
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of (±)-villosin C and (±)-teuvincenone B†
Jin-Xin
Zhou
ac,
Ming-Jie
Yan
ba,
Xiao-Yu
Shi
ba,
Yue-Qi
Wang
ba,
Hong-Yan
Shen
a,
Xu-Dong
Mao
*a and
Jie
Wang
 *abc
*abc
aState Key Laboratory of Chemical Biology, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China. E-mail: maoxd1993@163.com; jiewang@simm.ac.cn
bSchool of Chinese Materia Medica, Nanjing University of Chinese Medicine, Nanjing 210023, China
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 30th November 2023
Abstract
Herein, we report the first total synthesis of 17(15 → 16)-abeo-abietane diterpenoids (±)-villosin C (5) and (±)-teuvincenone B (4) in 11 steps. The A/B/C ring system was assembled via a modified three-step sequence on gram-scale, while the D ring was constructed by intramolecular iodoetherification. This synthesis relied largely on the rational design of the order for oxidation state escalation (C6/11/14 → C7 → C12 → C17), which was realized through sequential benzylic iodination/Kornblum oxidation, Siegel–Tomkinson C–H oxidation and iodoetherification. In addition, villosin C (5) and its epimer (5a) were found to have indistinguishable NMR data and the correct configuration for villosin C was elucidated by comparing HPLC trace with a natural sample.
Introduction
Abietanes represent a rich array of naturally occurring diterpenoids isolated from a variety of terrestrial plant sources.1 Skeleton rearrangements were frequently observed in this family, where 17(15 → 16)-abeo-abietane is amongst the most common one, as was found in various natural products (Fig. 1).2 Villosin C (5), characterized by this framework, was first isolated by Ulubelen in 1994 from Teucrium divaricatum subsp. Villosum.3 In the past decade, this natural product was reisolated from several other plant sources independently, including Teucrium polium,4 the Chinese medicinal plants Clerodendrum trichotomum5 and Clerodendrum bungei,6 and Akebia quinata.7 Multiple biological activities such as antioxidative,4 NO production inhibitory5c and antitumor activities5d were reported for villosin C (5), which could partly account for the therapeutic effects of Clerodendrum trichotomum (known as “Chou-Wu-Tong”) in traditional Chinese medicine. Teuvincenone B was first isolated from the root of Teucrium polium8 and was later found to exhibit antioxidant4 and antitumor activities.5d Nonetheless, these natural products are rather scarce from natural sources, with less than 1 mg kg−1 isolated from the corresponding dried plants. Thus, the development of a synthetic strategy allowing the access to teuvincenone B (4), villosin C (5), and their natural and unnatural analogs would be highly desired. Herein, we present our study on the total synthesis of (±)-villosin C (5) and (±)-teuvincenone B (4) by a convergent approach. | ||
| Fig. 1 Representative natural products bearing 17(15 → 16)-abeo-abietane skeleton. | ||
Structurally, villosin C (5) and teuvincenone B (4) were characterized by a 6/6/6/5 tetracyclic ring system with an aromatized C ring and a dihydrofuran D ring. The synthetic challenge is mainly embedded in the highly oxidized nature (9 out of 10 carbons of the B/C ring are unsaturated and 5 are directly attached to oxygen) and that the C ring is a fully substituted benzene moiety. Although there is a plethora of synthetic studies on C ring aromatized abietane diterpenoids,9 seldom has targeted those with high oxidation states and fully substituted benzene in this family,10 and the total synthesis of villosin C (5) and teuvincenone B (4) are yet to be reported. In an initial synthetic study, one of us (Mao) obtained an elaborated intermediate (8) with fully established carbon skeleton and ring system. Unfortunately, the late stage oxidation of the C6/C7 sites proved unsuccessful (Scheme 1A). Instead, oxidative cleavage of the strong electron rich benzene ring was observed. These results prompted us to sketch a new retrosynthetic design, where the α-hydroxy enone moiety in the B ring was constructed prior to installation of the C12 oxidation state (Scheme 1B). Thus, the D ring of 4/5 was envisaged to be built via a Claisen rearrangement/iodoetherification sequence from phenol 9. The C12 and C7 oxidation states could be introduced via sequential C–H oxidations from ketone 10, which was then disconnected to commercially available β-cyclocitral 12 and a known benzyl chloride 13.
 | ||
| Scheme 1 (A) Initial unsuccessful attempt and (B) Retrosynthetic plan. | ||
Results and discussion
Our synthesis commenced from the assembly of aldehyde 12 and benzyl chloride 13 through a similar three-step sequence previously utilized in other abietane natural product and analog synthesis (Scheme 2).11 In these studies, stoichiometric Li/naphthalene was used unexceptionally to activate benzyl chloride, which is problematic for scale-up. Here, the union of 12 and 13 through Grignard addition afforded 90% yield on multigram-scale. After oxidation with IBX, enone 11 was obtained in 93% yield. Friedel–Crafts cyclization of a similar type was previously reported for either enone11 or unconjugated alkene substrates12 under Lewis and Brønsted acid conditions. In our case, condition screening on 0.1 mmol scale revealed that AlCl3 and BF3·Et2O delivered cyclized products 10a and 10b in poor yields while TFA and Tf2NH proved ineffective (Table S1, see the ESI† for more details). The optimum result was obtained with 3 equivalents of TfOH in DCE at 0 °C on 0.1 mmol scale, furnishing 10a and 10b in 78% combined yield with 2.6/1 dr as determined by crude 1H NMR, and the efficiency could be well maintained on gram-scale (67% isolated yield).13 | ||
| Scheme 2 Synthesis of tricyclic α-methoxy enone 15. | ||
Both diastereoisomers could be used in the subsequent synthesis, however, under different conditions. Jones oxidation and O2 under basic conditions are two common methods reported in literature for converting ketone to the corresponding α-diketone or α-hydroxy enone moiety in abietane natural product synthesis.14,11c,12a In our attempt to oxidize ketone 10a, while the Jones oxidation was messy, base/O2 provided variable 30–70% yields depending on reaction scale. After optimization, a one-pot benzylic iodination/Kornblum oxidation using CuO and I2 in DMSO was found to deliver α-hydroxy enone 14 efficiently in 83% yield on gram-scale.15 Surprisingly, application of this condition to the other diastereomer 10b gave a messy reaction. Interestingly, this diastereomer was found to be easily oxidized by air, even when placed on bench without any reagent. MeONa/O2 was then used to achieve the desired oxidation in high conversion, however, affording a mixture of α-hydroxy enone 14 and α-diketone 14a. After treating the mixture with DBU, 14 could be obtained in 70% overall yield from 10b. Protection of the free enol with NaOH/MeI afforded the tricyclic α-methoxy enone 15 in 95% yield.
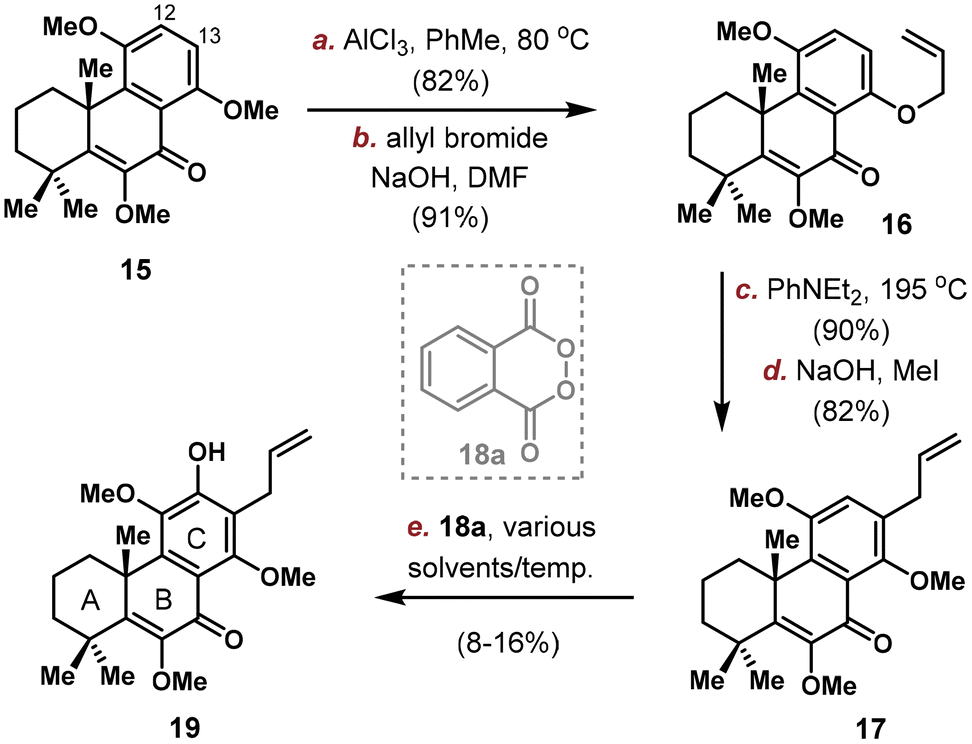
Quantities of 15 was produced through the implementation of the above-mentioned sequence on gram-scale and the stage is now setting for the introduction of C12/C13 substituents. Electronic analysis pointed C13 position as the more electron rich site, which was confirmed by exclusive electrophilic bromination at C13.16 Thus, we planned to first install an allyl group on C13 and then an oxygen atom at C12. Selective removal of 1 methyl group out of 3 was realized using AlCl3 in PhMe (Scheme 3). Allylation provided 16 in 91% yield, with the location of allyl group confirmed by NOESY spectrum analysis. Claisen rearrangement took place efficiently to position the allyl group on C13, and 17 was obtained after methyl protection. Unfortunately, the subsequent C(12)–H oxidation proved problematic. While electrophilic bromination/iodination were unsuccessful, radical based arene C–H oxidation developed by Siegel using 18a yielded the desired phenol 19 in very low productivity (8–16% yield under various solvents/temperatures), probably due to steric hindrance and incompatibility with the terminal alkene group.17
 | ||
| Scheme 3 Synthesis of the fully substituted C ring intermediate 19. | ||
In an optimized protocol, we tried to switch the order of C(12)–H oxidation and C13 allylation (Scheme 4, upper left). Direct oxidation of 15 with 18a provided a mixture of C12–OH (9) and C13–OH products in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.6 ratio. After optimization (Table S2†), cyclopropane malonyl peroxide 18c in TFE delivered the desired phenol 9 in 37% isolated yield along with the C13–OH isomer in 1:1.2 ratio.18,19 Subsequent allylation and Claisen rearrangement proceeded smoothly to provide phenol 19 in 79% yield.
:1.6 ratio. After optimization (Table S2†), cyclopropane malonyl peroxide 18c in TFE delivered the desired phenol 9 in 37% isolated yield along with the C13–OH isomer in 1:1.2 ratio.18,19 Subsequent allylation and Claisen rearrangement proceeded smoothly to provide phenol 19 in 79% yield.
 | ||
| Scheme 4 Modified synthesis of 19 and total synthesis of (±)-villosin C (5), (±)-teuvincenone B (4), and (±)-16-epi-villosin C (5a). | ||
Next, the D ring was constructed through iodoetherification uneventfully to give 20a and 20b in 50% combined yield with almost no selectivity at the newly generated stereocenter (1:1.1 dr at C16). The two diastereomers were isolated by flash column chromatography and the structure of the undesired isomer 20b was confirmed by X-ray crystallographic analysis unambiguously. Thus, the conversion of the primary iodide in 20a to alcohol was achieved using CF3CO2Cs in DMF at elevated temperature. After global deprotection with BBr3, (±)-villosin C (5) was obtained in 68% yield. In addition, iodide 20a was reduced with AIBN/Bu3SnH and globally deprotected with HBr in HOAc to afford (±)-teuvincenone B (4) in 50% yield over two steps. The spectroscopic data for 5 and 4 are all in good agreement with those reported from the isolation literature.
The same two-step sequence (hydroxylation, global deprotection) was also applied to 20b to deliver 16-epi-villosin C (5a) in 44% yield (Scheme 4). Surprisingly, the 1H and 13C NMR spectra of synthetic 5a and 5 were almost identical, and could not distinguish with the reported data in the isolation literature (C11–OH represented the only large difference: 5.12, 4.98 and 4.99 ppm for natural 5, synthetic 5 and 5a respectively, see the ESI† for detailed comparison).5b Although the C10/C16 relative stereochemistry of some 17(15 → 16)-abeo-abietane diterpenoids was elucidated by X-ray crystallographic analysis to be cis as drawn in 5, it is still uncertain in the case of villosin C. Therefore, we requested a sample of natural villosin C from Yang, and the HPLC trace of which was compared to that of synthetic 5 and 5a, confirming that the true structure of villosin C (5) was indeed as originally proposed (Scheme 4, left bottom).5c While the C16 stereochemistry in some 17(15 → 16)-abeo-abietane diterpenoids was proposed by comparing the chemical shifts and coupling constant values in NMR around C15–C17 to known ones, our work suggests that NMR data might not be sufficient to make a definite conclusion, at least in the category of 17(15 → 16)-abeo-abietane natural products.
Conclusions
In summary, total synthesis of 17(15 → 16)-abeo-abietane diterpenoids (±)-villosin C (5) and (±)-teuvincenone B (4) was accomplished for the first time employing a convergent approach. The synthesis features (i) five-step gram-scale assembly of A/B/C ring system with oxidation state of B ring fully positioned through one-pot benzylic iodination/Kornblum oxidation, (ii) Siegel–Tomkinson C–H oxidation and Claisen rearrangement to install all substituents required on C ring, and (iii) iodoetherification to construct D ring. The successful implementation of this route also relied largely on the rational design of the order for oxidation state escalation (C6/11/14 → C7 → C12 → C17). In addition, our finding that villosin C (5) and 16-epi-villosin C (5a) showed indistinguishable NMR spectroscopy called for more prudence during structural characterization of the 17(15 → 16)-abeo-abietane family.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Professor Li Yang for providing the natural sample of villosin C isolated from Clerodendrum trichotomum. Financial support for this work was provided by the National Natural Science Foundation of China (Grant No. 22101290), Chinese Academy of Sciences and the State Key Laboratory of Chemical Biology, Shanghai Institute of Materia Medica.References
- (a) M. Gaborova, K. Smejkal and R. Kubinova, Abietane Diterpenes of the Genus Plectranthus sensu lato, Molecules, 2022, 27, 166 CrossRef CAS PubMed; (b) L. Kuzma and J. Gomulski, Biologically Active Diterpenoids in the Clerodendrum Genus-A Review, Int. J. Mol. Sci., 2022, 23, 11001 CrossRef CAS PubMed; (c) A. Ulubelen, G. Topu and U. Sonmez, Chemical and biological evaluation of genus teucrium, Stud. Nat. Prod. Chem., 2000, 23, 591–648 CrossRef CAS.
- (a) M. J. S. Cuadrado, M. Bruno, M. C. De La Torre, F. Piozzi, G. Savona and B. Rodríguez, Rearranged Abietane Diterpenoids from the Root of Two Teucrium Species, Phytochemistry, 1992, 31, 1697–1701 CrossRef; (b) J. M. Schmid, P. Rüedi and C. H. Eugster, Diterpenoid Leaf-gland Pigments from Labiatae: 22 Novel Coleons and Royleanones from Plectranthus Lanuginosus, Helv. Chim. Acta, 2004, 65, 2136–2163 CrossRef; (c) B. Rodríguez, A. Robledo and M. J. Pascual-Villalobos, Rearranged Abietane Diterpenoids from the Root of Teucrium lanigerum, Biochem. Syst. Ecol., 2009, 37, 76–79 CrossRef; (d) M. Xu, L. Shen, K. Wang and Q. Du, Two New Abietane Diterpenoids from the Stems of Clerodendrum kaichianum P. S. Hsu, Helv. Chim. Acta, 2011, 94, 539–544 CrossRef CAS; (e) D. B. Pu, T. Wang, X. J. Zhang, J. B. Gao, R. H. Zhang, X. N. Li, Y. M. Wang, X. L. Li, H. Y. Wang and W. L. Xiao, Isolation, Identification and Bioactivities of Abietane Diterpenoids from Premna szemaoensis, RSC Adv., 2018, 8, 6425–6435 RSC; (f) B. Zhou, Y. H. Ren, Y. S. Han, T. Mesplede and J. M. Yue, Diverse Types of Diterpenoids with an Aromatized C Ring from the Twigs of Podocarpus imbricatus, J. Nat. Prod., 2020, 83, 2416–2424 CrossRef CAS PubMed; (g) J. Qi, Y. Zhang, Q. Liu, H. Liu, Y. Fan and J. Yue, Clerodenoids A—F: C–ring Aromatized and/or Rearranged Abietane Diterpenoids from Clerodendrum chinense var. simplex, Chin. J. Chem., 2021, 39, 1891–1897 CrossRef CAS.
- A. Ulubelen, G. Topcu and S. Olçal, Rearranged abietane diterpenes from Teucrium divaricatum subsp. Villosum, Phytochemistry, 1994, 37, 1371–1375 CrossRef CAS.
- A. Fiorentino, B. D'Abrosca, S. Pacifico, M. Scognamiglio, G. D'Angelo and P. Monaco, abeo-Abietanes from Teucrium polium Roots as Protective Factors against Oxidative Stress, Bioorg. Med. Chem., 2010, 18, 8530–8536 CrossRef CAS PubMed.
- (a) W. X. Wang, J. Xiong, Y. Tang, J. J. Zhu, M. Li, Y. Zhao, G. X. Yang, G. Xia and J. F. Hu, Rearranged Abietane Diterpenoids from the Roots of Clerodendrum trichotomum and Their Cytotoxicities against Human Tumor Cells, Phytochemistry, 2013, 89, 89–95 CrossRef CAS PubMed; (b) L. Li, L. Wu, M. Wang, J. Sun and J. Liang, Abietane Diterpenoids from Clerodendrum trichotomum and Correction of NMR Data of Villosin C and B, Nat. Prod. Commun., 2014, 9, 907–910 CAS; (c) H. J. Hu, Y. Zhou, Z. Z. Han, Y. H. Shi, S. S. Zhang, Z. T. Wang and L. Yang, Abietane Diterpenoids from the Roots of Clerodendrum trichotomum and Their Nitric Oxide Inhibitory Activities, J. Nat. Prod., 2018, 81, 1508–1516 CrossRef CAS PubMed; (d) L. Li, Y. Zhang, L. Chen, Y. Cen, Y. Tu, X. Yang and Y. Li, Chemical Constituents of the n-Butanol Fraction from the Stems of Clerodendrum trichotomum and Their Antitumor Activities in vitro, China Pharm., 2022, 33, 2578–2583, DOI:10.6039/j.issn.1001-0408.2022.21.04.
- Q. Liu, H. J. Hu, P. F. Li, Y. B. Yang, L. H. Wu, G. X. Chou and Z. T. Wang, Diterpenoids and Phenylethanoid Glycosides from the Toots of Clerodendrum bungei and Their Inhibitory Effects against Angiotensin Converting Enzyme and Alpha-glucosidase, Phytochemistry, 2014, 103, 196–202 CrossRef CAS PubMed.
- J. P. An, T. K. Ha, J. Kim, T. O. Cho and W. K. Oh, Protein Tyrosine Phosphatase 1B Inhibitors from the Stems of Akebia quinata, Molecules, 2016, 21, 1091 CrossRef PubMed.
- M. C. Carreiras, B. Rodriguez, M. C. de la Torre, A. Perales, M. R. Torres, G. Savona and F. Piozzi, Rearranged Abietane Diterpenoids from the Root of Teucrium polium subsp. vincentinum, Tetrahedron, 1990, 46, 847–860 CrossRef CAS.
- For seletcted reviews, see: (a) M. A. Gonzalez, Synthetic Derivatives of Aromatic Abietane Diterpenoids and Their Biological Activities, Eur. J. Med. Chem., 2014, 87, 834–842 CrossRef CAS PubMed; (b) M. A. Gonzalez, Aromatic Abietane Diterpenoids: Their Biological Activity and Synthesis, Nat. Prod. Rep., 2015, 32, 684–704 RSC; (c) M. A. González, Aromatic Abietane Diterpenoids: Total Syntheses and Synthetic Studies, Tetrahedron, 2015, 71, 1883–1908 CrossRef; (d) J. Kang, T. Quynh Le and C. H. Oh, Recent Advances in Abietane/Icetexane Synthesis, Tetrahedron Lett., 2022, 108, 154133 CrossRef CAS.
- (a) M. Li, P. Chen, H. Liu, J. Huang and Y. Chen, Review of Total Synthesis of Aromatic Abietane Diterpenoid: Ferruginol, Synthesis, 2023, DOI:10.1055/a-2186-7983; (b) M. Fischer, K. Harms and U. Koert, Total Syntheses of 7,20-Oxa-Bridged Dinorditerpenes: Antihepatitis C Virus Active (+)-Elevenol from Flueggea virosa and (+)-Przewalskin, Org. Lett., 2016, 18, 5692–5695 CrossRef CAS PubMed; (c) X. Li and R. G. Carter, Total Syntheses of Aromatic Abietane Diterpenoids Utilizing Advances in the Pummerer Rearrangement, Org. Lett., 2018, 20, 5546–5549 CrossRef CAS PubMed; (d) X.-D. Mao, T.-T. Du, Q. Gu, L. Yang, H.-L. Shi, R. Hong and G.-X. Chou, Synthesis and Bioactivity Evaluation of Nepetaefolin F and Its Analogues, ACS Omega, 2023, 8, 14830–14840 CrossRef CAS PubMed.
- (a) T. Matsumoto, S. Usui and T. Morimoto, A Convenient Synthesis of (±)-Taxodione, (±)-Ferruginol, and (±)-Sugiol, Bull. Chem. Soc. Jpn., 1977, 50, 1575–1579 CrossRef CAS; (b) J. Huang, D. Foyle, X. Lin and J. Yang, Total Synthesis and Biological Evaluation of an Antifungal Tricyclic o-Hydroxy-p-Quinone Methide Diterpenoid, J. Org. Chem., 2013, 78, 9166–9173 CrossRef CAS PubMed; (c) A. Crusco, C. Bordoni, A. Chakroborty, K. C. L. Whatley, H. Whiteland, A. D. Westwell and K. F. Hoffmann, Design, Synthesis and Anthelmintic Activity of 7-keto-Sempervirol Analogues, Eur. J. Med. Chem., 2018, 152, 87–100 CrossRef CAS PubMed.
- (a) P. Bie, C. Zhang, A. Li, X. Peng, T. Wu and X. Pan, First Total Synthesis of (±)-Celaphanol A, J. Chin. Chem. Soc., 2002, 49, 581–583 CrossRef CAS; (b) D. C. Behenna, J. L. Stockdill and B. M. Stoltz, Synthesis of the Carbocyclic Core of Zoanthenol: Implementation of an Unusual Acid-Catalyzed Cyclization, Angew. Chem., Int. Ed., 2007, 46, 4077–4080 CrossRef CAS PubMed.
- Attempt to realize this cyclization with enantioselective control was unsuccessful (see the ESI† for a list of conditions tried).
- T. Matsumoto and S. Takeda, Total Synthesis of Coleons, U and V, Chem. Lett., 1979, 8, 409–412 CrossRef.
- G. Yin, B. Zhou, X. Meng, A. Wu and Y. Pan, Efficient C-C Double-Bond Formation Reaction via a New Synthetic Strategy: a Self-Sorting Tandem Reaction, Org. Lett., 2006, 8, 2245–2248 CrossRef CAS PubMed.
- Z. Ruan, M. Wang, C. Yang, L. Zhu, Z. Su and R. Hong, Total Synthesis of (+)-Hinckdentine A: Harnessing Noncovalent Interactions for Organocatalytic Bromination, JACS Au, 2022, 2, 793–800 CrossRef CAS PubMed.
- C. Yuan, Y. Liang, T. Hernandez, A. Berriochoa, K. N. Houk and D. Siegel, Metal-free Oxidation of Aromatic Carbon-Hydrogen Bonds through a Reverse-Rebound Mechanism, Nature, 2013, 499, 192–196 CrossRef CAS PubMed.
- A. Dragan, T. M. Kubczyk, J. H. Rowley, S. Sproules and N. C. Tomkinson, Arene Oxidation with Malonoyl Peroxides, Org. Lett., 2015, 17, 2618–2621 CrossRef CAS PubMed.
- Further screening revealed that with Ts protecting group on C6–OH, 1.3:1 ratio of 9 to C13–OH isomer could be obtained. However, the overall yield was lower (23% for 9, see the ESI† for details).
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2305767. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qo01841e |
| This journal is © the Partner Organisations 2024 |