
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Light-mediated catalytic enantioselective difluoroalkylation of β-ketoesters via phase-transfer catalysis†
Callum E.
Adams
and
Craig P.
Johnston
 *
*
EaStCHEM, School of Chemistry, University of St Andrews, St Andrews, Fife KY16 9ST, UK. E-mail: cpj3@st-andrews.ac.uk
First published on 9th May 2024
Abstract
We report a phase-transfer-catalysed asymmetric α-difluoroalkylation of β-ketoesters through visible light-promoted radical generation. This methodology affords quaternary stereocentres with mild enantioenrichment and isolated yields up to 99%. Through mechanistic investigations, we have determined that the reaction proceeds via difluoroalkyl radical generation arising from an in situ electron donor–acceptor complex capable of absorbing visible light. Extensive optimisation of the reaction parameters was carried in an attempt to overcome a facile racemic background reaction.
1. Introduction
Owing to their growing role in materials science, agrochemicals, and pharmaceuticals the introduction of difluoroalkyl groups has become an area of particular interest.1–3 This is due to the well-established improvements in metabolic stability, lipophilicity, and bioavailability gained by incorporating fluorine-containing functional groups into organic molecules.4–6 To install difluoroalkyl moieties enantioselectively typically requires the use of transition metals either as photocatalysts or to provide a chiral environment with an enantiopure ligand.7–13 In the context of difluoroalkyl radicals, their enantioselective incorporation into a molecule has been limited to two examples, but both require the use of transition metals. First, Lu and Xiao harnessed a chiral nickel Lewis acid catalyst with an iridium photocatalyst for the asymmetric difluoroalkylation of β-ketoesters (Fig. 1A).12 Later, Xu synthesised enantioenriched gem-difluoroalkyl containing γ-keto amides using a chiral-at-rhodium(III) complex and an Ir photocatalyst (Fig. 1B).10 Therefore, due to the lack of enantioselective protocols for introducing difluoroalkyl groups and the current constraint of using unsustainable, toxic, and expensive transition metal catalysts we sought to explore an alternative strategy. Primarily we were intrigued by light-initiated methods that remove the need for exogenous photocatalysts. For example, a system developed by Melchiorre and co-workers realised the α-trifluoromethylation and perfluoroalkylation of β-ketoesters in the absence of transition metals (Fig. 1C).14 This transformation was rendered asymmetric through the use of an organocatalytic phase-transfer catalyst (PTC).15–21 Aside from providing a chiral environment the PTC also facilitates the formation of an electron donor–acceptor (EDA) complex by solubilising the enolate of the β-ketoester in a non-polar solvent so that it can interact with the electron-deficient perfluoroalkyl iodide. Formation of EDA species can manifest pronounced characteristics and they can be identified through a number of different methods.22–28 Importantly, an EDA complex is capable of absorbing visible light, and undergoing a subsequent single electron transfer process, to afford a desirable radical intermediate derived from the electron acceptor. We sought to build on this observation by exploring a range of ketones and β-ketoesters to stereoselectively incorporate the difluoroalkyl moiety under sustainable, metal-free conditions. Herein, we report our endeavour to harness organocatalytic phase-transfer conditions with the generation of tertiary radicals resulting from an EDA complex, to afford enantioenriched α-difluoroalkylation of β-ketoesters (Fig. 1D).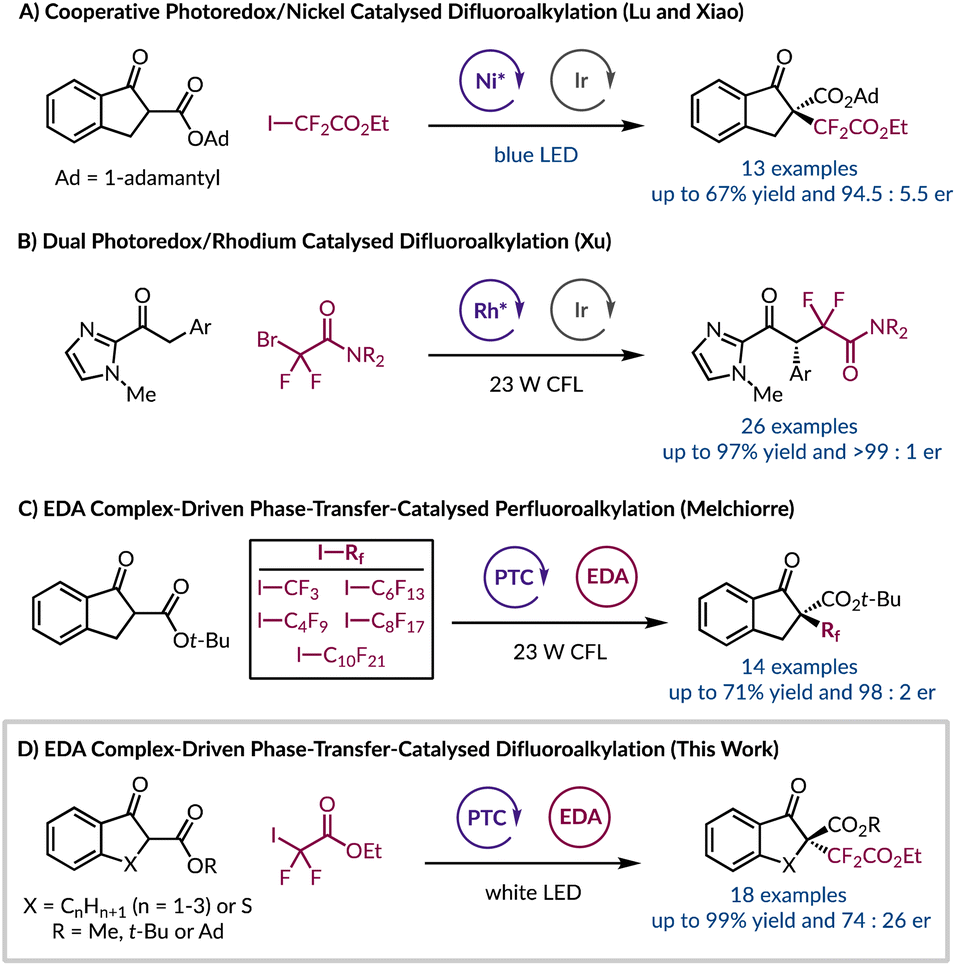 | ||
| Fig. 1 (A and B) Previous asymmetric difluoroalkylation reactions; (C) enantioselective perfluoroalkylation using a chiral phase-transfer catalyst; (D) organocatalytic enantioselective difluoroalkylation of β-ketoesters. | ||
2. Results and discussion
Initial reactivity and optimisation
For the initial investigations, β-ketoester 1a was selected as the model substrate for difluoroalkylation under asymmetric phase-transfer-catalysed conditions, as it has been shown to be applicable to both EDA complex formation and phase-transfer catalysis.14–16 Likewise, ester 2a is commercially available and when subjected to a variety of photochemical conditions is known to give the required difluoroalkyl radical.29–37 An initial result gave the desired product 3a in 10% yield under previously optimised reaction conditions (Table 1, entry 1),14 albeit with very poor asymmetric induction. Using this catalytic system an extensive solvent screen was undertaken (see ESI page S15†), and four solvents (CH2Cl2, CHCl3, t-BuOAc, and t-BuPh) showed a marginal improvement in enantioselectivity, as well as improved yields of product 3a (Table 1, entries 2–5). Initially, we thought the removal of catalyst C1 would impede the transformation, but we were surprised to find that a significant racemic background reaction occurs in both t-BuOAc and t-BuPh (Table 1, entries 6 and 7). At this stage, we hypothesised that the cesium cation was sufficiently solubilising to promote enolate formation and due to the less prominent background reactivity observed in t-BuOAc, optimisation continued using it as the solvent. Subsequently, a base screen was carried out in an attempt to minimise the uncatalysed pathway (see ESI page S16†). In general, alternative bases only resulted in lower yields of product 3a with reduced levels of enantioenrichment (Table 1, entries 8 and 9). During this phase of the optimisation process, a series of control reactions were undertaken. When the reaction was conducted in ambient light or in the dark no conversion of the starting materials was observed (Table 1, entries 10 and 11), highlighting that the process is initiated by LED light irradiation and ruling out a potential SN2 pathway between the in situ generated enolate and the difluoroalkyl halide 2a. Further experiments supported the involvement of radical intermediates as the presence of TEMPO (3.0 equiv.) completely inhibited the formation of product 3a with the TEMPO adduct of 2a being formed instead (see ESI for further information†).|
|
||||
|---|---|---|---|---|
| Entry | Deviationa | Solvent |
3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b [%] b [%] |
erc |
|
a Unless otherwise stated optimization reactions run on a 0.10 mmol scale of 1a, 2a (2.0 equiv.), C1 (20 mol%) and Cs2CO3 (2.0 equiv.) in stated solvent for 16 h at RT, irradiated with white LED (6200 K).
b Yield determined by 1H NMR analysis of the crude reaction with 1,3,5-trimethoxybenzene (0.33 equiv.).
c er [(S):(R)] determined by chiral HPLC.
d Reaction performed for 64 h. See ESI for further details.†
|
||||
| 1d | — | PhCl:C8F18 (2:1, 0.2 M) |
10 | 57:43 |
| 2 | — | CH2Cl2 (0.1 M) | 16 | 64:36 |
| 3 | — | CHCl3 (0.1 M) | 95 | 68:32 |
| 4 | — | t-BuOAc (0.1 M) | 88 | 65:35 |
| 5 | — | t-BuPh (0.1 M) | 64 | 69:31 |
| 6 | No C1 | t-BuOAc (0.1 M) | 52 | N/A |
| 7 | No C1 | t-BuPh (0.1 M) | 76 | N/A |
| 8 | K2CO3, instead of Cs2CO3 | t-BuOAc (0.1 M) | 81 | 52:48 |
| 9 | CsOAc, instead of Cs2CO3 | t-BuOAc (0.1 M) | 70 | 62:48 |
| 10 | Ambient light, instead of white LED | t-BuOAc (0.1 M) | 0 | N/A |
| 11 | Dark (vial wrapped in aluminium foil) | t-BuOAc (0.1 M) | 0 | N/A |
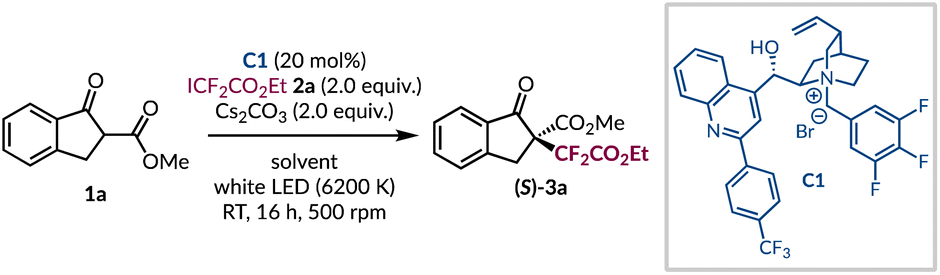
Phase-transfer catalyst evaluation
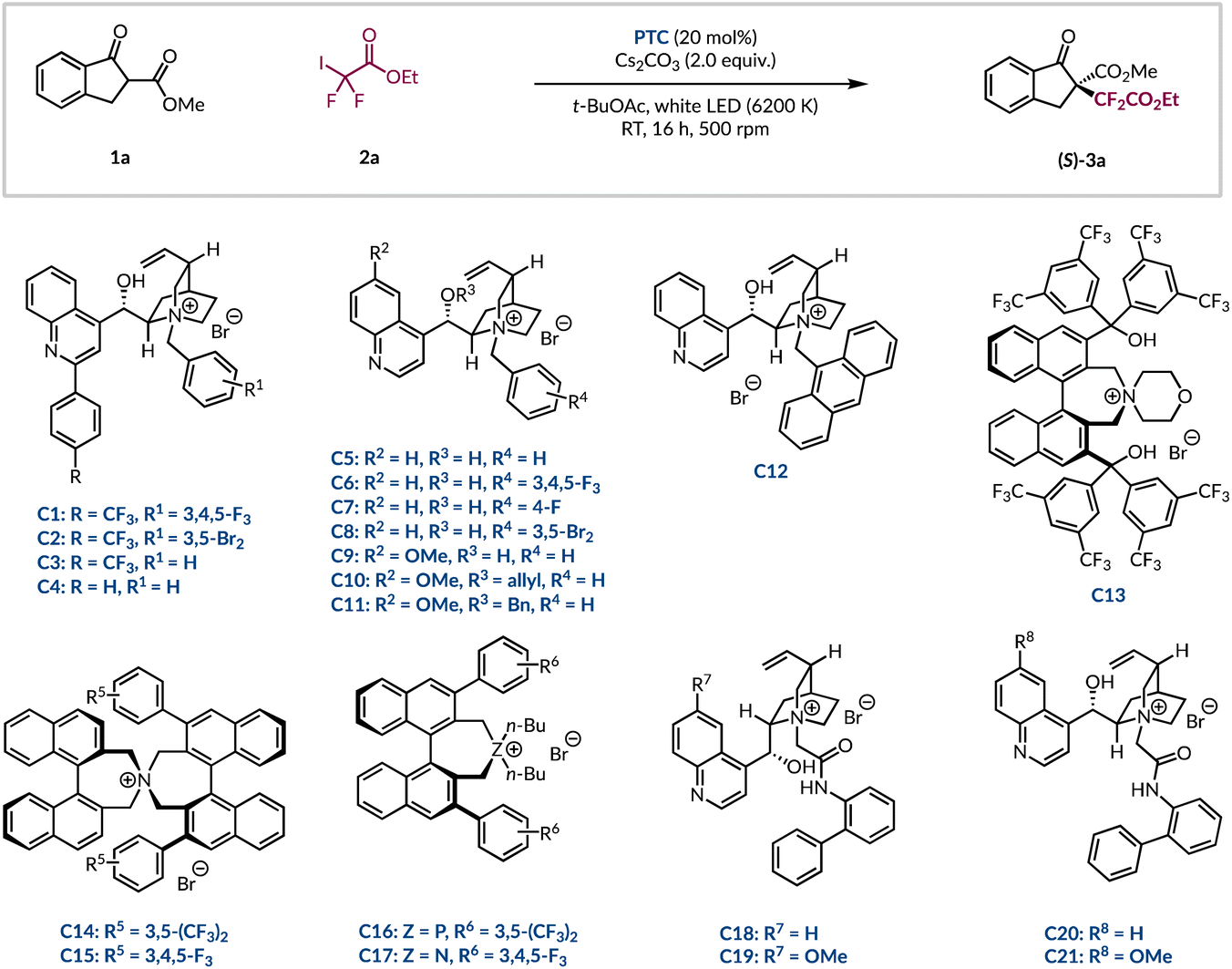
Subsequently, we investigated a range of alternative phase-transfer catalysts to determine if any would be able to outcompete the background reaction and improve the enantioselectivity of the difluoroalkylation process (Table 2). First, a series of 2-aryl quinoline substituted cinchonine catalysts (C1–4) were investigated. Replacing the 3,4,5-trifluorinated benzyl group with a 3,5-dibrominated analogue led to a noticeable drop in er (Table 2, entries 1 and 2).38,39 Related catalysts (C3–4), which contain a simple benzyl group, showed that the p-CF3 group on the other aryl ring was important as only racemic product (3a) was formed without it (Table 2, entries 3 and 4). Further investigations revealed that complete removal of the 2-aryl substituent on the quinoline core (C5–C11) was also detrimental as no enantioinduction was observed (Table 2, entries 5–11). Notably, this included catalyst C6, which has the same fluorinated benzyl group as C1. Even the introduction of the sterically cumbersome anthracenyl group in PTC C12 did not restore the stereoselectivity (Table 2, entry 12). At this stage, it was decided to investigate phase-transfer catalysts with greater structural diversity. Having been implemented for the asymmetric functionalisation of β-ketoesters, as well as a vast range of other carbonyl-containing compounds, the Maruoka-type structures (C13–17) were considered to be ideal candidates to realise the desired transformation with high stereocontrol.15,16,40–43 Unfortunately, from this selection of BINOL-derived catalysts no improvement over the N-benzylated cinchona alkaloid derivatives was found and only racemic 3a was generated (Table 2, entries 13–17). In 2017, Li and co-workers performed a DFT study that highlighted the extent to which a core network of H-bonding interactions dictated the high levels of asymmetric induction in the analogous perfluoroalkylation reaction.44 Inspired by this, it was decided to investigate more recently developed cinchona-based PTCs bearing arylated amides in lieu of benzyl groups from Jurczak.45 These organocatalysts have enjoyed broad applications with a range of substrates, including with β-ketoesters.46–48 The biphenyl amide was appended to the four common cinchona alkaloids to give catalysts C18–21. These all eclipsed PTC C1 giving the highest yields of enantioenriched product 3a seen thus far (Table 2, entries 1 vs. 18–21). In general, it was found that the amide motif, which provides additional hydrogen bonding capabilities, was superior to its direct benzylated analogues (e.g.C20vs.C5–8). The cinchonidine scaffold of PTC C18 marginally outperformed the other alkaloid architectures (C19–21) to give difluoroalkylated β-ketoester 3a in quantitative yield and 76:24 er (Table 2, entry 18). Further optimisation of the reaction parameters with PTC C18 was carried out, but any deviation from the standard conditions had a negative impact on both yield and stereocontrol (see ESI page S21†). As an exhaustive optimization process had been carried out, it was deemed appropriate to initiate an investigation into the scope of this methodology.
|
|
|||
|---|---|---|---|
| Entrya | Catalyst |
3ab [%] |
erc |
|
a Unless otherwise stated optimization reactions a 0.10 mmol scale of 1a, 2a (2.0 equiv.), PTC (20 mol%) and Cs2CO3 (2.0 equiv.) in t-BuOAc (0.1 M) for 16 h at RT, irradiated with white LED 6200 K.
b Yield determined by 1H NMR analysis of the crude reaction with 1,3,5-trimethoxybenzene (0.33 equiv.).
c er [(S):(R)] determined by chiral HPLC.
|
|||
| 1 | C1 | 88 | 65:35 |
| 2 | C2 | 82 | 54:46 |
| 3 | C3 | 69 | 63:37 |
| 4 | C4 | 66 | 50:50 |
| 5 | C5 | 75 | 50:50 |
| 6 | C6 | 83 | 51:49 |
| 7 | C7 | 79 | 51:49 |
| 8 | C8 | 71 | 50:50 |
| 9 | C9 | 67 | 51:49 |
| 10 | C10 | 37 | 50:50 |
| 11 | C11 | 55 | 50:50 |
| 12 | C12 | 22 | 50:50 |
| 13 | C13 | 73 | 50:50 |
| 14 | C14 | 43 | 50:50 |
| 15 | C15 | 72 | 50:50 |
| 16 | C16 | 55 | 50:50 |
| 17 | C17 | 61 | 50:50 |
| 18 | C18 | >99 | 26:74 |
| 19 | C19 | 92 | 27:73 |
| 20 | C20 | 78 | 68:32 |
| 21 | C21 | 81 | 72:28 |
Substrate scope
With the optimised conditions in hand, a variety of β-ketoesters with differing electronic and steric demands, as well as diverse aryl substitution patterns was explored (Scheme 1A). Interestingly, a clear trend did not emerge that would enable us to holistically rationalise how these changes impacted the yield and enantioselectivity of the reaction. Introducing a 5-fluoro substituent on the indanone ring had a minimal effect as the expected product 3b was isolated in 92% yield with 72:28 er. A chlorine atom was equally tolerated at this position (3c), but the larger brominated analogue (3d) did suffer from a drop in yield and er. An electron-rich methoxy group in the same site (3e) led to a further decline in yield but with moderate levels of asymmetric induction. Relocating the OMe substituent to the 4-position provided product 3f in good yield, although with 55:45 er. Replacing the 4-substituent with either a bromine atom (3g) or a methyl group (3h) restored the standard level of enantioenrichment. The yield and enantioselectivity were negatively influenced by the presence of an electron-deficient 4-trifluoromethyl group (3i). The bromine atom was best tolerated at the 4-site (3g) as lower yields and enantiomeric ratios were obtained at either the 5- or 6-position (3d & 3j). A similar slight detrimental effect was observed with the 6-methyl product 3k. 5,6-Disubstituted β-ketoesters were also evaluated with 5,6-diMeO (3l) and 5-Cl-6-MeO (3m) both giving similar enantioselectivities to those observed for 6-monosubstituted analogues. For product 3l comparable results were obtained on a 0.5 mmol scale. The introduction of a sulfur atom at the 3-position of the indanone motif afforded product 3n in 63% yield with 65:35 er, which when compared to the sulfur-free analogue 3a shows a lower asymmetric induction.
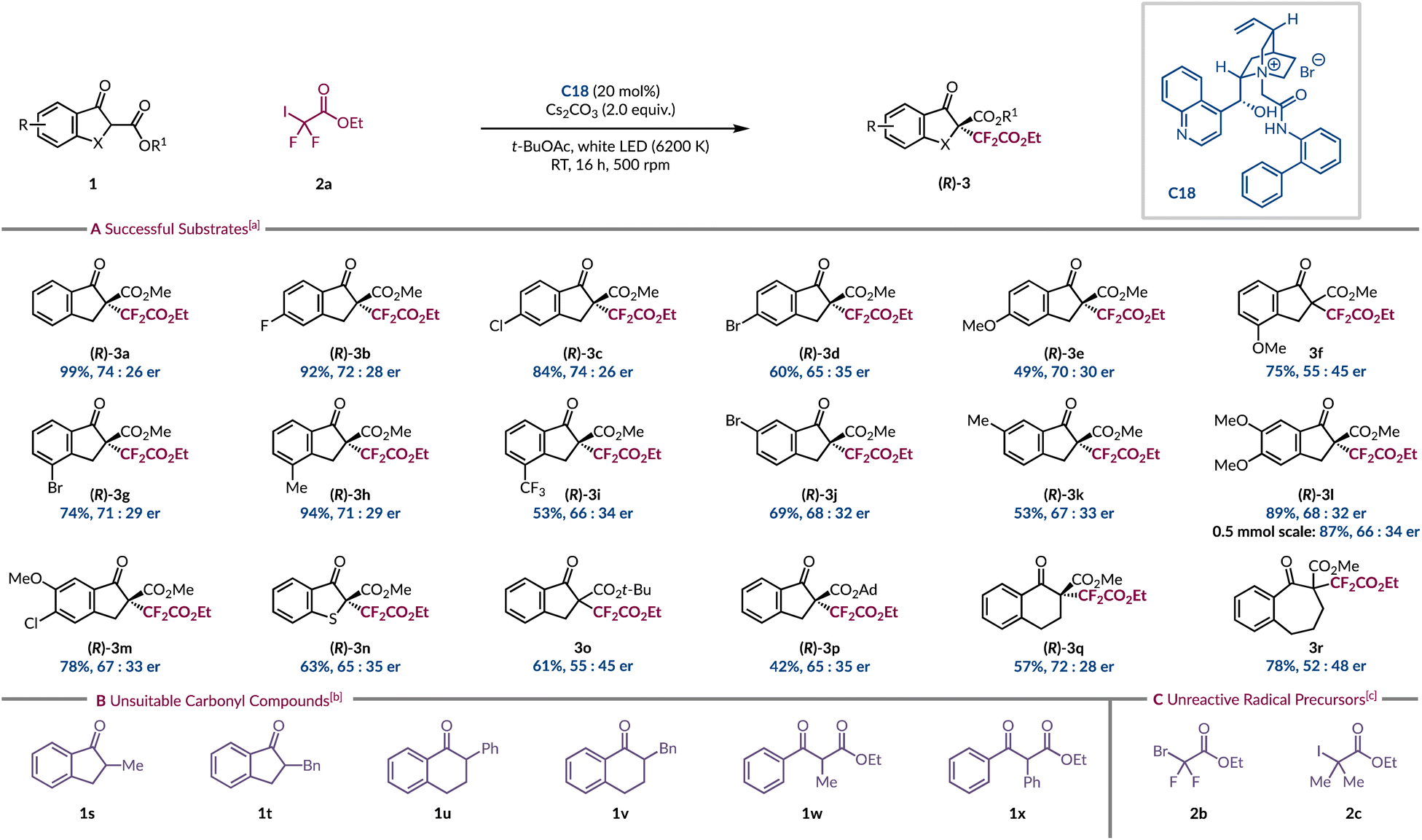 | ||
| Scheme 1 (A) Substrate scope for the light-initiated phase-transfer-catalysed asymmetric α-difluoroalkylation of β-ketoesters; (B) carbonyl compounds that were tested but unsuccessful; (C) alternative radical precursors that were unreactive. aUnless otherwise stated reactions were run on a 0.10 mmol scale of 1a, 2a (2.0 equiv.), C18 (20 mol%) and Cs2CO3 (2.0 equiv.) in t-BuOAc (0.1 M) for 16 h at RT, irradiated with white LED (6200 K), isolated yields are stated, er determined by chiral HPLC. Racemic reactions were ran by replacing C18 with Bu4NBr (20 mol%) bUnsuccessful cyclic and acyclic carbonyl substrates under both racemic and asymmetric protocols (see ESI for full details†). cRadical precursors that were unreactive under both racemic and asymmetric protocols with a variety of carbonyl substrates (see ESI for full details†). | ||
Previous studies have recognised that the steric bulk of the ester of β-ketoester 1a can result in excellent levels of stereocontrol during product formation when intercepting fluorinated carbon-centred radicals.12,14 To our surprise this strategy failed to yield the same benefit in our system as substrate 1o, bearing a t-butyl ester, led to a near racemic product (3o). Given this was the standard substrate for Melchiorre and co-workers in their related work, it again highlights the huge impact the modification of the radical precursor has had on the overall efficiency of the process.14 The larger 1-adamantyl ester (3p) was also tested, resulting in the lowest yield among the various esters. Although, the enantiocontrol was less affected compared to the t-butyl ester 3o. Two distinct substrate binding modes have been proposed for cyclic β-ketoesters with amide-containing cinchona alkaloids. One involves a hydrogen bond between the catalyst and the carbonyl of the ester, while the other does not invoke such a non-covalent interaction.47,49 Increasing the steric bulk of the ester may influence its ability to engage in hydrogen bonding with PTC C18. Product 3p enabled the absolute configuration of the major enantiomer to be determined by comparing its specific rotation to a literature value,12 and the remaining products have been assigned by analogy. Lastly, we explored modifying the ring size from indanone (3a) to tetralone (3q), which resulted in a lower yield, but with near identical stereoselectivity. Further ring expansion was not tolerated as 7-membered carbocyclic product 3r was afforded as a near racemate. Having explored a range of cyclic β-ketoesters we were keen to evaluate a wider array of carbonyl compounds to establish the differences in their reactivity and selectivity (Scheme 1B). A series of benzannulated α-alkyl and α-aryl substituted cyclic ketones (1s–v) all displayed no reactivity with full recovery of the starting materials. We believe that the formation of the EDA complex was not occurring with these substrates due to their higher pKa relative to β-ketoesters. To facilitate the formation of the required enolate we switched to a stronger inorganic base (CsOH·H2O) but to no avail. We also tested acyclic β-ketoesters (1w–x), but the desired products were not observed, alluding to the high substrate specificity for reactivity under these conditions. Ultimately, we redirected our focus towards exploring different radical precursors to determine whether they exhibited comparable restrictions (Scheme 1C). We hoped that the brominated form (2b) of the ethyl halodifluoroacetate reagent would quell undesired background activity, which might improve the enantioenrichment of the products. Unfortunately, no reactivity was witnessed at all even with the addition of common photocatalysts (see ESI page S27†). We also found that replacing the geminal fluorine atoms of 2a with methyl groups (2c) impeded the formation of the radical.
Mechanistic investigations into radical generation and background reactivity
Intrigued by the high efficiency of the reaction in non-polar solvents with an inorganic base in the absence of a phase-transfer catalyst (Table 1, entries 6 and 7), we sought to gain further insight into these reactions. First, we used UV-vis spectroscopy to determine if the photoexcitation of an individual reaction component or an EDA complex was responsible for the generation of the ethyl difluoroacetate radical (Fig. 2). Catalyst C1 was not used for these studies due to its evident absorption of visible light in solution. The spectra of both starting materials (1a and 2a) alone, do not exhibit significant absorbance at the shortest wavelengths (>400 nm) emitted by the white LED used (Fig. 2, blue and orange line). However, it should be noted that commercial samples of ester 2a tend to have a pink hue due to trace amounts of iodine. This has been identified as the reason for its apparent absorption of visible light as freshly purified material, as used for our UV-vis absorption studies, does not absorb appreciably in the visible region.30 The combination of 1a and 2a with Cs2CO3 in t-BuOAc did not generate any solution phase species with a different absorption profile from the starting materials (Fig. 2, grey line). However, the solid phase of the mixture was orange indicating the formation of an EDA complex on the surface of the inorganic base. The subsequent addition of the achiral phase-transfer catalyst, tetrabutylammonium bromide, led the previously colourless organic liquid layer to develop a yellow/orange hue – indicating the EDA complex had been successfully transported into the liquid phase. This was confirmed by UV-vis spectroscopy as a noticeable bathochromic shift towards the visible light region was observed (Fig. 2, red line). Therefore, in the presence of a phase-transfer catalyst, we believe that this EDA complex is contributing to the production of the ethyl difluoroacetate radical. However, the mode of radical initiation in the absence of a phase-transfer catalyst remained nebulous.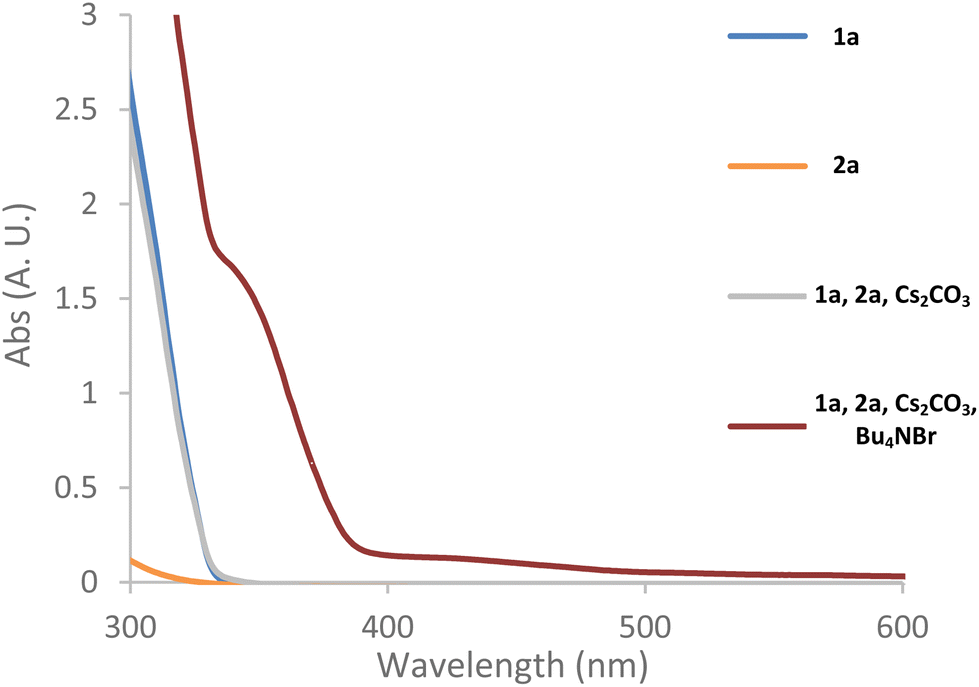 | ||
| Fig. 2 UV-vis absorption spectra of the typical reaction mixture and its components in t-BuOAc. See ESI page S22 for full experimental details and more in-depth discussion.† | ||
Therefore, to probe the radical formation step, and further our understanding of why the fluorinated radical precursors 2a and 2d give vastly different outcomes,14 a series of control experiments were conducted (Fig. 3A). Using styrene as the radical acceptor in the absence of base gave the expected product with ester-containing 2a, but not with perfluorohexyl iodide (2d).35,50 Under these conditions there is no spectroscopic evidence to suggest an EDA complex is forming, indicating that the direct homolysis of 2a occurs more readily than for 2d.35 This occurs despite poor spectral overlap between the emission spectrum of the white LED and the absorption spectrum of radical precursor 2a. However, this corroborates our observation that purified and colourless 2a turns pink in ambient light due to the liberation of iodine, but 2d remains colourless. In the context of comparing their background reactivity both radical progenitors were tested with β-ketoester 1a in the absence of a PTC. Both generated their respective products in 52% yield (2a) and 18% yield (2d), highlighting the greater extent of background reactivity with 2a. Given that direct homolysis of 2d was not previously witnessed implies that the cesium enolate of 1a can promote radical formation by excitation of its EDA complex with 2d. Using TIPS-protected 1a, which is an inferior electron donor, demonstrated lower reactivity and only afforded traces of product with precursor 2d. We rationalise that this is because the only route now available for radical initiation is direct homolysis of the C–I bond, which only occurs for 2a. Consequently, we can deduce that three different processes can account for the formation of the ethyl difluoroacetate radical from 2a under phase-transfer conditions (Fig. 3B). In the absence of the quaternary ammonium salt, two of these pathways remain available for reagent 2a, but only one route, an EDA complex with the Cs enolate, is feasible for precursor 2d.
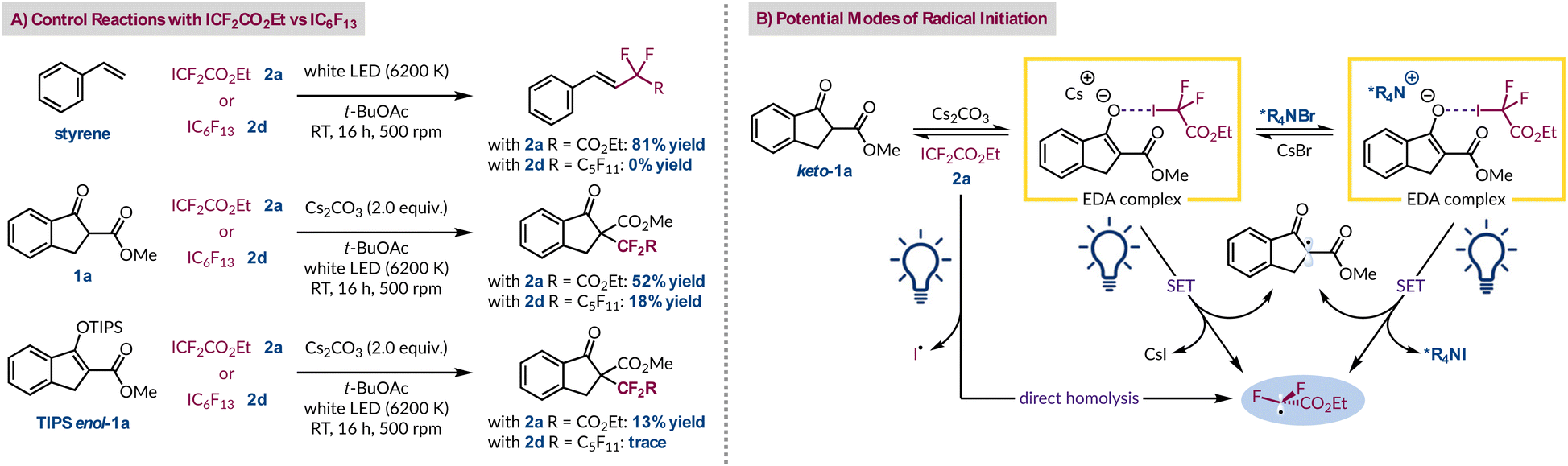 | ||
| Fig. 3 (A) A series of reactions that evaluate the differences between ICF2CO2Et and IC6F13 under identical reaction conditions (B) postulated pathways for the generation of the ethyl difluoroacetate radical in this system. | ||
Lastly, having considered the different pathways for the radical initiation step we also pondered the various mechanisms through which the new C–C bond could form (Fig. 4). Under asymmetric phase-transfer conditions the desired addition of the ethyl difluoroacetate radical to *R4N enolate-1a must be occurring due to the non-racemic production of product 3a. However, without the PTC the reaction still proceeds efficiently and must follow a distinct C–C bond forming event. One viable option is that the radical adds to enol-1a, which is observed by 1H NMR spectroscopy to varying degrees for the different β-ketoester substrates. Considering TIPSenol-1a as a model for this tautomeric form we have shown that it is a viable radical acceptor (Fig. 3A). Similarly, the fact that Cs enolate-1a was implicated in the formation of an EDA complex with perfluorohexyl iodide (2d) indicates that it is also present in solution. Radical addition to this species would also lead to racemic product 3a. A third possibility is radical–radical coupling between the α-keto radical and ethyl difluoroacetate radical formed after photoexcitation of the EDA complex. However, this would be polarity mismatched as both radicals are generally considered to be electrophilic.51 The heterogeneity of the reaction mixture thwarted attempts to determine the quantum yield under mechanistically relevant conditions, which would have helped clarify if radical–radical coupling or a chain process was dominant. Although, based on the philicity of the radicals involved and the fact that the fluorinated electrophilic radical is certainly adding to the electron-rich *R4N enolate-1a, and this could conceivably lead to a chain process as outlined previously,14 we do not think radical–radical coupling is operating appreciably under phase-transfer or PTC-free conditions. Thus, the more facile production of radicals from 2a compared to 2d is likely interacting with either the more abundant enol-1a or its more electron-rich deprotonated form (Cs enolate-1a). However, we cannot distinguish between these two pathways at present and our attempts to slow radical formation, for example, by changing the irradiation wavelength, were not beneficial.
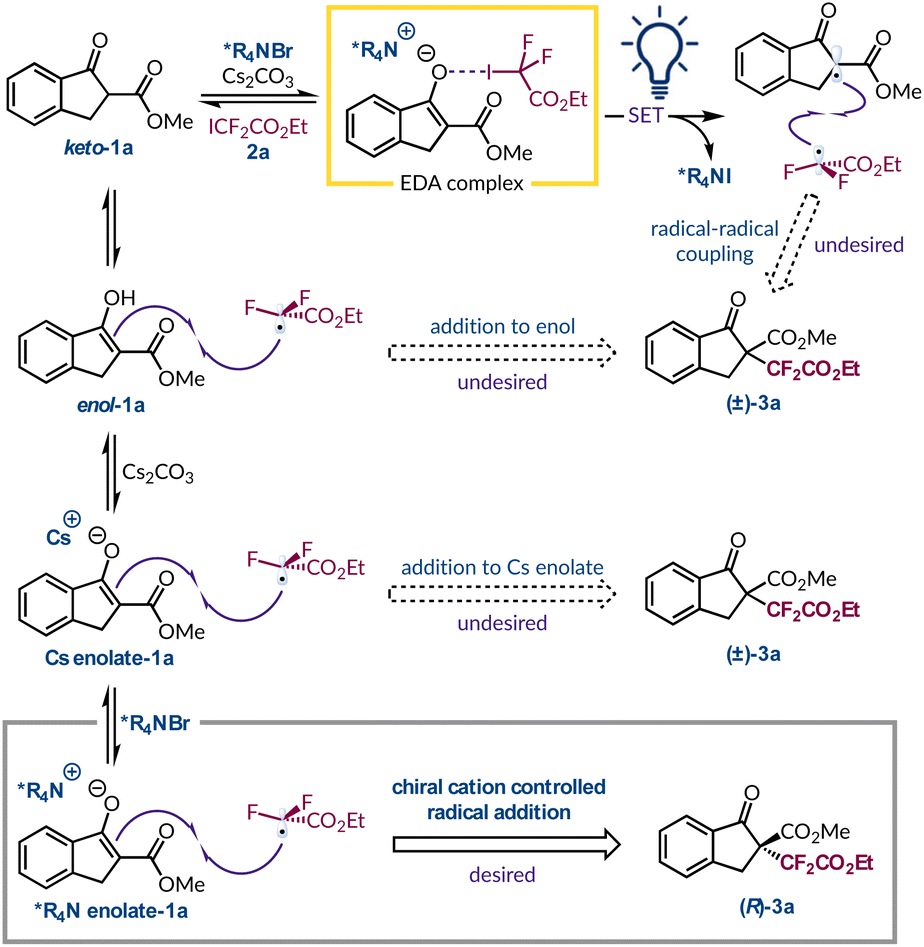 | ||
| Fig. 4 Outline of the different pathways for racemic and enantioselective product formation. | ||
3. Conclusion
In conclusion, we have developed a light-initiated asymmetric tertiary α-difluoroalkylation procedure that operates under mild phase-transfer conditions. Impressively, despite pronounced background reactivity moderate levels of asymmetric induction were still achievable. When using a PTC, UV-vis spectroscopic evidence suggests that a solution-phase visible light-absorbing EDA complex is responsible for radical initiation. However, contributions from other pathways, evident in the absence of a PTC, may also operate alongside to generate the ethyl difluoroacetate radical. We conducted several control experiments to determine why the behaviour of the two radical precursors (2a and 2d) was so different, but we were unable to come to a definite conclusion. Further studies will seek to address these challenges and broaden the scope of asymmetric radical reactions carried out under phase-transfer conditions.Data availability
The research data underpinning this publication can be accessed at https://doi.org/10.17630/301fa354-0637-4065-a526-fbc7c3a3f462.Author contributions
C. E. A.: methodology, investigation, writing. C. P. J.: conceptualization, methodology, writing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from the Royal Society (University Research Fellowship URF\R1\180017 (CPJ)) and the University of St Andrews (CEA). We thank Yitong Wang for his help in the synthesis of starting materials and Reece Hoogesteger for running scale-up experiments. We thank Prof. Andy Smith, Prof. Allan Watson, and their respective research groups for sharing their lab equipment and chemical inventories.References
- T. Lasing, A. Phumee, P. Siriyasatien, K. Chitchak, P. Vanalabhpatana, K.-K. Mak, C. H. Ng, T. Vilaivan and T. Khotavivattana, Synthesis and antileishmanial activity of fluorinated rhodacyanine analogues: The ‘fluorine-walk’ analysis, Bioorg. Med. Chem., 2020, 28, 115187 CrossRef CAS PubMed.
- A. Vulpetti and C. Dalvit, Fluorine local environment: from screening to drug design, Drug Discovery Today, 2012, 17, 890–897 CrossRef CAS PubMed.
- A. D. Dilman and V. V. Levin, Difluorocarbene as a Building Block for Consecutive Bond-Forming Reactions, Acc. Chem. Res., 2018, 51, 1272–1280 CrossRef CAS PubMed.
- K. Müller, C. Faeh and F. Diederich, Fluorine in Pharmaceuticals: Looking Beyond Intuition, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Fluorine in medicinal chemistry, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- T. Liang, C. N. Neumann and T. Ritter, Introduction of Fluorine and Fluorine-Containing Functional Groups, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed.
- M. Fornalczyk, K. Singh and A. M. Stuart, Enantioselective Reformatsky reaction of ethyl iododifluoroacetate with ketones, Org. Biomol. Chem., 2012, 10, 3332–3342 RSC.
- P. Zhang and C. Wolf, Catalytic Enantioselective Difluoroalkylation of Aldehydes, Angew. Chem., Int. Ed., 2013, 52, 7869–7873 CrossRef CAS PubMed.
- X. Gao, Y.-L. Xiao, X. Wan and X. Zhang, Copper-Catalyzed Highly Stereoselective Trifluoromethylation and Difluoroalkylation of Secondary Propargyl Sulfonates, Angew. Chem., Int. Ed., 2018, 57, 3187–3191 CrossRef CAS PubMed.
- H. Liang, G.-Q. Xu, Z.-T. Feng, Z.-Y. Wang and P.-F. Xu, Dual Catalytic Switchable Divergent Synthesis: An Asymmetric Visible-Light Photocatalytic Approach to Fluorine-Containing γ-Keto Acid Frameworks, J. Org. Chem., 2019, 84, 60–72 CrossRef CAS PubMed.
- X. Gao, R. Cheng, Y.-L. Xiao, X.-L. Wan and X. Zhang, Copper-Catalyzed Highly Enantioselective Difluoroalkylation of Secondary Propargyl Sulfonates with Difluoroenoxysilanes, Chem, 2019, 5, 2987–2999 CAS.
- J. Liu, W. Ding, Q.-Q. Zhou, D. Liu, L.-Q. Lu and W.-J. Xiao, Enantioselective Di-/Perfluoroalkylation of β-Ketoesters Enabled by Cooperative Photoredox/Nickel Catalysis, Org. Lett., 2018, 20, 461–464 CrossRef CAS PubMed.
- S. Huang, F.-F. Tong, D.-C. Bai, G.-P. Zhang, Y.-J. Jiang, B. Zhang, X. Leng, Y.-L. Guo, X.-L. Wan, X. Zhang, C.-H. Ding and X.-L. Hou, Regio- and enantioselective umpolung gem-difluoroallylation of hydrazones via palladium catalysis enabled by N-heterocyclic carbene ligand, Nat. Commun., 2021, 12, 6551 CrossRef CAS PubMed.
- Ł. Woźniak, J. J. Murphy and P. Melchiorre, Photo-organocatalytic Enantioselective Perfluoroalkylation of β-Ketoesters, J. Am. Chem. Soc., 2015, 137, 5678–5681 CrossRef PubMed.
- T. Ooi and K. Maruoka, Recent Advances in Asymmetric Phase-Transfer Catalysis, Angew. Chem., Int. Ed., 2007, 46, 4222–4266 CrossRef CAS PubMed.
- S. Shirakawa and K. Maruoka, Recent Developments in Asymmetric Phase-Transfer Reactions, Angew. Chem., Int. Ed., 2013, 52, 4312–4348 CrossRef CAS PubMed.
- R. S. J. Proctor, A. C. Colgan and R. J. Phipps, Exploiting attractive non-covalent interactions for the enantioselective catalysis of reactions involving radical intermediates, Nat. Chem., 2020, 12, 990–1004 CrossRef PubMed.
- T. E. Schirmer and B. König, Ion-Pairing Catalysis in Stereoselective, Light-Induced Transformations, J. Am. Chem. Soc., 2022, 144, 19207–19218 CrossRef CAS PubMed.
- S. Mondal, F. Dumur, D. Gigmes, M. P. Sibi, M. P. Bertrand and M. Nechab, Enantioselective Radical Reactions Using Chiral Catalysts, Chem. Rev., 2022, 122, 5842–5976 CrossRef CAS PubMed.
- M. J. Genzink, J. B. Kidd, W. B. Swords and T. P. Yoon, Chiral Photocatalyst Structures in Asymmetric Photochemical Synthesis, Chem. Rev., 2022, 122, 1654–1716 CrossRef CAS PubMed.
- D. C. M. Albanese and M. Penso, New Trends in Asymmetric Phase Transfer Catalysis, Eur. J. Org. Chem., 2023, e202300224 CrossRef CAS.
- R. S. Mulliken, Structures of Complexes Formed by Halogen Molecules with Aromatic and with Oxygenated Solvents1, J. Am. Chem. Soc., 1950, 72, 600–608 CrossRef CAS.
- R. S. Mulliken, Molecular Compounds and their Spectra. II, J. Am. Chem. Soc., 1952, 74, 811–824 CrossRef CAS.
- R. S. Mulliken, Molecular Compounds and their Spectra. III. The Interaction of Electron Donors and Acceptors, J. Phys. Chem., 1952, 56, 801–822 CrossRef CAS.
- R. Foster, Electron donor-acceptor complexes, J. Phys. Chem., 1980, 84, 2135–2141 CrossRef CAS.
- E. F. Hilinski, J. M. Masnovi, C. Amatore, J. K. Kochi and P. M. Rentzepis, Charge-transfer excitation of electron donor-acceptor complexes. Direct observation of ion pairs by time-resolved (picosecond) spectroscopy, J. Am. Chem. Soc., 1983, 105, 6167–6168 CrossRef CAS.
- S. V. Rosokha and J. K. Kochi, Fresh Look at Electron-Transfer Mechanisms via the Donor/Acceptor Bindings in the Critical Encounter Complex, Acc. Chem. Res., 2008, 41, 641–653 CrossRef CAS PubMed.
- G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, Synthetic Methods Driven by the Photoactivity of Electron Donor–Acceptor Complexes, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed.
- H.-R. Zhang, D.-Q. Chen, Y.-P. Han, Y.-F. Qiu, D.-P. Jin and X.-Y. Liu, Merging photoredox with copper catalysis: decarboxylative difluoroacetylation of α,β-unsaturated carboxylic acids with ICF2CO2Et, Chem. Commun., 2016, 52, 11827–11830 RSC.
- H. Lu, D. Wang and A. Zhang, Visible Light-Promoted Phosphine-Catalyzed Difluoroalkylation of Arenes and Heterocycles, J. Org. Chem., 2020, 85, 942–951 CrossRef CAS PubMed.
- X.-X. Liu, J. Jia, Z. Wang, Y.-T. Zhang, J. Chen, K. Yang, C.-Y. He and L. Zhao, Catalyst-Free and Visible Light Promoted Aminofluoroalkylation of Unactivated Alkenes: An Access to Fluorinated Aziridines, Adv. Synth. Catal., 2020, 362, 2604–2608 CrossRef CAS.
- T. Mao, M.-J. Ma, L. Zhao, D.-P. Xue, Y. Yu, J. Gu and C.-Y. He, A general and green fluoroalkylation reaction promoted via noncovalent interactions between acetone and fluoroalkyl iodides, Chem. Commun., 2020, 56, 1815–1818 RSC.
- Y. Li, M. Rao, Z. Fan, B. Nian, Y. Yuan and J. Cheng, A visible-light-irradiated electron donor-acceptor complex-promoted radical reaction system for the CH perfluoroalkylation of quinolin-4-ols, Tetrahedron Lett., 2019, 60, 151046 CrossRef CAS.
- A. Lemos, C. Lemaire and A. Luxen, Progress in Difluoroalkylation of Organic Substrates by Visible Light Photoredox Catalysis, Adv. Synth. Catal., 2019, 361, 1500–1537 CrossRef CAS PubMed.
- K. Li, X. Zhang, J. Chen, Y. Gao, C. Yang, K. Zhang, Y. Zhou and B. Fan, Blue Light Induced Difluoroalkylation of Alkynes and Alkenes, Org. Lett., 2019, 21, 9914–9918 CrossRef CAS PubMed.
- L. Li, Y.-N. Ma, M. Tang, J. Guo, Z. Yang, Y. Yan, X. Ma and L. Tang, Photoredox-Catalyzed Oxydifluoroalkylation of Styrenes for Access to Difluorinated Ketones with DMSO as an Oxidant, Adv. Synth. Catal., 2019, 361, 3723–3728 CrossRef CAS.
- W.-W. Xu, L. Wang, T. Mao, J. Gu, X.-F. Li and C.-Y. He, Visible-light Promoted Atom Transfer Radical Addition−Elimination (ATRE) Reaction for the Synthesis of Fluoroalkylated Alkenes Using DMA as Electron-donor, Molecules, 2020, 25, 508 CrossRef CAS PubMed.
- Y. Wang, Z. Zheng, M. Lian, H. Yin, J. Zhao, Q. Meng and Z. Gao, Photo-organocatalytic enantioselective α-hydroxylation of β-keto esters and β-keto amides with oxygen under phase transfer catalysis, Green Chem., 2016, 18, 5493–5499 RSC.
- Y. Wang, S. Wang, P. Qiu, L. Fang, K. Wang, Y. Zhang, C. Zhang and T. Zhao, Asymmetric α-electrophilic difluoromethylation of β-keto esters by phase transfer catalysis, Org. Biomol. Chem., 2021, 19, 4788–4795 RSC.
- T. Hashimoto and K. Maruoka, Recent Development and Application of Chiral Phase-Transfer Catalysts, Chem. Rev., 2007, 107, 5656–5682 CrossRef CAS PubMed.
- M. Kitamura, S. Shirakawa and K. Maruoka, Powerful Chiral Phase-Transfer Catalysts for the Asymmetric Synthesis of α-Alkyl- and α,α-Dialkyl-α-amino Acids, Angew. Chem., Int. Ed., 2005, 44, 1549–1551 CrossRef CAS PubMed.
- X. Wang, Q. Lan, S. Shirakawa and K. Maruoka, Chiral bifunctional phase transfer catalysts for asymmetric fluorination of β-keto esters, Chem. Commun., 2009, 46, 321–323 RSC.
- T. Ooi, Y. Arimura, Y. Hiraiwa, L. M. Yuan, T. Kano, T. Inoue, J. Matsumoto and K. Maruoka, Highly enantioselective monoalkylation of p-chlorobenzaldehyde imine of glycine tert-butyl ester under mild phase-transfer conditions, Tetrahedron: Asymmetry, 2006, 17, 603–606 CrossRef CAS.
- C. Yang, W. Zhang, Y.-H. Li, X.-S. Xue, X. Li and J.-P. Cheng, Origin of Stereoselectivity of the Photoinduced Asymmetric Phase-Transfer-Catalyzed Perfluoroalkylation of β-Ketoesters, J. Org. Chem., 2017, 82, 9321–9327 CrossRef CAS PubMed.
- M. Majdecki, P. Niedbala and J. Jurczak, Amide-Based Cinchona Alkaloids as Phase-Transfer Catalysts: Synthesis and Potential Application, Org. Lett., 2019, 21, 8085–8090 CrossRef CAS PubMed.
- M. Majdecki, A. Tyszka-Gumkowska and J. Jurczak, Highly Enantioselective Epoxidation of α,β-Unsaturated Ketones Using Amide-Based Cinchona Alkaloids as Hybrid Phase-Transfer Catalysts, Org. Lett., 2020, 22, 8687–8691 CrossRef CAS PubMed.
- P. Niedbała, M. Majdecki, P. Grodek and J. Jurczak, H-Bond Mediated Phase-Transfer Catalysis: Enantioselective Generating of Quaternary Stereogenic Centers in β-Keto Esters, Molecules, 2022, 27, 2508 CrossRef PubMed.
- Y. Wang, S. Wang, Y. Wu, T. Zhao, J. Liu, J. Zheng, L. Wang, J. Lv and T. Zhang, Fast, highly enantioselective, and sustainable fluorination of 4-substituted pyrazolones catalyzed by amide-based phase-transfer catalysts, Org. Chem. Front., 2023, 10, 2226–2233 RSC.
- M. Majdecki, P. Grodek and J. Jurczak, Stereoselective α-Chlorination of β-Keto Esters in the Presence of Hybrid Amide-Based Cinchona Alkaloids as Catalysts, J. Org. Chem., 2021, 86, 995–1001 CrossRef CAS PubMed.
- N. J. W. Straathof, S. E. Cramer, V. Hessel and T. Noël, Practical Photocatalytic Trifluoromethylation and Hydrotrifluoromethylation of Styrenes in Batch and Flow, Angew. Chem., Int. Ed., 2016, 55, 15549–15553 CrossRef CAS PubMed.
- F. Parsaee, M. C. Senarathna, P. B. Kannangara, S. N. Alexander, P. D. E. Arche and E. R. Welin, Radical philicity and its role in selective organic transformations, Nat. Rev. Chem., 2021, 5, 486–499 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qo00390j |
| This journal is © the Partner Organisations 2024 |