
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic insights into the rhodium catalysed dehydrogenative cycloaddition of cyano-yne-allene substrates†
Àlex
Díaz-Jiménez
 ,
Anna
Roglans
,
Miquel
Solà
* and
Anna
Pla-Quintana
*
,
Anna
Roglans
,
Miquel
Solà
* and
Anna
Pla-Quintana
*
Institut de Química Computacional i Catàlisi (IQCC) and Departament de Química, Universitat de Girona (UdG), Facultat de Ciències, C/Maria Aurèlia Capmany, 69, 17003-Girona, Catalunya, Spain. E-mail: miquel.sola@udg.edu; anna.plaq@udg.edu
First published on 16th May 2024
Abstract
The continued progress in creating novel acceptorless dehydrogenations for environmentally friendly and efficient chemical synthesis is anticipated to be significantly shaped by the design of efficient catalysts that can do the job. In this study, we present a comprehensive mechanistic investigation utilizing DFT calculations to elucidate the mechanism of the [RhCl(PPh3)3]-catalysed dehydrogenative cycloaddition of cyano-yne-allene substrates. Overall, the reaction can be described as a cycloaddition involving the three unsaturations, followed by a hydrogen shift, 6π electrocyclization and acceptorless H2 elimination. The crucial factors contributing to the observed reactivity are as follows: (i) the favoured cycloaddition pathway involves the external double bond of the allene in the formation of the initial metallacycle; (ii) the stabilization of the alkyne insertion intermediate that occurs through η3-coordination to the rhodium; and (iii) the presence of a weak base that assists in deprotonation.
1. Introduction
Acceptorless dehydrogenation processes represent a fascinating and environmentally friendly class of chemical transformations in which molecules undergo dehydrogenation reactions without the need for external hydrogen acceptors or oxidants.1 They are particularly significant in the context of catalysis and renewable energy applications, as they hold promise for the development of efficient and green methodologies for the production of fine chemicals, pharmaceuticals, and energy carriers like hydrogen.2–4The term acceptorless dehydrogenation encompasses a wide range of transformations. Common examples include the conceptually simple dehydrogenations5 of alkanes,6 alcohols,7 and N-containing compounds,8 as well as the coupling of alcohols, or alcohols with amines, to produce esters and amides,9 respectively. A less explored area involves catalytic dehydrogenative cycloadditions, including [4 + 2]10 and (3 + 2) cycloadditions.11
Some years ago, we disclosed the [RhCl(PPh3)3]-catalysed cycloaddition of cyano-yne-allene substrates 1, leading to the formation of tricyclic scaffolds 2 that can be easily transformed to 2,6-naphthyridine compounds (Scheme 1).12 A distinctive feature of this reaction is the formal elimination of hydrogen from the substrate throughout the process, allowing it to be described formally as a dehydrogenative [2 + 2 + 2] cycloaddition reaction. A previous example by Saito and co-workers described the intramolecular [2 + 2 + 2] cycloadditions of bis(propargylphenyl)carbodiimides, followed by dehydrogenation; however, it required an oxidant (MnO2) to generate aromatized L-shaped π-extended polycycles with pyrrolo[1,2-a][1,8]naphthyridine corner units,13 which is in contrast to our example.
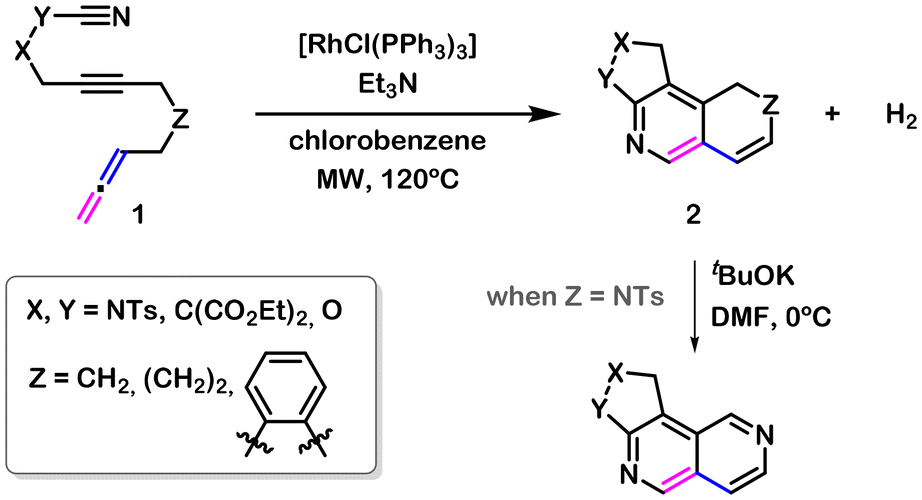 | ||
| Scheme 1 [RhCl(PPh3)3]-catalysed dehydrogenative cycloaddition of cyano-yne-allene substrates 1. | ||
The literature on the mechanism of transition metal-catalysed [2 + 2 + 2] cycloaddition reactions is extensive.14 However, there are very few studies that delve into the mechanism of dehydrogenative versions of cycloaddition reactions, and none on dehydrogenative [2 + 2 + 2] cycloadditions. As understanding and advancing the mechanisms behind acceptorless dehydrogenation reactions is crucial for harnessing the potential of these processes towards a more sustainable and cleaner future, we present here a comprehensive mechanistic study, focusing on our previously reported dehydrogenative cycloaddition.
2. Computational details
DFT calculations were performed with the Gaussian16 software package.15 The B3LYP hybrid exchange-correlation functional was used,16–18 together with the Grimme D3 correction term for the electronic energy.19,20 The all-electron cc-pVDZ basis set21,22 was employed for non-metal atoms and the cc-pVDZ-PP basis set containing an effective core relativistic pseudopotential was used for Rh.23 The electronic energy was improved by performing single point energy calculations with the cc-pVTZ (cc-pVTZ-PP for Rh) basis set and the M06 functional24 and including solvent effects computed with the solvent model based on density (SMD) continuum solvation (chlorobenzene as solvent).25 Frequency calculations were performed to ensure the nature of located stationary points. In addition, the Intrinsic Reaction Coordinate (IRC) procedure was used to confirm the two minima connected by each transition state.26 The reported Gibbs energies include electronic energies obtained at the M06-D3(SMD, chlorobenzene)/cc-pVTZ-PP//B3LYP-D3/cc-pVDZ-PP level of theory corrected with zero-point energies, thermal corrections, and entropy effects evaluated at 393.15 K (the temperature at which the reaction is carried out experimentally) computed with the B3LYP-D3/cc-pVDZ-PP method.3. Results and discussion
3.1. Oxidative cyclization
Due to the ability of transition metals, including rhodium, to promote metallacycle formation through oxidative cyclization reactions,14,27 we initiated our investigation by analysing the various possibilities for such a process in our cyano-yne-allene substrates 1 using Wilkinson's complex as catalyst. We selected substrate model 1a as our starting point, which features an oxygen tether between the alkyne and nitrile unsaturations, and a mesylamide group tethering between the alkyne and the allene. In our experimental study, we successfully reacted a substrate analogous to this one but with a tosylamide in place of the mesylamide. The substitution of tosylamide with mesylamide was done to reduce the computational cost. We explored three different oxidative cyclizations, all of them reckoning on the entropic gain associated with reacting consecutive 2π components. We conducted a throughout analysis, considering varying numbers of phosphines attached to rhodium and different relative ligand orientations. Scheme 2 gathers the most favourable reaction paths (in the case of A2 exchanging the ligand orientation leads to almost isoenergetic transition states, see Fig. 1 for full discussion). Starting from the Wilkinson's catalyst, two PPh3 ligands are released to generate the [RhCl(PPh3)] complex that interacts with 1a to form the 16e− square planar coordination intermediates A1, B1, and C1 (Scheme 2). The transition states leading to allene-yne oxidative coupling (TSA1A2 and TSB1B2) exhibited lower energy barriers than the one leading to cyano-yne coupling (TSC1C2). Furthermore, the reaction through the inner double bond of the allene, resulting in the 5-exo intermediate B2, required a barrier of 29.5 kcal mol−1, which is 3.9 kcal mol−1 higher than the barrier for the reaction involving the outer double bond of the allene, forming the 6-endo intermediate A2. It is important to note that the 5-exo intermediate B2 could not evolve into the formation of 2a, corresponding to the experimentally isolated scaffold. Instead, it could lead to the formation of 3a,28 which corresponds to a scaffold detected when using cationic rhodium catalysts during the reaction optimization. The formation of 3a can be explained through a canonical [2 + 2 + 2] cycloaddition and isomerization sequence (see the ESI† for the complete reaction profile). Consistent with this analysis, oxidative cyclization exclusively occurs with the alkyne and the external double bond of the allene to generate A2 (Fig. 1).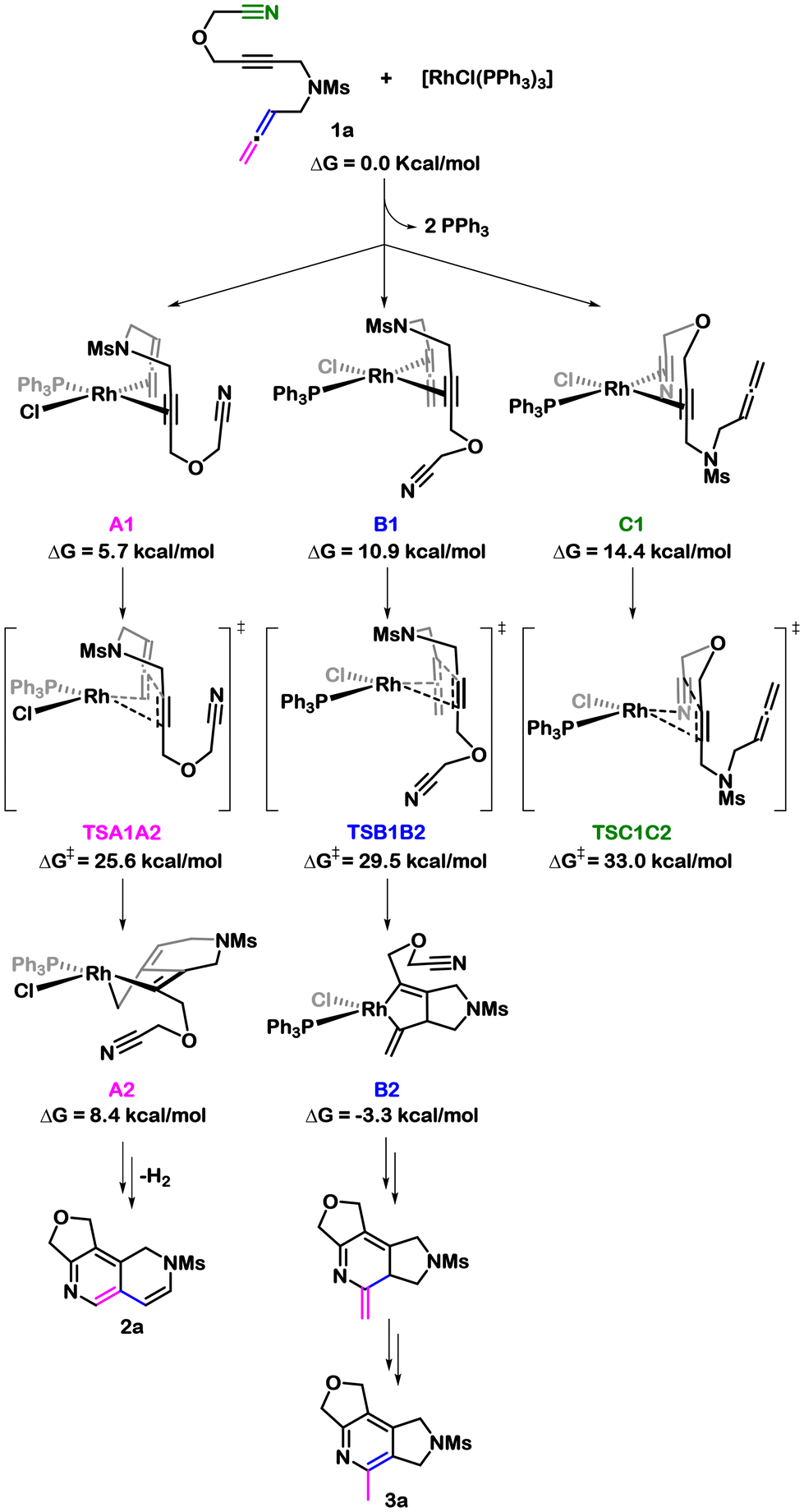 | ||
| Scheme 2 Most favourable pathways for the oxidative coupling of 1a. | ||
 | ||
| Fig. 1 Gibbs energy changes of [RhCl(PPh3)3]-catalysed oxidative cyclization and nitrile insertion with cyano-yne-allene 1a. Gibbs energies (393.15 K) with respect to 1a and [RhCl(PPh3)3] are shown in kcal mol−1. | ||
To gain a better understanding of the preferential reactivity of the external double bond of the allene, we calculated deformation energies for TSA1A2 and TSB1B2 (see Table S1†). The deformation energy was computed considering two fragments, 1a and [RhCl(PPh3)]. Our results show that the deformation energy of the TSB1B2 is 3.5 kcal mol−1 higher than that of TSA1A2. This difference can be attributed mainly to the larger deformation of 1a in TSB1B2 than in TSA1A2. During the oxidative cyclization the central carbon of the former allene rehybridizes from sp to sp2. In TSB1B2, the angles around this carbon significantly deviate from the expected 120° for an sp2 carbon, primarily due to the strained angle in the forming rhodacyclopentene moiety. This strain becomes evident when comparing the angles with those of intermediate B2 (see Scheme S1†). Conversely, the corresponding deformations around this carbon in TSA1A2 are notably minor, consistent with the decreased computed deformation energy.
3.2. Nitrile insertion
From the rhodacylopentene intermediate A2, we computationally analysed nitrile coordination and subsequent insertion (Fig. 1). Nitrile coordination to A2 is exergonic by 11.9 kcal mol−1 and leads to A3, a 16e− complex exhibiting a distorted square-based pyramid geometry. The insertion of the nitrile requires a barrier of 8.8 kcal mol−1 with respect to A3 and produces A4, a Rh(III) complex in which the three carbons from the reacted allene are η3-coordinated to the rhodium (dRh–C = 2.076, 2.201, and 2.342 Å, Fig. 2) forming a π-allyl metallacycle. π-Allyl metallacycle intermediates have previously been spectroscopically characterized, including X-ray diffraction, in cycloaddition reactions involving alkylidenecyclopropanes29 or dienes.10a They have also been postulated, by means of DFT calculations, in cycloaddition reactions involving allenes.30 | ||
| Fig. 2 Intermediate A4; selected distances in Å. | ||
It should be noted that when PPh3 and Cl ligands exchange position, the π-allyl metallacycle intermediate is no longer formed. From A′2, the nitrile coordinates to the rhodium in trans to the Cl (similar to A2) and then it is inserted in a process that is endergonic by 6.4 kcal mol−1, surpassing TSA′3A′4 with an energy barrier of 17.6 kcal mol−1. Interestingly, in contrast to A4, in which rhodium coordinates in an η3 fashion to the allyl, in A′4 the rhodium coordinates to the allyl in an η1 fashion and completes the coordination sphere through interaction with one of the oxygen atoms of the tethering mesylamide group (dRh–O = 2.455 Å). Moreover, analysis of the %Vbur![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31 show that in A′4 the ligands are more compressed and this could explain in part the lower stability of A′4 as compared to A4 (see Fig. S10†).
31 show that in A′4 the ligands are more compressed and this could explain in part the lower stability of A′4 as compared to A4 (see Fig. S10†).
To confirm that the different coordination is not due to steric hindrance from the sulfonamide moiety, analogous optimizations were conducted with a tosylamide group as in the experimental conditions. The same conclusion was reached suggesting that other factors induced divergent coordination modes in A4 and A4′. To better understand the steric requirements for the formation of π-allyl metallacycle intermediate A4, a predictive study was carried out by introducing methyl groups either at the terminus of the allene (A4ext) or at the internal allene carbon (A4int). After optimization, intermediate A4ext (with two methyl groups in the terminal position of the former allene moiety) still exhibited rhodium coordinated in an η3 fashion, whereas A4int (with the methyl at the internal position of the allene) isomerized to η1-allyl coordination with the Rh completing the coordination sphere by interaction with one of the oxygen atoms of the mesylamide, forming a structure analogous to A′4. Thus, the formation of η3-allyl coordinated intermediates is influenced by steric factors at the internal carbon of the allene but not at the external one (refer to the ESI† for a comparison of the intermediates).
Based on the conventional mechanism of a [2 + 2 + 2] cycloaddition, the reductive elimination from A4 and A′4 would yield the cycloaddition product without the extrusion of hydrogen. Our calculations reveal that the energy barrier for reductive elimination from both intermediates results in transition states with barriers exceeding 31 kcal mol−1 (31.1 kcal mol−1 for TSA′4A′5 and 31.4 kcal mol−1 for TSA4RE). These barriers are deemed excessively high, prompting the exploration of alternative pathways. It should be noted that once A4′ is formed, the barrier to return to A1 is less than the one to proceed through TSA′4A′5, and, therefore, since the equilibrium is shifted towards A1, this pathway is ineffective.
3.3. Cyclization and H2 elimination
The experimentally isolated product (2, Scheme 1) features a double bond in the newly created non-aromatic 6-membered ring, an enamine motif when the tether between the allene and the alkyne is based on nitrogen (Z, Scheme 1). Previous studies on the cyclization of 1,5-bisallenes with alkenes to afford dihydroazepine derivatives,32 showed that β-H elimination from rhodacycle intermediates could explain the formation of this scaffold. We thus analysed the possibility of a direct β-H elimination from Rh η3-coordinated intermediate A4 (TSA4BH, Fig. 1). However, such a transformation needs to overcome a reaction barrier of 42 kcal mol−1, rendering it unfavored at the reaction conditions experimentally used.During the optimization of the reaction, we found that the addition of a catalytic amount of triethylamine not only improved the yield but also decreased the reaction time. This observation led to the hypothesis that the base plays a crucial role in the catalytic reaction. Indeed, when triethylamine approaches A4, it forms complex A5, from which deprotonation at the α-N position yields rhodium complex A6, overcoming a barrier of 18.2 kcal mol−1 (Fig. 1 and 3). This barrier is notably lower than that for the reductive elimination from A4 and A′4, as well as the direct β-H elimination from A4. Consequently, the triethylamine-assisted pathway through A6 is the sole pathway we further investigated. Et3N could deprotonate in an analogous manner intermediates A2, A′2, A3 and A′3, however DFT calculations showed that these pathways are not energetically favoured (see ESI†).
 | ||
| Fig. 3 Gibbs energy changes of proton shift, cyclization and hydrogen elimination. Relative Gibbs energies (393.15 K) with respect to 1a and [RhCl(PPh3)3] are shown in kcal mol−1. | ||
Complex A6 is a 16e− complex exhibiting distorted square planar geometry where the ammonium salt is stabilized by interaction with one of the sulphonamide oxygens. This complex readily engages in a H transfer to the imine nitrogen, displacing the rhodium and generating penta-2,4-dien-1-imine intermediate A7, interacting with the rhodium through the terminal double bond of the conjugated system and the newly formed N–H bond (Fig. 3). Therefore, it can be concluded that triethylamine catalyses the observed hydrogen atom rearrangement.
From A7, a triphenylphosphine ligand displaces the amine from the rhodium catalyst coordination sphere and the metal fragment relocates towards the endocyclic double bond of the six-membered ring, leading to the much more thermodynamically stable intermediate A8. During this process the triethylamine molecule exits the catalytic cycle. From A8 a 6π-electrocyclization leads to tricyclic product A9 (Fig. 3). As expected, this 6π-electrocyclization is disrotatory, although there is no stereochemical implication.33 Electrocyclization reactions have previously been postulated as key steps in transition metal-catalysed cycloaddition reactions that occur with double bond reorganization.34 Upon electrocyclization, the rhodium catalyst migrates once again towards the N atom in the central ring leading to dihydropyridine intermediate A9, which has the carbonated skeleton of the isolated product although in a reduced form. Formation of the fused unsaturated pyridine scaffold in 2 is explained by acceptorless dehydrogenation catalysed by rhodium. The already coordinated Rh system undergoes an oxidative addition into the N–H bond, surpassing a reaction barrier of 23.0 kcal mol−1, to generate Rh(III) intermediate A10. Subsequent β-H elimination releases 2a and yields the rhodium intermediate A11 in a highly exergonic process. Finally, reductive elimination of H2 from the rhodium center, overcoming a reaction barrier of 3.0 kcal mol−1, with concomitant coordination of a new substrate molecule displacing one of the PPh3 on the rhodium, recovers the catalytic species A1 and restarts the catalytic cycle. The possibility of triethylamine-mediated deprotonation, whether with rhodium coordinated to the amine or not, was computed but disregarded due to the notably higher energies required (see Fig. S6†).
Taken together, the rhodium catalysed [2 + 2 + 2] cycloaddition of cyano-yne-allene substrates operates through the catalytic cycle shown in Scheme 3. The process is exothermic by 66.5 kcal mol−1 and has, as the rate-determining step, the rhodium catalysed acceptorless dehydrogenation with a barrier of 26.0 kcal mol−1, although the oxidative coupling between the alkyne and allene units has a barrier of 25.6 kcal mol−1 very close in energy. Several aspects allow us to theoretically explain the selective formation of the dehydro-2,6-naphthyridine scaffold. On one hand, the preferential formation of rhodacyclopentene intermediate A2 over B2 occurs through the reaction of the external double bond of the allene, generating the 6-membered ring fused to the rhodacyclopentene. Then, insertion of the nitrile in A2 selectively generates Rh η3-coordinated intermediate A4, from which reductive elimination is not favored. The increased stability provided by the Rh η3-coordination in intermediate A4 allows for the Et3N assisted H shift35 that installs the enamine moiety. From this point, the 6π-electrocyclization of penta-2,4-dien-1-imine intermediate A7 forges the second 6-membered ring in A9, which is obtained in the form of a rhodium coordinated dihydropyridine and sets the stage for the metal catalysed dehydrogenation.
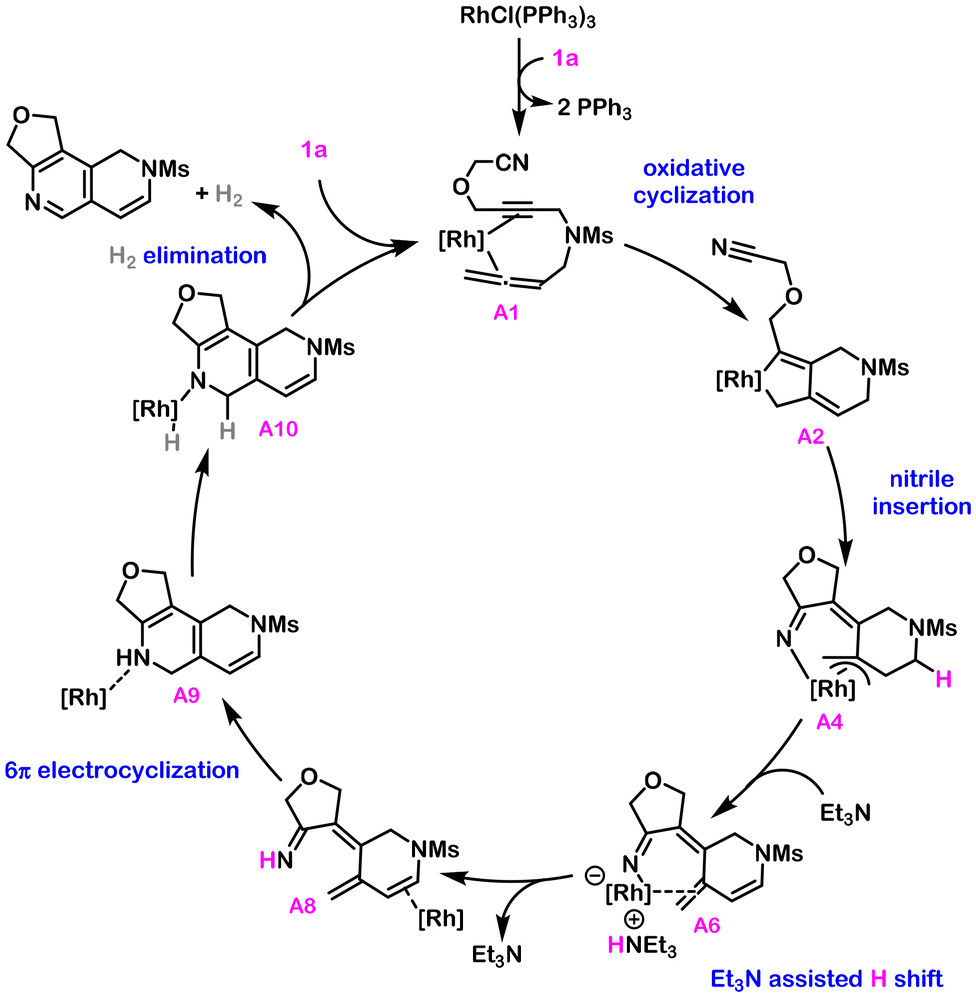 | ||
| Scheme 3 Overall mechanism for the dehydrogenative [2 + 2 + 2] cycloaddition. [Rh] = RhClPPh3. | ||
4. Conclusions
In light of the numerous innovative reactions stemming from metallacycle intermediates, the identification of unique reaction pathways holds the potential to unveil novel transformations. This is exemplified in the case of the rhodium-catalysed cycloaddition of cyano-yne-allene linear substrates under investigation here. The selective reaction on either the internal or external double bond of the allene during the initial oxidative cyclization step diverges the reactivity from a [2 + 2 + 2] cycloaddition, followed by isomerization, to a formal acceptorless dehydrogenative [2 + 2 + 2] cycloaddition. The detailed mechanistic study of this latter transformation yields a wealth of intriguing data. First, it underscores the pivotal role of Rh η3-coordination in 7-membered metallacycles, which disfavors reductive elimination. Secondly, the presence of weak bases is found to facilitate an H-shift process that produces a penta-2,4-dien-1-imine intermediate that readily engages in 6π-electrocyclization, yielding the cyclized core. Finally, rhodium catalyses the acceptorless dehydrogenation, a step that yields the final tricyclic compound 2a.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the Ministerio de Ciencia, Innovación y Universidades (Projects PID2020-113711GB-I00 MCIN/AEI/10.13039/50110001103, RED2022-134074-T, RED2022-134331-T, and predoctoral fellowship FPU18/02912 to A. D.-J.) and the Generalitat de Catalunya (Project 2021-SGR-623). Open access funding provided by the University of Girona.References
- C. Gunanathan and D. Milstein, Applications of Acceptorless Dehydrogenation and Related Transformations in Chemical Synthesis, Science, 2013, 341, 1229712 CrossRef PubMed.
- M. Trincado, J. Bösken and H. Grützmacher, Homogeneously Catalyzed Acceptorless Dehydrogenation of Alcohols: A Progress Report, Coord. Chem. Rev., 2021, 443, 213967 CrossRef CAS.
- P. K. Verma, Advancement in Photocatalytic Acceptorless Dehydrogenation Reactions: Opportunity and Challenges for Sustainable Catalysis, Coord. Chem. Rev., 2022, 472, 214805 CrossRef CAS.
- K. Oshiro and S. Fujimori, Role of Hydrogen-Based Energy Carriers as an Alternative Option to Reduce Residual Emissions Associated with Mid-Century Decarbonization Goals, Appl. Energy, 2022, 313, 118803 CrossRef CAS.
- S. Budweg, K. Junge and M. Beller, Catalytic Oxidations by Dehydrogenation of Alkanes, Alcohols and Amines with Defined (non)-Noble Metal Pincer Complexes, Catal. Sci. Technol., 2020, 10, 3825–3842 RSC.
- For selected recent examples, see: (a) A. I. McKay, A. J. Bukvic, B. E. Tegner, A. L. Burnage, A. J. Martínez-Martínez, N. H. Rees, S. A. Macgregor and A. S. Weller, Room Temperature Acceptorless Alkane Dehydrogenation from Molecular σ-Alkane Complexes, J. Am. Chem. Soc., 2019, 141, 11700–11712 CrossRef CAS PubMed; (b) M. J. Zhou, L. Zhang, G. Liu, C. Xu and Z. Huang, Site-Selective Acceptorless Dehydrogenation of Aliphatics Enabled by Organophotoredox/Cobalt Dual Catalysis, J. Am. Chem. Soc., 2021, 143, 16470–16485 CrossRef CAS PubMed.
- For selected recent examples, see: (a) T. Wang, J. Sha, M. Sabbe, P. Sautet, M. Pera-Titus and C. Michel, Identification of Active Catalysts for the Acceptorless Dehydrogenation of Alcohols to Carbonyls, Nat. Commun., 2021, 12, 5100 CrossRef CAS PubMed; (b) H. Fuse, H. Mitsunuma and M. Kanai, Catalytic Acceptorless Dehydrogenation of Aliphatic Alcohols, J. Am. Chem. Soc., 2020, 142, 4493–4499 CrossRef CAS PubMed.
- For selected recent examples, see: (a) L. V. A. Hale, T. Malakar, K.-N. T. Tseng, P. M. Zimmerman, A. Paul and N. Szymczak, The Mechanism of Acceptorless Amine Double Dehydrogenation by N,N,N-Amide Ruthenium(II) Hydrides: A Combined Experimental and Computational Study, ACS Catal., 2016, 6, 4799–4813 CrossRef CAS; (b) N. O. Balayeva, Z. Mamiyev, R. Dillert, N. Zheng and D. W. Bahnemann, Rh/TiO2-Photocatalyzed Acceptorless Dehydrogenation of N-Heterocycles upon Visible-Light Illumination, ACS Catal., 2020, 10, 5542–5553 CrossRef CAS; (c) J. A. Luque-Urrutia, M. Solà, D. Milstein and A. Poater, Mechanism of the Manganese-Pincer Catalyzed Acceptorless Dehydrogenative Coupling of Nitriles and Alcohols, J. Am. Chem. Soc., 2019, 141, 2398–2403 CrossRef CAS PubMed; (d) J. A. Luque-Urrutia, T. Pèlachs, M. Solà and A. Poater, Double-Carrousel Mechanism for Mn-Catalyzed Dehydrogenative Amide Synthesis from Alcohols and Amines, ACS Catal., 2021, 11, 6155–6161 CrossRef CAS.
- (a) J. Luo, Q.-Q. Zhou, M. Montag, Y. Ben-David and D. Milstein, Acceptorless Dehydrogenative Synthesis of Primary Amides from Alcohols and Ammonia, Chem. Sci., 2022, 13, 3894–3901 RSC; (b) M. Montag and D. Milstein, Sustainable Amidation through Acceptorless Dehydrogenative Coupling by Pincer-Type Catalysts: Recent Advances, Pure Appl. Chem., 2023, 95, 109–124 CrossRef CAS.
- (a) M. Ohashi, I. Takeda, M. Ikawa and S. Ogoshi, Nickel-Catalyzed Dehydrogenative [4 + 2] Cycloaddition of 1,3-Dienes with Nitriles, J. Am. Chem. Soc., 2011, 133, 18018–18021 CrossRef CAS PubMed; (b) E. M. Stang and M. C. White, Molecular Complexity via C-H Activation: A Dehydrogenative Diels-Alder Reaction, J. Am. Chem. Soc., 2011, 133, 14892–14895 CrossRef CAS PubMed; (c) A. Collado, M. A. Esteruelas, M. Gulías, J. L. Mascareñas and E. Oñate, Reactions of an Osmium(IV) Complex with Allenedienes: Coordination and Intramolecular Cycloadditions, Organometallics, 2012, 31, 4450–4458 CrossRef CAS.
- W.-Q. Wu, Y. Lin, Y. Li and H. Shi, Catalytic Dehydrogenative (3 + 2) Cycloaddition of Alkylbenzenes via π-Coordination, J. Am. Chem. Soc., 2023, 145, 9464–9470 CrossRef CAS PubMed.
- E. Haraburda, A. Lledó, A. Roglans and A. Pla-Quintana, Dehydrogenative [2 + 2 + 2] Cycloaddition of Cyano-yne-allene Substrates: Convenient Access to 2,6-Naphthyridine Scaffolds, Org. Lett., 2015, 17, 2882–2885 CrossRef CAS PubMed.
- T. Otani, T. Saito, R. Sakamoto, H. Osada, A. Hirahara, N. Furukawa, N. Kutsumura, T. Matsuo and K. Tamao, Intramolecular [2 + 2 + 2] Cycloaddition of Bis(propargylphenyl)carbodiimides: Synthesis of L-Shaped π-Extended Compounds with Pyrrolo[1,2-a][1,8]naphthyridine Corner Units, Chem. Commun., 2013, 49, 6206–6208 RSC.
- A. Roglans, A. Pla-Quintana and M. Solà, Mechanistic Studies of Transition-Metal-Catalyzed [2 + 2 + 2] Cycloaddition Reactions, Chem. Rev., 2021, 121, 1894–1979 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- A. D. Becke, Density–functional Thermochemistry. III. The Role of Exact Exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H–Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the Damping Function in Dispersion Corrected Density Functional Theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- T. H. Dunning Jr., Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- D. E. Woon and T. H. Dunning Jr., Gaussian Basis Sets for Use in Correlated Molecular Calculations. III. The Atoms Aluminum through Argon, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- K. A. Peterson, D. Figgen, M. Dolg and H. Stoll, Energy-Consistent Relativistic Pseudopotentials and Correlation Consistent Basis Sets for the 4d Elements Y-Pd, J. Chem. Phys., 2007, 126, 124101 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 other Functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- C. Gonzalez and H. B. Schlegel, An improved Algorithm for Reaction Path Following, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS.
- U. Rosenthal, V. V. Burlakov, M. A. Bach and T. Beweries, Five-Membered Metallacycles of Titanium and Zirconium – Attractive Compounds for Organometallic Chemistry and Catalysis, Chem. Soc. Rev., 2007, 36, 719–728 RSC.
- For the complete mechanism towards the formation of 3a see the ESI.†.
- (a) P. A. Inglesby, J. Bacsa, D. E. Negru and P. A. Evans, The Isolation and Characterization of a Rhodacycle Intermediate Implicated in Metal-Catalyzed Reactions of Alkylidenecyclopropanes, Angew. Chem., Int. Ed., 2014, 53, 3952–3956 CrossRef CAS PubMed; (b) E. Da Concepción, I. Fernández, J. L. Mascareñas and F. López, Highly Enantioselective Cobalt-Catalyzed (3 + 2) Cycloadditions of Alkylidenecyclopropanes, Angew. Chem., Int. Ed., 2021, 60, 8182–8188 CrossRef PubMed.
- (a) S. Ma, P. Lu, L. Lu, H. Hou, J. Wei, Q. He, Z. Gu, X. Jiang and X. Jin, What Can a Metal Catalyst Do with Allenes? One-Step Formation of Steroid Scaffolds from Readily Available Starting Materials, Angew. Chem., Int. Ed., 2005, 44, 5275–5278 CrossRef CAS PubMed; (b) X. Deng, L.-Y. Shi, J. Lan, Y.-Q. Guan, X. Zhang, H. Lv, L. W. Chung and X. Zhang, Enantioselective Rhodium-Catalyzed Cycloisomerization of 1,6-Allenynes to Access 5/6-Fused Bicycle[4.3.0]nonadienes, Nat. Commun., 2019, 10, 949 CrossRef PubMed; (c) J. Vila, M. Solà, T. Achard, S. Bellemin-Laponnaz, A. Pla-Quintana and A. Roglans, Rh(I) Complexes with Hemilabile Thioether-Functionalized NHC Ligands as Catalysts for [2 + 2 + 2] Cycloaddition of 1,5-Bisallenes and Alkynes, ACS Catal., 2023, 13, 3201–3210 CrossRef CAS PubMed.
- A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, SambVca: A Web Application for the Calculation of the Buried Volume of N-Heterocyclic Carbene Ligands, Eur. J. Inorg. Chem., 2009, 13, 1759–1766 CrossRef.
- A. Artigas, J. Vila, A. Lledó, M. Solà, A. Pla-Quintana and A. Roglans, A Rh-Catalyzed Cycloisomerization/Diels-Alder Cascade Reaction of 1,5-Bisallenes for the Synthesis of Polycyclic Heterocycles, Org. Lett., 2019, 21, 6608–6613 CrossRef CAS PubMed.
- A. Patel, G. A. Barcan, O. Kwon and K. N. Houk, Origins of 1,6-Stereoinduction in Torquoselective 6π Electrocyclizations, J. Am. Chem. Soc., 2013, 135, 4878–4883 CrossRef CAS PubMed.
- (a) J. A. Varela, S. G. Rubín, L. Castedo and C. Saá, “Formal” and Standard Ruthenium-Catalyzed [2 + 2 + 2] Cycloaddition Reaction of 1,6-Diynes to Alkenes: A Mechanistic Density Functional Study, J. Org. Chem., 2008, 73, 1320–1332 CrossRef CAS PubMed; (b) D. Leboeuf, L. Iannazzo, A. Geny, M. Malacria, K. P. C. Vollhardt, C. Aubert and V. Gandon, Cobalt-Mediated Linear 2:1 Co-oligomerization of Alkynes with Enol Ethers to Give 1-Alkoxy-1,3,5-Trienes: A Missing Mode of Reactivity, Chem. – Eur. J., 2010, 16, 8904–8913 CrossRef CAS PubMed; (c) W. Yuan, Y. Wei and M. Shi, Ruthenium-Catalyzed Intramolecular [2 + 2 + 2] Cycloaddition and Tandem Cross-Metathesis of Triynes and Enediynes, ChemistryOpen, 2013, 2, 63–68 CrossRef CAS PubMed; (d) S. Alvarez, S. Medina, G. Domínguez and J. Pérez-Castells, Ruthenium Alkylidene-Catalyzed Reaction of 1,6-Heptadiynes with Alkenes, J. Org. Chem., 2015, 80, 2436–2442 CrossRef CAS PubMed.
- The reaction also proceeds in the absence of triethylamine (for substrate 1 (X = Z = NTs and Y = (CH2)2), Scheme 1, without Et3N cycloadduct was obtained in 50% yield in 30 min at 120 °C, whereas upon addition of catalytic amounts (0.05 equiv.) of Et3N a 66% yield was obtained in 10 min at 120 °C). In the absence of triethylamine, this proton shift can be assisted by traces of water present in the reaction mixture at the beginning of the reaction and by the pyridine ring generated in the cycloadduct as the reaction progresses.
Footnote |
| † Electronic supplementary information (ESI) available: Complete mechanism for the formation of 3a. All xyz coordinates and absolute energies of all computed species. See DOI: https://doi.org/10.1039/d4qo00378k |
| This journal is © the Partner Organisations 2024 |