
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polyenolate-mediated reaction cascade initiated by higher-order-cycloaddition for the construction of polycarbocyclic scaffolds†
Adam
Cieśliński
,
Anna
Skrzyńska
,
Artur
Przydacz
and
Łukasz
Albrecht
 *
*
Institute of Organic Chemistry, Faculty of Chemistry, Lodz University of Technology, Żeromskiego 116, 90-924 Łódź, Poland. E-mail: lukasz.albrecht@p.lodz.pl; anna.skrzynska@p.lodz.pl; Web: https://www.a-teamlab.p.lodz.pl/
First published on 19th January 2024
Abstract
In this manuscript, the application of polyenolate chemistry for the activation of higherenes towards a higher-order-cycloaddition-initiated cascade reaction is demonstrated. This has been made possible by using alkylidenemalononitriles derived from indene-2-carbaldehydes as the higherene precursors. Their reaction with an iminium ion, generated in situ in the presence of a suitable amino catalyst, allowed for a cascade reaction involving [10 + 2]-higher-order cycloaddition followed by Michael addition and aldol condensation to proceed in a highly stereoselective fashion.
The introduction of two electron-withdrawing substituents on a double bond constitutes a common strategy that enables the transformation of its properties from nucleophilic into electrophilic.1,2 If an enolizable alkyl group is present in a suitable position of such compounds, it becomes a pronucleophilic species that serves as a precursor of a polyenolate intermediate (Scheme 1, top).2 This group of reactive intermediates, generated under Brønsted base catalysis, has recently proven its usefulness for asymmetric synthesis. Such an activation strategy of pronucleophilic species constitutes an interesting alternative to aminocatalytic polyenamine chemistry.3 It is noteworthy that many reactions involving polyenolate or polyenamine intermediates proceed in a cascade manner allowing for the construction of multiple carbon–carbon and carbon–heteroatom bonds via a single chemical procedure.4
 | ||
| Scheme 1 Various activation strategies for higher-order cycloadditions and synthetic objectives of our studies. | ||
Chemical transformations that enable the construction of a new ring system starting from acyclic precursors are commonly referred to as cycloaddition reactions.5 According to Huisgen “the concept of cycloaddition gives a formal description of an overall reaction but not a mechanistic interpretation”.6 As a consequence, their mechanism can be either concerted, concerted asynchronous or stepwise, with all these transformations belonging to this specious group of chemical reactions. Cycloadditions constitute a highly reliable synthetic tool commonly employed for the preparation of both carbo- and heterocyclic systems with the Diels–Alder reaction and its hetero-versions being the most prominent examples.7
Higher-order cycloadditions have been introduced already in 1970 by Woodward and Houk.8 They can be described as transformations that take place with the participation of overall more than 6π-electrons. Recently, this field of research has regained the attention of the chemical community with interesting new reports appearing in the literature, thus indicating the potential for further research.9–13 In particular, the application of organocatalytic activation modes opened new possibilities in this area.10 Notably, two main strategies have been adopted for the activation of higherenes towards higher-order cycloadditions (Scheme 1, middle): (1) LUMO-lowering approach (via introduction of electron-withdrawing substituents into their structure)11 and (2) HOMO-rising approach (via aminocatalytic formation of polyenamine intermediates).12 The application of the related HOMO-rising activation of higherenes based on polyenolate formation remains elusive.
Recently, we demonstrated that polyenolates, generated under Brønsted base catalysis, serve as a reactive group of higherenophiles for higher-order cycloadditions.13 Dienolates generated from either 2-alkyl-1,4-naphthoquinones or 5-substituted-2(3H)-furanones reacted with 8,8-dicyanoheptafulvenes via an approach involving LUMO-lowering activation of higherenes. Given the lack of higherene activation strategies based on polyenolate formation, studies on such a reactivity pattern were undertaken. It was anticipated that by conversion of indene-2-carbaldehydes into alkylidenemalononitrile derivatives, access to effective polyenolate precursors should be possible. Their deprotonation under basic conditions and reaction with suitable higherenophiles should create a unique reaction manifold for the development of new reactivities.
Herein, we present our studies on the development of a cascade reaction involving alkylidenemalononitriles derived from indene-2-carbaldehydes as higherene precursors (Scheme 1, bottom). Their reaction with α,β-unsaturated aldehydes realized under aminocatalytic conditions proceeded in a cascade manner and involved [10 + 2]-higher-order cycloaddition followed by Michael addition and aldol condensation.
Studies were initiated by testing the reaction between 2-((3-phenyl-1H-inden-2-yl)methylene)malononitrile 1a and cinnamaldehyde 2a for the synthesis of the desired 3a. The model reaction was performed using several commercially available aminocatalysts 4 in dichloromethane as a solvent (Table 1, entries 1–3). Gratifyingly, the use of 4a provided the expected product 3a, but the conversion of the starting material 1a was unsatisfactory. To improve the reaction outcome, the screening of additives was performed. Better results were observed when an organic acid was used as a co-catalyst (Table 1, entries 4 and 5). Then, a thorough evaluation of the solvent effect showed that 1,2-dichloroethane improved the conversion and afforded product 3a in 43% yield (Table 1, entries 6–10). Additionally, the use of a chiral acidic co-catalyst increased the diastereoselectivity of the process. R-Mandelic acid co-catalyzed this reaction, providing product 3a in 45% yield with excellent stereoselectivity (Table 1, entry 11). The use of S-mandelic acid in combination with catalyst 4a diminished the efficiency of the reaction (Table 1, entry 12). Then, the change of the reagent's ratio showed that the application of a 2-fold excess of aldehyde 2a did not bring a significant improvement in terms of conversion (Table 1, entry 13). However, reducing the excess of 2a allowed us to obtain 3a with excellent selectivity and the yield increased to 65% (Table 1, entry 14), indicating the optimized conditions for the synthesis of 3avia a higher-order-cycloaddition-initiated cascade reaction. Furthermore, product 3a could also be effectively accessed when the reaction was carried out on a 20-fold higher scale under the optimized conditions (Table 1, entry 15).
|
|
|||||
|---|---|---|---|---|---|
| Solvent (catalyst) | Additive (40 mol%) | Conv. (yield)b [%] | drc | erd | |
| a Reactions performed at the 0.05 mmol scale using 1a (0.05 mmol), 2a (0.12 mmol) and catalyst 4 (20 mol%) in 0.2 mL of the solvent for 24 h at rt. b Determined by 1H NMR of the crude reaction mixture. The yield of the isolated product 3a after column chromatography is given in parentheses. c Determined by 1H NMR of the crude reaction mixture. d Determined by chiral UPC2. e The reaction performed using 1a (0.05 mmol) and 2a (0.2 mmol). f The reaction performed using 1a (0.1 mmol) and 2a (0.1 mmol). g Reaction performed on a 1 mmol scale for 48 h. | |||||
| 1 | CH2Cl2 (4a) | — | 20 | n.d. | n.d. |
| 2 | CH2Cl2 (4b) | — | Trace | n.d. | n.d. |
| 3 | CH2Cl2 (4c) | — | Trace | n.d. | n.d. |
| 4 | CH2Cl2 (4a) | PhCO2H | 52 | 4.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
>99.1 |
| 5 | CH2Cl2 (4a) | NaOAc | Trace | n.d. | n.d. |
| 6 | CHCl3 (4a) | PhCO2H | 49 | 3:1 |
>99.1 |
| 7 | Et2O (4a) | PhCO2H | 64 | 4:1 |
>99.1 |
| 8 | Toluene (4a) | PhCO2H | 36 | 4.5:1 |
n.d. |
| 9 | PhCF3 (4a) | PhCO2H | 47 | 8:1 |
n.d. |
| 10 | Cl(CH2)2Cl (4a) | PhCO2H | >95 (43) | 5:1 |
>99.1 |
| 11 | Cl(CH2)2Cl (4a) | R-Mandelic acid | >95 (45) | 15:1 |
>99.1 |
| 12 | Cl(CH2)2Cl (4a) | S-Mandelic acid | 35 | 20:1 |
>99.1 |
| 13e | Cl(CH2)2Cl (4a) | R-Mandelic acid | >95 (50) | 20:1 |
>99.1 |
| 14f | Cl(CH2)2Cl (4a) | R-Mandelic acid | >95 (65) | >20:1 |
>99.1 |
| 15g | Cl(CH2)2Cl (4a) | R-Mandelic acid | >95 (63) | >20:1 |
>99:1 |

To verify the application scope of this polyenolate-mediated reaction cascade, a series of α,β-unsaturated aldehydes 2 and alkylidenemalononitrile derivatives 1 were investigated (Table 2 and Scheme 2). As shown in Table 2, a variety of α,β-unsaturated aldehydes 2b–h bearing different aromatic groups successfully participated in the cascade, delivering the desired polycarbocyclic scaffolds 3b–h in generally good yields with excellent stereoselectivities. In detail, trans-cinnamaldehydes 2b–f bearing either electron-rich or electron-deficient substituents at the para- or meta-positions of the phenyl moiety underwent facile transformation into the desired products 3b–f in moderate to good yields with high diastereo- and enantioselectivities. It is noteworthy that in the case of aldehyde 2f, the reaction with alkylidenemalononitrile derivative 1a provided the desired product 3f but in a lower yield. Besides, doubly substituted 2g was successfully utilized as a reaction partner to give the corresponding product 3g in 50% yield and >99:1 er. Finally, the reactivity and stereoselectivity were hardly affected by the incorporation of the furan ring in 2h, as the target polycarbocyclic scaffold 3h was effectively obtained. Moreover, under the standard conditions, the aliphatic α,β-unsaturated aldehydes were not tolerated in this cascade process as the formation of the desired products 3 was not detected. Unfortunately, the reaction with ortho-substituted aromatic α,β-unsaturated aldehydes 2 did not lead to the desired product 3.
 | ||
| Scheme 2 Polyenolate-mediated reaction cascade initiated by the higher-order-cycloaddition – scope of alkylidenemalononitrile derivatives 1. Reactions were performed using 1 (0.2 mmol), 2a (0.2 mmol), R-mandelic acid (0.04 mmol) and catalyst 4 (0.02 mmol) in 1,2-dichloroethane (0.4 mL) at rt. Diastereomeric ratio (dr) was determined by 1H NMR of the crude reaction mixture. Enantiomeric ratio (er) was determined by chiral UPC2. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | 3 (2) | Ar | Yield [%] | drb | erc |
| a Reactions were performed using 1a (0.2 mmol), 2 (0.2 mmol), R-mandelic acid (0.04 mmol) and catalyst 4 (0.02 mmol) in 1,2-dichloroethane (0.4 mL) at rt. b Determined by 1H NMR of the crude reaction mixture. c Determined by chiral UPC2. | |||||
| 1 | 3a (2a) | Ph | 65 | >20:1 |
>99:1 |
| 2 | 3b (2b) | 4-MeOC6H4 | 60 | 17:1 |
>99:1 |
| 3 | 3c (2c) | 3-MeOC6H4 | 44 | 20:1 |
>99:1 |
| 4 | 3d (2d) | 4-MeC6H4 | 59 | 20:1 |
>99:1 |
| 5 | 3e (2e) | 4-ClC6H4 | 55 | 20:1 |
>99:1 |
| 6 | 3f (2f) | 4-CF3C6H4 | 36 | 20:1 |
>99:1 |
| 7 | 3g (2g) | 2-Naphthyl | 50 | 20:1 |
>99:1 |
| 8 | 3h (2h) | 2-Furyl | 52 | 20:1 |
>99:1 |
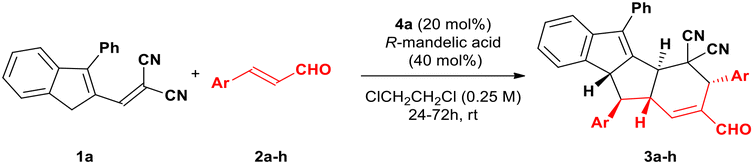
Next, the generality of the cascade reaction for alkylidenemalononitrile derivatives 1 as higherene precursors was investigated (Scheme 2). The reaction proved applicable for a series of substituted substrates 1 with different R1 substituents at the C5–C7 positions of the phenyl ring. Several representative substrates 1i–m with methyl, methoxy and bromine substituents successfully took part in the cascade reaction, providing the target products 3i–m in moderate to good yields with perfect enantioselectivity. However, the steric hindrance of the bromine atom at the C7 position had an effect on the reactivity of 1m and delivered a relatively low yield of product 3m compared to other substrates. Similarly, alkylidenemalononitrile derivatives 1n–q bearing aromatic groups of varying electronic nature at the 3-position were employed for this reaction, affording the target products 3n–q in overall good yields. Furthermore, the unsubstituted derivative 1r could also be effectively used as a competent higherene precursor in the presented cascade. Notably, all the products 3i–r were generated with uniformly excellent diastereoselectivity (dr > 20:1). Disappointingly, when the aliphatic group was introduced at the R2 position, no reaction was observed.
The absolute configuration of the polycarbocyclic products 3a–r was unequivocally confirmed by single crystal X-ray analysis of 3m (for details, see the ESI†).14 The stereochemistry of other products was assigned by analogy. Scheme 3 shows the detailed mechanism of the developed transformation, based on the assigned configuration of the final polycyclic products 3. The reaction cascade begins with the formation of two key intermediates: (1) polyenolate 5a (through deprotonation of indene derivative 2a) and (2) iminium ion 6a (by condensation of aldehyde 2a and catalyst 4a). For the first cyclization to occur, higherene 5a approaches higherenophile 6a from the less sterically-hindered side and in an endo-like fashion. The latter may be explained by the attractive interactions between the oppositely charged dicyanomethine scaffold and the pyrrolidine nitrogen atom. The resulting zwitterionic intermediate 7a undergoes a 1,4-addition to another iminium ion 6a followed by an intermolecular Mannich reaction. Similarly to the periselective [10 + 2]-cycloaddition step, the spatial orientation of both substrates and hence the stereochemical outcome of the second reaction sequence are controlled by the bulky substituents originating from the catalyst. The catalytic cycle is then completed by the release of two catalyst molecules though elimination and hydrolysis.
 | ||
| Scheme 3 Polyenolate-mediated reaction cascade initiated by the higher-order-cycloaddition – mechanistic considerations. | ||
To demonstrate the potential utility of the cascade reaction products, selected transformations of 3 were attempted. Chemoselective reduction of the carbonyl group in 3a led to the formation of alcohol 10a in an excellent 94% yield (Scheme 4, top). The reaction was realized using NaBH4 in the presence of cerium chloride as a reducing reagent. Moreover, the Suzuki coupling involving aldehyde 3l and phenylboronic acid 11 was performed to give the desired product 12l in a good 62% yield (Scheme 4, bottom). Both reactions occurred with the preservation of the optical purity of 3a,l as all products were obtained as single diastereoisomers.
 | ||
| Scheme 4 Polyenolate-mediated reaction cascade initiated by the higher-order-cycloaddition – selected transformations. | ||
In conclusion, we have demonstrated that polyenolate chemistry constitutes a powerful strategy, enabling the formation of higherenes for a higher-order-cycloaddition-initiated cascade reaction. Alkylidenemalononitriles derived from indene-2-carbaldehyde derivatives upon deprotonation underwent the reaction cascade involving [10 + 2]-higher-order cycloaddition followed by Michael addition and aldol condensation that proceeded in a highly stereoselective fashion.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was realized within the Opus programme (grant number: UMO-2021/41/B/ST4/03385) from the National Science Centre, Poland. Thanks are expressed to Dr Lesław Sieroń (Lodz University of Technology) for the X-ray analysis. This contribution has been completed while the first author (AC) was a Doctoral Candidate in the Interdisciplinary Doctoral School of Lodz University of Technology, Poland.References
- For selected reviews, see: (a) H.-L. Cui and Y.-C. Chen, α,α-Dicyanoalkenes: versatile vinylogous nucleophiles for organic synthesis, Chem. Commun., 2009, 4479 RSC; (b) Y. Zhanga and W. Wang, Recent advances in organocatalytic asymmetric Michael reactions, Catal. Sci. Technol., 2012, 2, 42 RSC; (c) G. Casiraghi, L. Battistini, C. Curti, G. Rassu and F. Zanardi, The Vinylogous Aldol and Related Addition Reactions: Ten Years of Progress, Chem. Rev., 2011, 111, 3076 CrossRef CAS PubMed; (d) L. Battistini, C. Curti, G. Rassu, A. Sartori and F. Zanardi, Enolizable Alkylidene Heterocyclic and Carbocyclic Carbonyl Systems: Valuable Vinylogous Donor Substrates in Synthesis, Synthesis, 2017, 2297 CAS; (e) C. Curti, L. Battistini, A. Sartori and F. Zanardi, New Developments of the Principle of Vinylogy as Applied to π-Extended Enolate-Type Donor Systems, Chem. Rev., 2020, 120, 2448 CrossRef CAS PubMed.
- For selected examples, see: (a) C. Curti, G. Rassu, V. Zambrano, L. Pinna, G. Pelosi, A. Sartori, L. Battistini, F. Zanardi and G. Casiraghi, Bifunctional Cinchona Alkaloid/Thiourea Catalyzes Direct and Enantioselective Vinylogous Michael Addition of 3-Alkylidene Oxindoles to Nitroolefins, Angew. Chem., Int. Ed., 2012, 51, 6200 CrossRef CAS PubMed; (b) G. Rassu, V. Zambrano, L. Pinna, C. Curti, L. Battistini, A. Sartori, G. Pelosi, F. Zanardi and G. Casiraghi, Direct Regio-, Diastereo-, and Enantioselective Vinylogous Michael Addition of Prochiral 3-Alkylideneoxindoles to Nitroolefins, Adv. Synth. Catal., 2013, 355, 1881 CrossRef CAS; (c) L. Dell'Amico, Ł. Albrecht, T. Naicker, P. H. Poulsen and K. A. Jørgensen, Beyond Classical Reactivity Patterns: Shifting from 1,4- to 1,6-Additions in Regio- and Enantioselective Organocatalyzed Vinylogous Reactions of Olefinic Lactones with Enals and 2,4-Dienals, J. Am. Chem. Soc., 2013, 135, 8063 CrossRef PubMed; (d) Q. Chen, G. Wang, X. Jiang, Z. Xu, L. Lin and R. Wang, Highly Enantioselective Organocatalyzed Vinylogous Michael-Type Reaction for the Construction of Trifluoromethylated All-Carbon Quaternary Stereocenters, Org. Lett., 2014, 16, 1394 CrossRef CAS PubMed; (e) T.-P. Gao, J.-B. Lin, X.-Q. Hu and P.-F. Xu, A catalytic asymmetric hetero-Diels–Alder reaction of olefinic azlactones and isatins: facile access to chiral spirooxindole dihydropyranones, Chem. Commun., 2014, 50, 8934 RSC; (f) Y. Zhong, S. Ma, Z. Xu, M. Chang and R. Wang, Organocatalytic asymmetric vinylogous Michael addition of 3-alkylidene oxindoles to α-substituted β-nitroacrylates: facile construction of a chiral all-carbon quaternary center, RSC Adv., 2014, 4, 49930 RSC; (g) G. Rassu, V. Zambrano, L. Pinna, C. Curti, L. Battistini, A. Sartori, G. Pelosi, G. Casiraghi and F. Zanardi, Direct and Enantioselective Vinylogous Michael Addition of α-Alkylidenepyrazolinones to Nitroolefins Catalyzed by Dual Cinchona Alkaloid Thioureas, Adv. Synth. Catal., 2014, 356, 2330 CrossRef CAS; (h) N. Di Iorio, P. Righi, S. Ranieri, A. Mazzanti, R. G. Margutta and G. Bencivenni, Vinylogous Reactivity of Oxindoles Bearing Nonsymmetric 3-Alkylidene Groups, J. Org. Chem., 2015, 80, 7158 CrossRef CAS PubMed; (i) X. Xiao, H. Mei, Q. Chen, X. Zhao, L. Lin, X. Liu and X. Feng, Direct asymmetric vinylogous Michael addition of 3-alkylidene oxindoles to chalcones catalyzed by a chiral N,N′-dioxide ytterbium(III) complex, Chem. Commun., 2015, 51, 580 RSC; (j) J.-L. Han and C.-H. Chang, An asymmetric assembly of spirooxindole dihydropyranones through a direct enantioselective organocatalytic vinylogous aldol-cyclization cascade reaction of 3-alkylidene oxindoles with isatins, Chem. Commun., 2016, 52, 2322 RSC.
- For selected reviews, see: (a) H. Jiang, Ł. Albrecht and K. A. Jørgensen, Aminocatalytic remote functionalization strategies, Chem. Sci., 2013, 4, 2287 RSC; (b) S. Reboredo, A. Parra and J. Alemán, Trienamines: Their Key Role in Extended Organocatalysis for Diels-Alder Reactions, Asymmetric Catal., 2013, 1, 24 Search PubMed; (c) V. Marcos and J. Alemán, Old tricks, new dogs: organocatalytic dienamine activation of α,β-unsaturated aldehydes, Chem. Soc. Rev., 2016, 45, 6812 RSC; (d) A. Przydacz, A. Skrzyńska and Ł. Albrecht, Breaking Aromaticity with Aminocatalysis: A Convenient Strategy for Asymmetric Synthesis, Angew. Chem., Int. Ed., 2019, 58, 63 CrossRef CAS PubMed; (e) T. J. Pawar, S. B. Mitkari, E. Peña-Cabrera, C. V. Gómez and D. C. Cruz, Polyenals and Polyenones in Aminocatalysis: A Decade Building Complex Frameworks from Simple Blocks, Eur. J. Org. Chem., 2020, 6044 CrossRef CAS.
- For selected reviews, see: (a) C. Grondal, M. Jeanty and D. Enders, Organocatalytic cascade reactions as a new tool in total synthesis, Nat. Chem., 2010, 2, 167 CrossRef CAS PubMed; (b) D. B. Ramachary and S. Jain, Sequential one-pot combination of multi-component and multi-catalysis cascade reactions: an emerging technology in organic synthesis, Org. Biomol. Chem., 2011, 9, 1277 RSC; (c) R. C. Wende and P. R. Schreiner, Evolution of asymmetric organocatalysis: multi- and retrocatalysis, Green Chem., 2012, 14, 1821 RSC; (d) P. Chauhan, S. Mahajan, U. Kaya, D. Hack and D. Enders, Bifunctional Amine-Squaramides: Powerful Hydrogen-Bonding Organocatalysts for Asymmetric Domino/Cascade Reactions, Adv. Synth. Catal., 2015, 357, 253 CrossRef CAS; (e) S. Nayak, P. Panda, S. Bhakta, S. K. Mishraa and S. Mohapatra, Current advances of organocatalytic Michael–Michael cascade reaction in the synthesis of highly functionalized cyclic molecules, RSC Adv., 2016, 6, 96154 RSC; (f) T. Parvin, R. Yadav and L. H. Choudhury, Recent applications of thiourea-based organocatalysts in asymmetric multicomponent reactions (AMCRs), Org. Biomol. Chem., 2020, 18, 5513 RSC.
- For selected reviews on cycloadditions, see: (a) H.-W. Frühauf, Metal-Assisted Cycloaddition Reactions in Organotransition Metal Chemistry, Chem. Rev., 1997, 97, 523 CrossRef PubMed; (b) L. Yet, Metal-Mediated Synthesis of Medium-Sized Rings, Chem. Rev., 2000, 100, 2963 CrossRef CAS PubMed; (c) Cycloaddition Reactions in Organic Synthesis, ed. S. Kobayashi and K. A. Jørgensen, Wiley-VCH, Weinheim, 2001 Search PubMed; (d) E. M. Carreira and L. Kvaerno, Classics in Stereoselective Synthesis, Wiley-VCH, Weinheim, 2009 Search PubMed; (e) A. Moyano and R. Rios, Asymmetric Organocatalytic Cyclization and Cycloaddition Reactions, Chem. Rev., 2011, 111, 4703 CrossRef CAS PubMed; (f) F. E. Held and S. B. Tsogoeva, Asymmetric cycloaddition reactions catalyzed by bifunctional thiourea and squaramide organocatalysts: recent advances, Catal. Sci. Technol., 2016, 6, 645 RSC; (g) L. Klier, F. Tur, P. H. Poulsen and K. A. Jørgensen, Asymmetric cycloaddition reactions catalysed by diarylprolinol silyl ethers, Chem. Soc. Rev., 2017, 46, 1080 RSC.
- R. Huisgen, Kinetics and Mechanism of 1,3-Dipolar Cycloadditions, Angew. Chem., Int. Ed. Engl., 1963, 2, 633 ( Angew. Chem. , 1963 , 75 , 742 ) CrossRef.
- For reports on Diels–Alder reactions, see: (a) O. Diels and K. Alder, Syntheses in the hydroaromatic series. I. Addition of “diene” hydrocarbons, Justus Liebigs Ann. Chem., 1928, 460, 98 CrossRef CAS; (b) O. Diels and K. Alder, Synthesen in der hydro-aromatischen Reihe, II. Mitteilung: Über Cantharidin, Ber. Dtsch. Chem. Ges., 1929, 62, 554 CrossRef; (c) R. B. Woodward and R. Hoffmann, Stereochemistry of Electrocyclic Reactions, J. Am. Chem. Soc., 1965, 87, 395 CrossRef CAS; (d) R. Hoffmann and R. B. Woodward, Orbital Symmetries and endo-exo Relationships in Concerted Cycloaddition Reactions, J. Am. Chem. Soc., 1965, 87, 4388 CrossRef CAS; (e) R. Hoffmann and R. B. Woodward, Selection Rules for Concerted Cycloaddition Reactions, J. Am. Chem. Soc., 1965, 87, 2046 CrossRef CAS; (f) R. B. Woodward and R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1969, 81, 797 CrossRef; Frontier Orbitals and Reaction Paths: Selected Papers of Kenichi Fukui, ed. H. Fujimoto and K. Fukui, World Scientific, Singapore, 1997 Search PubMed. For selected reviews on Diels–Alder and related reactions, see: (g) D. L. Boger, Diels-Alder Reactions of Heterocyclic Azadienes: Scope and Applications, Chem. Rev., 1986, 86, 781 CrossRef CAS; (h) K. A. Jørgensen, Catalytic Asymmetric Hetero-Diels–Alder Reactions of Carbonyl Compounds and Imines, Angew. Chem., Int. Ed., 2000, 39, 3558 ( Angew. Chem. , 2000 , 112 , 3702 ) CrossRef; (i) E. J. Corey, Catalytic Enantioselective Diels–Alder Reactions: Methods, Mechanistic Fundamentals, Pathways, and Applications, Angew. Chem., Int. Ed., 2002, 41, 1650 ( Angew. Chem. , 2002 , 114 , 1724 ) CrossRef CAS; (j) H. Pellissier, Asymmetric hetero-Diels–Alder reactions of carbonyl compounds, Tetrahedron, 2009, 65, 2839 CrossRef CAS; (k) S. Reymond and J. Cossy, Copper-Catalyzed Diels–Alder Reactions, Chem. Rev., 2008, 108, 5359 CrossRef CAS PubMed; (l) S. Goudedranche, W. Raimondi, X. Bugaut, T. Constantieux, D. Bonne and J. Rodriguez, Enantioselective Organocatalyzed Domino Synthesis of Six-Membered Carbocycles, Synthesis, 2013, 1909 CAS; (m) X. Jiang and R. Wang, Recent Developments in Catalytic Asymmetric Inverse-Electron-Demand Diels–Alder Reaction, Chem. Rev., 2013, 113, 5515 CrossRef CAS PubMed.
- K. N. Houk and R. B. Woodward, Cycloaddition Reactions of Cycloheptatriene and 2,5-DimethyI-3,4-diphenylcyclopenta-dienone, J. Am. Chem. Soc., 1970, 92, 4143 CrossRef CAS.
- For selected reviews, see: (a) T. A. Palazzo, R. Mose and K. A. Jørgensen, Cycloaddition Reactions: Why Is It So Challenging To Move from Six to Ten Electrons?, Angew. Chem., Int. Ed., 2017, 56, 10033 ( Angew. Chem. , 2017 , 129 , 10165 ) CrossRef CAS PubMed; (b) X. Liu, H. Zheng, Y. Xia, L. Lin and X. Feng, Asymmetric Cycloaddition and Cyclization Reactions Catalyzed by Chiral N,N′-Dioxide–Metal Complexes, Acc. Chem. Res., 2017, 50, 2621 CrossRef CAS PubMed; (c) T. A. Palazzo and K. A. Jørgensen, Higher-order cycloaddition reactions: A computational perspective, Tetrahedron, 2018, 74, 7381 CrossRef CAS. For selected metal-catalyzed higher-order cycloadditions, see: (d) M. Xie, X. Liu, X. Wu, Y. Cai, L. Lin and X. Feng, Catalytic Asymmetric [8 + 2] Cycloaddition: Synthesis of Cycloheptatriene-Fused Pyrrole Derivatives, Angew. Chem., Int. Ed., 2013, 52, 5604 ( Angew. Chem. , 2013 , 125 , 5714 ) CrossRef CAS PubMed; (e) A. R. Rivero, I. Fernández and M. Sierra, Regio- and Diastereoselective Stepwise [8 + 3]-Cycloaddition Reaction between Tropone Derivatives and Donor–Acceptor Cyclopropanes, Org. Lett., 2013, 15, 4928 CrossRef CAS PubMed; (f) R. Tejero, A. Ponce, J. Adrio and J. C. Carretero, Ni-Catalyzed [8 + 3] cycloaddition of tropones with 1,1-cyclopropanediesters, Chem. Commun., 2013, 49, 10406 RSC; (g) T.-L. Liu, Q.-H. Li, L. Wei, Y. Xiong and C.-J. Wang, Copper(I)-Catalyzed One-Pot Sequential [3 + 2]/[8 + 2] Annulations for the (Z)-Selective Construction of Heterocyclic Diazabicyclo[5.3.0]decatrienes, Adv. Synth. Catal., 2017, 359, 1854 CrossRef CAS; (h) J. Zhang, W. Xiao, H. Hu, L. Lin, X. Liu and X. Feng, Catalytic Asymmetric [8 + 3] Annulation Reactions of Tropones or Azaheptafulvenes with meso-Aziridines, Chem. – Eur. J., 2018, 24, 13428 CrossRef CAS PubMed.
- For selected reviews, see: (a) S. Frankowski, M. Romaniszyn, A. Skrzyńska and Ł. Albrecht, The Game of Electrons: Organocatalytic Higher-Order Cycloadditions Involving Fulvene- and Tropone-Derived Systems, Chem. – Eur. J., 2020, 26, 2120 CrossRef CAS PubMed; (b) N. I. Jessen, D. McLeod and K. A. Jørgensen, Higher-order cycloadditions in the age of catalysis, Chem, 2022, 8, 20 CrossRef CAS.
- For selected examples, see: (a) R. Mose, G. Preegel, J. Larsen, S. Jakobsen, E. H. Iversen and K. A. Jørgensen, Organocatalytic stereoselective [8 + 2] and [6 + 4] cycloadditions, Nat. Chem., 2017, 9, 487 CrossRef CAS PubMed; (b) M. Romaniszyn, K. Gronowska and Ł. Albrecht, 2-Substituted 1,4-Naphthoquinones in [6 + 4]-Cycloaddition with 8,8-Dicyanoheptafulvene, J. Org. Chem., 2019, 84, 9929 CrossRef CAS PubMed; (c) M. Romaniszyn, L. Sieroń and Ł. Albrecht, 5-Substituted-furan-2(3H)-ones in [8 + 2]-Cycloaddition with 8,8-Dicyanoheptafulvene, J. Org. Chem., 2022, 87, 5296 CrossRef CAS PubMed.
- For examples, see:
(a) B. S. Donslund, A. Monleón, T. A. Palazzo, M. L. Christensen, A. Dahlgaard, J. D. Erickson and K. A. Jørgensen, Organocatalytic Enantioselective Higher-Order Cycloadditions of In Situ Generated Amino Isobenzofulvenes, Angew. Chem., Int. Ed., 2018, 57, 1246 CrossRef CAS PubMed;
(b) B. S. Donslund, N. I. Jessen, G. Bertuzzi, M. Giardinetti, T. A. Palazzo, M. L. Christensen and K. A. Jørgensen, Catalytic Enantioselective [10 + 4]Cycloadditions, Angew. Chem., Int. Ed., 2018, 57, 13182 CrossRef CAS PubMed;
(c) M. Giardinetti, N. I. Jessen, M. L. Christensen and K. A. Jørgensen, Organocatalytic [10 + 4] cycloadditions for the synthesis of functionalised benzo[a]azulenes, Chem. Commun., 2019, 55, 202 RSC.
- A. Cieśliński, S. Frankowski and Ł. Albrecht, Pentaenolate activation in the organocatalytic allylic alkylation of indene-2-carbaldehydes, Chem. Commun., 2023, 59, 7655 RSC.
- CCDC 2279932† contains the supplementary crystallographic data for this paper.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2279932. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qo02040a |
| This journal is © the Partner Organisations 2024 |