. Finally, dissociation of the copper(II) center from IM5 produces the final α-glycoside.
C π-ally system from either face resulted in optimized structures with the metal center dissociated from the CC system and precluded the formation of stable Sc(OTf)3 coordinated intermediates (see ESI†). Therefore, starting from the only stable copper(II) coordinated intermediate IM4α and passing through a transition state of directed nucleophilic attack TS4α (ΔG‡TS4α = +9.7 kcal mol−1) with moderate energy barrier gives rise to the α-glycosylation product intermediate IM5. It is important to note that in TS4α, the direction is not directly mediated by the coordination of MeOH to the α-face metal center, but rather the H-bond-like attractive non-covalent interaction between the α-face MeOH and metal bound triflate at the same face (Fig. 2c right). Therefore, DFT results suggest that the preferential α-face metal-triflate mediated direction results in the observed α-stereoselectivity in the glycosylation, which is absent when Sc(OTf)3 with a poorer capability to coordinate to the π-allyl-system. Finally, dissociation of the copper(II) center from IM5 produces the final α-glycoside.
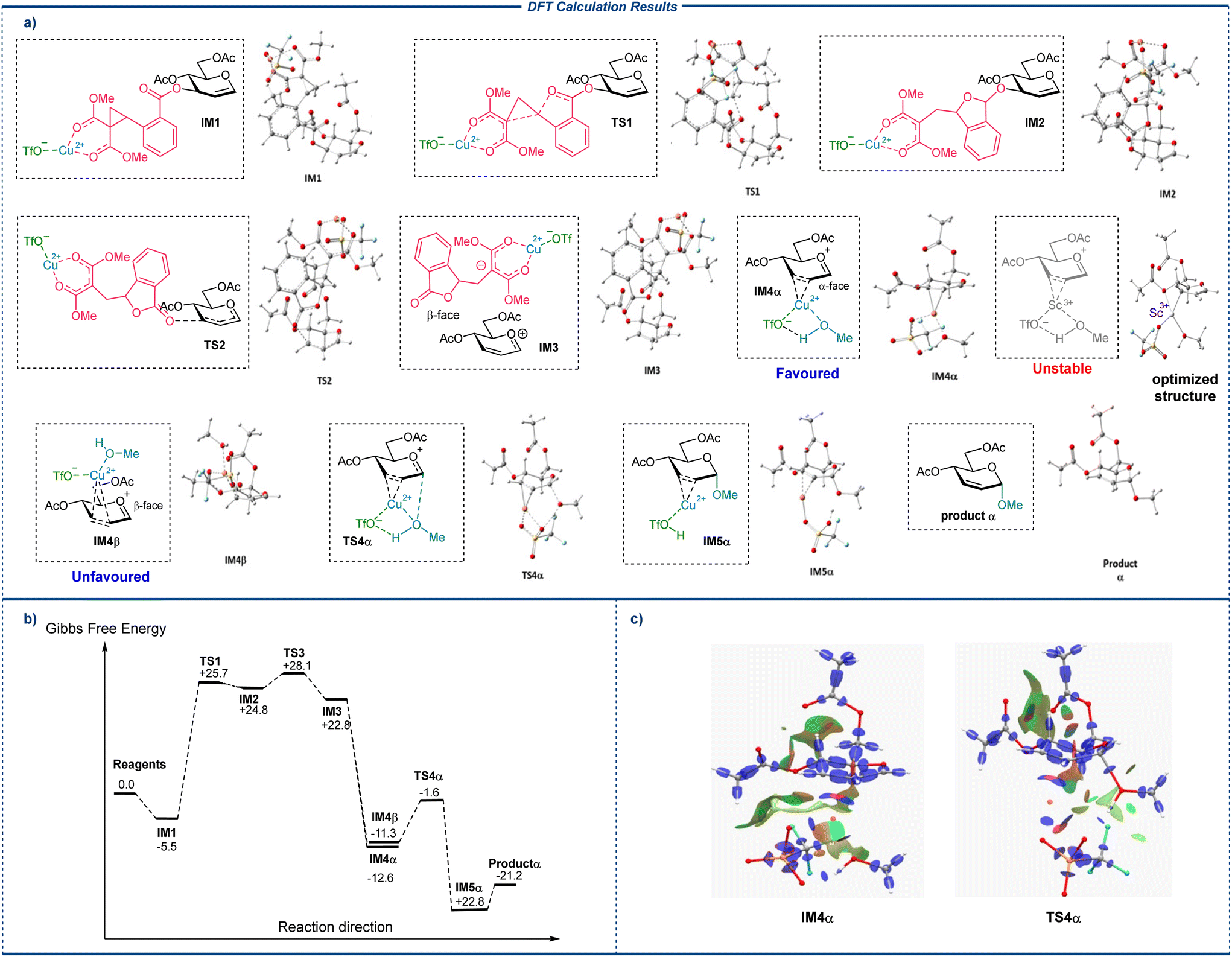 | ||
| Fig. 2 DFT simulation of the reaction paths. (a) Optimized structures of important intermediates (IMs) and transition states (TSs) along the main reaction route. (b) The free energy profile along the fastest-rate reaction route from the reagents 1a and MeOH to the α-glycoside product. The Gibbs free energies are reported in kcal mol−1, with the starting materials taken as reference (0.0). (c) Interaction region indicator (IRI) surfaces of IM4α (left) and TS4α (right).43 | ||
Conclusion
We have developed a novel Ferrier rearrangement of rationally designed glycal donors with C3-position CCBz, promoted by low-cost and environment-friendly Cu(OTf)2 or Fe(OTf)3 catalysts, enabling efficient synthesis of 2,3-unsaturated glycosides under mild conditions. The new rearrangement approach exhibits high α-stereoselectivity and applies to a wide scope of nucleophile acceptors, affording 2,3-unsaturated O-, S-, N-, and C-glycosides with excellent yields. We demonstrate the synthetic potential of our method through the facile construction of a library of natural products of pharmaceutical interest. DFT calculations were performed to elucidate the reaction mechanism, revealing that the high α-stereoselectivity arises from the preferential and directed nucleophilic attack from the α-face mediated by the H-bond interactions between the incoming acceptor and the metal-bound triflate anion, demonstrating the dual role of the catalyst along with the ability of bulky leaving group in directing the stereoselective glycosylation reactions.Author contributions
X.-W. L. and H. D. conceived the idea. H. Z. established the standard reaction condition. H. Z., H. D., Y. X., Y. Z., and D. Y. conducted the experiments. A. G. performed the DFT calculations. X.-W. L., H. Z., A. G., and H. D. drafted the manuscript. All authors commented on the manuscript. X.-W. L. supervised the whole project.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Nanyang Technological University and the Ministry of Education (MOE-T2EP30120-0007, Tier-1 RG107/23), and the National Research Foundation (NRF-CRP22-2019-0002) from Singapore for the financial support.References
- (a) N. E. Zachara and G. W. Hart, The Emerging Significance of O-GlcNAc in Cellular Regulation, Chem. Rev., 2002, 102, 431–438 CrossRef CAS PubMed; (b) R. A. Dwek, Glycobiology: Toward Understanding the Function of Sugars, Chem. Rev., 1996, 96, 683–720 CrossRef CAS PubMed.
- M. IzuMi, H. Miyazawa, T. Kamakura, I. Yamaguchi, T. Endo and F. Hanaoka, Blasticidin S-resistance Gene (bsr): A Novel Selectable Marker for Mammalian Cells, Exp. Cell Res., 1991, 197, 229–233 CrossRef CAS PubMed.
- K. Polkowski and A. P. Mazurek, Cytostatic and Cytotoxic Activity of Synthetic Genistein Glycosides Against Human Cancer Cell Lines, Cancer Lett., 2004, 203, 59–69 CrossRef CAS PubMed.
- J. Popiołkiewicz, K. Polkowski, J. S. Skierski and A. P. Mazurek, In Vitro Toxicity Evaluation in the Development of New Anticancer Drugs—Genistein Glycosides, Cancer Lett., 2005, 229, 67–75 CrossRef PubMed.
- C. Meier, T. Knispel, E. De Clercq and J. Balzarini, ADA-Bypass by Lipophilic CycloSal-ddAMP Pro-nucleotides A Second Example of the Efficiency of the CycloSal-Concept, Bioorg. Med. Chem. Lett., 1997, 7, 1577–1582 CrossRef CAS.
- S. J. Danishefsky and M. T. Bilodeau, Glycals in Organic Synthesis: the Evolution of Comprehensive Strategies for the Assembly of Oligosaccharides and Glycoconjugates of Biological Consequence, Angew. Chem., Int. Ed. Engl., 1996, 35, 1380–1419 CrossRef CAS.
- (a) P. Chen and S. Wang, CF3SO3H–SiO2 as Catalyst for Ferrier Rearrangement: An Efficient Procedure for the Synthesis of Pseudoglycosides, Tetrahedron, 2013, 69, 583–588 CrossRef CAS; (b) G. Narasimha, B. Srinivas, P. R. Krishna and S. Kashyap, Zn(OTf)2-c atalyzed Glycosylation of Glycals: Synthesis of 2,3-Unsaturated Glycosides via a Ferrier Reaction, Synlett, 2014, 523–526 CAS; (c) P. Chen and S. Li, Y(OTf)3 a s A Highly Efficient Catalyst in Ferrier Rearrangement for the Synthesis of O-and S-2,3-Unsaturated Glycopyranosides, Tetrahedron Lett., 2014, 55, 5813–5816 CrossRef CAS; (d) P. Chen and S. Wang, Iron(III) triflate, a New Efficient Catalyst for Type I Ferrier Rearrangement, Tetrahedron, 2012, 68, 5356–5262 CrossRef CAS; (e) B. G. Williams, S. B. Simelane and H. H. Kinfe, Aluminium Triflate Catalysed O-glycosidation: Temperature-switched Selective Ferrier rearrangement or Direct Addition with Alcohols, Org. Biomol. Chem., 2012, 10, 5636–5642 RSC.
- (a) A. M. Gómez, F. Lobo, C. Uriel and J. C. López, Recent Developments in the Ferrier Rearrangement, Eur. J. Org. Chem., 2013, 7221–7262 CrossRef; (b) N. Jiang, Z. Wu, Y. Dong, X. Xu, X. Liu and J. Zhang, Progress in the Synthesis of 2,3-unsaturated Glycosides, Curr. Org. Chem., 2020, 24, 184–199 CrossRef CAS; (c) C. S. Bennett and M. C. Galan, Methods for 2-Deoxyglycoside Synthesis, Chem. Rev., 2018, 118, 7931–7985 CrossRef CAS PubMed; (d) A. M. Gómez, F. Lobo, S. Miranda and J. C. López, A Survey of Recent Synthetic Applications of 2,3-Dideoxy-Hex-2-enopyranosides, Molecules, 2015, 20, 8357–8394 CrossRef PubMed.
- M. J. McKay and H. M. Nguyen, Recent Advances in Transition Metal-Catalyzed Glycosylation, ACS Catal., 2012, 2, 1563–1595 Search PubMed.
- E. B. Bauer, Transition Metal Catalyzed Glycosylation Reactions—an Overview, Org. Biomol. Chem., 2020, 18, 9160–9180 RSC.
- E. Rafiee, S. Tangestaninejad, M. H. Habibi and V. Mirkhani, A Mild, Efficient and α-Selective Glycosidation by Using Potassium Dodecatungstocobaltate Trihydrate as Catalyst, Bioorg. Med. Chem. Lett., 2004, 14, 3611–3614 CrossRef CAS PubMed.
- (a) K. Toshima, T. Ishizuka, G. Matuo, M. Nakata and M. Kinoshita, Glycosidation of Glycals by 2,3-Dichloro-5,6-dicyano-p-benzoquinone (DDQ) as A Catalytic Promoter, J. Chem. Soc., Chem. Commun., 1993, 704–706 RSC; (b) A. A. Ansari, Y. S. Reddy and Y. D. Vankar, Efficient Carbon-Ferrier Rearrangement on Glycals Mediated by Ceric Ammonium Nitrate: Application to the Synthesis of 2-Deoxy-2-amino-C-glycoside, Beilstein J. Org. Chem., 2014, 10, 300–306 CrossRef PubMed.
- (a) N. Bhuma, L. Lebedel, H. Yamashita, Y. Shimizu, Z. Abada, A. Ardá, J. Désiré, B. Michelet, A. Martin-Mingot, A. Abou-Hassan, M. Takumi, J. Marrot, J. Jiménez Barbero, A. Nagaki, Y. Blériot and S. Thibaudeau, Insight into the Ferrier Rearrangement by Combining Flash Chemistry and Superacids, Angew. Chem., Int. Ed., 2021, 60, 2036–2041 ( Angew. Chem. , 2021 , 133 , 2064–2069 ) CrossRef CAS PubMed; (b) M. Takumi, H. Sakaue and A. Nagaki, Flash Electrochemical Approach to Carbocations, Angew. Chem., Int. Ed., 2022, 61, e202116177 ( Angew. Chem. , 2022 , 134 , e2021161 ) CrossRef CAS PubMed.
- R. J. Ferrier and G. H. Sankey, Unsaturated Carbohydrates. Part VII. The Preference Shown by Allylic Ester Groupings on Pyranoid Rings for the Quasi-axial Orientation, J. Chem. Soc. C, 1966, 2345–2349 RSC.
- B. Liberek, D. Tuwalska, A. Konitz and A. Sikorski, X-ray Diffraction and High-resolution NMR Spectroscopy of Methyl 3,4-di-O-acetyl-1,5-anhydro-2-deoxy-D-arabino-hex-1-enopyranuronate, Carbohydr. Res., 2007, 342, 1280–1284 CrossRef CAS PubMed.
- W.-L. Leng, H. Yao, J.-X. He and X.-W. Liu, Venturing Beyond Donor-controlled Glycosylation: New Perspectives Toward Anomeric Selectivity, Acc. Chem. Res., 2018, 51, 628–639 CrossRef CAS PubMed.
- I. V. Alabugin, L. Kuhn, M. G. Medvedev, N. V. Krivoshchapov, V. A. Vil’, I. A. Yaremenko, P. Mehaffy, M. Yarie, A. O. Terent'ev and M. A. Zolfigol, Stereoelectronic Power of Oxygen in Control of Chemical Reactivity: the Anomeric Effect is Not Alone, Chem. Soc. Rev., 2021, 50, 10253–10345 RSC.
- For our group's recent reports on Ferrier-type glycosylation methodologies, see: (a) S. Xiang, Z. Lu, J. He, K. Le Mai Hoang, J. Zeng and X.-W. Liu, β-Type Glycosidic Bond Formation by Palladium-Catalyzed Decarboxylative Allylation, Chem. Eur. J., 2013, 19, 14047–14051 Search PubMed; (b) J. Zeng, J. Ma, S. Xiang, S. Cai and X.-W. Liu, Stereoselective β-C-Glycosylation by a Palladium-Catalyzed Decarboxylative Allylation: Formal Synthesis of Aspergillide A, Angew. Chem., Int. Ed., 2013, 52, 5134–5137 CrossRef CAS PubMed; (c) F. Ding, R. William and X.-W. Liu, Ferrier-Type N-Glycosylation: Synthesis of N-Glycosides of Enone Sugars, J. Org. Chem., 2013, 78, 1293–1299 Search PubMed; (d) S. Xiang, K. Le Mai Hoang, J. He, Y. J. Tan and X.-W. Liu, Reversing the Stereoselectivity of a Palladium-Catalyzed O-Glycosylation through an Inner-Sphere or Outer-Sphere Pathway, Angew. Chem., Int. Ed., 2015, 54, 604–607 ( Angew. Chem. , 2015 , 127 , 614–661 ) CrossRef CAS PubMed; (e) L. Ji, S.-H. Xiang, W.-L. Leng, K. Le Mai Hoang and X.-W. Liu, Palladium-Catalyzed Glycosylation: Novel Synthetic Approach to Diverse N-Heterocyclic Glycosides, Org. Lett., 2015, 17, 1357–1360 CrossRef CAS PubMed; (f) H. Yao, S. Zhang, W.-L. Leng, M.-L. Leow, S. Xiang, J. He, H. Liao, K. Le Mai Hoang and X.-W. Liu, Catalyst-Controlled Stereoselective O-Glycosylation: Pd(0) vs Pd(II), ACS Catal., 2017, 7, 5456–5460 CrossRef CAS; (g) N. Huang, H. Liao, H. Yao, T. Xie, S. Zhang, K. Zou and X.-W. Liu, Diastereoselective Synthesis of C-Vinyl Glycosides via Gold(I)-Catalyzed Tandem 1,3-Acyloxy Migration/Ferrier Rearrangement, Org. Lett., 2018, 20, 16–19 CrossRef CAS PubMed; (h) K. B. Pal, J. Lee, M. Das and X.-W. Liu, Palladium(II)-catalyzed Stereoselective Synthesis of C-glycosides from Glycals with Diaryliodonium Salts, Org. Biomol. Chem., 2020, 18, 2242–2251 RSC; (i) K. B. Pal, A. Guo, M. Das, J. Lee, G. Báti, B. R. P. Yip, T.-P. Loh and X.-W. Liu, Iridium-promoted Deoxyglycoside Synthesis: Stereoselectivity and Mechanistic Insight, Chem. Sci., 2021, 12, 2209–2216 RSC.
- H. Ding, J. Lyu, X.-L. Zhang, X. Xiao and X.-W. Liu, Efficient and Versatile Formation of Glycosidic Bonds via Catalytic Strain-release Glycosylation with Glycosyl Ortho−2,2-dimethoxycarbonylcyclopropylbenzoate Donors, Nat. Commun., 2023, 14, 4010 CrossRef CAS PubMed.
- X. Xiao, H. Ding, L.-C. Peng, X.-Y. Fang, Y.-Y. Qin, Q.-Q. Mu and X.-W. Liu, Sweet Strain Release: Donor–Acceptor Cyclopropane Mediated Glycosylation, CCS Chem., 2023, 5, 2910–2921 CrossRef CAS.
- V. Pirenne, B. Muriel and J. Waser, Catalytic Enantioselective Ring-Opening Reactions of Cyclopropanes, Chem. Rev., 2021, 121, 227–263 CrossRef CAS PubMed.
- G. Lian, X. Zhang and B. Yu, Thioglycosides in Carbohydrate Research, Carbohydr. Res., 2015, 403, 13–22 CrossRef CAS PubMed.
- H. Herzner, T. Reipen, M. Schultz and H. Kunz, Synthesis of Glycopeptides Containing Carbohydrate and Peptide Recognition Motifs, Chem. Rev., 2000, 100, 4495–4538 CrossRef CAS PubMed.
- R. Sangwan, A. Khanam and P. K. Mandal, An Overview on the Chemical N-Functionalization of Sugars and Formation of N-Glycosides, Eur. J. Org. Chem., 2020, 2020, 5949–5977 CrossRef CAS.
- H. Fukuo, T. Suzuki, J. Shimabukuro, N. Komura, H.-N. Tanaka, A. Imamura, H. Ishida and H. Ando, Synthesis of Diverse Seleno-Glycolipids via the Transacetalization of Selenoacetals, Eur. J. Org. Chem., 2021, 5455–5467 CrossRef CAS.
- (a) J. Giljanović and A. Prkić, Determination of Coenzyme A (CoASH) in the Presence of Different Thiols by Using Flow-Injection with a UV/Vis Spectrophotometric Detector and Potentiometric Determination of CoASH Using an Iodide ISE, Molecules, 2010, 15, 100–113 CrossRef PubMed; (b) O. C. M. Sibon and E. Strauss, Coenzyme A: to Make it or Uptake it?, Nat. Rev. Mol. Cell Biol., 2016, 17, 605–606 CrossRef CAS PubMed.
- (a) S. Dharuman, P. Gupta, P. K. Kancharla and Y. D. Vankar, Synthesis of 2-Nitroglycals from Glycals Using the Tetrabutylammonium Nitrate–Trifluoroacetic Anhydride–Triethylamine Reagent System and Base-Catalyzed Ferrier Rearrangement of Acetylated 2-Nitroglycals, J. Org. Chem., 2013, 78, 8442–8450 CrossRef CAS PubMed; (b) L. V. Dunkerton, N. K. Adair, J. M. Euske, K. T. Brady and P. D. Robinson, Regioselective Synthesis of Substituted 1-Thiohex-2-enopyranosides, J. Org. Chem., 1988, 53, 845–850 CrossRef CAS; (c) P. Chen, S. Guo, J. Zuo, R. Chu, X. He and G. Zhu, Synthesis of 3-S- and 3-Se-glycals by Using R-S-S-R and R-Se-Se-R as the Nucleophile Precursors Promoted by Hf(OTf)4 and the Temperature-dependent Formation of the Above-mentioned 3-S- and 3-Se Products, Tetrahedron Lett., 2020, 61, 151648 CrossRef CAS.
- S. Danishefsky and J. F. Kerwin Jr., On the Addition of Allyltrimethylsilane to Glycal Acetates, J. Org. Chem., 1982, 47, 3803–3805 CrossRef CAS.
- V. Denavit, D. Lainé, J. St-Gelais, P. A. Johnson and D. Giguère, A Chiron Approach Towards the Stereoselective Synthesis of Polyfluorinated Carbohydrates, Nat. Commun., 2018, 9, 4721 CrossRef PubMed.
- R. Hevey, The Role of Fluorine in Glycomimetic Drug Design, Chem. – Eur. J., 2021, 27, 2240–2253 CrossRef CAS PubMed.
- B. Linclau, A. Ardá, N.-C. Reichardt, M. Sollogoub, L. Unione, S. P. Vincent and J. Jiménez-Barbero, Fluorinated Carbohydrates as Chemical Probes for Molecular Recognition Studies. Current Status and Perspectives, Chem. Soc. Rev., 2020, 49, 3863–3888 RSC.
- L. Wu, Z. Zhou, D. Sathe, J. Zhou, S. Dym, Z. Zhao, J. Wang and J. Niu, Precision Native Polysaccharides from Living Polymerization of Anhydrosugars, Nat. Chem., 2023, 15, 1276–1284 CrossRef CAS PubMed.
- S.-C. Hung, S. R. Thopate, F.-C. Chi, S.-W. Chang, J.-C. Lee, C.-C. Wang and Y.-S. Wen, 1,6-Anhydro-β-L-hexopyranoses as Potent Synthons in the Synthesis of the Disaccharide Units of Bleomycin A2 and Heparin, J. Am. Chem. Soc., 2001, 123, 3153–3154 CrossRef CAS PubMed.
- W. Meng, B. A. Ellsworth, A. A. Nirschl, P. J. McCann, M. Patel, R. N. Girotra, G. Wu, P. M. Sher, E. P. Morrison, S. A. Biller, R. Zahler, P. P. Deshpande, A. Pullockaran, D. L. Hagan, N. Morgan, J. R. Taylor, M. T. Obermeier, W. G. Humphreys, A. Khanna, L. Discenza, J. G. Robertson, A. Wang, S. Han, J. R. Wetterau, E. B. Janovitz, O. P. Flint, J. M. Whaley and W. N. Washburn, Discovery of Dapagliflozin: A Potent, Selective Renal Sodium-Dependent Glucose Cotransporter 2 (SGLT2) Inhibitor for the Treatment of Type 2 Diabetes, J. Med. Chem., 2008, 51, 1145–1114 CrossRef CAS PubMed.
- C. Tohda, T. Urano, M. Umezaki, I. Nemere and T. Kuboyama, Diosgenin is an Exogenous Activator of 1,25D3-MARRS/Pdia3/ERp57 and Improves Alzheimer's Disease Pathologies in 5XFAD Mice, Sci. Rep., 2012, 2, 535 CrossRef PubMed.
- A. D. Becke, Density-Functional Thermochemistry., III. The Role of Exact Exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of The Damping Function in Dispersion Corrected Density Functional Theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- H. S. Yu, X. He and D. G. Truhlar, MN15-L: A New Local Exchange-Correlation Functional for Kohn–Sham Density Functional Theory with Broad Accuracy for Atoms, Molecules, and Solids, J. Chem. Theory Comput., 2016, 12, 1280–1293 CrossRef CAS PubMed.
- R. Weigend and F. Ahlrichs, Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- J. Zheng, X. Xu and D. G. Truhlar, Minimally Augmented Karlsruhe Basis Sets, Theor. Chem. Acc., 2011, 128, 295–305 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- T. Lu and Q. Chen, Interaction Region Indicator: A Simple Real Space Function Clearly Revealing Both Chemical Bonds and Weak Interactions, Chem.: Methods, 2021, 1, 231–239 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qo00410h |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2024 |