
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxidative cyclization and enzyme-free deiodination of thyroid hormones†
Julian
Spils
 a,
Lucien D.
Caspers
a,
Pim
Puylaert
b and
Boris J.
Nachtsheim
*a
a,
Lucien D.
Caspers
a,
Pim
Puylaert
b and
Boris J.
Nachtsheim
*a
aInstitute for Organic and Analytical Chemistry, University of Bremen, Leobener Straße 7, 28359 Bremen, Germany. E-mail: nachtsheim@uni-bremen.de
bInstitute for Inorganic Chemistry and Crystallography, University of Bremen, Leobener Straße 7, 28359 Bremen, Germany
First published on 18th March 2024
Abstract
We introduce the first non-enzymatic deiodination of thyroid hormones from a so far unknown hypervalent iodaoxinium state. After developing oxidative processes for thyroxine (T4)-derived model cyclic diaryliodonium salts, we successfully produced an iodaoxinium salt through the direct oxidation of O- and N-protected T4. DFT calculations revealed a novel halogen bonding-based deiodination mechanism, circumventing the traditional selenium-dependent pathways. Our findings open new avenues in thyroid hormone chemistry, suggesting alternative mechanisms for their involvement in metabolic processes, regulation of oxidative stress, and gene expression.
Introduction
Thyroid hormones (TH) play a vital role in regulating metabolic processes and are crucial for the development and metabolic homeostasis of higher organisms.1 They are in particular capable of modulating gene expression through binding to TH-specific transcription factors, which are present in almost all vertebrate tissues.1–3 From thyroxine (T4) as the main TH initially secreted by the mammalian thyroid gland, other THs such as 3,5,3′-triiodothyronine (T3) or 3,3′,5′-triiodothyronine (rT3) are formed via selective deiodination by one of three selenoenzymes DIO1, DIO2 and DIO3 (Scheme 1).4–6 Of all THs, T3 is the most active derivative with up to 40 times higher affinity to thyroid receptors than its parent compound T4.7,8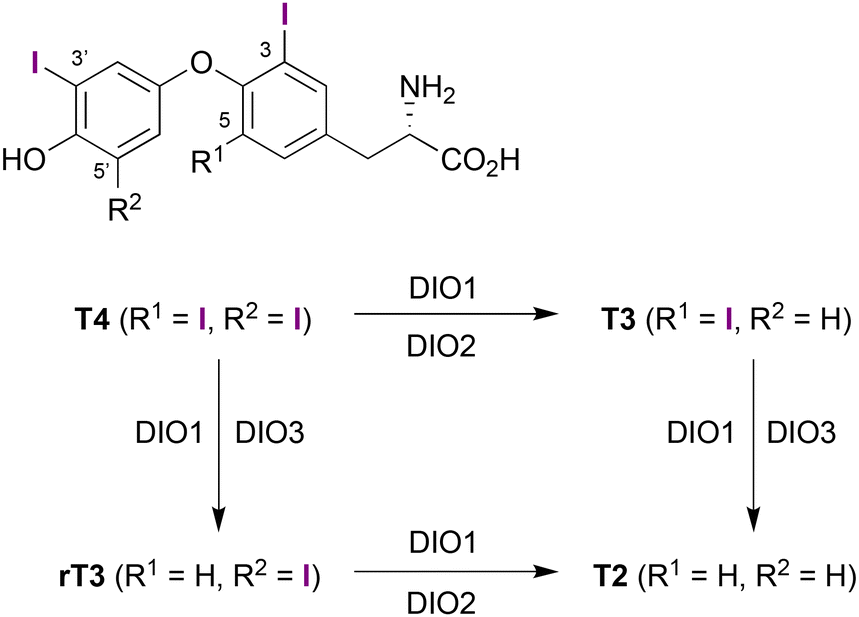 | ||
| Scheme 1 Formation of different THs via deiodination of thyroxine (T4). | ||
The exact mechanism of the reductive deiodination is still under investigation, but it is evident that selenocysteines are present in all related enzymes and recent literature has shown that halogen bond activation plays an important role in this process.9–12 Nobody described a selenium-independent deiodination mechanism so far in biological systems that results in a regioselective deiodination.
We found such a potential pathway by serendipity as part of an ongoing research program that is intended to elaborate and understand a potential biologically relevant non-enzymatic oxidation of thyroxines to hypervalent iodine reagents ox-T3/T4 or cyclic iodaoxinium salts 1 (cyclo-ox-T3/T4) derived from them (Scheme 2).13–18 In the future these substrates could also enable diverse further derivations based on previous work by Chen and co-workers.19,20
 | ||
| Scheme 2 Proposed oxidation of THs to hypervalent iodine reagents and their cyclization to iodaoxinium salts 1. | ||
Results and discussion
In our initial experiments, we oxidized commercially available thyroxine (T4) using common oxidants such as peracids, hypochlorite solutions, SelectFluor®, and Oxone®. However, these attempts only led to the decomposition of the starting material.Notably, the solubility issues and cleavage of the central diaryl ether, due to quinone formation, posed significant challenges.
Consequently, we shifted our focus from THs to simplified structures, where the α-amino acid is replaced with a simple methyl group and the free phenol group is protected as a methyl ether. We employed a straightforward reaction sequence, beginning with a nucleophilic aromatic substitution followed by a Sandmeyer iodination. This approach enabled us to isolate the desired simplified TH analogues 2a–c in yields of 50–61%, starting from 4-methoxyphenol and the corresponding nitroarenes, as shown in Scheme 3. We then investigated their oxidation using literature-known conditions.18,21,22
 | ||
| Scheme 3 Synthesis of iodoarenes 2 and their oxidative cyclization to iodaoxinium salts 3 (50 μmol scale). Yields in brackets are upscaled (500 μmol). A detailed synthetic procedure is shown in the ESI.† | ||
In our initial oxidation experiments, as outlined in Scheme 2, we aimed to isolate an acyclic aryl-λ3-iodane derivative from either ox-T3 or ox-T4. However, these attempts were unsuccessful. Instead, when using SelectFluor® as the oxidant and subsequently treating the reaction with TfOH, we successfully isolated the desired cyclic diaryliodonium salts, 3a and 3c. These compounds were obtained in yields of 73% and 23%, respectively.
Treating compound 2b under similar oxidative conditions proved more challenging, as we only detected trace amounts of product 3b and its isomers via HPLC-MS. Attempts with other polyiodinated diaryl ethers did not yield any products, as detailed in the ESI, Table 1.† Our subsequent objective was to synthesize the free phenol of type 4, as depicted in Scheme 4. A recently published one-step method has demonstrated the stability of similar acyclic iodonium salts,23 yet only one meta-hydroxy-substituted derivative closely related to our target structure has been described.24
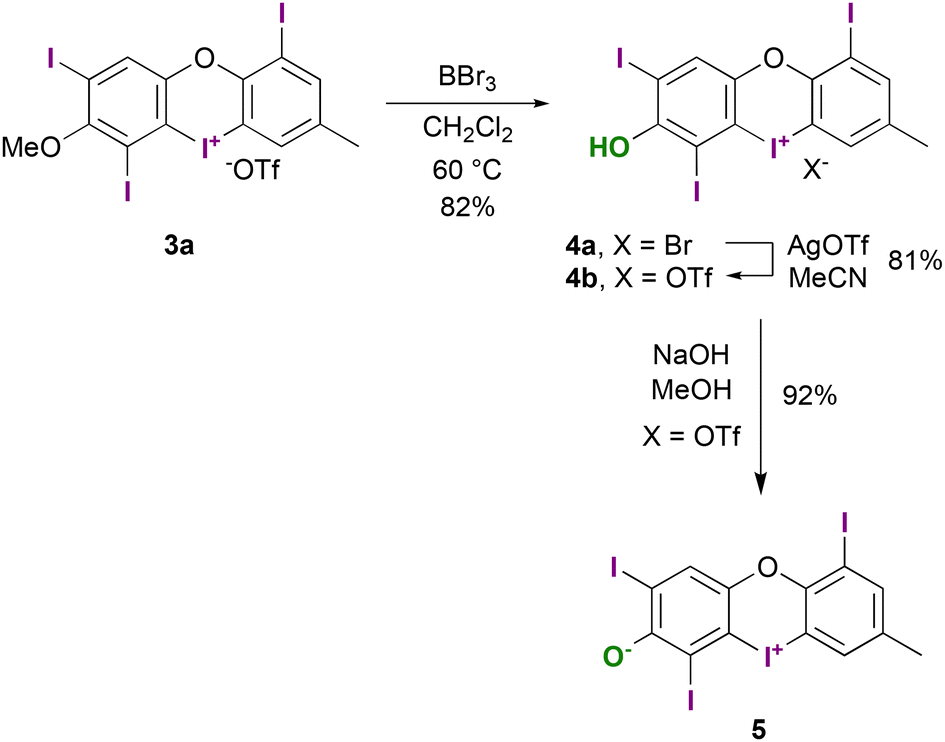 | ||
| Scheme 4 Deprotection of 3a towards 4a and subsequent anion exchange to the triflate salt 4b, followed by deprotonation to the zwitterion 5. | ||
For the deprotection of the methyl ether, we found that typical conditions using BBr3 were effective. However, due to the low solubility of 3a and its bromide salt, we had to employ elevated temperatures. Ultimately, we successfully obtained the unprotected bromide salt 4a in 82% yield. We then achieved anion exchange to the triflate salt 4b using AgOTf with an 81% yield. Further deprotonation with NaOH gave the orange-coloured zwitterion 5 with a 92% yield.
After confirming the stability of cyclic iodaoxinium salts 4, we confidently proceeded to work with thyroid hormones (THs) directly. We began with thyroxine, converting its carboxylic acid into a methyl ester and acylating both the amino and phenol groups. However, using our previously developed method, we only observed trace amounts of the desired product. Switching to well-established oxidative conditions with mCPBA and TfOH proved successful. This approach enabled the direct synthesis of the iodaoxinium salt 6a through in situ deprotection of the phenol group, yielding a moderate efficiency with an isolated yield of 53% (as shown in Scheme 5). We also noted that phenol 6 could be deprotonated to produce the zwitterion 7, which was stable at room temperature in its solid form achieving a yield of 72%, although decomposition was observed in highly polar solvents such as DMSO, DMF, and NMP.
 | ||
| Scheme 5 Synthesis of TH-derived iodaoxinium salts 6 and 7 from protected thyroxine T4. | ||
The solid state structure of the iodaoxinium salt 6a was determined by X-ray diffraction analysis, revealing it had crystallised as a hydrogen-bridged dimer (Fig. 1). The geometry around the iodonium centre is as expected in a T-shape with C–I–C angles of 90.0° and 89.7° and triflates coordinating at angles of 175.2° and 172.69°. The I–O bond between triflate and iodine is at 2.757 Å and 2.752 Å, comparable to known iodaoxinium salts.22 Interestingly the dimer shows hydrogen bonding between the phenol and the amide carbonyl indicated by O–O distances of 2.563 Å and 2.606 Å. We also observed significant π–π-interactions between the two phenol-bearing aromatic rings with a centroid-to-centroid distance of 3.650 Å.
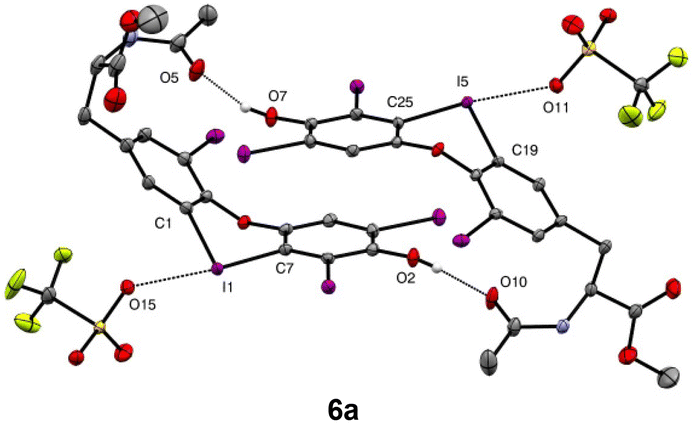 | ||
| Fig. 1 Single crystal structure (ORTEP drawing; hydrogen atoms, except those participating in hydrogen bonding, were omitted) of 6a as a dimer (CCDC 2291802†) with aromatic ring interaction and hydrogen bonding visible inside the dimer. Thermal ellipsoids displayed with 50% probability. Selected parameters [Å, °]: I1–O15: 2.757(2); I5–O11: 2.752(2); O5–O7: 2.563(4); O2–O10: 2.606(4); C1–I1–C7: 90.0(1); C19–I5–C25: 89.7(1); C7–I1–O15: 175.2(1); C25–I5–O11: 172.69(9). | ||
Our objective was to create a thyroid hormone (TH)-derived substrate, with the sole modification being a cyclized iodonium centre. To achieve this, we needed to deprotect the amino acid. Treating 6a with aqueous HCl at 120 °C led to the formation of the desired T4-derived iodaoxinium salt 1a, with a yield of 57% (Scheme 6). However, during a side-product analysis of this reaction using HPLC-MS, we unexpectedly discovered a deiodination product. On lowering the reaction temperature and increasing the reaction time, this side product was formed exclusively. We identified this compound as the T3-derived iodaoxinium salt 1b, which was isolated in 65% yield.
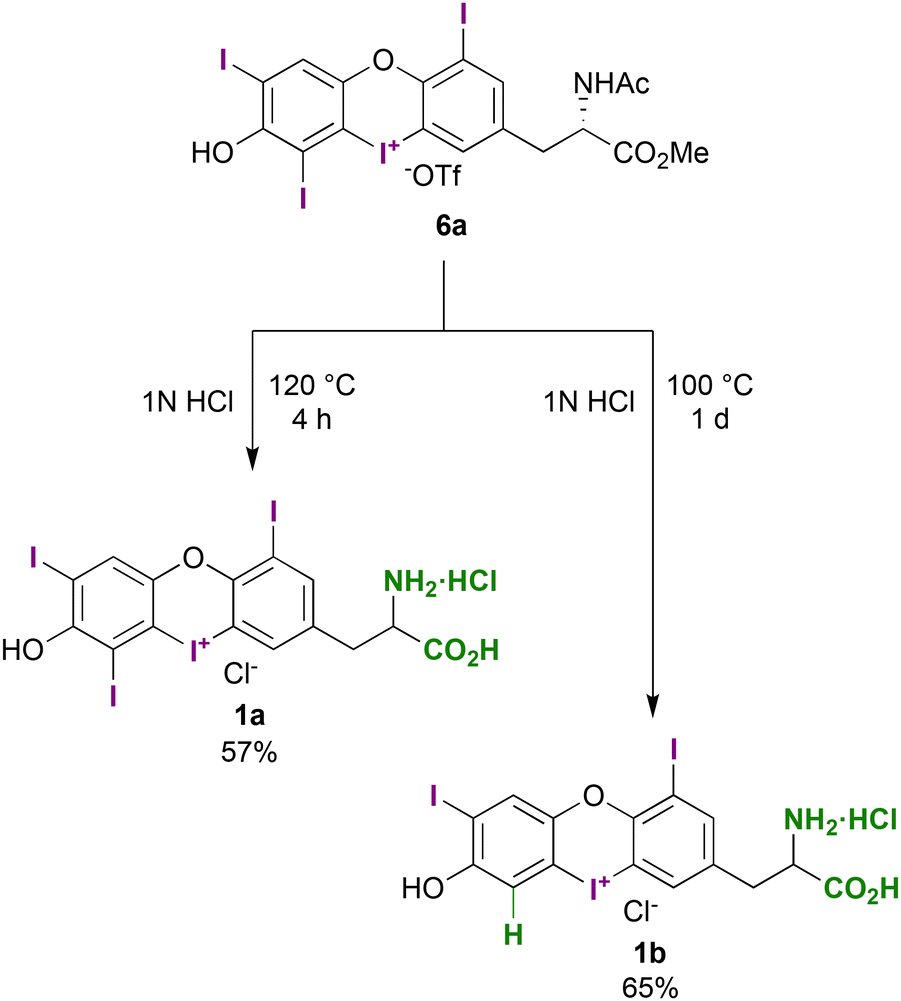 | ||
| Scheme 6 Deprotection of the amino acid functionality of salt 7a to generate the salt 1a and via selective protic deiodination 1b. | ||
To gain a deeper understanding of the unexpected deiodination, especially its high selectivity for the C6 position ortho to the iodonium centre, we conducted Density Functional Theory (DFT) calculations. These were performed at the PBE0-D3(BJ)/def2-TZVP+CPCM level of theory, using geometries optimized at the PBE0-D3(BJ)/def2-SVP+CPCM level. For guidance in elucidating the correct reaction pathway, we referred to a study by Rafferty and co-workers. This research explored the deiodination of thyroid hormones by selenols, employing a simplified catalytic triad.9 Following this path, in the presence of HCl as a Brønsted-acid, a trimolecular reaction through TS-A should be induced to generate ICl and the deiodinated arene B (Scheme 7).
 | ||
| Scheme 7 A: Selenocysteine-mediated deiodination mechanism as proposed by Rafferty (red) vs. the mechanism via enolization (green). B: Corresponding chloride/HCl-mediated deiodination mechanism. | ||
Building on this mechanism, we explored three separate pathways, each beginning with an XB interaction between a chloride ion and one of the three monovalent iodine atoms located at positions C1, C6, and C8. These calculated reaction pathways are depicted in Scheme 8. Initially, the addition of HCl led to the formation of XB adducts 1a-6-HCl (with an energy of +9.23 kcal mol−1), 1a-8-HCl (+10.09 kcal mol−1), and 1a-1-HCl (+10.49 kcal mol−1). In our notation, the middle index (1a-X-HCl) specifies the position of the coordinated iodine. These XB complexes then transitioned into the states TS6-1, TS6-2, TS8, and TS1. The difference between TS6-1 and TS6-2 lies in the varying attack sites of the chloride anion. A comparison of the relative energies of these transition states revealed that TS6-1 and TS6-2, leading to the formation of the T3-derived iodaoxinium salt, possessed the lowest activation barriers at 24.92 kcal mol−1 and 24.26 kcal mol−1, respectively. In contrast, TS8 and TS1 exhibited significantly higher barriers, at 28.32 kcal mol−1 and 28.54 kcal mol−1, respectively. Moreover, deiodination at C6 uniquely resulted in the transformation of the bent iodaoxinium 1a into a less bent geometry 1b, accompanied by a notable twist angle change of +6.7° between the two phenyl rings. Additionally, the dihedral angle between C6–C5a–I–Cl relaxed by −23.8° during the transition from 1a to 1b. Transitions to other deiodinated iodaoxinium salts 1b′ and 1b′′, did not exhibit such significant changes in ground state geometry.
 | ||
| Scheme 8 Energy profile for the deiodination of 1avia the addition of HCl to generate 1b, 1b′ and 1b′′. And a simplified schematic representation of the mechanism. | ||
To gain further insight into this reaction and to verify the proposed halogen bond interaction, we looked at the binding of compound 6a with tetra-n-butylammonium chloride (TBACl). The initial addition of one equivalent of TBACl resulted in a general downfield shift, caused by anion exchange from triflate to chloride, matching spectral data for 6b. Upon further addition of TBACl (5 eq.) only for one carbon a shift (downfield) is observed (Fig. 2). This signal likely corresponds to position C6 as there is no correpsonding 13C-signal in the NMR of C6-deiodinated 1b. When we added a huge excess of TBACl (50 eq.), further downfield shifts are observed for C6 as well as for the other two iodinated carbons (C1 and C8).
 | ||
| Fig. 2 NMR observations upon addition of different amounts of tetra-n-butylammonium chloride (TBACl) to iodaoxinium salt 6a and comparison to 1b. | ||
As previously mentioned, the iodaoxinium salt 1a possesses an asymmetric bent geometry, allowing it to be protonated by HCl from either a convex or a concave face. In Scheme 9, we present a direct comparison of both pathways. Protonation through the convex face, viaTS6-1, reveals a weak XB-interaction between the chlorine atom of the HCl molecule and the iodonium ion, measured at a distance of 3.22 Å and an angle of 158.49°. On the other hand, protonation from the concave face leads to a slightly more favourable transition state, TS6-2, with an energy difference of −0.66 kcal mol−1. In this state, a shorter than van der Waals distance between the iodine atom at C6 and the iodonium ion still suggests a weak XB-bond, however with an even more distorted angle of 146.83°. This interaction appears to be energetically slightly more favourable compared to the Cl–I interaction observed in TS6-1. Given the minimal energy difference between the two transition states, it's likely that both pathways will occur under the applied elevated reaction temperature of 100 °C.
 | ||
| Scheme 9 Distinct transition states from a convex and concave protonation of 1a, both leading to 1b. | ||
However, both transition states represent a unique selenoenzyme-free deiodination mechanism of thyroxines based on intrinsic XB interactions. This discovery not only broadens the understanding of thyroid hormone chemistry but also challenges the conventional belief that enzymatic pathways are the sole mediators for the selective deiodination of THs. These XB interactions, driving the deiodination process, highlight a novel aspect of thyroxine's molecular behaviour, potentially reshaping our approach to thyroid hormone-related (bio)chemistry.
Conclusions
In summary, our research demonstrates that oxidized thyroxine (T4), in the form of a hypervalent iodaoxinium salt (1a), is a stable entity that can be directly synthesized from O-acylated thyroxine under oxidative and acidic conditions. Interestingly, we discovered that 1a can undergo selective deiodination at the C6 position, resulting in an oxidized T3-derivative. This deiodination process, which occurs without the involvement of selenoenzymes, is not only unprecedented but also features a unique activation mechanism driven by XB interactions with the central iodonium ion. To further explore the possibility of these thyroxine-derived iodaoxinium salts acting as reactive intermediates under oxidative stress in biological systems, it is essential to confirm their presence in vivo. Additionally, we aim to investigate their potential reduction to T3 or reverse T3 (rT3) by natural reducing agents, such as flavins or dihydropyridines.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. P. acknowledges the Central Research Development Fund of the University of Bremen for the postdoctoral fellowship.References
- R. Mullur, Y. Y. Liu and G. A. Brent, Thyroid hormone regulation of metabolism, Physiol. Rev., 2014, 94, 355–382 CrossRef CAS PubMed.
- A. C. Bianco and B. W. Kim, Deiodinases: implications of the local control of thyroid hormone action, J. Clin. Invest., 2006, 116, 2571–2579 CrossRef CAS PubMed.
- G. A. Brent, Mechanisms of thyroid hormone action, J. Clin. Invest., 2012, 122, 3035–3043 CrossRef CAS PubMed.
- J. Köhrle, Iodothyronine deiodinases, Methods Enzymol., 2002, 347, 125–167 Search PubMed.
- A. C. Bianco, D. Salvatore, B. Gereben, M. J. Berry and P. R. Larsen, Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases, Endocr. Rev., 2002, 23, 38–89 CrossRef CAS PubMed.
- G. G. Kuiper, M. H. Kester, R. P. Peeters and T. J. Visser, Biochemical mechanisms of thyroid hormone deiodination, Thyroid, 2005, 15, 787–798 CrossRef CAS PubMed.
- H. H. Samuels, J. S. Tsai, J. Casanova and F. Stanley, Thyroid hormone action: in vitro characterization of solubilized nuclear receptors from rat liver and cultured GH1 cells, J. Clin. Invest., 1974, 54, 853–865 CrossRef CAS PubMed.
- J. W. Apriletti, N. L. Eberhardt, K. R. Latham and J. D. Baxter, Affinity chromatography of thyroid hormone receptors. Biospecific elution from support matrices, characterization of the partially purified receptor, J. Biol. Chem., 1981, 256, 12094–12101 CrossRef CAS PubMed.
- C. A. Bayse and E. R. Rafferty, Is halogen bonding the basis for iodothyronine deiodinase activity?, Inorg. Chem., 2010, 49, 5365–5367 CrossRef CAS PubMed.
- D. Manna, S. Mondal and G. Mugesh, Halogen bonding controls the regioselectivity of the deiodination of thyroid hormones and their sulfate analogues, Chemistry, 2015, 21, 2409–2416 CrossRef CAS PubMed.
- C. A. Bayse, Halogen Bonding from the Bonding Perspective with Considerations for Mechanisms of Thyroid Hormone Activation and Inhibition, New J. Chem., 2018, 42, 10623–10632 RSC.
- S. Mondal, D. Manna, K. Raja and G. Mugesh, Halogen Bonding in Biomimetic Deiodination of Thyroid Hormones and their Metabolites and Dehalogenation of Halogenated Nucleosides, ChemBioChem, 2020, 21, 911–923 CrossRef CAS PubMed.
- B. Olofsson, in Hypervalent Iodine Chemistry, ed. T. Wirth, Springer International Publishing, Cham, 2015/10/27 edn, 2016, pp. 135–166 Search PubMed.
- A. Yoshimura and V. V. Zhdankin, Advances in Synthetic Applications of Hypervalent Iodine Compounds, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed.
- R. Robidas, D. L. Reinhard, C. Y. Legault and S. M. Huber, Iodine(III)-Based Halogen Bond Donors: Properties and Applications, Chem. Rec., 2021, 21, 1912–1927 CrossRef CAS PubMed.
- N. Chatterjee and A. Goswami, Synthesis and Application of Cyclic Diaryliodonium Salts: A Platform for Bifunctionalization in a Single Step, Eur. J. Org. Chem., 2017, 3023–3032 CrossRef CAS.
- H. C. Cheng, J. L. Ma and P. H. Guo, Cyclic Diaryliodonium Salts: Eco–Friendly and Versatile Building Blocks for Organic Synthesis, Adv. Synth. Catal., 2023, 365, 1112–1139 CrossRef CAS.
- X. Peng, A. Rahim, W. Peng, F. Jiang, Z. Gu and S. Wen, Recent Progress in Cyclic Aryliodonium Chemistry: Syntheses and Applications, Chem. Rev., 2023, 123, 1364–1416 CrossRef CAS PubMed.
- C. Chen, P. Wu, J. Zhou and Z. Bao, The Preparation and Application of Diaryliodonium Salts Derived from Gemfibrozil and Gemfibrozil Methyl Ester, Synthesis, 2021, 1388–1394 Search PubMed.
- J. Zhou, Z. Bao, P. Wu and C. Chen, Preparation and Synthetic Application of Naproxen-Containing Diaryliodonium Salts, Molecules, 2021, 26, 3240 CrossRef CAS PubMed.
- D. L. Reinhard, F. Heinen, J. Stoesser, E. Engelage and S. M. Huber, Tuning the Halogen Bonding Strength of Cyclic Diaryliodonium Salts, Helv. Chim. Acta, 2021, 104, e2000221 CrossRef CAS.
- M. Damrath, L. D. Caspers, D. Duvinage and B. J. Nachtsheim, One-Pot Synthesis of Heteroatom-Bridged Cyclic Diaryliodonium Salts, Org. Lett., 2022, 24, 2562–2566 CrossRef CAS PubMed.
- A. Yoshimura, M. T. Shea, O. Guselnikova, P. S. Postnikov, G. T. Rohde, A. Saito, M. S. Yusubov, V. N. Nemykin and V. V. Zhdankin, Preparation and structure of phenolic aryliodonium salts, Chem. Commun., 2018, 54, 10363–10366 RSC.
- K. Zhu, K. Xu, Q. Fang, Y. Wang, B. Tang and F. Zhang, Enantioselective Synthesis of Axially Chiral Biaryls via Cu-Catalyzed Acyloxylation of Cyclic Diaryliodonium Salts, ACS Catal., 2019, 9, 4951–4957 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2291802. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qo00220b |
| This journal is © the Partner Organisations 2024 |