
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mo-catalyzed cyclization of N-vinylindoles and skatoles: synthesis of dihydroindolo[1,2-c]-quinazolines and dihydroindolo[3,2-b]-indoles, and evaluation of their anticancer activities†
Shannon
Pecnard‡
a,
Xinya
Liu‡
a,
Olivier
Provot
*a,
Pascal
Retailleau
b,
Christine
Tran
*a and
Abdallah
Hamze
 *a
*a
aUniversité Paris-Saclay, CNRS, BioCIS, 91400 Orsay, France. E-mail: abdallah.hamze@universite-paris-saclay.fr
bUniversité Paris-Saclay, CNRS, Institut de Chimie des Substances Naturelles, UPR 2301, 91198, Gif-sur-Yvette, France
First published on 8th February 2024
Abstract
A novel approach has been developed for synthesizing unexpected dihydroindolo[1,2-c]-quinazolines (DINQ) and dihydroindolo[3,2-b]indole (DINI) compounds using N-vinylazoles as starting materials. The presence of a Mo-catalyst was found to be essential for the formation of DINQs from skatole derivatives and for accelerating the cyclization of DINI precursors. Biological evaluation revealed that compound 3o, a 2-phenyl-1-(1-phenylvinyl)-1H-indole derivative and DINI analog 6g exhibited nanomolar cytotoxic effects in vitro against a human colon cancer cell line (HCT-116).
Introduction
Heterocyclic compounds play a pivotal role in medicinal chemistry due to their wide range of applications in drug design and discovery.1 More than 85% of all biologically active chemical entities contain a heterocycle unit,2 with prevalent nitrogen-containing heterocycles usually found in the form of five- or six-membered rings. Therefore, developing new synthetic methodologies that enable rapid access to diverse functionalized heterocyclic compounds holds great significance to medicinal chemists. Such advancements not only broaden the chemical space available for drug development but also streamline drug discovery programs. Due to their diverse biological activities, nitrogen-containing heterocycles have consistently captivated the interest of synthetic organic chemists.3–6 In cancer therapy, microtubules crucial for cell division serve as pivotal targets. Clinically used antimicrotubule agents include vinca alkaloids and taxanes (e.g., paclitaxel and docetaxel). Paclitaxel (Taxol), an antimicrotubule agent, is employed in the treatment of malignant solid tumors such as breast, lung, head and neck, and ovarian cancer.7 Common side effects encompass cardiotoxicity, myelosuppression, and peripheral neuropathy.8Vinca alkaloids are approved for clinical use; vincristine, vinblastine, vinorelbine, vindesine, and vinflunine serve as agents in the treatment of hematological and lymphatic neoplasms.9 The principal toxicity symptom of vincristine is neurotoxicity, while neutropenia is the main toxicity sign of the other vinca alkaloids.10
Compounds derived from combretastatin, acting as anti-tubulin agents, exhibit the potential to inhibit tubulin polymerization into microtubules, offering a promising avenue in cancer treatment.11 Over the past decade, our research group has dedicated efforts to designing and synthesizing anti-tubulin agents.12,13 Despite the notable anticancer activity of natural combretastatin A-4 (CA-4), its efficacy is hampered by the presence of a Z double bond, resulting in the formation of an inactive E isomer (Scheme 1A).
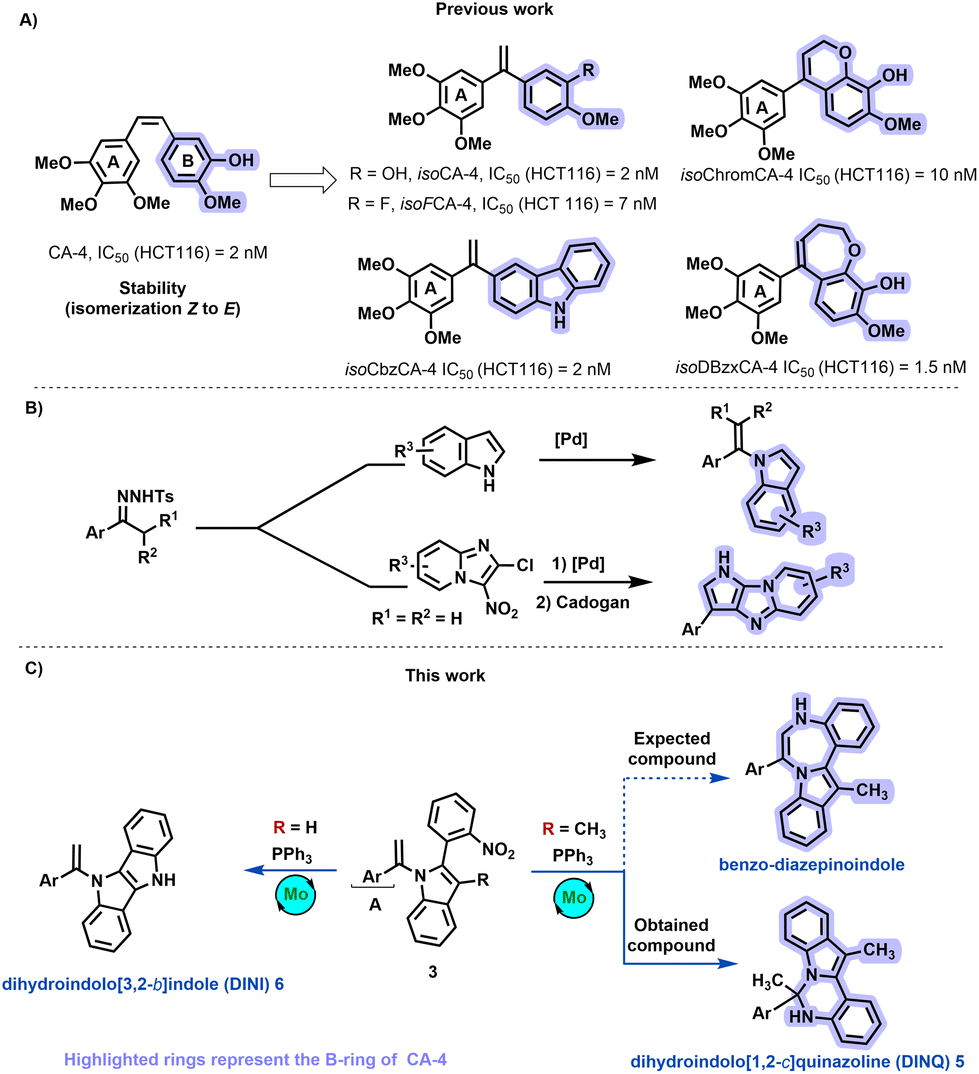 | ||
| Scheme 1 (A) Structure of CA-4, isoCA-4 analogs, (B) Our previous work, and (C) Targeted structures. | ||
Furthermore, the isomerization of CA-4's active Z-double bond to the less active E-CA-4 is readily observed during administration and metabolism, presenting a challenge to its clinical application.14 To address this limitation, our group identified isoCA-415 and isoFCA-4,16 1,1′-diarylethylenes compounds with antiproliferative activity equivalent to CA-4 but with enhanced stability. Additionally, we demonstrated that derivatives such as isoCbzCA-4,17–19 isoChromCA-4,20 and isoDBzxCA-4,21 featuring carbazole, arylchromene, and dihydrobenzoxepine scaffolds, respectively, exhibit outstanding antiproliferative activities against colon cancer cell lines HCT-116 in the low nanomolar range (Scheme 1A). Recently, we synthesized N-vinylazoles by coupling N-tosylhydrazones derivatives and N–H azoles,22 enabling the construction of a compound library with a pyrrolo-imidazo[1,2-a]pyridine backbone (Scheme 1B).23
Building on our comprehension of the structure–activity relationships of isoCA-4 and our advancements in the chemistry of N-vinylazoles, we aimed to create compound collections based on privileged scaffolds (Scheme 1C). Consequently, our investigation focused on synthesizing novel isoCA-4 analogs featuring seven-membered ring heterocycles, specifically benzo-diazepinoindole, using N-vinylazoles as starting materials.
In this study, we will investigate the synthesis of various terminal N-vinylazoles 3, and their cyclization under Mo-Cadogan-reductive cyclization conditions. This cyclization could afford benzo-diazepinoindole (Scheme 1).
Results and discussion
Our investigation commenced with the preparation of N-vinylazole substrates (3) (Table 1). In accordance with our prior work,22 we performed the coupling reaction between indole derivative (1a) and N-tosylhydrazone (2a) using Pd2dba3·CHCl3 as the catalyst and iodobenzene as the oxidant agent. Unfortunately, under these conditions, the desired product (3a) was obtained but in a low 25% yield (entry 1, Table 1). The presence of a hindered ortho-nitrophenyl substituent on the C-2 position of (1a) resulted in decreased reactivity of the indole. Changing the oxidant from iodobenzene to oxygen and gradually introducing compound (2a) over 1 h using a syringe, led to a slight increase in yield (entry 2). Increasing the catalytic load to 20 mol% resulted in a yield of 45% (entry 3). The use of the precatalyst PdG3(XPhos) did not provide conclusive results (entry 4), and (3a) was obtained in a moderate 55% yield.|
|
|||||||
|---|---|---|---|---|---|---|---|
| [Cat.] | 2 | L | Base | Solvent | Time (h) temp °C | Yieldb (%) | |
| a Reaction conditions: indole 1a (0.2 mmol), reagent 2 (0.4 mmol), catalyst (10 mol%), ligand (20 mol%), base (0.3 mmol) and 0.8 mL of solvent in a sealed tube under argon for 20 h. b Isolated yield. c Reaction was performed in the presence of iodobenzene (1.2 equiv.) as oxidant. d Reaction was performed under O2 balloon, and the N-tosylhydrazone was added with a push syringe in 1 h. e Catalyst 20 mol%, ligand 40 mol%. | |||||||
| 1 | Pd2dba3·CHCl3 | 2a | — | NaOtBu | CPME | 5/115 | 25c |
| 2 | Pd(OAc)2 | 2a | L1 | K3PO4 | DMF | 5/100 | 36d |
| 3 | Pd(OAc)2 | 2a | L1 | K3PO4 | DMF | 5/100 | 45d,e |
| 4 | PdG3(XPhos) | 2a | L1 | K3PO4 | DMF | 5/100 | 55d |
| 5 | Cu powder | 2b | L2 | K 2 CO 3 | Toluene | 20/135 | 80 |
| 6 | Cu powder | 2c | L2 | K 2 CO 3 | Toluene | 20/135 | 85 |
| 7 | Cu powder | 2c | L2 | K2CO3 | Toluene | 20/110 | 25 |
| 8 | Cu powder | 2c | L3 | K2CO3 | Toluene | 20/135 | 55 |
| 9 | Cu powder | 2c | L2 | K2CO3 | Dioxane | 20/135 | 45 |
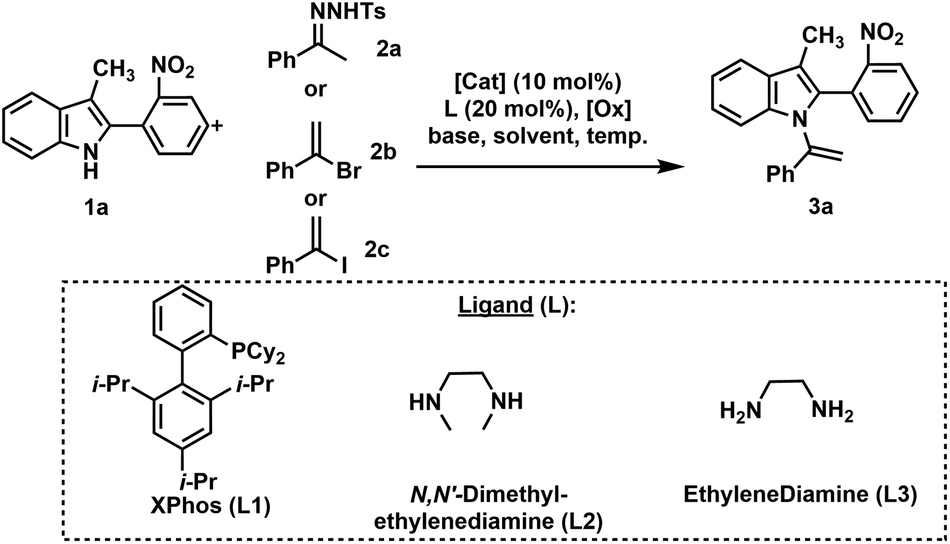
Copper demonstrates remarkable versatility, efficiently catalyzing reactions involving both radical and polar mechanisms. In the context of an extended reaction time (20 hours), copper's unique reactivity profile, cost-effectiveness, and sustainability. Accordingly, our focus shifted to the evaluation of copper catalysts (entries 5–9).24 Employing Cu(0) and N,N-dimethylethylenediamine (L2) as a ligand (entry 5) in toluene for the coupling reaction between (1a) and 1-bromovinyl benzene (2b) resulted in an 80% yield of (3a). Encouraged by this favorable outcome, we explored additional reaction conditions employing copper as the catalyst. Substituting 1-bromovinyl benzene with 1-iodovinyl benzene led to a slight increase in yield to 85% (entry 6). However, reducing the temperature to 110 °C resulted in a significant drop in yield to 25% (entry 7). Switching the ligand to ethylenediamine (L3) yielded a lower yield of (3a) compared to L2 ligand (entry 8 vs. entry 6). Furthermore, replacing toluene with dioxane as the solvent led to a substantial decrease in the yield of (3a) (entry 9 vs. entry 6).
Using the optimized conditions outlined in entries 5 and 6 of Table 1, we proceeded with the synthesis of N-vinylazole-2-(2-nitrophenyl) substrates (3) (Scheme 2). Depending on the availability of starting materials, specifically vinylhalides, and in some cases, due to the instability of (1-iodovinyl)phenyl derivatives, we mainly employed (1-bromovinyl)phenyl derivatives for the coupling reaction. The cross-coupling reaction exhibited good compatibility with electron-rich vinyl halides when paired with skatole or indole derivatives.
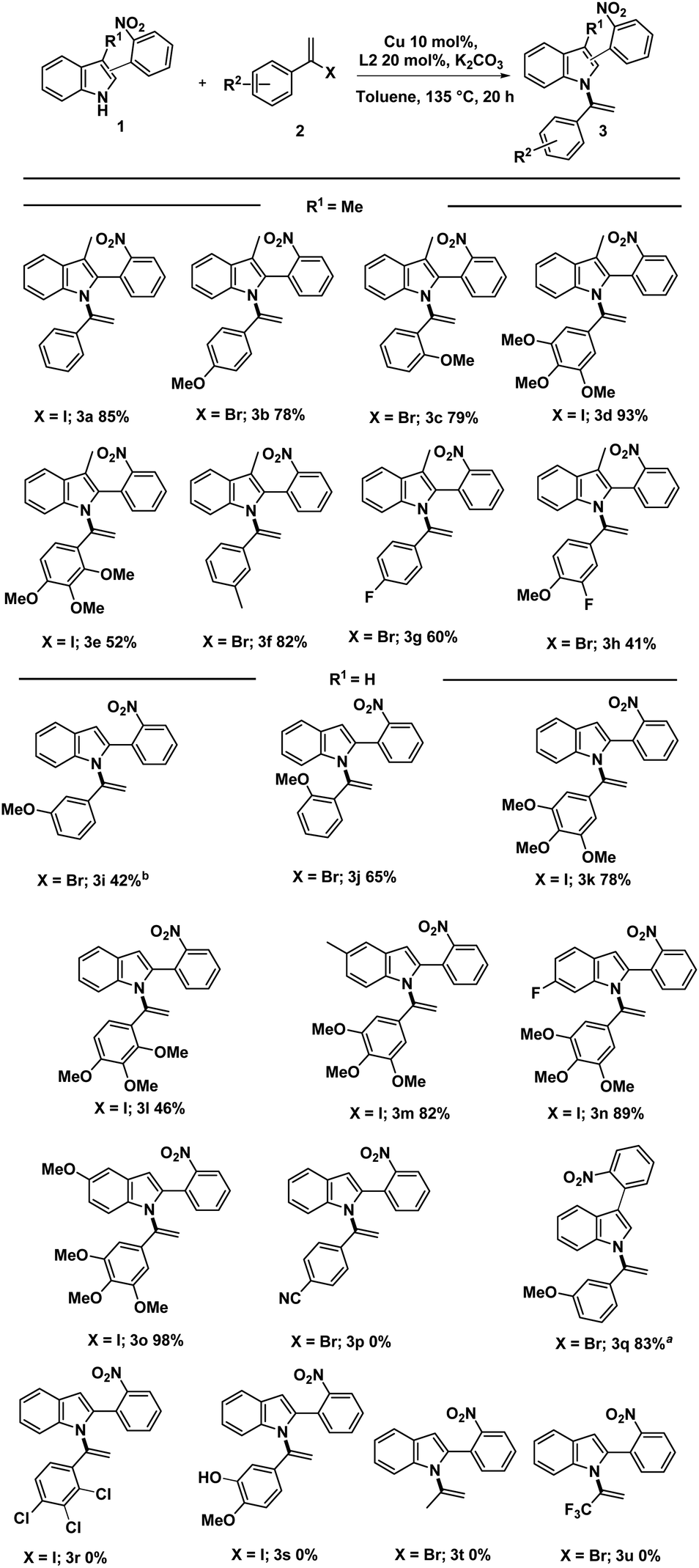 | ||
Scheme 2 Substrate scope for the synthesis of N-vinylazole-2-(2-nitrophenyl) substrates 3a. Reaction conditions: all reactions were conducted with 0.2 mmol of indole 1, vinylhalide 2 (0.4 mmol), Cu(0) (10 mol%), N,N′-dimethyl-ethylenediamine (L2) (20 mol%), K2CO3 (1.5 equiv.), 0.8 mL of dry toluene in a sealed tube under argon for 20 hours at 135 °C. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) The reactions were conducted with 1.9 mmol of indole 1, vinylhalide 2 (2.4 mmol), Cu(0) (20 mol%), L2 (40 mol%), K2CO3 (1.5 equiv.), 7.2 mL of dry toluene in a sealed tube under argon for 20 hours at 135 °C. The reactions were conducted with 1.9 mmol of indole 1, vinylhalide 2 (2.4 mmol), Cu(0) (20 mol%), L2 (40 mol%), K2CO3 (1.5 equiv.), 7.2 mL of dry toluene in a sealed tube under argon for 20 hours at 135 °C. | ||
These conditions proved efficient even with sterically hindered vinyl halides, resulting in the successful synthesis of compounds (3c, 3e, 3j, and 3l) with good yields. While the coupling reaction was successful in synthesizing unsubstituted indoles on the C3 position (3i–o), slightly lower yields were obtained for (3i, 3j, and 3l) due to partial vinylation at the C3 position of (1, R1 = H).
Notably, the structure of (3i) was confirmed by 2D NMR analysis, including COSY, HSQC, HMBC, and NOESY experiments. Except for compounds (3g) and (3h) containing a fluorine atom, the standard conditions were less effective in synthesizing terminal N-vinylazoles bearing an electron-withdrawing group (e.g., R2 = CN, 3p or R2 = 2′, 3′,4′-Cl, 3r). Also, no reaction was observed with free phenol derivative (3s). However, to our delight, we successfully attempted the reaction with a C3-arylated indole, affording compound (3q) with a good 83% yield.
Applying our optimal conditions to alkyl substrates such as 2-bromopropene and 2-bromo-3,3,3-trifluoro-1-propene, does not provide the corresponding coupling products 3t and 3u.
After obtaining N-vinylazoles (3), our investigation shifted towards exploring the cyclization process. Our initial focus was on converting N-vinylazole derivatives derived from skatole, such as 3a. However, when the reaction was conducted using the standard Cadogan reaction conditions (4 equiv. of PPh3 in dioxane),25 no desired product 4a was observed, and the starting material (3a) was fully recovered (Table 2, entry 1). It appears that the presence of steric hindrance in the structure of skatole hampers the reaction compared to the standard deoxygenation of ortho-nitrostyrenes. Arnáiz et al., developed a Mo(VI)/PPh3 catalyst system that is compatible with labile substrates.26
|
|
|||||
|---|---|---|---|---|---|
| Entry | [Mo] Cat. | PR3 (equiv.) | Solvent | Time (h) | Yieldb (%) |
| a General conditions: reactions were conducted with 0.1 mmol of N-vinylazole (3a), PPh3, MoO2Cl2(DMF)2, solvent (1 mL) in a sealed tube, 160 °C. b Isolated yield. c Reaction was conducted at 135 °C. d No reaction. | |||||
| 1 | PPh3 (4) | Dioxane | 16 | 0 | |
| 2 | 10 mol% | PPh3 (4) | Dioxane | 16 | 77 |
| 3 | 5 mol% | PPh3 (4) | Dioxane | 16 | 78 |
| 4 | 5 mol% | PPh3 (4) | DMF | 16 | 75 |
| 5 | 5 mol% | PPh3 (4) | Toluene | 16 | 94 |
| 6 | 5 mol% | PPh3 (4) | Toluene | 16 | 72c |
| 7 | 5 mol% | PPh3 (4) | Toluene | 6 | 95 |
| 8 | 5 mol% | PPh 3 (2.4) | Toluene | 6 | 95 |
| 9 | 5 mol% | PCy3 (2.4) | Toluene | 6 | n.r.d |
| 10 | 5 mol% | P(o-tol)3 (2.4) | Toluene | 6 | n.r.d |
| 11 | 5 mol% | TFP (2.4) | Toluene | 6 | 35 |
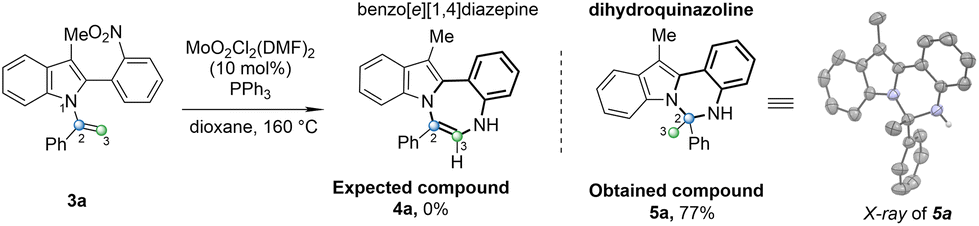
In our recent work, we successfully applied molybdenum-catalyzed cyclization conditions to synthesis pyrrolo-imidazo[1,2-a]pyridines.23 Adapting these conditions to N-vinylazole (3a) involved utilizing MoO2Cl2(DMF)2 (10 mol%) in the presence of PPh3 (4 equiv.) in dioxane at 160 °C in a sealed tube, resulting in complete conversion of the starting material (entry 2). However, a careful analysis of the crude product's 1H NMR spectrum did not reveal the characteristic proton signal of the anticipated 7-membered ring (proton H3). Instead, a singlet was observed for 3 protons at 2.17 ppm in deuterated acetone. Additionally, mass spectrometry (ESI+) analysis displayed a major peak of (M + H)+ 325.1705, deviating from the expected mass of 323.1548 for (4a). To unequivocally confirm the structure of the newly obtained compound, we successfully obtained a crystalline solid suitable for X-ray analysis.27 This analysis confirmed the formation of the six-membered ring dihydroindolo-quinazoline (5a), isolated with a yield of 77% (Table 2). Encouraged by this outcome, we continued optimizing the reaction conditions to enhance the formation of this novel heterocycle (5a). Initially, reducing the catalytic amount of the Mo catalyst to 5 mol% did not impact the yield, and (5a) was obtained with a good yield of 78% (entry 3). Subsequently, exploring of different solvents revealed that employing toluene increased the yield (entries 4–6). Altering the temperature to 135 °C resulted in a slight decrease in yield (entry 6). Ultimately, fine tuning of the reaction time and the amount of PPh3 allowed us to identify the optimal conditions for the formation of (5a), which involved the use of 2.4 equivalents of PPh3 for 6 h (entry 8). Using other reductant phosphine sources gave no reaction with tricyclohexylphosphine or tri-(o-tolyl)phosphine (entries 9 and 10), and only 35% yield was obtained with tri-(2-furyl)phosphine (entry 11).
Using the optimized reaction conditions outlined in Table 2, we investigated the substrate diversity of this protocol. As depicted in Scheme 3, various substrates with different electronic nature and steric properties underwent the reaction under the standardized conditions, affording the dihydroindolo-quinazoline derivatives (5a–h) in good yields. Notably, the reaction displayed tolerance towards steric hindrance substrates, as evidenced by the successful formation of compounds (5c) and (5e). Similarly, electronic factors did not impede the reaction, as demonstrated by the effective synthesis of compounds (5b) and (5g). We also successfully obtained a crystalline solid suitable for X-ray analysis for 5f.
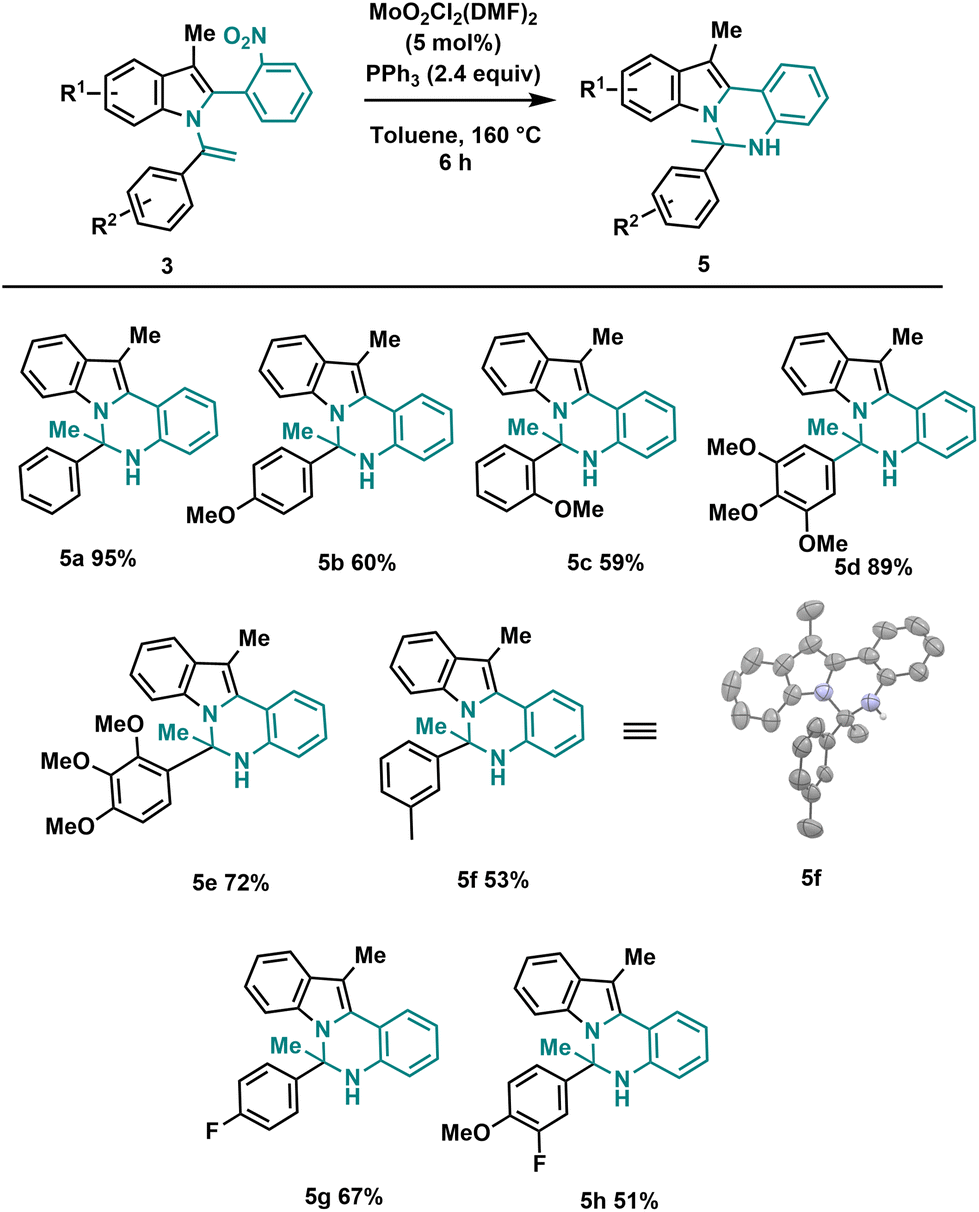 | ||
| Scheme 3 General conditions: 3 (0.2 mmol), MoO2Cl2(DMF)2 (5 mol%), PPh3 (2.4 equiv.), toluene (3 mL), sealed tube 160 °C, 6 h. | ||
Having explored the scope of the Mo/PPh3 cyclization process with skatole derivatives, next, we proceeded to investigate the standard cyclization conditions using unsubstituted C3-indole substrates (3i–o). This cyclization process can occur either on the terminal double bond of N-vinylazole (3) as observed with skatole derivatives, or on the C3 position of the indole moiety. By conducting the reaction under the standard conditions (Table 1), we achieved complete selectivity for cyclization at the C3 position of the indole, resulting in the formation of dihydroindolo[3,2-b]indole (DINI) derivatives (Scheme 4). The absence of the methyl group on the skatole allows C–H insertion on the carbon of the C3 position of the indole, preferentially over the enamine terminal carbon (see infra for the mechanism). Owing to the electron-rich and ease of oxidation of the DINI scaffold, materials incorporating DINI structures28 have found extensive applications in organic electronics, including organic photovoltaics29 and organic light-emitting diodes (LEDs).30 Also, compounds 6a–g were successfully synthesized using our standard conditions in good yields. It is worth noting that the aryl groups of these compounds do not contain electron-withdrawing groups (EWGs). However, we have not yet been able to obtain the corresponding N-vinylazole derivatives for this type of compounds. Interestingly, when utilizing 3-(2-nitrophenyl)-indole (3q), we obtained 5,10-dihydroindolo[3,2-b]indole 8a with a yield of 79%. The structure of this novel heterocycle was confirmed through X-ray analysis.
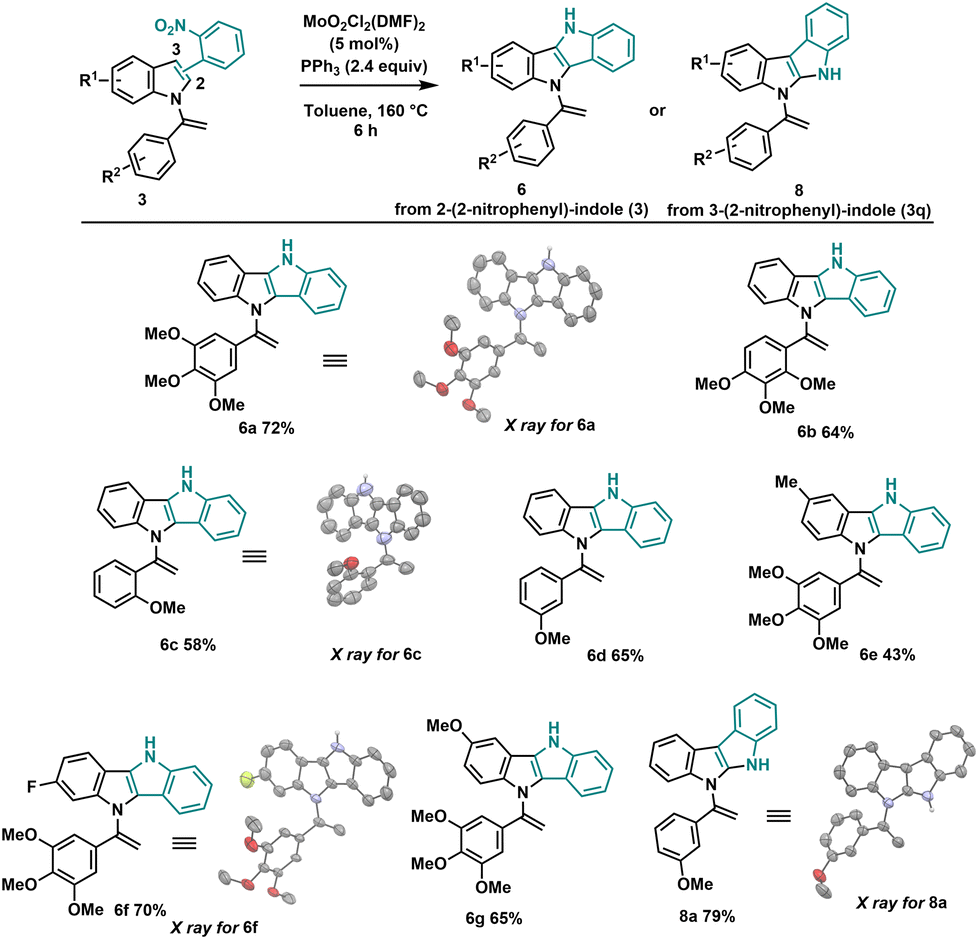 | ||
| Scheme 4 General conditions: 3 (0.2 mmol), MoO2Cl2(DMF)2 (5 mol%), PPh3 (2.4 equiv.), toluene (3 mL), sealed tube 160 °C, 6 h. | ||
To gain insights into the mechanism of these reactions, we conducted several controlled experiments (Scheme 5). Initially, we investigated the formation of dihydroindolo[1,2-c]quinazoline compounds 5. In the absence of the Mo-catalyst, no conversion of N-vinylazole 3a to 5a was observed even after 6 h (Scheme 5b), indicating the crucial role of the Mo-catalyst in this transformation.
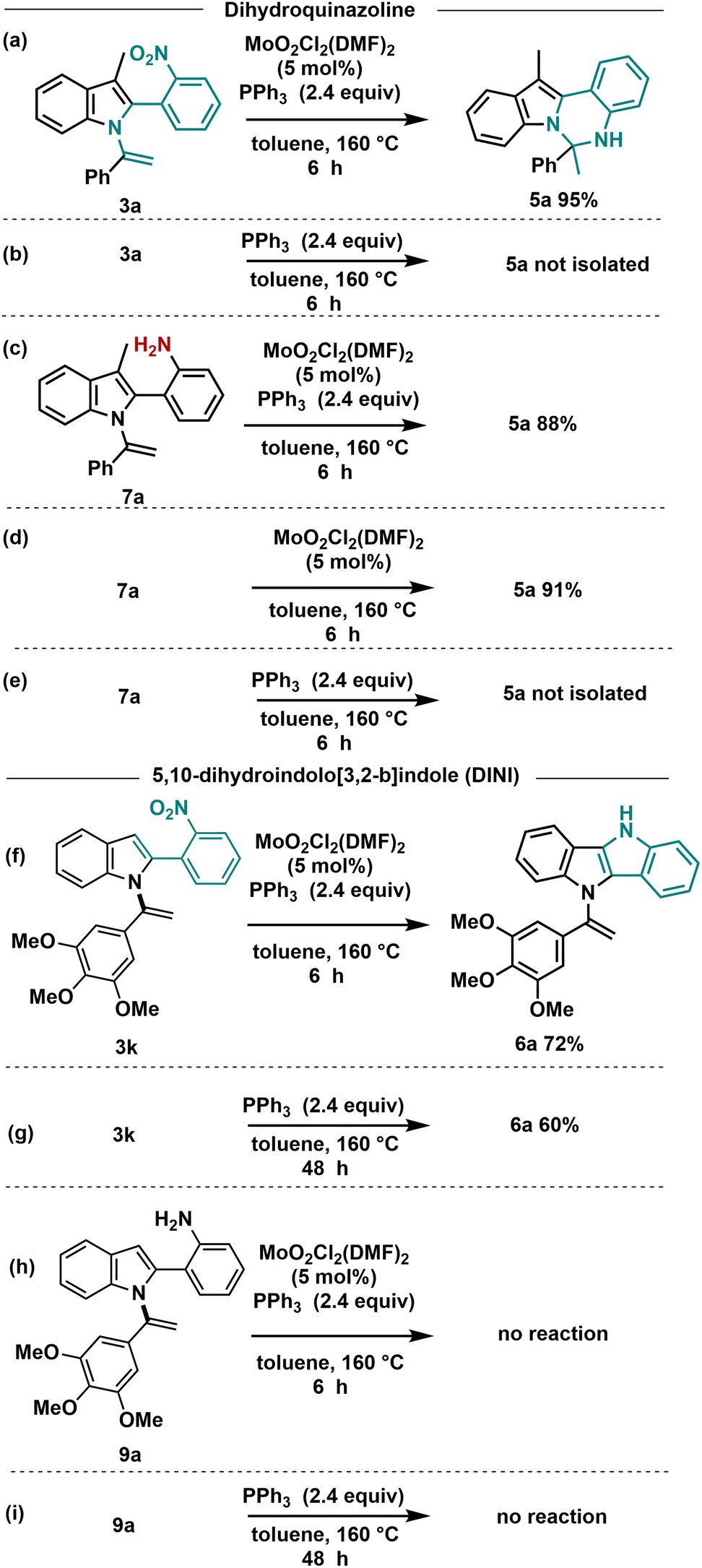 | ||
| Scheme 5 Control experiments. (a) Reaction with 3a under standard conditions. (b) Reaction with 3a without Mo catalyst. (c) Reaction starting from amine derivative 7a under standard conditions. (d) Reactions starting from amine derivative 7a with Mo catalyst and without PPh3. (e) Reactions starting from amine derivative 7a with PPh3 without Mo catalyst. (f) Reaction with 3k under standard conditions. (g) Reaction with 3k with PPh3 without Mo catalyst. (h) Reaction with amine 9a under standard conditions. (i) Reaction with amine 9a with PPh3 without Mo catalyst. | ||
To determine whether the cyclization reaction proceeds through an amine or a nitrene intermediate, we subsequently reduced the nitro group of 3a to the corresponding amine 7a using an iron catalyst (Fe/HCl cat.).
We then carried out the cyclization reaction using 7a as the starting material (Scheme 5c), and under the standard conditions, we successfully obtained 5a with an 88% yield. This experiment provided evidence that the reaction proceeds through the formation of an ortho-aniline intermediate. In the Cadogan reaction, PPh3 facilitates the deoxygenation of o-nitrostyrenes derivatives to nitrenes intermediates. As this transformation proceeded with the amine 7a, we decided to carry out the reaction without the addition of PPh3. As depicted in Scheme 5d, complete conversion of 7a to 5a was achieved in a 91% yield, suggesting that PPh3 does not play a significant role in this cyclization process starting from the amine. Finally, in the absence of Mo-catalyst, no conversion of the amine derivative to 5a was observed. These experiments led to the conclusion that the formation of dihydroindolo[1,2-c]quinazoline 5a from 3a necessitates the presence of both Mo and PPh3. Additionally, it was observed that the formation of the amine intermediate occurred in the presence of a wet solvent, as previously reported in our work.31
Concerning the formation of DINI derivatives, the nitro compound 3k underwent conversion to the DINI derivative in a 72% yield within 6 h under the standard conditions (Scheme 5f). In the absence of Mo-catalyst, the reaction proceeded, albeit at a slower rate, requiring 48 hours for completion (Scheme 5g). To further validate the nature of the intermediate involved in this cyclization process, we conducted the reaction using the amine derivative 9a. No reaction was observed when starting from 9a in the presence of Mo-catalyst and PPh3, nor in the absence of Mo-catalyst (Scheme 5h and i). These experimental results strongly support a Cadogan-type reaction mechanism.
Based on the control experiments and previous literature,32 a mechanism is proposed in Scheme 6. Initially, in the presence of PPh3, the Mo(VI) catalyst A undergoes a reduction step, leading to Mo(IV) complex B. Subsequently, N-vinylazole 3 coordinates with complex B, forming complex C, which undergoes oxidative addition to provide the Mo(VI) metallacycle D. Then, D transforms into complex E, through oxygen atom transfer to the metal center leading to the loss of an oxygen atom from the initial nitro group. Complex E proceeds to generate the nitroso intermediate F and Mo-complex G. The departure of the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PPh3 ligand from complex G regenerates the initial Mo-catalyst A, ready to initiate a new cycle.
PPh3 ligand from complex G regenerates the initial Mo-catalyst A, ready to initiate a new cycle.
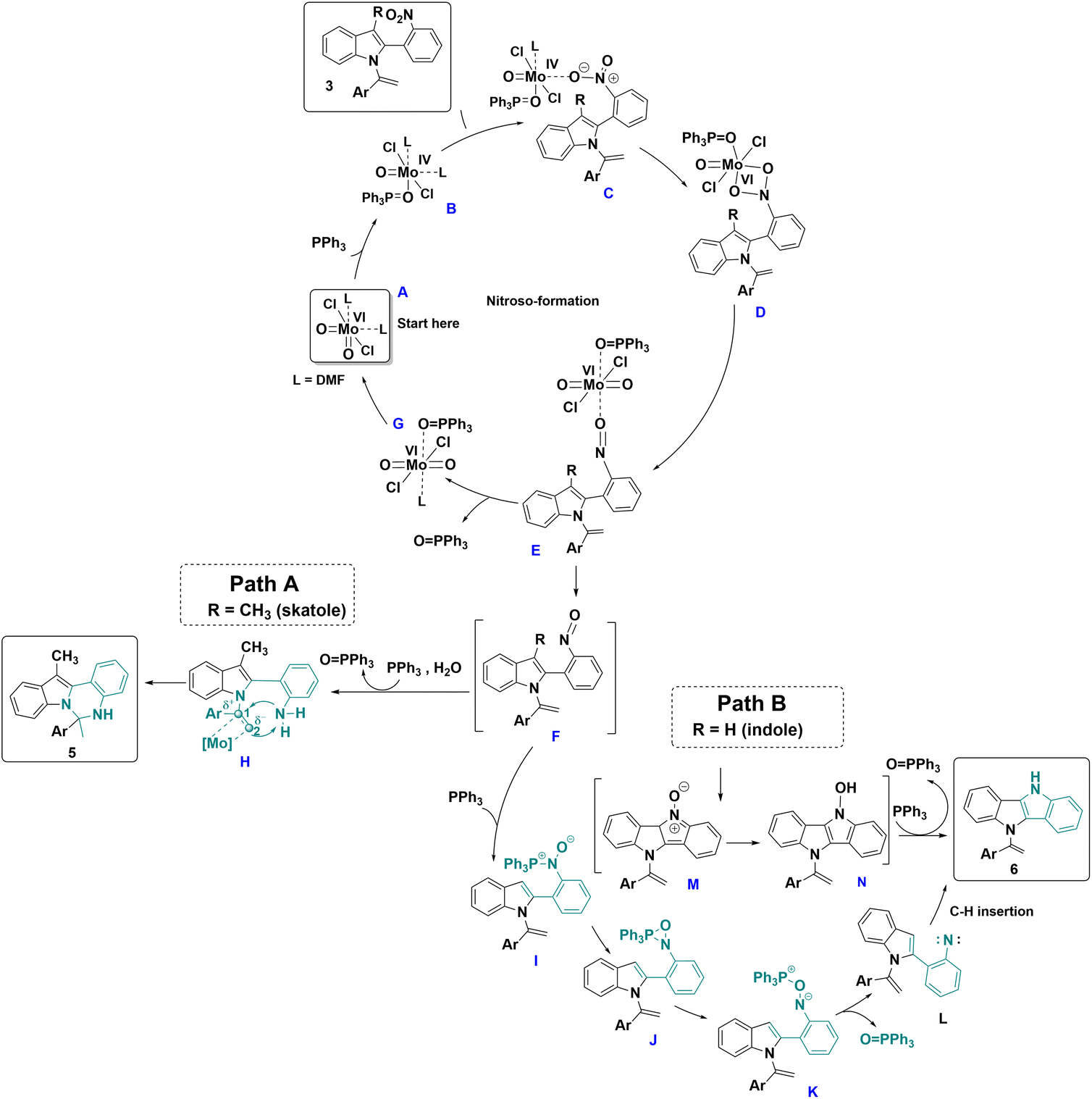 | ||
| Scheme 6 Proposed mechanism for the formation of 5 and 6. | ||
In the case of skatole derivatives (R = Me), where the formation of the seven-membered ring is unfavorable, the nitroso intermediate F is reduced to the corresponding amine H in the presence of PPh3 and a wet solvent.33 The coordination of molybdenum to the ethylene moiety activates the double bond of the N-vinylazole and results in the formation of compound 5.
When R = H (indoles derivatives), a second equivalent of PPh3 reacted with complex F, resulting in the formation of intermediate I. Intermediate I can then undergo further transformation to yield oxazaphosphiridine intermediate J. The nitrogen–phosphore bond of J is subsequently cleaved, generating intermediate K. This intermediate undergoes deoxygenation, leading to the formation of nitrene L and triphenylphosphine oxide. Finally, nitrene L undergoes C–H insertion,34 resulting in cyclization and the formation of the dihydroindolo[3,2-b]indole derivative 6. Alternatively, nitroso intermediates F could undergo direct 6π electrocyclization,35 resulting in the formation of nitronene M and then the N-oxide derivative N. Finally, N would be further deoxygenated to 6 in the presence of PPh3.
Biological results
To assess the potential of the newly synthesized compounds to inhibit cell growth, an in vitro cytotoxicity assay was conducted using the human colon cancer cell line HCT116. The CellTiter-Glo® cell viability assay was employed, utilizing luminescence to measure the number of viable cells based on the quantification of ATP, an indicator of metabolically active cells (the transformation of luciferin to oxyluciferin in the presence of ATP). This assay enables the determination of the number of living cells in culture based on the amount of ATP, which is directly proportional to cell viability.In this study, all the synthesized compounds were tested at a concentration of 10−6 M in triplicate on the HCT-116 cell line. The results are expressed in Fig. 1 as cytotoxicity %. A higher percentage of cytotoxicity indicates a higher level of compound activity against the cancer cells. Among the tested compounds, two heterocycles (3o and 6g) showed significant cytotoxicity against the HCT116 cell line. (3o) compound exhibited a cytotoxicity percentage of 66%, while (6g), also possesses a methoxy group on the indole ring and demonstrated a notably higher cytotoxicity percentage of 93%.
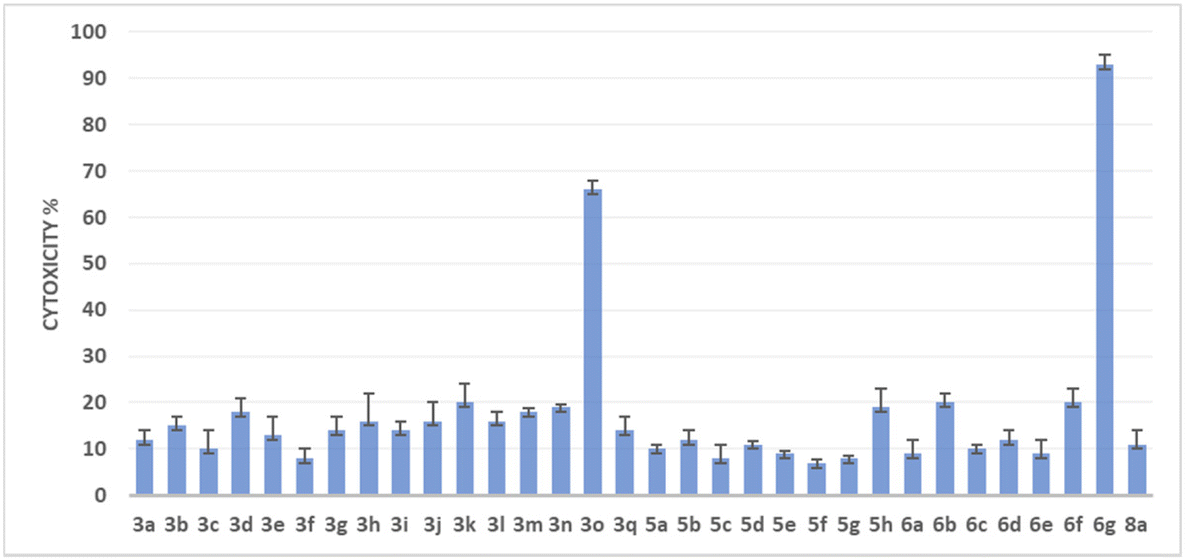 | ||
| Fig. 1 Screening of generated analogs 3, 5, 6 and 8 in a human colon cancer cell line (HCT116). The cell proliferation inhibition was plotted for each of the analogs at 10−6 M concentration in HCT-116 cells. Three sets of experiments were carried out. Error bars represent standard deviation. | ||
These results highlight the importance of the 3,4,5-trimethoxy phenyl moiety and the presence of the methoxy group on either the indole or the dihydroindolo[3,2-b]indole scaffold for achieving significant cytotoxic activity against the HCT-116 cell line. It is worth noting that compounds belonging to the DINQ family did not exhibit any significant cytotoxic activity against the HCT-116 cell line in this study. Next, we determined the required concentrations of (3o) and (6g) for the reduction of the respective cell viability by 50% (IC50) using a luminescent assay. After 24 h of culture, the cells were treated with the tested compounds at 10 different final concentrations. After a 72 h treatment of the HCT-116 cell line with the respective compound, CellTiter Glo Reagent containing luceferin was added and luminescence was recorded with a spectrophotometric plate reader.
The results revealed that compounds (3o) and (6g) exhibited potent antiproliferative activity, with IC50 values of 298 nM and 62 nM, respectively (Fig. 2).
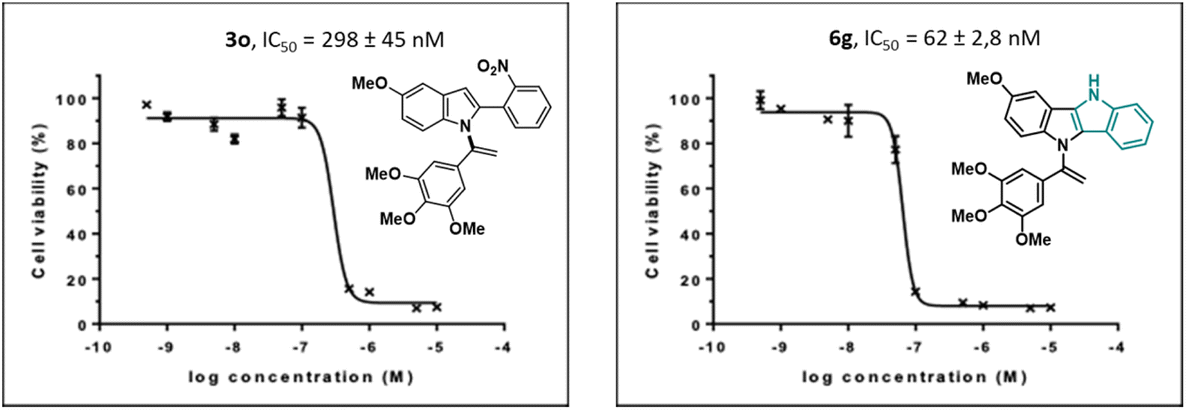 | ||
| Fig. 2 Effect of compounds 3o and 6g on the HCT-116 cells viability. | ||
As isoCA-4 derivatives act as antitubulin,13,16,18,21,36,37 to elucidate the potential binding mode of compound (6g) in tubulin, a docking study was conducted using the X-ray structure of tubulin (PDB code: 6H9B).18 Compound (6g), which exhibited the highest cytotoxicity in this series, was docked and compared to isoCA-4. The docking results and superimposition of (6g) and isoCA-4 are shown in Fig. 3.
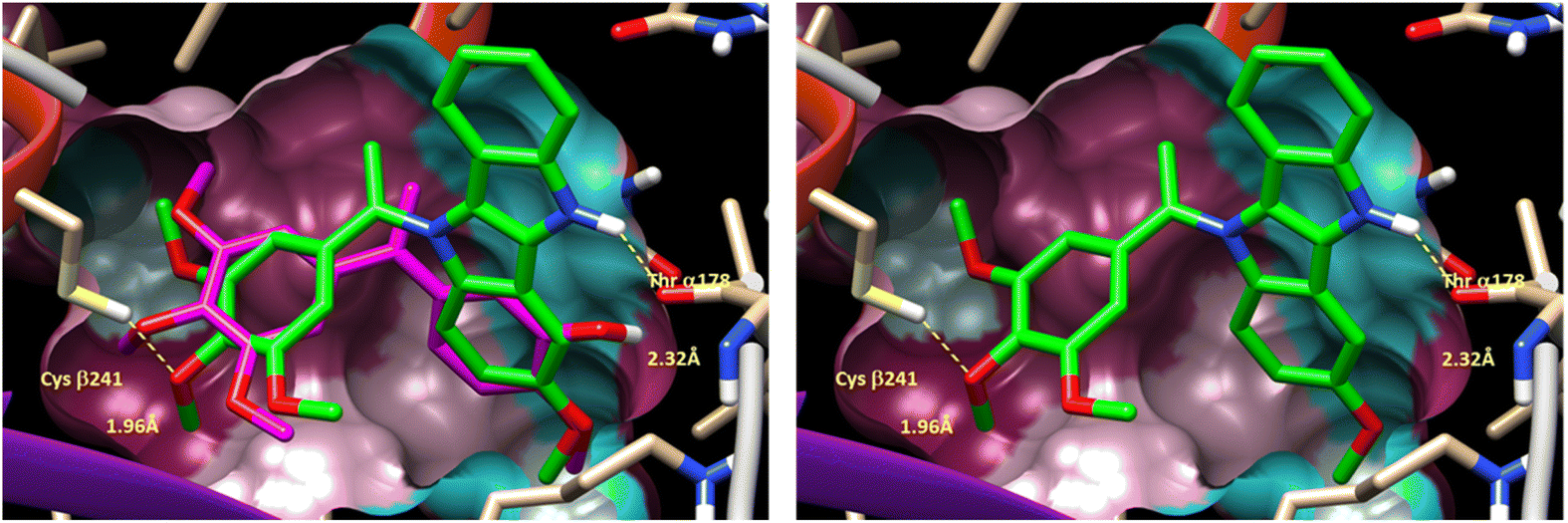 | ||
| Fig. 3 (Left) Putative binding mode of DINI derivative 6 g (green color) within colchicine binding site of tubulin X-ray structure (accession code 6H9B) (left) with previously reported binding mode of reference compound isoCA-4 in overlay (magenta color). (Right) Same figure (without isoCA-4 reference binding mode). | ||
In the docking study, compound (6g) demonstrated a binding pose similar to that observed with isoCA-4. Similar to CA-4 and isoCA-4, the trimethoxyphenyl ring of (6g) was positioned in close proximity to Cysβ241, while the 3-methoxy-5,10-dihydroindolo[3,2-b]indole ring was situated near Thrα178, forming a hydrogen bond between its NH group and the oxygen atom of Thrα178. This binding arrangement suggests that compound (6g) interacts with tubulin in a similar manner to isoCA-4, which is known to act as an antitubulin agent.
Conclusions
In conclusion, this study presents a copper-catalyzed approach for the synthesis of various terminal N-vinylazoles. Through optimization of the Cadogan-reductive cyclization conditions, unexpected but welcome dihydroindolo[1,2-c]quinazoline compounds 5 were obtained from skatole derivatives. The mechanism of the formation of these compounds involves the formation of an aniline intermediate, which undergoes intramolecular cyclization in the presence of Mo-catalyst. This cyclization occurs through a nucleophilic aniline attack on the terminal double bond of the N-vinylazole substrate, resulting in the formation of compound 5. However, when N-vinylazoles bearing a hydrogen atom at the C3 position of the indole were used, the double bond of the indole reacted preferentially over the terminal double bond of the N-vinylindole. This preference led to the trapping of the nitrene intermediate and the formation of dihydroindolo[3,2-b]indole derivatives 6. Biological evaluation of the synthesized derivatives revealed for compounds 3o and 6g their potential as cytotoxic agents against the HCT-116 human colon cancer cell line. These compounds exhibited significant cytotoxicity, with IC50 values of 298 nM and 62 nM, respectively. Furthermore, docking studies suggested that DINI derivative 6g interacts with tubulin in a similar manner to the antitubulin agent isoCA-4, supporting its potential as an antitubulin agent. These findings highlight the promising nature of the synthesized analogs as potential candidates for further development as anticancer agents.Experimental
General
Full experimental procedures for the synthesis and spectroscopic characterization of all compounds and intermediates can be found in the ESI.†General procedure for preparation of N-vinylazole compounds 3a–o
In a sealed tube under argon atmosphere, 2-(2-nitrophenyl)-1H-indole derivative (1) (0.2 mmol, 1.0 equiv.), copper powder (10 mol%), and K2CO3 (1.5 equiv.) were solubilized in dry toluene (0.8 mL). Then, N,N′-dimethylethylenediamine (DMEDA) (20 mol%), and the corresponding styrene (2) (2.0 equiv.) were added. The sealed tube was stirred at 135 °C for 20 hours.After completion, the mixture reaction was filtered through a Celite© pad and concentrated under reduced pressure. Finally, the crude product was purified by silica gel chromatography with cyclohexane/ethyl acetate (0 to 15% of ethyl acetate) as eluent to give the corresponding N-vinylazole.
General procedure for Cadogan-like cyclization: synthesis of compounds 5a–h, 6a–g and 8a
In a sealed tube, 2-(2-nitrophenyl)-1-(1-aryllvinyl)-1H-indole (3) (0.1 mmol, 1.0 equiv.), MoO2Cl2(DMF)2 (5 mol%) and PPh3 (2.4 equiv.) were dissolved in toluene (3.0 mL). Next, the solution was degassed with an argon balloon from −196 °C in liquid nitrogen to room temperature before being warmed at 160 °C for 6 hours. After completion, the mixture reaction was filtered through a Celite© pad, and concentrated under reduced pressure. Finally, the crude product was purified by silica gel chromatography with cyclohexane/ethyl acetate as eluent (0 to 20% of ethyl acetate) to give the corresponding compounds 5,6-dihydroindolo[1,2-c]quinazoline derivatives (5).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support of this project by CNRS, Paris-Saclay University and La Fondation ARC pour la recherche sur le cancer ARCPJA2022060005209. The authors also thank the China Scholarship Council for a fellowship (CSC) to X. L.References
- A. P. Taylor, R. P. Robinson, Y. M. Fobian, D. C. Blakemore, L. H. Jones and O. Fadeyi, Modern advances in heterocyclic chemistry in drug discovery, Org. Biomol. Chem., 2016, 14, 6611–6637 RSC.
- J. Jampilek, Heterocycles in Medicinal Chemistry, Molecules, 2019, 24, 3839 CrossRef CAS PubMed.
- K. Zhang, C. Tran, M. Alami, A. Hamze and O. Provot, Synthesis and Biological Activities of Pyrazino[1,2-a]indole and Pyrazino[1,2-a]indol-1-one Derivatives, Pharmaceuticals, 2021, 14, 779 CrossRef CAS PubMed.
- S. Pecnard, A. Hamze, J.-L. Pozzo, M. Alami and O. Provot, Synthesis of Oxazino[4,3-a]indoles and biological applications, Eur. J. Med. Chem., 2021, 224, 113728 CrossRef CAS PubMed.
- Y. Yao, M. Alami, A. Hamze and O. Provot, Recent advances in the synthesis of pyrido[1,2-a]indoles, Org. Biomol. Chem., 2021, 19, 3509–3526 RSC.
- M. M. Heravi and V. Zadsirjan, Prescribed drugs containing nitrogen heterocycles: an overview, RSC Adv., 2020, 10, 44247–44311 RSC.
- B. Mittal, S. Tulsyan, S. Kumar, R. D. Mittal and G. Agarwal, in Advances in Clinical Chemistry, ed. G. S. Makowski, Elsevier, 2015, vol. 71, pp. 77–139 Search PubMed.
- T. Tejada and M. F. El-Chami, in Cardio-Oncology, ed. R. A. Gottlieb and P. K. Mehta, Academic Press, Boston, 2017, pp. 321–330, DOI:10.1016/B978-0-12-803547-4.00022-7.
- E. Martino, G. Casamassima, S. Castiglione, E. Cellupica, S. Pantalone, F. Papagni, M. Rui, A. M. Siciliano and S. Collina, Vinca alkaloids and analogues as anti-cancer agents: Looking back, peering ahead, Bioorg. Med. Chem. Lett., 2018, 28, 2816–2826 CrossRef PubMed.
- V. Kuete, in Toxicological Survey of African Medicinal Plants, ed. V. Kuete, Elsevier, 2014, pp. 611–633, DOI:10.1016/B978-0-12-800018-2.00021-2.
- C. Dumontet and M. A. Jordan, Microtubule-binding agents: a dynamic field of cancer therapeutics, Nat. Rev. Drug Discovery, 2010, 9, 790–803 CrossRef CAS PubMed.
- C. Mousset, A. Giraud, O. Provot, A. Hamze, J. Bignon, J.-M. Liu, S. Thoret, J. Dubois, J.-D. Brion and M. Alami, Synthesis and antitumor activity of benzils related to combretastatin A-4, Bioorg. Med. Chem. Lett., 2008, 18, 3266–3271 CrossRef CAS PubMed.
- J. Aziz, E. Brachet, A. Hamze, J.-F. Peyrat, G. Bernadat, E. Morvan, J. Bignon, J. Wdzieczak-Bakala, D. Desravines, J. Dubois, M. Tueni, A. Yassine, J.-D. Brion and M. Alami, Synthesis, biological evaluation, and structure-activity relationships of tri- and tetrasubstituted olefins related to isocombretastatin A-4 as new tubulin inhibitors, Org. Biomol. Chem., 2013, 11, 430–442 RSC.
- S. Aprile, E. Del Grosso, G. C. Tron and G. Grosa, In Vitro Metabolism Study of Combretastatin A-4 in Rat and Human Liver Microsomes, Drug Metab. Dispos., 2007, 35, 2252–2261 CrossRef CAS PubMed.
- S. Messaoudi, B. Treguier, A. Hamze, O. Provot, J.-F. Peyrat, J. R. De Losada, J.-M. Liu, J. Bignon, J. Wdzieczak-Bakala, S. Thoret, J. Dubois, J.-D. Brion and M. Alami, Isocombretastatins A versus Combretastatins A: The Forgotten isoCA-4 Isomer as a Highly Promising Cytotoxic and Antitubulin Agent, J. Med. Chem., 2009, 52, 4538–4542 CrossRef CAS PubMed.
- T. Naret, J. Bignon, G. Bernadat, M. Benchekroun, H. Levaique, C. Lenoir, J. Dubois, A. Pruvost, F. Saller, D. Borgel, B. Manoury, V. Leblais, R. Darrigrand, S. Apcher, J.-D. Brion, E. Schmitt, F. R. Leroux, M. Alami and A. Hamze, A fluorine scan of a tubulin polymerization inhibitor isocombretastatin A-4: Design, synthesis, molecular modelling, and biological evaluation, Eur. J. Med. Chem., 2018, 143, 473–490 CrossRef CAS PubMed.
- T. Bzeih, T. Naret, A. Hachem, N. Jaber, A. Khalaf, J. Bignon, J.-D. Brion, M. Alami and A. Hamze, A general synthesis of arylindoles and (1-arylvinyl)carbazoles via a one-pot reaction from N-tosylhydrazones and 2-nitro-haloarenes and their potential application to colon cancer, Chem. Commun., 2016, 52, 13027–13030 RSC.
- T. Naret, I. Khelifi, O. Provot, J. Bignon, H. Levaique, J. Dubois, M. Souce, A. Kasselouri, A. Deroussent, A. Paci, P. F. Varela, B. Gigant, M. Alami and A. Hamze, 1,1-Diheterocyclic Ethylenes Derived from Quinaldine and Carbazole as New Tubulin-Polymerization Inhibitors: Synthesis, Metabolism, and Biological Evaluation, J. Med. Chem., 2019, 62, 1902–1916 CrossRef CAS PubMed.
- I. Khelifi, T. Naret, A. Hamze, J. Bignon, H. Levaique, M. C. Garcia Alvarez, J. Dubois, O. Provot and M. Alami, N,N-bis-heteroaryl methylamines: Potent anti-mitotic and highly cytotoxic agents, Eur. J. Med. Chem., 2019, 168, 176–188 CrossRef CAS PubMed.
- D. Renko, O. Provot, E. Rasolofonjatovo, J. Bignon, J. Rodrigo, J. Dubois, J.-D. Brion, A. Hamze and M. Alami, Rapid synthesis of 4-arylchromenes from ortho-substituted alkynols: A versatile access to restricted isocombretastatin A-4 analogues as antitumor agents, Eur. J. Med. Chem., 2015, 90, 834–844 CrossRef CAS PubMed.
- E. Rasolofonjatovo, O. Provot, A. Hamze, J. Rodrigo, J. Bignon, J. Wdzieczak-Bakala, C. Lenoir, D. Desravines, J. Dubois, J.-D. Brion and M. Alami, Design, synthesis and anticancer properties of 5-arylbenzoxepins as conformationally restricted isocombretastatin A-4 analogs, Eur. J. Med. Chem., 2013, 62, 28–39 CrossRef CAS PubMed.
- M. Roche, G. Frison, J.-D. Brion, O. Provot, A. Hamze and M. Alami, Csp2-N Bond formation via ligand-free Pd-catalyzed oxidative coupling reaction of N-tosylhydrazones and indole derivatives, J. Org. Chem., 2013, 78, 8485–8495 CrossRef CAS PubMed.
- K. Zhang, A. El Bouakher, H. Levaique, J. Bignon, P. Retailleau, M. Alami and A. Hamze, Pyrrolo-imidazo[1,2-a]pyridine Scaffolds through a Sequential Coupling of N-Tosylhydrazones with Imidazopyridines and Reductive Cadogan Annulation, Synthetic Scope, and Application, J. Org. Chem., 2019, 84, 13807–13823 CrossRef CAS PubMed.
- S. R. Chemler, Copper catalysis in organic synthesis, Beilstein J. Org. Chem., 2015, 11, 2252–2253 CrossRef CAS PubMed.
- J. I. G. Cadogan and M. Cameron-Wood, Reduction of Nitro-compounds by Triethyl Phosphite: A New Cyclisation Reaction, Proc. Chem. Soc., 1962, 361–361 CAS.
- R. Sanz, J. Escribano, M. R. Pedrosa, R. Aguado and F. J. Arnáiz, Dioxomolybdenum(VI)-Catalyzed Reductive Cyclization of Nitroaromatics. Synthesis of Carbazoles and Indoles, Adv. Synth. Catal., 2007, 349, 713–718 CrossRef CAS.
- CCDC 2310238 (5a), 2310239 (5f), 2310240 (6a), 2310241 (6c), 2310242 (6f) and 2310243 (8a) contain the supporting crystallographic data for this paper.†.
- C. Hauguel, J.-L. Pozzo, A. Hamze and O. Provot, Recent Advances in Synthesis of Pyrrolo[3,2-b]indole and Indolo[3,2-b]indole Derivatives, Asian J. Org. Chem., 2022, 11, e202200306 CrossRef CAS.
- Z. R. Owczarczyk, W. A. Braunecker, A. Garcia, R. Larsen, A. M. Nardes, N. Kopidakis, D. S. Ginley and D. C. Olson, 5,10-Dihydroindolo[3,2-b]indole-Based Copolymers with Alternating Donor and Acceptor Moieties for Organic Photovoltaics, Macromolecules, 2013, 46, 1350–1360 CrossRef CAS.
- Y.-E. Jin, K.-H. Kim, S.-H. Song, J.-W. Kim, J.-H. Kim, S.-H. Park, K.-H. Lee and H.-S. Suh, New Conjugated Polymer Based on Dihydroindoloindole for LEDs, Bull. Korean Chem. Soc., 2006, 27, 1043–1047 CrossRef CAS.
- K. Zhang, A. El Bouakher, H. Lévaique, J. Bignon, P. Retailleau, M. Alami and A. Hamze, Imidazodipyridines via DMAP Catalyzed Domino N–H Carbonylation and 6π-Electrocyclization: Synthetic Scope and Application, Adv. Synth. Catal., 2020, 362, 3243–3256 CrossRef CAS.
- For the mechanism of Mo-mediated Cadogan reaction, see: M. Castiñeira Reis, M. Marín-Luna, C. Silva López and O. N. Faza, Mechanism of the Molybdenum-Mediated Cadogan Reaction, ACS Omega, 2018, 3, 7019–7026 CrossRef PubMed.
- S. Liu, J. I. Amaro-Estrada, M. Baltrun, I. Douair, R. Schoch, L. Maron and S. Hohloch, Catalytic Deoxygenation of Nitroarenes Mediated by High-Valent Molybdenum(VI)–NHC Complexes, Organometallics, 2021, 40, 107–118 CrossRef CAS.
- R. Sanz, J. Escribano, M. R. Pedrosa, R. Aguado and F. J. Arnáiz, Dioxomolybdenum(VI)-Catalyzed Reductive Cyclization of Nitroaromatics. Synthesis of Carbazoles and Indoles, Adv. Synth. Catal., 2007, 349, 713–718 CrossRef CAS.
- I. W. Davies, V. A. Guner and K. N. Houk, Theoretical Evidence for Oxygenated Intermediates in the Reductive Cyclization of Nitrobenzenes, Org. Lett., 2004, 6, 743–746 CrossRef CAS PubMed.
- A. Hamze, E. Rasolofonjatovo, O. Provot, C. Mousset, D. Veau, J. Rodrigo, J. Bignon, J.-M. Liu, J. Wdzieczak-Bakala, S. Thoret, J. Dubois, J.-D. Brion and M. Alami, B-Ring-Modified isoCombretastatin A-4 Analogues Endowed with Interesting Anticancer Activities, ChemMedChem, 2011, 6, 2179–2191 CrossRef CAS PubMed.
- E. Rasolofonjatovo, O. Provot, A. Hamze, J. Rodrigo, J. Bignon, J. Wdzieczak-Bakala, D. Desravines, J. Dubois, J.-D. Brion and M. Alami, Conformationally restricted naphthalene derivatives type isocombretastatin A-4 and isoerianin analogues: Synthesis, cytotoxicity and antitubulin activity, Eur. J. Med. Chem., 2012, 52, 22–32 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization data, and copies of 1H, 13C, and 19F NMR spectra. CCDC 2310238–2310243. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qo01973j |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2024 |