
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoredox catalyzed release of carbon-based radicals from 2-substituted-1,3-imidazolidines†
Adrián
Luguera Ruiz
 ,
Elena
Mariani
,
Stefano
Protti
* and
Maurizio
Fagnoni
*
,
Elena
Mariani
,
Stefano
Protti
* and
Maurizio
Fagnoni
*
PhotoGreen Lab, Department of Chemistry, University of Pavia, Viale Taramelli 12, 27100 Pavia, Italy. E-mail: fagnoni@unipv.it; stefano.protti@unipv.it
First published on 7th December 2023
Abstract
In the frame of developing easily oxidizable compounds for the photorelease of carbon-based radicals, we describe herein the use of 2-substituted-1,3-imidazolidines. These compounds (Eoxca. 1 V vs. SCE) were used to generate (substituted) alkyl radicals under photoredox conditions. The radicals smoothly added to electron-poor C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds for the forging of C(sp3)–C(sp3) bonds under metal-free conditions. Acridinium salts and even 4-CzIPN could be used as photocatalysts thanks to the favorable redox properties of these heterocycles.
C bonds for the forging of C(sp3)–C(sp3) bonds under metal-free conditions. Acridinium salts and even 4-CzIPN could be used as photocatalysts thanks to the favorable redox properties of these heterocycles.
Introduction
Carbon based radicals were probably the most used intermediates in synthesis in the past decades.1 In this context, photoredox catalysis has become the elective method for their generation in a more sustainable and fancy manner than classical synthetic methodologies.2,3 During the last few years, photoredox generation of alkyl radicals has allowed for the development of protocols for the formation of C(sp3)–C(sp3) bonds which is currently a hot topic in organic synthetic chemistry.2,4 Such a strategy made use of radical precursors bearing redox active moieties that allow their easy oxidation/reduction by a suitable photoexcited catalyst (PC).2d Several PCs exhibiting variable oxidation/reduction capability have been designed and investigated for this purpose, but in the case of substrates difficult to oxidize/reduce, the number of suitable PCs strongly decreased.For these reasons, the redox active moiety is often charged, to shift the radical precursors to a “comfort zone” that allows testing several visible light absorbing PCs. The preparation of these precursors may be in some cases troublesome or impose restrictions to the reaction conditions (the choice of solvent, the compatibility of additives and so on).
The obvious (but not trivial) strategy to overcome such limitations is having recourse to easily oxidizable/reducible uncharged derivatives, and recently, we reported that substituted oxazolidines (Eoxca. 1.3 V vs. SCE) perfectly fulfill this requirement.5 We were, however, intrigued to search for further uncharged super electron donors (SEDs)6 able to release a set of radical intermediates upon oxidation, including tertiary, α-oxy and α-amino carbon-based radicals. In this context, we focused our attention on aminals. The most famous compounds belonging to this class are N,N-dimethyldihydrobenzoimidazoles (I, Scheme 1). 1,3-Dimethyl-2-phenylbenzimidazoline (DMBI) was extensively used to promote a single electron transfer reaction (SET) acting as a reducing agent on compounds R–X with no need of any additives (Scheme 1, path a).7,8 When R–X is a haloketone, a reductive dehalogenation occurred via the fragmentation of the resulting radical anion and hydrogen incorporation from radical V (paths a′ and a′′).7d,8b The radical cation (II) formed from the aminal may lose a proton to give radical III (path b) again prone to reduce R–X leading to the corresponding benzimidazolium salt IV (path c).9 To strengthen the reducing power of compounds I the process was later promoted by light via a photoinduced electron transfer reaction (Scheme 1).10 Interestingly, aminals I may also act as efficient hydrogen atom donors to radicals (e.g.V, path d)9 More recently, even radical III has found application as a surrogate of an acyl radical VI in the reaction with styrenes.11
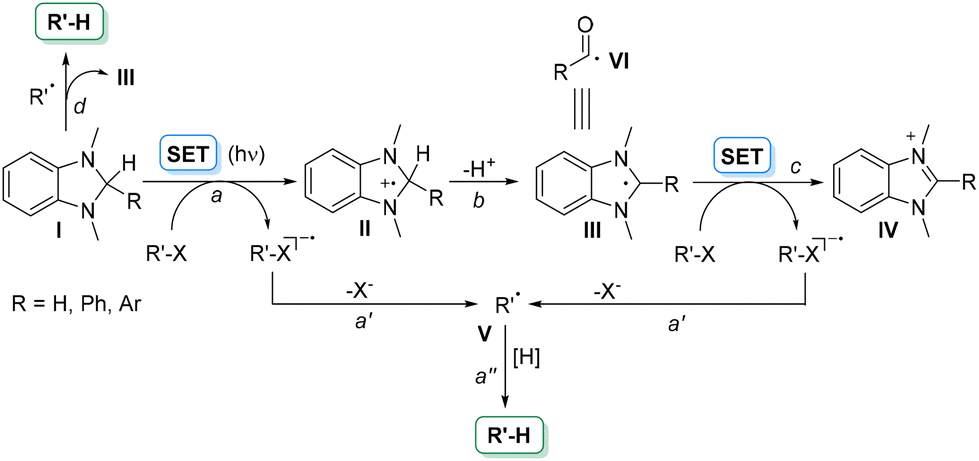 | ||
| Scheme 1 Various pathways in the (photo)chemistry of N,N-dimethyldihydrobenzoimidazoles I. | ||
In this context, a modified DMBI structure (dihydroquinazolinones, VII) has emerged as a radical precursor (Scheme 2a).12 The strategy relies on the aromatic stabilization energy (ASE) of the redox moiety VIII when released from the fragmentation of the radical cation.13
 | ||
| Scheme 2 Photocatalyzed generation of carbon-based radicals from the single electron transfer oxidation of (a) dihydroquinazolinones VII and (b) 2-substituted imidazolidines IX (this work). | ||
In such a way, the efficiency in the fragmentation of the radical cation pushed the liberation of a plethora of different radicals by C–C bond cleavage. However, the strategy suffers from some drawbacks such as the poor atom economy of the process (due to the weight of the aromatized fragment VIII) and the fact that pre-aromatic derivatives VII must be obtained only from methyl or phenyl ketones. This is probably due to the fact that the C–C bond cleavage of the other substituents present in position 2 in compounds VII must be suppressed thanks to the instability of the methyl/phenyl radicals with respect to the desired radical released (mostly tertiary or, however, stabilized). The radical may, however, be liberated even under photosensitized conditions.14 Related imidazolidinones have likewise an increased role in dual photocatalytic reactions as chiral bases while remaining, however, photochemically inactive.15
Inspired by these studies and by our achievement in the light induced visible light generation of radical intermediates5 we envisioned that simple aminals such as 2-substituted 1,3-dimethylimidazolidines IX (Scheme 2b) could be valid candidates to test the release of carbon radicals via C–C bond cleavage by a photoredox SET process. We thus reasoned that the presence of two nitrogen atoms may induce good oxidizability, and noteworthily, the stability of the resulting cation X may drive the fragmentation even if this does not evolve into an aromatic derivative. In this work, we report that readily prepared imidazolidines act as more atom-economical (with respect to quinazolidinones) uncharged alkyl radical precursors as detailed below.
Results and discussion
Different imidazolidines 1a–j (Fig. S2, see the ESI†) were smoothly prepared by condensation of the corresponding aldehyde with N,N′-dimethylethylenediamine. These heterocycles exhibit an oxidation potential of ca. 1.0 V vs. SCE as shown in Fig. S6† for compound 1c (Eox = 1.16 V vs. SCE). The more accessible Eox allowed us to investigate the use of several colored PCs for the radical addition onto electron-deficient CC double bonds.
With the aim of investigating the feasibility of our proposal, we initially focused on the tertbutylation of dimethylmaleate 2a (Fig. S1, see the ESI†) by using imidazolidine 1a as the model reactant. Different reaction parameters were investigated, and these included the nature of the photocatalyst, the reaction media, the stoichiometric ratio, and the influence of oxygen on the reaction outcome (see Table S1 in the ESI† for a detailed description of the experiments). The most representative control experiments are described in Table 1.
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | 3 (% yield) |
| 1 | None | 99 |
| 2 | 4CzIPN (10 mol%), N2 atmosphere | 98 |
| 3 | 3CzClIPN (10 mol%), N2, dimethylcarbonate | 22 |
| 4 | Ph3pyrylium+ BF4− (10 mol%) | — |
| 5 | Ir(ppy)3 (5 mol%), N2 atmosphere | — |
| 6 | N2 atmosphere | 90 |
| 7 | MeCN as solvent | — |
| 8 | DCM as solvent | 84 |
| 9 | No light | — |

Gratifyingly, the desired product 3 was isolated in almost quantitative amount, 99% yield (Table 1, entry 1), when [Acr-Mes]+(BF4)− ( vs. SCE
vs. SCE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16) was used as a PC. Similar results were found by employing 4CzIPN (
16) was used as a PC. Similar results were found by employing 4CzIPN ( vs. SCE16) under a N2 atmosphere (entry 2). The 3CzClIPN photocatalyst (
vs. SCE16) under a N2 atmosphere (entry 2). The 3CzClIPN photocatalyst ( vs. SCE16) showed a poor performance (entry 3) whereas the use of both pyrylium salts (entry 4) and Ir(ppy)3 (entry 5) did not lead to the desired product. A slight decrease in the reaction yield was observed when performing the reaction under a N2 atmosphere (entry 6). No product formation occurred when the reaction was carried out in polar solvents (entry 7). Alternative halogenated media (e.g. DCM) gave a lower terbutylation yield (entry 8). On-off experiments (Table S3, Fig. S7†) and the absence of light (entry 9) confirmed the photochemical nature of the process. In addition, a decrease of the overall yield to 50% occurred when TEMPO (1 equiv., Table S1,† entry 15) was present in the reaction media.
vs. SCE16) showed a poor performance (entry 3) whereas the use of both pyrylium salts (entry 4) and Ir(ppy)3 (entry 5) did not lead to the desired product. A slight decrease in the reaction yield was observed when performing the reaction under a N2 atmosphere (entry 6). No product formation occurred when the reaction was carried out in polar solvents (entry 7). Alternative halogenated media (e.g. DCM) gave a lower terbutylation yield (entry 8). On-off experiments (Table S3, Fig. S7†) and the absence of light (entry 9) confirmed the photochemical nature of the process. In addition, a decrease of the overall yield to 50% occurred when TEMPO (1 equiv., Table S1,† entry 15) was present in the reaction media.
In view of these results, we adopted the followed conditions: an air-equilibrated DCE solution of 1a (1.5 equiv.), 2a (0.05 M), and Acr-Mes+ BF4− (10 mol%), under irradiation at 405 nm (EvoluChem 18 W LED) for 24 h (Fig. S3, ESI†). In some cases (see below), the use of 4CzIPN was considered as a convenient alternative to [Acr-Mes]+(BF4)− as the PC.
We thus explored the scope of the reaction by adding the generated alkyl radicals to electron-poor alkenes and vinyl heteroarenes (Fig. S1†) as sketched in Scheme 3. Notably, all tert-butylated derivatives 3–11 (including the alkylated 2,3-dihydronaphthoquinone 11) were obtained in satisfactory yields, as well as adamantyl derivatives 12–19 that were isolated in up to 99% yield. Interestingly, to the best of our knowledge, the tert-butylation of 1,4-naphthoquinone 2i was previously reported only by using rather toxic organomercury derivatives.17
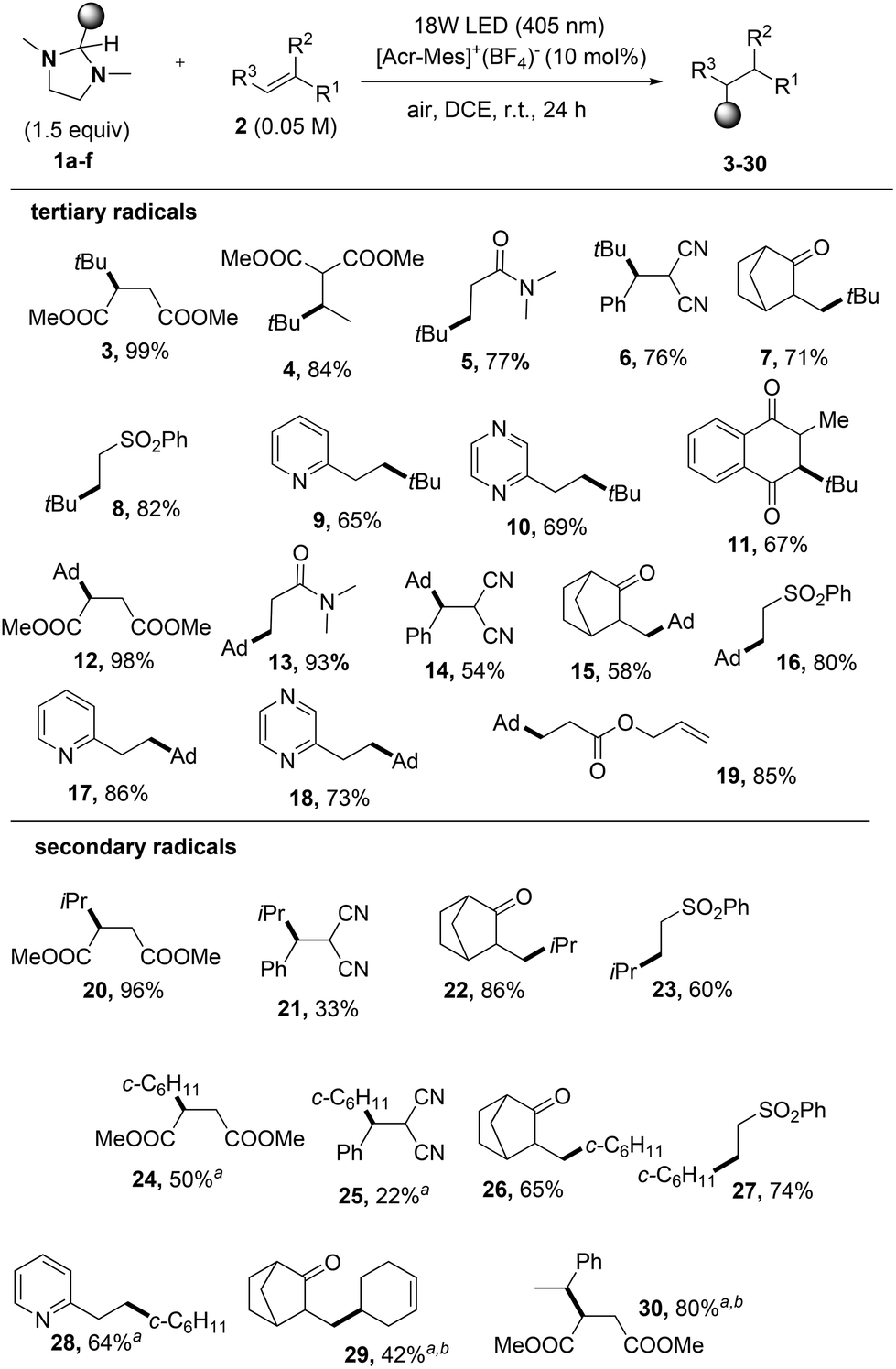 | ||
| Scheme 3 Imidazolidines for the photocatalyzed alkylation of CC bonds. Conditions: 1a–f (0.075 M, 1.5 equiv., 0.375 mmol), 2 (0.25 mmol), [Acr-Mes]+ (BF4−) (10 mol%), DCE (5 mL), air, under 18 W LED irradiation (405 nm) at r.t. for 24 h. Isolated yields. Ad = adamantyl. a4CzIPN (10 mol%) was used as the PC under a nitrogen atmosphere. bMixture of two diastereoisomers (dr 1:1). | ||
A complete regioselectivity was observed when using allyl methacrylate 2j as the electron-poor olefin, where the electrophilic CC bond was exclusively derivatized to form product 19.
The release of secondary radicals (iso-propyl, cyclohexyl and 3-cyclohexenyl) from imidazolidines 1c–e was next tested. Even in this case, the corresponding alkylated compounds 20–29 were obtained in a satisfactory yield with the only exception of dicyano derivatives 21 and 25. When releasing secondary radicals, the adoption of 4CzIPN as the photoredox catalyst was beneficial for the successful outcome of the reaction (see the results obtained for compounds 24, 28, and 29). The 4CzIPN photocatalyzed benzylation of dimethyl maleate by using 1f afforded the benzylated derivative 30 as a mixture of diastereoisomers. Unfortunately, any attempts to perform alkylation of olefins with primary alkyl radicals starting from imidazolidine 1g were ineffective.
The synthesis of 3 was also optimized under flow conditions by using a lab-made flow photoreactor (see Fig. S4 and S5, ESI,† for details). In this case, we were able to use less toxic dichloromethane (DCM) in place of DCE as the solvent. To this aim, different parameters were considered, including the light source and its intensity, the residence time and the concentration of the substrates. A complete list of the experiments is included in Table S2† but the most representative results are shown in Table 2 (accompanied by the corresponding Space Time Yield (STY) values).
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | 3, % yield (STY, mmol L−1 min−1) |
| 1 | None | 99 (0.825) |
| 2 | 390 nm (100% power), flow rate: 0.8 mL min−1, 2 h | 96 (0.80) |
| 3 | 2a (0.4 M), 1a (1.5 equiv.), 405 nm (100% power), flow rate: 0.8 mL min−1, 2 h | 92 (3.07) |
| 4 | Flow rate: 1.6 mL min−1, 1 h | 43 (0.72) |
| 5 | 390 nm (Kessil 40 W LED) (50% power), flow rate: 1.2 mL min−1, 1.5 h | 65 (0.72) |
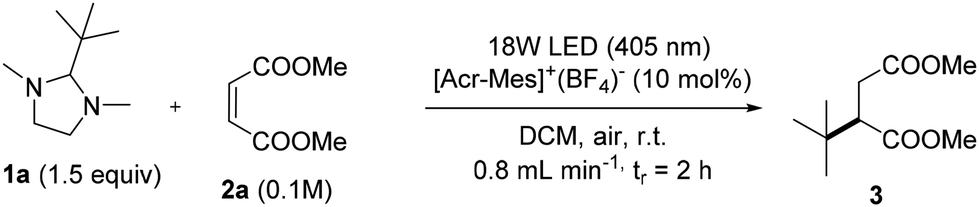
Gratifyingly, as depicted in entry 1, when the reaction was carried out in DCM, compound 3 was isolated in quantitative yield with a residence time of only 2 h. Interestingly, the light source could be replaced with a 390 nm Kessil lamp without a significant decrease of the yield (entry 2). It is also possible to scale up the reaction from 0.1 M to 0.4 M (1 mmol 2a) with no clogging problems and no precipitate formation inside the reactor tubing (entry 3, 92% yield with a STY of 3.07 mmol L−1 min−1).
On the other hand, an increase in the flow rate (when using either a 405 nm or a 390 nm LED lamp) resulted in a significant lowering of the reaction yield (entries 4 and 5).
We then turned our attention to electron-rich carbon centered radicals bearing heteroatom-based substituents (N, O) starting from suitably functionalized imidazolidines 1h–j and the results are summarized in Scheme 4. Thus, when using the dioxolane derivative 1h, the desired products 31–34 were isolated in up to 97% yield (mostly as a 1:1 mixture of diasteroisomers). Analogously, α-amido radicals (photogenerated from 1i and 1j) were exploited to incorporate cyclic and acyclic NBoc protected amines 35–43 in a different range of electron-poor olefins with yields ranging from 50 to 86%.
 | ||
| Scheme 4 Functionalization of CC bonds via photogenerated α-oxy and α-amido alkyl radicals. Conditions: 1h–j (0.075 M, 1.5 equiv., 0.375 mmol), 2 (0.25 mmol), [Acr-Mes]+ (BF4−) (10 mol%), DCE (5 mL), air, under 18 W LED irradiation (405 nm) at r.t. for 24 h. Isolated yields. aMixture of two diastereoisomers (dr 1:1). | ||
In the mechanism suggested, reductive quenching of the photoexcited acridinium catalyst [Acr-Mes]+* (or 4CzIPN*) with the concomitant oxidation of imidazolidines 1a–j to generate the corresponding radical cations 1a–j˙+ occurred (Scheme 5, path a). The radical species R˙ was formed upon fragmentation of the C–C bond in 1a–f, h–j˙+ releasing a stable iminium ion A+ (path b). The nucleophilic R˙ was trapped by electron-poor olefins 2a–j (path c) and the resulting adduct radical B˙ underwent monoelectronic reduction by Acr-Mes˙ to generate the corresponding anion C− while restoring the starting photocatalyst. Protonation of C− (path e) by adventitious water afforded the desired products 3–43. Hydrogen abstraction from the solvent by radical B˙ was safely excluded by deuteration experiments (see the ESI, section 4†).
 | ||
| Scheme 5 Proposed mechanism for the synthesis of compounds 3–43. | ||
The key role of the carbon centered intermediate R˙ has been further evidenced by the detrimental effect of TEMPO when present in the reaction mixture (Table S1, entry 15†).
As hinted above, the forging of C(sp3)–C(sp3) bonds is a current hot topic in organic synthesis. In the present work, we highlighted the potential of imidazolidines 1 (smoothly prepared from commercially available aldehydes and 1,2-diamines) as appealing alternatives to both uncharged (including among others, 1,3-dioxolanes,18 1,3-oxazolidines,5 Barton esters,19N-(acyloxy)phthalimides,4f,20a substituted Hantzsch ester)20b,c and charged (alkyl tetrafluoro borates,2d alkyl carboxylates,2d alkyl N-phthalimidoyl oxalates2d) known radical precursors.
The protocol presented here offers the chance to generate a wide range of carbon-based radicals, including tertiary, secondary, benzyl, α-oxy and α-amido, by exploiting the easy oxidazability of the radical precursors. The latter point has the consequence of both allowing the use of metal-free PCs having poor oxidizing power in the excited state (e.g. 4CzIPN) and liberating a stable iminium ion in the fragmentation of the thus formed radical cation, which is the driving force of the reaction. Contrary to dihydroquinazolinones, there is no possible competitive release of other radicals upon fragmentation, overcoming the requirement to have an aromatic leaving group that strongly affected the atom economy of the process.
Furthermore, such a methodology exhibited excellent functional group tolerance, allowing for the alkylation of differently decorated olefins bearing carbonyl and carboxylic groups, sulfones and even of heteroarenes.
Conclusions
Summing up, we developed the application of 2-substituted-N,N-imidazolidines as uncharged carbon centered precursors under photoredox catalyzed conditions, for the versatile functionalization of a large variety of CC bonds. The adoption of super-donors for the liberation of radicals is of urgent importance since it may allow the use of mild oxidative conditions limiting the interference of other reagents/additives that may be oxidized as well in the process when using powerful oxidizing PCs. Furthermore, the data presented herein also evidenced the chance of applying imidazolidines in alkylation strategies under continuous flow conditions.
Experimental
General procedure for the synthesis of 2-substituted N,N′-dimethylimidazolidines 1a–j
The desired imidazolidines 1a–j (Fig. S2†) were synthesized by adapting a procedure previously described for the synthesis of 1,3-oxazolidines.5N,N′-Dimethylethylenediamine (1 equiv.) was added to a suspension of MgSO4 (25 mg mmol−1) and the corresponding aldehyde (1 equiv.) in Et2O (1.7 mL mmol−1). The reaction mixture was refluxed and stirred overnight, and the resulting residue was diluted with DCM, filtered, and concentrated in vacuo to yield the desired imidazolidine that was employed for the photocatalytic step without any further purification.General procedure for the photoredox catalyzed preparation of compounds 3–43
A solution of the chosen imidazolidine 1a–j (1.5 equiv.), olefin 2a–j (1 equiv.) and [Acr-Mes]+(BF4)− in DCE (5 mL) was prepared in a Pyrex glass vessel (see Fig. S3†). The solution was irradiated for 24 h at 405 nm by means of an 18 W EvoluChem lamp. The resulting solution was then concentrated in vacuo and the residue was purified by flash column chromatography using an Isolera apparatus (Biotage) and a SiO2 cartridge (eluant: cyclohexane/ethyl acetate mixture except where indicated).Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. L. R. acknowledges the European Union for a PhD fellowship. This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 956324. The authors acknowledge support from the Ministero dell'Università e della Ricerca (MUR) and the University of Pavia through the program “Dipartimenti di Eccellenza 2023–2027”.References
- (a) M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, Radicals: Reactive Intermediates with Translational Potential, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS PubMed; (b) A. Studer and D. P. Curran, Catalysis of Radical Reactions: A Radical Chemistry Perspective, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef CAS PubMed; (c) K. J. Romero, M. S. Galliher, D. A. Pratt and C. R. J. Stephenson, Radicals in natural product synthesis, Chem. Soc. Rev., 2018, 47, 7851–7866 RSC; (d) Free Radicals: Fundamentals and Applications in Organic Synthesis, ed. L. Fensterbank and C. Ollivier, Georg Thieme Verlag KG, Stuttgart, Germany, 2021 Search PubMed.
- (a) D. Ravelli, S. Protti and M. Fagnoni, Carbon–Carbon Bond Forming Reactions via Photogenerated Intermediates, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed; (b) J.-P. Goddard, C. Ollivier and L. Fensterbank, Photoredox Catalysis for the Generation of Carbon Centered Radicals, Acc. Chem. Res., 2016, 49, 1924–1936 CrossRef CAS PubMed; (c) S. P. Pitre, N. A. Weires and L. E. Overman, Forging C(sp3)–C(sp3) Bonds with Carbon-Centered Radicals in the Synthesis of Complex Molecules, J. Am. Chem. Soc., 2019, 141, 2800–2813 CrossRef CAS PubMed; (d) S. Crespi and M. Fagnoni, Generation of Alkyl Radicals: From the Tyranny of Tin to the Photon Democracy, Chem. Rev., 2020, 120, 9790–9833 CrossRef CAS PubMed; (e) M. Fagnoni, D. Ravelli and S. Protti, in Science of Synthesis: Free Radicals: Fundamentals and Applications in Organic Synthesis, ed. L. Fensterbank and C. Ollivier, Thieme, Stuttgart, 2021, vol. 2, pp. 339–357 Search PubMed; (f) F. Juliá, T. Constantin and D. Leonori, Applications of Halogen-Atom Transfer (XAT) for the Generation of Carbon Radicals in Synthetic Photochemistry and Photocatalysis, Chem. Rev., 2022, 122, 2292–2352 CrossRef PubMed; (g) L. Capaldo, D. Ravelli and M. Fagnoni, Direct Photocatalyzed Hydrogen Atom Transfer (HAT) for Aliphatic C–H Bonds Elaboration, Chem. Rev., 2022, 121, 1875–1924 CrossRef PubMed.
- (a) D. Ravelli, S. Protti, M. Fagnoni and A. Albini, Visible Light Photocatalysis. A Green Choice?, Curr. Org. Chem., 2013, 17, 2366–2373 CrossRef CAS; (b) Visible Light Photocatalysis in Organic Chemistry, ed. C. R. J. Stephenson, T. P. Yoon and D. W. C. MacMillan, Wiley-VCH, Weinheim, Germany, 2018 Search PubMed; (c) M. Fagnoni, S. Protti and D. Ravelli, Photoorganocatalysis in organic synthesis, World Scientific Publishing Europe Ltd., Singapore, 2019 CrossRef; (d) Chemical Photocatalysis, ed. B. König and De Gruyter, 2nd edn, 2020 Search PubMed.
- For recent reviews on the photogeneration of alkyl radicals see: (a) J. K. Matsui, S. B. Lang, D. R. Heitz and G. A. Molander, Photoredox-Mediated Routes to Radicals: The Value of Catalytic Radical Generation in Synthetic Methods Development, ACS Catal., 2017, 7, 2563–2575 CrossRef CAS PubMed; (b) S. Roslin and L. R. Odell, Visible-Light Photocatalysis as an Enabling Tool for the Functionalization of Unactivated C(sp3)-Substrates, Eur. J. Org. Chem., 2017, 1993–2007 CrossRef CAS; (c) P. Niu, J. Li, Y. Zhang and C. Huo, One-Electron Reduction of Redox-Active Esters to Generate Carbon-Centered Radicals, Eur. J. Org. Chem., 2020, 5801–5814 CrossRef CAS; (d) J. T. M. Correia, V. A. Fernandes, B. T. Matsuo, J. A. C. Delgado, W. C. de Souza and M. W. Paixão, Photoinduced deaminative strategies: Katritzky salts as alkyl radical precursors, Chem. Commun., 2020, 56, 503–514 RSC; (e) S. Karmakar, A. Silamkoti, N. A. Meanwell, A. Mathur and A. K. Gupta, Utilization of C(sp3)-Carboxylic Acids and Their Redox-Active Esters in Decarboxylative Carbon-Carbon Bond Formation, Adv. Synth. Catal., 2021, 363, 3693–3736 CrossRef CAS; (f) S. K. Parida, T. Mandal, S. Das, S. K. Hota, S. De Sarkar and S. Murarka, Single Electron Transfer-Induced Redox Processes Involving N-(Acyloxy)phthalimides, ACS Catal., 2021, 11, 1640–1683 CrossRef CAS; (g) S. Protti, D. Ravelli and M. Fagnoni, Designing Radical Chemistry by Visible-Light Promoted Homolysis, Trends Chem., 2022, 4, 305–317 CrossRef CAS.
- A. Luguera Ruiz, M. La Mantia, D. Merli, S. Protti and M. Fagnoni, Alkyl Radical Generation via C–C Bond Cleavage in 2-Substituted Oxazolidines, ACS Catal., 2022, 12, 12469–12476 CrossRef CAS PubMed.
- J. A. Murphy, Discovery and Development of Organic Super-Electron-Donors, J. Org. Chem., 2014, 79, 3731–3746 CrossRef CAS PubMed.
- (a) H. Chikashita, S. Nishida, M. Miyazaki and K. Itoh, 2-Phenylbenzimidazoline as a reducing agent in the preparation of malononitriles from α,β-unsaturated dinitriles, Synth. Commun., 1983, 13, 1033–1039 CrossRef CAS; (b) H. Chikashita, Y. Morita and K. Itoh, An efficient method for the selective reduction of 2-aryl-1-nitroalkenes to 2-aryl-1-nitroalkanes by 2-phenylbenzimidazoline, Synth. Commun., 1985, 15, 527–533 CrossRef CAS; (c) H. Chikashita and K. Itoh, Reduction of α,β-unsaturated carbonyls, Bull. Chem. Soc. Jpn., 1986, 59, 1747–1752 CrossRef CAS; (d) H. Chikashita, H. Ide and K. Itoh, 1,3-Dimethyl-2-phenylbenzimidazoline as a Novel and Efficient Reagent for Mild Reductive Dehalogenation of -Halo Carbonyl Compounds and Acid Chlorides, J. Org. Chem., 1986, 51, 5400–5405 CrossRef CAS.
- (a) J. Chen and D. D. Tanner, New Method for the Facile Reduction of -Nitro Sulfones to Nitroalkanes via an Electron-Transfer-Hydrogen Atom Abstraction Mechanism, J. Org. Chem., 1988, 53, 3897–3900 CrossRef CAS; (b) D. D. Tanner and J. J. Chen, On the Mechanism of the Reduction of α-Halo Ketones by l,3-Dimethyl-2-phenylbenzimidazoline. Reduction by a SET-Hydrogen Atom Abstraction Chain Mechanism, J. Org. Chem., 1989, 54, 3842–3846 CrossRef CAS; (c) D. D. Tanner, J. J. Chen, C. Luelo and P. M. Peters, Reversible Cyclopropyl Ring Opening of 1 -Aroyl-2-phenylcyclopropane Radical Anions. Determination of the Ring Opening and Closure Rates of the Intermediate Ketyls, J. Am. Chem. Soc., 1992, 114, 713–717 CrossRef CAS; (d) D. D. Tanner and J. J. Chen, Thiophenol-Promoted Radical Chain Reduction of “-Substituted Isobutyrophenones by l,3-Dimethyl-2-phenylbenzimidazoline, J. Org. Chem., 1992, 57, 662–666 CrossRef CAS.
- S. Rohrbach, S. R. Shah, T. Tuttle and J. A. Murphy, Neutral Organic Super Electron Donors Made Catalytic, Angew. Chem., Int. Ed., 2019, 58, 11454–11458 CrossRef CAS PubMed.
- (a) E. Hasegawa, T. Kato, T. Kitazume, K. Yanagi, K. Hasegawa and T. Horaguchi, Photoinduced Electron Transfer Reactions of α,β-Epoxy Ketones with 2-Phenyl-N,N-dimethylbenzimidazoline 0PDMBI): Significant Water Effect on the Reaction Pathway, Tetrahedron Lett., 1996, 37, 7079–7082 CrossRef CAS; (b) E. Hasegawa, A. Yoneoka, K. Suzuki, T. Kato, T. Kitazume and K. Yanagi, Reductive Transformation of α,β-Epoxy Ketones and Other Compounds Promoted through Photoinduced Electron Transfer Processes with 1,3-Dimethyl-2-phenylbenzimidazoline (DMPBI), Tetrahedron, 1999, 55, 12957–12968 CrossRef CAS; (c) Y.-S. Feng, C.-Y. Yang, Q. Huang and H.-J. Xu, Study on comparison of reducing ability of three organic hydride compounds, Tetrahedron, 2012, 68, 5053–5059 CrossRef CAS; (d) E. Hasegawa, T. Ohta, S. Tsuji, K. Mori, K. Uchida, T. Miura, T. Ikoma, E. Tayama, H. Iwamoto, S. Takizawa and S. Murata, Aryl-substituted dimethylbenzimidazolines as effective reductants of photoinduced electron transfer reactions, Tetrahedron, 2015, 71, 5494–5505 CrossRef CAS; (e) E. Hasegawa, Y. Nagakura, N. Izumiya, K. Matsumoto, T. Tanaka, T. Miura, T. Ikoma, H. Iwamoto and K. Wakamatsu, Visible Light and Hydroxynaphthylbenzimidazoline Promoted Transition-Metal-Catalyst-Free Desulfonylation of N-Sulfonylamides and N-Sulfonylamines, J. Org. Chem., 2018, 83, 10813–10825 CrossRef CAS PubMed; (f) E. Hasegawa, N. Yoshioka, T. Tanaka, T. Nakaminato, K. Oomori, T. Ikoma, H. Iwamoto and K. Wakamatsu, Sterically Regulated α–Oxygenation of α–Bromocarbonyl Compounds Promoted Using 2–Aryl-1,3-dimethylbenzimidazolines and Air, ACS Omega, 2020, 5, 7651–7665 CrossRef CAS PubMed; (g) R. Miyajima, Y. Ooe, T. Miura, T. Ikoma, H. Iwamoto, S.-a Takizawa and E. Hasegawa, Triarylamine-Substituted Benzimidazoliums as Electron Donor–Acceptor Dyad-Type Photocatalysts for Reductive Organic Transformations, J. Am. Chem. Soc., 2023, 145, 10236–10248 CrossRef CAS PubMed; (h) S. Okumura, T. Takahashi, K. Torii and Y. Uozumi, Photocatalytic Cross-Pinacol Coupling Promoted by Carbon Dioxide, Chem. – Eur. J., 2023, 29, e202300840 CrossRef CAS PubMed.
- Y. Saga, Y. Nakayama, T. Watanabe, M. Kondo and S. Masaoka, Visible-Light-Driven Hydroacylation of Unactivated Alkenes Using Readily Available Acyl Donors, Org. Lett., 2023, 25, 1136–1141 CrossRef CAS PubMed.
- (a) L. Li, L. Fang, W. Wu and J. Zhu, Visible-Light-Mediated Intermolecular Radical Conjugate Addition for the Construction of Vicinal Quaternary Carbon Centers, Org. Lett., 2020, 22, 5401–5406 CrossRef CAS PubMed; (b) S.-C. Lee, L.-Y. Li, Z.-N. Tsai, Y.-H. Lee, Y.-T. Tsao, P.-G. Huang, C.-K. Cheng, H.-B. Lin, T.-W. Chen, C.-H. Yang, C.-C. Chiu and H.-H. Liao, Aromatization as an Impetus to Harness Ketones for Metallaphotoredox-Catalyzed Benzoylation/Benzylation of (Hetero)arenes, Org. Lett., 2022, 24, 85–89 CrossRef CAS PubMed; (c) P. P. Mondal, S. Das, S. Venugopalan, M. Krishnan and B. Sahoo, Visible-Light-Photocatalyzed Dicarbofunctionalization of Conjugated Alkenes with Ketone-Based Dihydroquinazolinones, Org. Lett., 2023, 25, 1441–1446 CrossRef CAS PubMed; (d) P. P. Mondal, A. Pal, S. Das, S. M. Vijayan, A. V. Nair, S. Ojha and B. Sahoo, Organophotoredox-Catalyzed Oxidative C(sp2)–H Alkylation of N-Heteroarenes with Dihydroquinazolinones by C–C Cleavage, Synlett, 2023, 34, 1241–1246 CrossRef CAS; (e) X. Yang, R. Abrams and R. Martin, Dihydroquinazolinones as adaptative C(sp3) handles in arylations and alkylations via dual catalytic C–C bond-functionalization, Nat Commun., 2022, 13, 2394–2402 CrossRef PubMed; (f) Z. M. Rubanov, V. I. Supranovich, V. V. Levin and A. D. Dilman, BF2-Chelates of N-Acylhydrazones as Versatile Coupling Partners in Photoredox Promoted Reactions, Eur. J. Org. Chem., 2023, 26, e202300247 CrossRef CAS.
- (a) P. von Ragué-Schleyer and F. Puhlhofer, Recommendations for the Evaluation of Aromatic Stabilization Energies, Org. Lett., 2002, 4, 2873–2876 CrossRef PubMed; (b) A. Bhunia and A. Studer, Recent Advances in Radical Chemistry Proceeding through Pro-aromatic Radicals, Chem, 2021, 7, 2060–2100 CrossRef CAS.
- T. Uchikura, H. Nakamura, H. Sakai and T. Akiyama, 2-Silylated Dihydroquinazolinone as a Photocatalytic Energy Transfer Enabled Radical Hydrosilylation Reagent, Chem. – Eur. J., 2023, 29, e202301090 CrossRef CAS PubMed.
- (a) D. A. Nicewicz and D. W. C. MacMillan, Merging photoredox catalysis with organocatalysis: the direct asymmetric alkylation of aldehydes, Science, 2008, 322, 77–80 CrossRef CAS PubMed; (b) K. L. Skubi, T. R. Blum and T. P. Yoon, Dual Catalysis Strategies in Photochemical Synthesis, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (c) W. Yao, E. A. Bazan-Bergamino and M.-Y. Ngai, Asymmetric Photocatalysis Enabled by Chiral Organocatalysts, ChemCatChem, 2022, 14, e202101292 CrossRef CAS PubMed; (d) C. C. Malakar, L. Dell'Amico and W. Zhang, Dual Catalysis in Organic Synthesis: Current Challenges and New Trends, Eur. J. Org. Chem., 2023, 26, e202201114 CrossRef CAS.
- Y. Wu, D. Kim and T. S. Teets, Photophysical Properties and Redox Potentials of Photosensitizers for Organic Photoredox Transformations, Synlett, 2022, 33, 1154–1179 CrossRef CAS.
- G. A. Russell, B. H. Kim and S. V. Kulkarni, Electron transfer processes. 48. Free-radical alkylations of enones involving proton transfers, J. Org. Chem., 1989, 54, 3768–3770 CrossRef CAS.
- M. Mella, E. Fasani and A. Albini, Electron Transfer Photoinduced Cleavage of Acetals. A Mild Preparation of Alkyl Radicals, J. Org. Chem., 1992, 57, 3051–3057 CrossRef CAS.
- M. F. Saraiva, M. R. C. Couri, M. Le Hyaric and M. V. de Almeida, The Barton ester free-radical reaction: a brief review of applications, Tetrahedron, 2009, 65, 3563–3572 CrossRef CAS.
- (a) X. Zhu and H. Fu, Photocatalytic cross-couplings via the cleavage of N–O bonds, Chem. Commun., 2021, 57, 9656–9671 RSC; (b) W. Chen, Z. Liu, J. Tian, J. Li, J. Ma, X. Cheng and G. Li, Building Congested Ketone: Substituted Hantzsch Ester and Nitrile as Alkylation Reagents in Photoredox Catalysis, J. Am. Chem. Soc., 2016, 138, 12312–12315 CrossRef CAS PubMed; (c) P.-Z. Wang, J.-R. Chen and W.-J. Xiao, Hantzsch esters: an emerging versatile class of reagents in photoredox catalyzed organic synthesis, Org. Biomol. Chem., 2019, 17, 6936–6951 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, detailed synthetic procedures, and 1H and 13C NMR spectra of all compounds. See DOI: https://doi.org/10.1039/d3qo01856c |
| This journal is © the Partner Organisations 2024 |