
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iodine radical mediated cascade [3 + 2] carbocyclization of ene-vinylidenecyclopropanes with thiols and selenols via photoredox catalysis†
Zhe
Meng
a,
Min
Shi
 *ab and
Yin
Wei
*b
*ab and
Yin
Wei
*b
aKey Laboratory for Advanced Materials and Institute of Fine Chemicals, School of Chemistry & Molecular Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China
bState Key Laboratory of Organometallic Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: mshi@mail.sioc.ac.cn; weiyin@sioc.ac.cn
First published on 8th January 2024
Abstract
An iodine radical mediated cascade [3 + 2] carbocyclization of ene-vinylidenecyclopropanes with thiols and selenols via photoredox catalysis has been reported in this paper. With this visible light-induced photocatalytic protocol, an efficient synthetic methodology for the rapid construction of sulfur- and selenium-containing polycyclic derivatives in moderate to good yields has been realized with broad substrate scope. Mechanistic investigations were also performed using control experiments, deuterium labeling and Stern–Volmer analysis as well as DFT calculations, suggesting that this cascade cyclization reaction stems from an iodine radical addition to the allenyl moiety of ene-vinylidenecyclopropane along with a cascade cyclization. Then, the reaction proceeds through a cyclopropane-ring opening pathway along with a HAT process and an intramolecular substitution. Further transformation of the obtained product has also been disclosed.
Introduction
Sulfur-containing polycyclic compounds play a significant bioactive role in natural products, pharmaceuticals and polymeric materials.1 Moreover, in the field of life sciences, disulfide bonds can enhance the stability of polypeptides and realize biological functions of proteins.1c For example, gliocladine C is a fungal metabolite that possesses a unique molecular structure and potent activities against parasites, viruses, bacteria and cancer cells (Scheme 1a).2 With regard to selenium, which is in the same main group as sulfur, selenium-containing compounds generally exhibit more potent biological activity than their sulfur-containing counterparts.3 In addition, a prominent feature of selenium compounds is the fact that they are more easily absorbed by cancer cells, although the mechanism of selective selenium uptake in cancer cells is still not completely understood.4 For instance, β-lapachone-selenide can exert submicromolar inhibitory activity toward tumor cells but weak cytotoxic activity toward normal cells (Scheme 1a).3d Considering the above factors, the design and synthesis of chalcogen-containing polycyclic compounds have attracted great interest in recent years.5 | ||
| Scheme 1 Chalcogen-containing cyclic compounds, previous work and this work. | ||
The synthesis of complex polycyclic molecules has always been a momentous research subject in synthetic chemistry, especially for the preparation of multiple cyclic systems within one step.6 As an eco-friendly and cost-effective accelerator, iodine and its derivatives present undisputed advantages in most cases, including being easily operated and metal-free, as well as affording economies of resource, time, and labor. It is well known that the coordination between the electrophilic iodine cation and the unsaturated bond can generate the corresponding iodinium ion,7 which would be subsequently attacked by other unsaturated groups within the molecule, triggering a cascade cyclization reaction to obtain the iodocyclization products (Scheme 1b).7 For example, our group reported a novel cascade process for the synthesis of 3,3′-diphenyl-1,1′-spirobi[indene] derivatives from propargyl alcohol-tethered alkylidenecyclopropanes in the presence of I2.7e In addition, Liang's group presented iodine promoted cascade cycloisomerization of 1-en-6,11-diynes for the easy preparation of tetrahydrobenzo[f]isoquinolines.7j On the other hand, recently with the rapid development of visible light-induced photocatalysis in organic synthesis, iodine and its derivatives have been widely utilized in a variety of photo-induced transformations.8 It has been known that I− can be oxidized by photosensitizers through a SET process to form the iodine radical; moreover, I2 can also undergo a homolytic cleavage reaction through photoexcitation to give the iodine radical.8g–8i Thus, the in situ generated iodine radical tends to be subsequently captured by unsaturated groups, leading to an intramolecular cascade cyclization reaction (Scheme 1c).
We previously investigated a series of cyclization reactions of ene-vinylidenecyclopropanes (ene-VDCPs)9 with radical-based reagents under different conditions, providing a variety of nitrogen-containing heterocycles.9e In view of the aforementioned research circumstances, we wish to report in this paper a novel iodine radical mediated cascade [3 + 2] carbocyclization of ene-vinylidenecyclopropanes via photoredox catalysis upon visible light irradiation, delivering sulfur- or selenium-containing polycyclic derivatives under mild conditions (Scheme 1d).
Results and discussion
We first utilized substrate 1a as the model substrate for the initial investigation and subsequently optimized the reaction conditions. The results are presented in Table 1. After several initial experimental examinations, the optimal reaction conditions were identified as the following: ene-vinylidenecyclopropane (ene-VDCP) 1a (0.2 mmol, 1.0 equiv.) was used as the substrate, 4-methylbenzenethiol 2a (0.4 mmol, 2.0 equiv.) was employed as a reagent, TBAI (0.4 mmol, 2.0 equiv.) was used as an additive, and Ir[(dtbbpy)ppy2]PF6 was utilized as a photosensitizer in acetonitrile (MeCN) (10.0 mL) under irradiation with a 30 W blue LED for 12 h, affording the desired product 3aa in 78% NMR yield and 73% isolated yield (Table 1, entry 1). In addition, other photosensitizers such as Ir[(dFCF3ppy)2dtbbpy]PF6, 4CzIPN, and Ir(ppy)3PF6 afforded 3aa in moderate yields of 55%–64% (entries 2–4) (see Tables S1–S7 in the ESI† for more information). We further examined solvent effects in this photochemical transformation and found that the use of other solvents, such as DCM, acetone and DMF, afforded 3aa in lower yields ranging from 0% to 47%, demonstrating that the best solvent choice for the reaction was MeCN (entries 5–7) (see Table S2 in the ESI† for more information). When the solvent volume was changed to 2.0 mL, the yield of 3aa decreased to 60% (entry 8) (see Table S6 in the ESI† for more information). In addition, when lowering the employed amount of TBAI to 0.2 equiv., the reaction efficiency decreased significantly (entry 9) (see Table S4 in the ESI† for more information). Moreover, we observed that other iodine anion sources or bromine anion sources gave 3aa in lower yields (entries 10 and 11) (see Table S3 in the ESI† for more information). When using disulfide (Ph2S2) instead of thiol, no reaction occurred, indicating that disulfides or diselenides cannot be used instead of thiols or selenols (entry 12). Furthermore, the control experiments revealed that a photosensitizer, light, and TBAI were essential for this reaction (entries 13–15).|
|
||
|---|---|---|
| Entrya | Variation from the standard conditions | 3aa, Yieldb [%] |
| a Reaction was carried out with 1a (0.1 mmol), 2a (2.0 equiv.), TBAI (2.0 equiv.), [Ir(dtbpy)ppy2]PF6 (3.0 mol%) in MeCN (10.0 mL) at ambient temperature using 30 W blue LED irradiation for 12 hours. b 1H NMR yield using dimethyl terephthalate as an internal standard. c Isolated yield. | ||
| 1 | None | 78 (73)c |
| 2 | [lr(dFCF3ppy)2dtbpy]PF6 as PC | 62 |
| 3 | 4CzlPN as PC | 64 |
| 4 | fac-lr(ppy)3 as PC | 55 |
| 5 | DCM instead of MeCN | 47 |
| 6 | Acetone instead of MeCN | 51 |
| 7 | DMF instead of MeCN | 0 |
| 8 | 2.0 ml MeCN instead of 10.0 ml MeCN | 60 |
| 9 | 0.2 equiv. TBAI instead of 2.0 equiv. TBAI | 50 |
| 10 | Kl instead of TBAI | 53 |
| 11 | TBAB instead of TBAI | 58 |
| 12 | Disulfide instead of thiol | 0 |
| 13 | Without light | 0 |
| 14 | Without photocatalyst | 0 |
| 15 | Without TBAI | 0 |

With the reaction conditions being optimized, we explored the generality of this cascade annulation reaction, and the results are summarized in Scheme 2. It was found that most of the substrates successfully underwent these reactions smoothly, providing the desired products in moderate to good yields. In order to investigate the electronic effect of the substituent R2, the reactions were conducted using the substrates by altering the substituent R located at the para position on the benzene ring. The VDCPs 1b–1h having either electron-withdrawing or -donating groups present on the benzene ring were all compatible in this reaction, and the desired products 3ba–3ha were obtained in 57%–79% yields, revealing that the electronic property of R2 had an impact on the yields of the product. When CF3-substituted VDCP 1f was utilized as the substrate, the desired product 3fa was not produced in this reaction, probably due to the influence of its electronic effect. The structure of 3ga was unambiguously determined by X-ray crystallographic analysis and its ORTEP drawing is shown in Scheme 2. In addition, its CIF data are summarized in the ESI.† When a meta-substituted methyl group was present at the benzene ring of VDCP 1i, the corresponding product 3ia was afforded in 61% yield. Product 3ja, bearing a benzo[d][1,3]dioxole moiety, could be obtained in 60% yield under the standard conditions. Moreover, substrate 1k, replacing the aryl group with a five-membered heterocyclic ring, could also afford the corresponding product 3ka in 46% yield. However, the reaction of a methyl-substituted substrate 1l proceeded sluggishly, affording a complex mixture. As for VDCPs 1m and 1n, in which a cyclohexyl or a benzyl group was introduced, the corresponding products 3ma–3na were produced in 53% and 43% yields, respectively. Next, we shifted our attention to examine the R1 group in VDCPs 1 and found that introducing the substituents R′ at the ortho-, meta-, and para-position on the benzene ring in VDCPs 1o–1s gave the desired products 3oa–3sa in moderate to good yields ranging from 61% to 87%, which is similar to those of R substituents. When a methyl group was present at R1 in substrate 1t, the corresponding product 3ta was afforded in 41% yield. To our delight, upon changing the linker to an oxygen atom, the desired product 3ua was obtained in 43% yield. Furthermore, when the (C(SO2Ph)2)-linked substrate 1v was used to carry out the reaction, the desired product 3va was furnished in 45% yield. Next, the R3 moiety of thiol reactants 2 was investigated. For the thiols containing a para-substituted benzene ring, the reactions took place smoothly, affording the target products 3ab–3ae in 61–81% yields. However, when pentachlorothiophenol 2f and alkyl-substituted thiol 2g were used as the reactants, the reaction did not afford the desired products.
 | ||
Scheme 2
a
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Standard conditions: substrate 1 (0.1 mmol, 1.0 equiv.), 2 (2.0 equiv.), TBAI (2.0 equiv.), [Ir(dtbpy)ppy2]PF6 (3.0 mol%) in MeCN (10.0 mL) at ambient temperature using 30 W blue LED irradiation for 12 hours. bNo reaction. cThe desired product was obtained in a complex mixture. Standard conditions: substrate 1 (0.1 mmol, 1.0 equiv.), 2 (2.0 equiv.), TBAI (2.0 equiv.), [Ir(dtbpy)ppy2]PF6 (3.0 mol%) in MeCN (10.0 mL) at ambient temperature using 30 W blue LED irradiation for 12 hours. bNo reaction. cThe desired product was obtained in a complex mixture. | ||
In order to further demonstrate the generality of this synthetic strategy, we then turned our attention to examining the substrate scope of 4 using selenol 2h as the cyclization reactant (Scheme 3). Similarly, using VDCPs 1 as the substrates, the desired selenium-containing polycyclic products 4a–4f were produced in moderate to good yields ranging from 35% to 67% under the standard conditions, indicating that this photochemical transformation is suitable for chalcogen-containing cyclic compounds. The substrates 1 having either electron-withdrawing or electron-donating groups present on the benzene ring were all tolerated in this reaction.
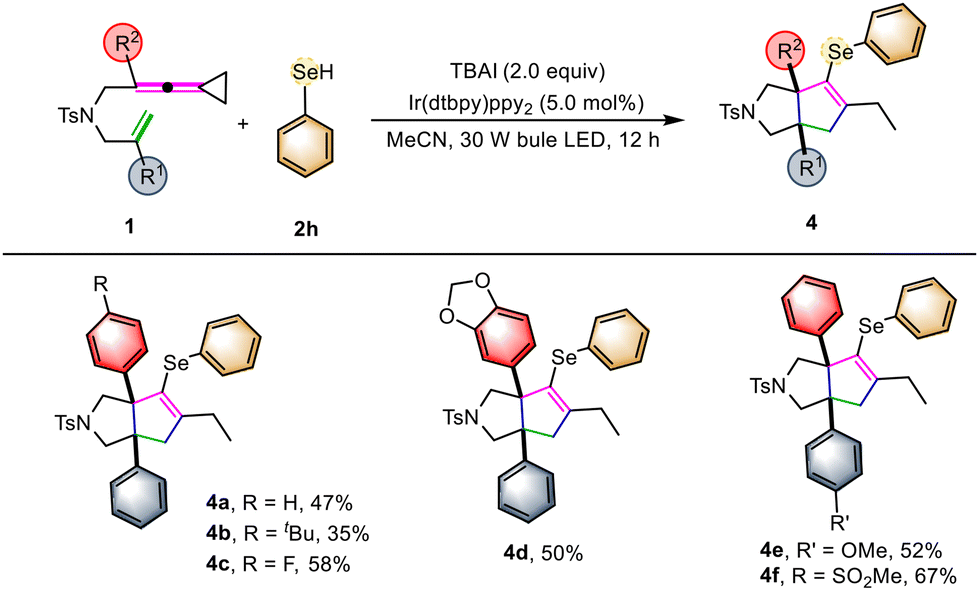 | ||
| Scheme 3 Standard conditions: substrate 1 (0.1 mmol, 1.0 equiv.), 2h (2.0 equiv.) TBAI (2.0 equiv.), [Ir(dtbpy)ppy2]PF6 (3.0 mol%) in MeCN (10.0 mL) at ambient temperature using 30 W blue LED irradiation for 12 hours. | ||
To gain more insights into the reaction mechanism, we carried out several control experiments (Scheme 4). First, the Stern–Volmer luminescence quenching analysis using TBAI and 2a showed that both species can quench the emission of Ir[(dtbbpy)ppy2]PF6. However, the quenching efficiency of TBAI was much stronger than that of 2a (Scheme 4A).10 The above result revealed that TBAI was an effective quencher for the excited state of Ir[(dtbbpy)ppy2]PF6. In order to investigate whether a free radical chaining process occurs in the reaction, we conducted the exclusive light-dependence of the reaction, in which the reaction basically stopped under dark conditions and continued when light was restored, indicating that visible light irradiation is a necessary condition for this reaction (Scheme 4B)11 and the quantum yield was found to be Φ = 0.005 in this reaction (see S18 in the ESI†), also suggesting that the intervention of a radical chain mechanism is unlikely. In addition, when TEMPO was added to the reaction system as a radical scavenger, the reaction was inhibited completely, and the desired product 3aa was not obtained (Scheme 4C). Subsequently, the reaction using deuterium-labeled thiophenol d1-2a (90% D content) was conducted under the standard conditions, and we found that the deuterium atom was incorporated exclusively at the terminal carbon atom of d1-3aa, showing that SET and HAT processes were both involved in the last step (Scheme 4D).12 We also performed kinetic isotope experiments under the standard conditions (Scheme 4E). The parallel kinetic isotope effect was determined as kH/kD = 2.02 using 2a and d1-2a as the substrates; this result demonstrated that the initial HAT process might be involved in the rate-determining step following the rationale that C–D bond dissociation is harder than C–H bond dissociation.13 Furthermore, when acetic acid (2.0 equiv.) as a proton source was added to the reaction system in the presence of 0.2 equiv. of thiophenol 2a, the intermediate 3aa′ was detected by LC-MS in 34% yield along with 3aa in 12% yield (see Fig. S7 in the ESI† for more information). Then, we utilized 2e as a nucleophilic reagent to react with intermediate 3aa′ in the presence of potassium carbonate, giving the desired product 3ea in 53% yield. Therefore, we believed that the desired product 3aa was obtained via intermediate 3aa′ through an intermolecular substitution reaction (Scheme 4F).14 We subsequently embarked on DFT calculations to gain insight into the reaction mechanism. All calculations were performed at the SMD(acetonitrile)/B3LYP/6-311+G(d,p)//B3LYP/6-31G(d) level using the Gaussian 16 program.15 The solvation Gibbs free energy profile in acetonitrile for the suggested reaction pathway is shown in Scheme 4G (see Table S8 in the ESI† for more information). We investigated the reaction pathway starting from the radical intermediate INT1 shown in Scheme 4G. The intermediate INT1 undergoes cyclization viaTS1 with an energy barrier of 15.0 kcal mol−1 to form an intermediate INT2, which produces the cyclized radical intermediate INT3 through another intramolecular addition reaction with an energy barrier of 12.9 kcal mol−1. We also investigated another possible cyclization process: cyclization of intermediate INT1viaTS1′ with an energy barrier of 26.8 kcal mol−1 forming an intermediate INT2′ containing a five-membered ring (Scheme 4G); however, the energy barrier is relatively high and the transition state TS2′ could not be located theoretically. Therefore, we excluded the pathway from INT1 to INT3viaINT2′. The intermediate INT3 undergoes a cyclopropane ring-opening process to give the radical intermediate INT4 with an energy barrier of 9.7 kcal mol−1. Subsequently, the intermediate INT4 undergoes a HAT process with thiol 2a to deliver the intermediate INT5 and a sulfur radical with an energy barrier of 15.3 kcal mol−1. Based on the DFT calculations, the HAT process is the rate-determining step which agrees with the KIE result shown in Scheme 4E.
 | ||
| Scheme 4 Mechanistic studies. (A) Luminescence quenching experiments (Stern–Volmer Studies). (B) Light on/off experiment. (C) Radical trapping experiments. (D) Deuterium labeling experiments. (E) Kinetic isotope effect experiment. (F) Control experiment. (G) DFT calculation. | ||
On the basis of control experiments and DFT calculations, we proposed a plausible mechanism to elucidate this visible light-induced photochemical reaction (Scheme 5). Upon irradiation with blue light, the ground state of the photosensitizer Ir[(dtbbpy)ppy2]PF6 is converted into its excited state, which can further oxidize I− through the SET process to afford the iodine radical, which tends to react with the allenyl moiety of VDCP 1 to furnish an iodinated radical intermediate A. The intermediate A undergoes cyclization viaTS1 with an energy barrier of 15.0 kcal mol−1 to form an intermediate B, which produces a cyclized radical intermediate C through another intramolecular addition reaction. Based on the calculation results, cyclization of intermediate AviaTS1′ with an energy barrier of 26.8 kcal mol−1 forms the intermediate B′. Thus, we exclude Path A from A to CviaB′. The intermediate C undergoes a cyclopropane ring-opening process to furnish the radical intermediate D, which subsequently undergoes a HAT process with thiol 2a13 to deliver intermediate E and a sulfur radical. Then, the in situ generated IrII species reduces the sulfur radical to the corresponding sulfur anion, which can substitute the iodine atom in intermediate E to give the desired product 3 and close the catalytic cycle.14
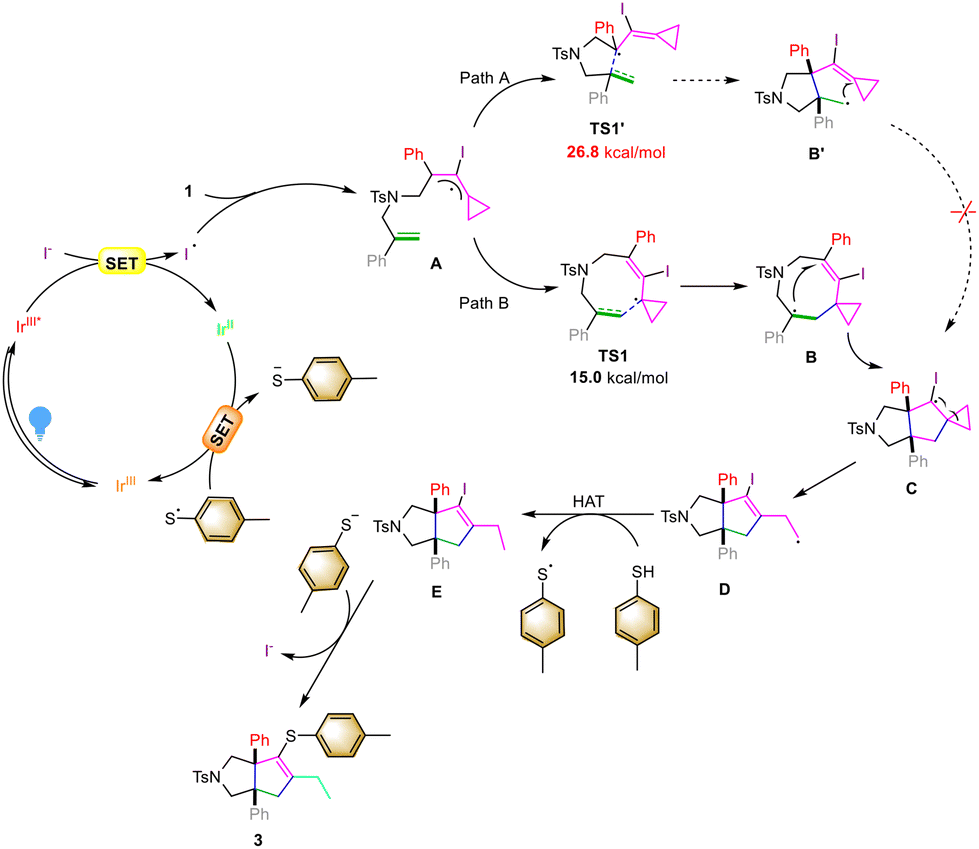 | ||
| Scheme 5 The proposed reaction mechanism. | ||
To demonstrate the synthetic applicability of this protocol, a gram-scale reaction was conducted by using 0.88 g (2.0 mmol) of 1a, obtaining the desired product 3aa in 80% yield (0.9 g) under the standard conditions (Scheme 6A). Hydrogenation of the obtained product 3aa effectively afforded the corresponding product 5 in 83% yield (Scheme 6B). Moreover, epoxidation of 3aa with m-CPBA as an oxidant furnished the product 6 in 78% yield (Scheme 6C).
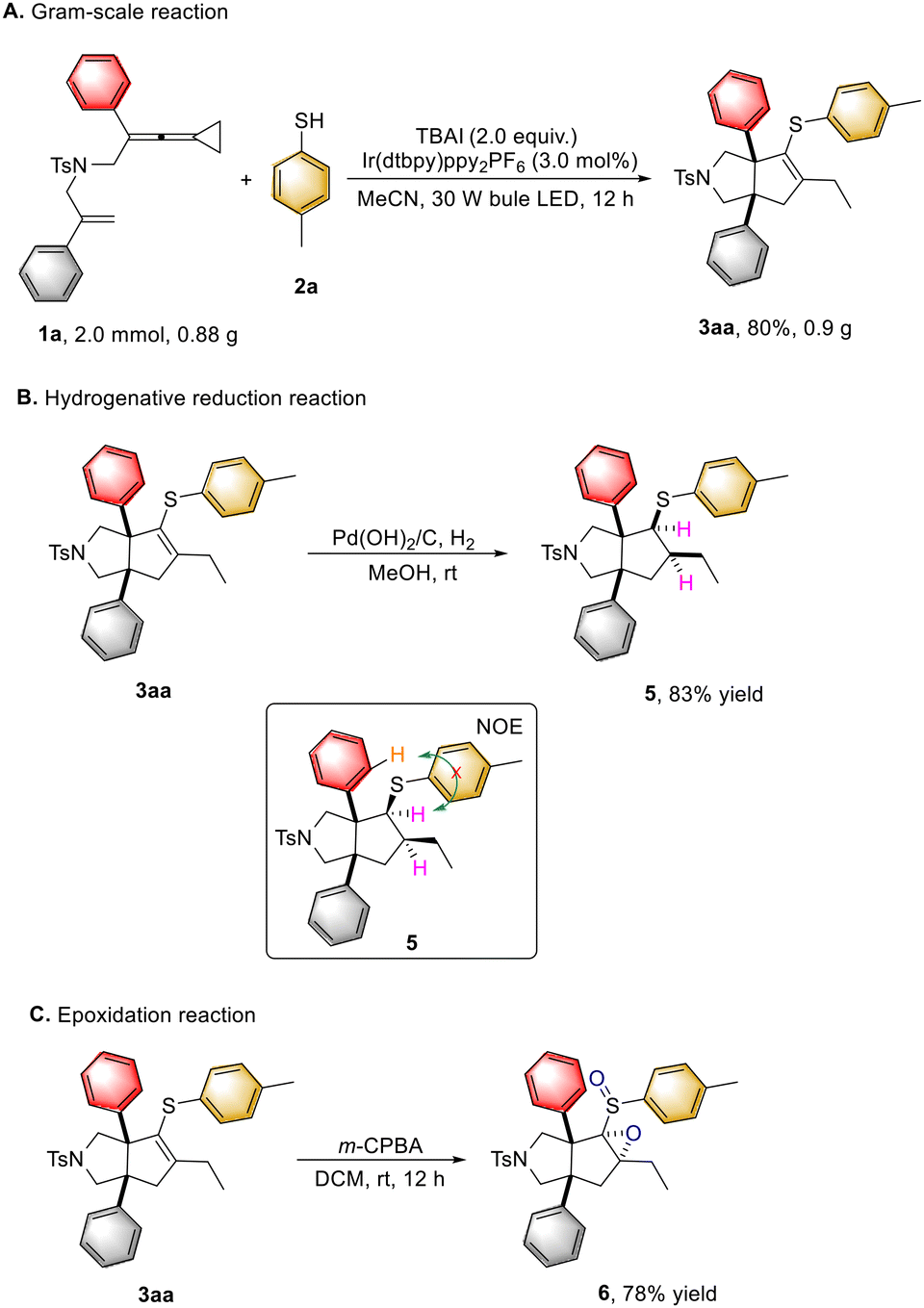 | ||
| Scheme 6 Synthetic transformations. (A) 1a (2.0 mmol, 1.0 equiv.), 2a (2.0 equiv.), TBAI (2.0 equiv.), [Ir(dtbpy)ppy2]PF6 (3.0 mol%) in MeCN (60.0 mL) at ambient temperature using 30 W blue LED irradiation for 12 hours; (B) Pd/C, MeOH, rt, H2; (C) m-CPBA (2.5 equiv.). | ||
On the other hand, when we utilized ethyl iododifluoroacetate as the iodine source for the reaction, a new iodocyclization product 7 was obtained in 20% yield upon direct visible light irradiation (Scheme 7). Its structure has been unequivocally identified by X-ray diffraction and the ORTEP drawing is depicted in Scheme 7. A plausible reaction mechanism is also shown in Scheme 7. Upon direct irradiation with blue light, the iodine radical is generated from homolytic cleavage of ethyl iododifluoroacetate, which tends to react with 1a to furnish an iodinated radical intermediate I. Subsequently, similar intramolecular cascade cyclization and cyclopropane ring-opening take place to afford radical intermediate IV through radical intermediates II and III, which reacts with ethyl iododifluoroacetate again to afford product 7 through a radical chain process.9e In our previous paper, we have described that some unknown side reaction products could be formed upon photoirradiation of ethyl iododifluoroacetate.16
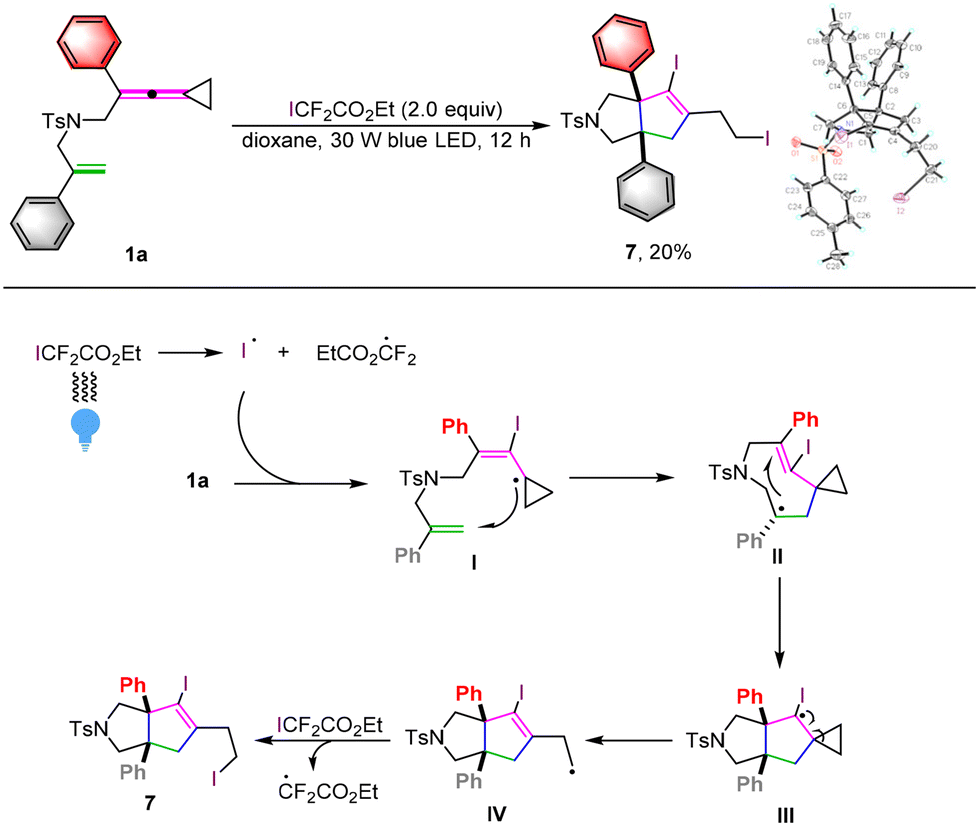 | ||
| Scheme 7 1a (0.1 mmol, 1.0 equiv.), ICF2CO2Et (2.0 equiv.), Ir(ppy)3 (5.0 mol%) in dioxane (10.0 mL) at ambient temperature using 30 W blue LED irradiation for 12 hours. | ||
Conclusions
In summary, we have developed a novel and practical photoredox catalytic methodology for iodine radical mediated cascade carbocyclization of ene-vinylidenecyclopropanes with thiols and selenols, delivering the chalcogen-containing polycyclic derivatives in moderate to good yields with broad substrate scope and good functional group tolerance under mild conditions. Moreover, this reaction could be achieved on a gram scale, and the products could be further functionalized to afford other novel polycyclic compounds. The reaction mechanism paradigm has been proposed on the basis of control experiments, deuterium labeling and photophysical analysis as well as DFT calculations. Further exploration of this visible light photoinduced synthetic strategy for the synthesis of medicinally useful products is underway.Data availability
Experimental and computational data have been made available in the ESI.†Author contributions
Z. Meng contributed to the investigation. Z. Meng, Y. Wei and M. Shi contributed to conceptualization and writing the original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Key R & D Program of China (2022YFC2303100), the National Natural Science Foundation of China (21372250, 21121062, 21302203, 21772037, 21772226, 21861132014, 91956115 and 22171078) and the Fundamental Research Funds for the Central Universities 222201717003.References
- (a) R. A. Bragg, S. Brocklehurst, F. Gustafsson, J. Goodman, K. Hickling, P. A. MacFaul, S. Swallow and J. Tugwood, Aortic Binding of AZD5248: Mechanistic Insight and Reactivity Assays To Support Lead Optimization, Chem. Res. Toxicol., 2015, 10, 1991–1999 Search PubMed; (b) N. Z. Wang, P. Saidhareddy and X. F. Jiang, Construction of sulfur-containing moieties in the total synthesis of natural products, Nat. Prod. Rep., 2020, 37, 246–275 Search PubMed; (c) R. Takeuchi, J. Shimokawa and T. Fukuyama, Development of a route to chiral epidithiodioxopiperazine moieties and application to the asymmetric synthesis of (+)-hyalodendrin, Chem. Sci., 2014, 5, 2003–2006 Search PubMed; (d) T. C. Adams, J. N. Payette, J. H. Cheah and M. Movassaghi, Concise Total Synthesis of (+)-Luteoalbusins A and B, Org. Lett., 2015, 17, 4268–4271 Search PubMed; (e) K. L. Rinehart, T. G. Holt, N. L. Fregeau, J. G. Stroh, P. A. Keifer, F. Sun, L. H. Li and D. G. Martin, Ecteinascidins 729, 743, 745, 759A, 759B, and 770: potent antitumor agents from the Caribbean tunicate Ecteinascidia turbinata, J. Org. Chem., 1990, 15, 4512–4515 Search PubMed; (f) B. R. Beno, K. S. Yeung, M. D. Bartberger, L. D. Pennington and N. A. Meanwell, A Survey of the Role of Noncovalent Sulfur Interactions in Drug Design, J. Med. Chem., 2015, 11, 4383–4438 Search PubMed.
- J. E. DeLorbe, D. Horne, R. Jove, S. M. Mennen, S. Nam, F. L. Zhang and L. E. Overman, General Approach for Preparing Epidithiodioxopiperazines from Trioxopiperazine Precursors: Enantioselective Total Syntheses of (+)- and (-)-Gliocladine C, (+)-Leptosin D, (+)-T988C, (+)-Bionectin A, and (+)-Gliocladin A, J. Am. Chem. Soc., 2013, 135, 4117–4128 Search PubMed.
- (a) W. Hou and H. T. Xu, Incorporating Selenium into Heterocycles and Natural Products-From Chemical Properties to Pharmacological Activities, J. Med. Chem., 2022, 65, 4436–4456 Search PubMed; (b) Y. Ogra, K. Ishiwata, H. Takayama, N. Aimi and K. T. Suzuk, Identification of a novel selenium metabolite Se-methyl-N-acetylselenohexosamine, in rat urine by high-performance liquid chromatography-inductively coupled plasma mass spectrometry and -electrospray ionization tandem mass spectrometry, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2002, 767, 301–312 Search PubMed; (c) P. Arsenyan, E. Paegle, I. Domracheva, A. Gulbe, I. K. Lapsa and I. Shestakova, Selenium analogues of raloxifene as promising antiproliferative agents in treatment of breast cancer, Eur. J. Med. Chem., 2014, 87, 471–483 Search PubMed; (d) A. A. Vieira, I. R. Brandao, W. O. Valenca, C. A. D. Simone, B. C. Cavalcanti, C. Pessoa, T. R. Carneiro, A. L. Braga and E. N. D. S. Júnior, Hybrid compounds with two redox centres: Modular synthesis of chalcogen-containing lapachones and studies on their antitumor activity, Eur. J. Med. Chem., 2015, 101, 254–265 Search PubMed.
- (a) R. P. Spencer, G. Montana, G. T. Scanlon and O. R. Evans, Uptake of selenomethionine by mouse and in human lymphomas, with observations on selenite and selenate, J. Nucl. Med., 1967, 8, 197–208 Search PubMed; (b) J. Esteban, D. Lasa and S. P. Modrego, Detection of Cartilaginous Tumors with Selenium 75, Radiology, 1965, 85, 149–150 Search PubMed; (c) R. R. Cavalieri and K. G. Scott, Sodium Selenite Se 75: A More Specific Agent for Scanning Tumors, J. Am. Med. Assoc., 1968, 206, 591–595 Search PubMed; (d) R. R. Cavalieri, K. G. Scott and E. Sairenji, Selenite (75(Se) as a tumor-localizing agent in man, J. Nucl. Med., 1966, 7, 197–208 Search PubMed.
- (a) C. X. Li, R. J. Liu, K. Yin, L. R. Wen and M. Li, Synthesis of disulfides tethered pyrroles from β-ketothioamides via a bicyclization/ring-opening/oxidative coupling reaction, Org. Biomol. Chem., 2017, 15, 5820–5823 Search PubMed; (b) Y. A. Zhang, Z. Ding, P. Liu, W. S. Guo, L. R. Wen and M. Li, Access to SCN-containing thiazolines via electrochemical regioselective thiocyanothiocyclization of N-allylthioamides, Org. Chem. Front., 2020, 7, 1321–1326 Search PubMed; (c) B. B. Liu, H. W. Bai, H. Liu, S. Y. Wang and S. J. Ji, Cascade Trisulfur Radical Anion (S3˙−) Addition/Electron Detosylation Process for the Synthesis of 1,2,3-Thiadiazoles and Isothiazoles, J. Org. Chem., 2018, 17, 10281–10288 Search PubMed.
- (a) M. Randić, Aromaticity of Polycyclic Conjugated Hydrocarbons, Chem. Rev., 2003, 103, 3449–3606 Search PubMed; (b) T. Nakata, Total Synthesis of Marine Polycyclic Ethers, Chem. Rev., 2005, 105, 4314–4347 Search PubMed; (c) R. A. Craig and B. M. Stoltz, Polycyclic Furanobutenolide-Derived Cembranoid and Norcembranoid Natural Products: Biosynthetic Connections and Synthetic Efforts, Chem. Rev., 2017, 117, 7878–7909 Search PubMed; (d) M. Hirai, N. Tanaka, M. Sakaiand and S. Yamaguchi, Structurally Constrained Boron-, Nitrogen-, Silicon-, and Phosphorus-Centered Polycyclic π-Conjugated Systems, Chem. Rev., 2019, 119, 8291–8331 Search PubMed.
- (a) K. G. Ji, H. T. Zhu, F. Yang, X. Z. Shu, S. C. Zhao, X. Y. Liu, A. Shaukat and Y. M. Liang, A Novel Iodine-Promoted Tandem Cyclization: An Efficient Synthesis of Substituted 3,4-Diiodoheterocyclic Compounds, Chem. – Eur. J., 2010, 16, 6151–6154 Search PubMed; (b) C. Wan, J. Zhang, S. Wang, J. Fan and Z. Wang, Facile Synthesis of Polysubstituted Oxazoles via A Copper-Catalyzed Tandem Oxidative Cyclization, Org. Lett., 2010, 12, 2338–2341 Search PubMed; (c) H. Batchu, S. Bhattacharyya and S. Batra, Iodine-Mediated Intramolecular Electrophilic Aromatic Cyclization in Allylamines: A General Route to Synthesis of Quinolines, Pyrazolo[4,3-b]pyridines, and Thieno[3,2-b]pyridines, Org. Lett., 2012, 14, 6330–6333 Search PubMed; (d) J. Zhang, Z. Wang, Y. Wang, C. Wan, X. Zheng and Z. Wang, A metal-free catalytic system for the oxidation of benzylic methylenes and primary amines under solvent-free conditions, Green Chem., 2009, 11, 1973–1978 Search PubMed; (e) Q. Z. Li, L. Z. Yu, Y. Wei and M. Shi, Synthesis of Diiodinated All-Carbon 3,3′-Diphenyl-1,1′-spirobiindene Derivatives via Cascade Enyne Cyclization and Electrophilic Aromatic Substitution, J. Org. Chem., 2019, 84, 9282–9296 Search PubMed; (f) J. Tang, C. Q. Zhao, S. Li, J. Zhang, X. L. Zheng, M. L. Yuan and H. Y. Fu, Tandem Ring-Contraction/Regioselective C-H Iodination Reaction of Pyridinium Salts, J. Org. Chem., 2023, 88, 2809–2821 Search PubMed; (g) T. Khan and S. Yaragorla, Iodocyclization of Propargyl Alcohols: Highly Facile Approach to Hetero/Carbocyclic Iodides, Eur. J. Org. Chem., 2019, 25, 3989–4012 Search PubMed; (h) U. P. N. Tran, G. Oss, M. Breugst, E. Detmar, D. P. Pace, K. Liyanto and T. V. Nguyen, Carbonyl-Olefin Metathesis Catalyzed by Molecular Iodine, ACS Catal., 2019, 9, 912–919 Search PubMed; (i) M. Breugst, E. Detmar and D. von der Heiden, Origin of the Catalytic Effects of Molecular Iodine: A Computational Analysis, ACS Catal., 2016, 6, 3203–3212 Search PubMed; (j) Y. F. Qiu, Y. J. Niu, X. R. Song, X. Wei, H. Chen, S. X. Li, X. C. Wang, C. D. Huo, Z. J. Quan and Y. M. Liang, Iodine promoted cascade cycloisomerization of 1-en-6,11-diynes, Chem. Commun., 2020, 56, 1421 Search PubMed; (k) Z. Meng, J. Yan, C. Ning, M. Shi and Y. Wei, Construction of pyrroles, furans and thiophenes via intramolecular cascade desulfonylative/dehydrogenative cyclization of vinylidenecyclopropanes induced by NXS (X = I or Br), Chem. Sci., 2023, 14, 7648–7655 Search PubMed.
- (a) S. Maejima, E. Yamaguchi and A. Itoh, trans-Diastereoselective Syntheses of γ-Lactones by Visible Light-Iodine-Mediated Carboesterification of Alkenes, ACS Omega, 2019, 3, 4856–4870 Search PubMed; (b) K. S. Kanyiva, T. Marina, S. Nishibe and T. Shibata, Metal-Free Aminoiodination of Alkynes Under Visible Light Irradiation for the Construction of a Nitrogen-Containing Eight-Membered Ring System, Adv. Synth. Catal., 2021, 363, 2746–2751 Search PubMed; (c) Y. K. Li, D. E. Wise, J. K. Mitchell and M. Parasram, Cascade Synthesis of Phenanthrenes under Photoirradiation, J. Org. Chem., 2023, 88, 717–721 Search PubMed; (d) H. Liu, X. Fan, J. K. Hu, T. T. Ma, F. Wang, J. H. Yang and D. J. Li, Visible-Light-Enabled Ph3P/LiI-Promoted Tandem Radical Trifluoromethylation/Cyclization/Iodination of 1,6-Enynes with Togni's Reagent, J. Org. Chem., 2022, 87, 12877–12889 Search PubMed; (e) Y. L. Lu, C. M. Chen, H. C. Zhu, Z. W. Luo and Y. H. Zhang, Highly efficient and fast synthesis of di-iodinated succinimide derivatives from 1,6-enyne and I2 under air at room temperature, Green Chem., 2022, 24, 8021–8028 Search PubMed; (f) Q. Cheng, F. R. Zhang, X. Y. Chen, Y. Han, C. G. Yan, Y. C. Shi, H. Hou and S. Q. Zhu, Visible-Light-Mediated Three-Component Radical Iodosulfonylative Cyclization of Enynes, Org. Lett., 2022, 24, 2515–2519 Search PubMed; (g) M. Takeda, S. Maejima, E. Yamaguchi and A. Itoh, Direct lactonization from 1,3-dienes and malonate esters mediated by a combination of iodine and visible light, Tetrahedron Lett., 2019, 60, 151284 Search PubMed; (h) S. Maejima, E. Yamaguchi and A. Itoh, Three-Component Iminolactonization Reaction via Bifunctionalization of Olefins Using Molecular Iodine and Visible Light, J. Org. Chem., 2020, 85, 10709–10718 Search PubMed; (i) S. Maejima, E. Yamaguchi and A. Itoh, Intermolecular Tandem Addition/Esterification Reaction of Alkenes with Malonates Leading to γ-Lactones Mediated by Molecular Iodine under Visible Light Irradiation, Adv. Synth. Catal., 2017, 359, 3883–3887 Search PubMed; (j) A. Maity, B. L. Frey and D. C. Powers, Iodanyl Radical Catalysis, Acc. Chem. Res., 2023, 14, 2026–2036 Search PubMed; (k) S. M. Hyun, M. B. Yuan, A. Maity, O. Gutierrez and D. C. Powers, The Role of Iodanyl Radicals as Critical Chain Carriers in Aerobic Hypervalent Iodine Chemistry, Chem, 2019, 5, 2388–2404 Search PubMed; (l) C. P. Dong, K. Nakamura, T. Taniguchi, S. Mita, S. Kodama, S. Kawaguchi, A. Nomoto, A. Ogawa and T. Mizuno, Synthesis of Aryl Iodides from Arylhydrazines and Iodine, ACS Omega, 2018, 8, 9814–9821 Search PubMed; (m) S. Maejima, E. Yamaguchi and A. Itoh, Visible Light/Molecular-Iodine-Mediated Intermolecular Spirolactonization Reaction of Olefins with Cyclic Ketones, J. Org. Chem., 2019, 15, 9519–9531 Search PubMed; (n) Y. Sudo, E. Yamaguchi and A. Itoh, , Photo-oxidative Cross-Dehydrogenative Coupling-Type Reaction of Thiophenes with α-Position of Carbonyls Using a Catalytic Amount of Molecular Iodine, Org. Lett., 2017, 7, 1610–1613 Search PubMed; (o) K. Oe, M. Goto, S. Maejima, E. Yamaguchi and A. Itoh, Front Cover: Cyanomethylation of β-Alkoxyaldehydes: Toward a Short Synthesis of Atorvastatin, Asian J. Org. Chem., 2020, 9, 1–5 Search PubMed; (p) K. Usami, Y. Nagasawa, E. Yamaguchi, N. Tada and A. Itoh, Intermolecular Cyclopropanation of Styrenes Using Iodine and Visible Light via Carbon-Iodine Bond Cleavage, Org. Lett., 2016, 1, 8–11 Search PubMed.
- (a) M. Shi, L. X. Shao, J. M. Lu, Y. Wei, K. Mizuno and H. Maeda, Chemistry of Vinylidenecyclopropanes, Chem. Rev., 2010, 110, 5883–5913 Search PubMed; (b) S. Yang and M. Shi, Recent Advances in Transition-Metal-Catalyzed/Mediated Transformations of Vinylidenecyclopropanes, Acc. Chem. Res., 2018, 51, 1667–1680 Search PubMed; (c) D. H. Zhang, X. Y. Tang and M. Shi, Gold-Catalyzed Tandem Reactions of Methylenecyclopropanes and Vinylidenecyclopropanes, Acc. Chem. Res., 2014, 47, 913–924 Search PubMed; (d) K. H. Rui and M. Shi, Rh(i)-Catalyzed intramolecular [3 + 2] cycloaddition reactions of yne-vinylidenecyclopropanes, Org. Chem. Front., 2019, 6, 1816–1820 Search PubMed; (e) Z. Meng, X. Y. Zhang and M. Shi, Visible-light mediated cascade cyclization of ene-vinylidenecyclopropanes: access to fluorinated heterocyclic compounds, Org. Chem. Front., 2021, 8, 3796–3801 Search PubMed; (f) C. Xu, C. Ning, S. Yang, Y. Wei and M. Shi, Rhodium-Catalyzed Asymmetric Cycloisomerization of 1,3-Diketones with Keto-Vinylidenecyclopropanes: Synthesis of Enantiomerically Enriched Cyclic β-Amino Alcohols, Adv. Synth. Catal., 2021, 363, 1727–1732 Search PubMed; (g) C. Ning, K. H. Rui, Y. Wei and M. Shi, Rh(i)-catalyzed dimerization of ene-vinylidenecyclopropanes for the construction of spiro[4,5]decanes and mechanistic studies, Chem. Sci., 2022, 13, 7310–7317 Search PubMed.
- X. Y. Zhang, X. Y. Wu, B. Zhang, Y. Wei and M. Shi, Silyl Radical-Mediated Carbocyclization of Acrylamide-/Vinyl Sulfonamide-Attached Alkylidenecyclopropanes via Photoredox Catalysis with a Catalytic Amount of Silane Reagent, ACS Catal., 2021, 11, 4372–4380 Search PubMed.
- Y. Luo, Q. Wei, L. K. Yang, Y. Q. Zhou, W. D. Cao, Z. S. Su, X. H. Liu and X. M. Feng, Enantioselective Radical Hydroacylation of α,β-Unsaturated Carbonyl Compounds with Aldehydes by Triplet Excited Anthraquinone, ACS Catal., 2022, 12, 12984–12992 Search PubMed.
- (a) Q. Z. Li, X. T. Gu, Y. Wei and M. Shi, Visible-light-induced indole synthesis via intramolecular C-N bond formation: desulfonylative C(sp2)-H functionalization, Chem. Sci., 2022, 13, 11623–11632 Search PubMed; (b) S. Z. Nie, A. r Lu, E. L. Kuker and V. M. Dong, Enantioselective Hydrothiolation: Diverging Cyclopropenes through Ligand Control, J. Am. Chem. Soc., 2021, 16, 6176–6184 Search PubMed.
- (a) J. M. Mayer, Understanding Hydrogen Atom Transfer: From Bond Strengths to Marcus Theory, Acc. Chem. Res., 2011, 44, 36–46 Search PubMed; (b) W. G. Zhang, Z. L. Zou, Y. H. Wang, Y. Wang, Y. Liang, Z. G. Wu, Y. X. Zheng and Y. Pan, Leaving Group Assisted Strategy for Photoinduced Fluoroalkylations Using N-Hydroxybenzimidoyl Chloride Esters, Angew. Chem., Int. Ed., 2019, 58, 624–627 Search PubMed.
- (a) Z. Rappoport and A. Topol, Nucleophilic attacks on carbon-carbon double bonds 26 Stereoconvergence in nucleophilic vinylic substitution of an activated nitro olefin, J. Am. Chem. Soc., 1980, 102, 406–407 Search PubMed; (b) Z. Rappoport and A. Topol, Nucleophilic attacks on carbon-carbon double bonds. Part 39 Nucleophile and nucleofuge effects, catalysis and stereochemistry in vinylic substitution of electrophilic nitro olefins, J. Org. Chem., 1989, 54, 5967–5977 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Z. Liu, Y. Wei and M. Shi, Visible-light-mediated interrupted Cloke-Wilson rearrangement of cyclopropyl ketones to construct oxy-bridged macrocyclic framework, Tetrahedron Org., 2022, 1, 10001 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of new compounds. CCDC 2073868 and 2244043. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qo01866k |
| This journal is © the Partner Organisations 2024 |