
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydantoin hexameric rosettes: harnessing H-bonds for supergelation and liquid crystals†
Lucía
González
a,
Iván
Marín
a,
Rosa M.
Tejedor
 b,
Joaquín
Barberá
a,
Pilar
Romero
a,
Alberto
Concellón
a,
Santiago
Uriel
*a and
José L.
Serrano
*a
b,
Joaquín
Barberá
a,
Pilar
Romero
a,
Alberto
Concellón
a,
Santiago
Uriel
*a and
José L.
Serrano
*a
aDepartamento de Química Orgánica, Instituto de Nanociencia y Materiales de Aragón (INMA), CSIC-Universidad de Zaragoza, 50009 Zaragoza, Spain. E-mail: joseluis@unizar.es; suriel@unizar.es
bCentro Universitario de la Defensa, Academia General Militar, 50090 Zaragoza, Spain
First published on 4th December 2023
Abstract
We have synthesized two geometric isomers of a cyclohexane-5-spirohydantoin derivative (1,3-diazaspiro[4.5]decane-2,4-dione) incorporating a hydrophobic phenyl 3,4,5-tris(dodecyloxy)benzoate unit at position 8. Separation of these diastereomers was accomplished through silica gel flash chromatography. The interplay of intermolecular hydrogen bonding and micro-segregation between the polar hydantoin unit and nonpolar aliphatic chains within the molecule endows them with remarkable self-assembly capabilities, both in solution and in the solid state. These hydantoin derivatives spontaneously form rosette-shaped structures composed of six molecules. In the solid state, these compounds display hexagonal columnar liquid crystal phases, with hydrogen-bonded disks as their fundamental building blocks. Similarly, when exposed to apolar solvents such as cyclohexane or dodecane, they adopt a columnar arrangement, resulting in gel formation comprising nanoscale fibers that intricately interlace to form a network. Remarkably, the two isomers exhibit markedly different properties. The major isomer behaves as a glassy liquid crystalline material, while the minor one exhibits liquid crystalline behavior with a high propensity to crystallize. Our experimental findings, in combination with theoretical studies, underscore the fundamentally distinct supramolecular organizations present in these isomers, shedding light on their unique self-assembling properties.
Introduction
Liquid crystals (LCs) are multimolecular supramolecular organizations that combine the characteristics of fluidity found in the liquid state with the crystalline order.1,2 Due to these unique characteristics, LCs serve as a powerful tool for precisely positioning molecules in specific and well-defined arrangements, enabling the construction of nanostructured materials with potential applications in fields such as optoelectronics, sensing, or electron/ion transport.3–8 LC molecules typically consist of a series of rigid covalent nuclei with terminal groups, usually composed of flexible alkyl chains. However, mesogenic organizations can also be formed from supramolecules through non-covalent intermolecular interactions, such as hydrogen bonds (H-bonds), halogen bonds, or electrostatic interactions.9–11 In particular, H-bonds offer an excellent means to construct various nanostructures due to their high selectivity and directionality. Pioneering works by Kato and Fréchet12,13 and Lehn et al.14 were followed by growing interest in these supramolecular systems. The dynamic behavior of H-bonds in such systems presents unique opportunities for developing a new generation of smart functional materials, including nanoporous polymers, optical materials, soft actuators, and chemical sensors.15–19The novelty of this supramolecular approach lies in the simplicity of molecular geometry and the versatility of structural modifications. Indeed, a wide variety of LC organizations can be constructed by combining a small number of components, which typically include both rod-like (calamitic) and disc-like (discotic) mesogenic complexes. Many examples of supramolecular macrocycles have been reported based on self-complementary units such as melamine, cyanuric acid, folic acid, barbituric acid derivatives, or nucleobases.20–29 Specifically, “rosettes” (i.e., H-bonded macrocycles) have garnered significant attention recently as they enable the formation of complex columnar architectures with responsive behaviors.30–34 In this context, our research group has undertaken pioneering work on H-bonded rosette structures, consisting of six melamines and six carboxylic acids, which self-assemble into highly ordered columnar organizations.35,36
Nonetheless, hydantoin derivatives, or imidazoline-2,4-dione compounds, which feature a structure with two H-bond accepting carbonyl groups and two H-bond donating amino groups (two carbonyl and two N–H acceptors and donors), have been less frequently considered for building supramolecular architectures. These molecules are essential intermediates in amino acid synthesis and have been demonstrated to possess various biological properties, making them valuable compounds.37 This has led to intense research activity and the development of novel synthetic methods for accessing these valuable molecules.38 The unique geometry of hydantoins has also been exploited to prepare organometallic derivatives39–42 and to create different crystalline architectures linked by hydrogen bonds.43–46 In some of these derivatives, the introduction of a cyclopentane or cyclohexane unit at position 5 of the hydantoin heterocycle introduces additional steric factors that influence the supramolecular organization and properties of these molecules.47,48
Herein, we present two novel hydantoin derivatives with unprecedented self-assembly properties. Two geometric isomers of 1,3 diazaspiro[4.5]decane 2,4 dione ((Z)-4 and (E)-4 in Scheme 1) have been prepared bearing a hydrophobic phenyl 3,4,5-tris(dodecyloxy)benzoate unit at position 8. The intermolecular H-bonding and microsegregation between the polar (hydantoin unit) and nonpolar (aliphatic chains) regions of the molecule lead to an excellent self-assembly capability in solution and in the solid state. These hydantoin derivatives self-organize into rosette-shaped structures showing columnar hexagonal mesophases, in which six molecules form the hydrogen-bonded disk. In addition, in the presence of apolar solvents such as cyclohexane or dodecane, they form columnar-type organizations that give rise to gels made up of fibers of several tens of nanometers that entangle to form a network.
 | ||
Scheme 1 Synthesis and chemical structure of the two diastereomers of hydantoin intermediate derivative 2 [(Z)-2 and (E)-2] and final hydantoin 4 [(Z)-4 and (E)-4]. (Z)-4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4-((5s,8s)-2,4-dioxo-1,3-diazaspiro[4.5]decan-8-yl)phenyl3,4,5 tris(dodecyloxy)benzoate, (E)-4:4-((5r,8r)-2,4-dioxo-1,3-diazaspiro[4.5]decan-8-yl)phenyl 3,4,5-tris(dodecyloxy)benzoate. :4-((5s,8s)-2,4-dioxo-1,3-diazaspiro[4.5]decan-8-yl)phenyl3,4,5 tris(dodecyloxy)benzoate, (E)-4:4-((5r,8r)-2,4-dioxo-1,3-diazaspiro[4.5]decan-8-yl)phenyl 3,4,5-tris(dodecyloxy)benzoate. | ||
Results and discussion
Synthesis of hydantoins
The synthesis of hydantoins 4 was conducted through a modification of the Bucherer–Bergs method (Scheme 1 and section 2 in the ESI†).49,50 The Bucherer–Bergs reaction, when employing substituted cyclohexanones, exhibits stereoselectivity and yields compound 2 (55% yield) as a mixture of two diastereomers, namely (Z)-2 and (E)-2. This outcome depends on the configuration of the phenyl ring with respect to the NH group of hydantoin, which is in close proximity to cyclohexane (Scheme 1). The diastereomeric mixture of 2 then undergoes a reaction with 3,4,5-tris(decyloxy)benzoic anhydride (3), resulting in the formation of 4 as a diastereomeric mixture in a 3:1 ratio (as determined by 1H-NMR spectroscopy; see Fig. 2a). The separation of the two diastereomers, (Z)-4 and (E)-4, was accomplished using silica gel flash chromatography. Furthermore, each diastereomer has two potential conformers, corresponding to two distinct chair conformations of the cyclohexane ring (Fig. 1). The absolute identification of the diastereomers and conformers was achieved through NMR spectroscopy.
 | ||
| Fig. 1 Chemical structures of the diastereomers (Z)-4 and (E)-4 and the possible corresponding two conformers depending on the cyclohexane chair conformations. | ||
 | ||
| Fig. 2 (a) 1H-NMR spectrum (CDCl3, 298 K) of mixture 4 of diastereomers 4 diastereomers at a concentration of 40 mM. (b) Signals corresponding to the 1H-NMR spectrum (CDCl3, 298 K) of mixture 4 at different concentrations. The signals corresponding to the major isomer appear, linked by the red line in Fig. 2b and surrounded in red in Fig. 2a. In the case of the minor isomer, these lines and circles appear in blue. | ||
Fig. 2a shows the 1H NMR spectrum of the diastereomeric mixture of 4 in CDCl3 (40 mM), in which the two protons of the NH groups of the hydantoin ring appear as broad signals at different chemical shifts depending on the diastereomer. In particular, the NH protons of the main diastereomer appear at around 9.36 and 8.21 ppm, while in the minor diastereomer, the NH signals appear at around 9.09 and 6.71 ppm. The integration of these signals shows a 75 and 25% ratio. Moreover, we observed a concentration-dependent behaviour in the 1H-NMR spectra, wherein the NH signals originating from the hydantoin unit sharpen and shift to higher field values as the concentration decreases. In contrast, the other signals remain unaffected (see Fig. 2b). This phenomenon arises due to intermolecular hydrogen bonding.
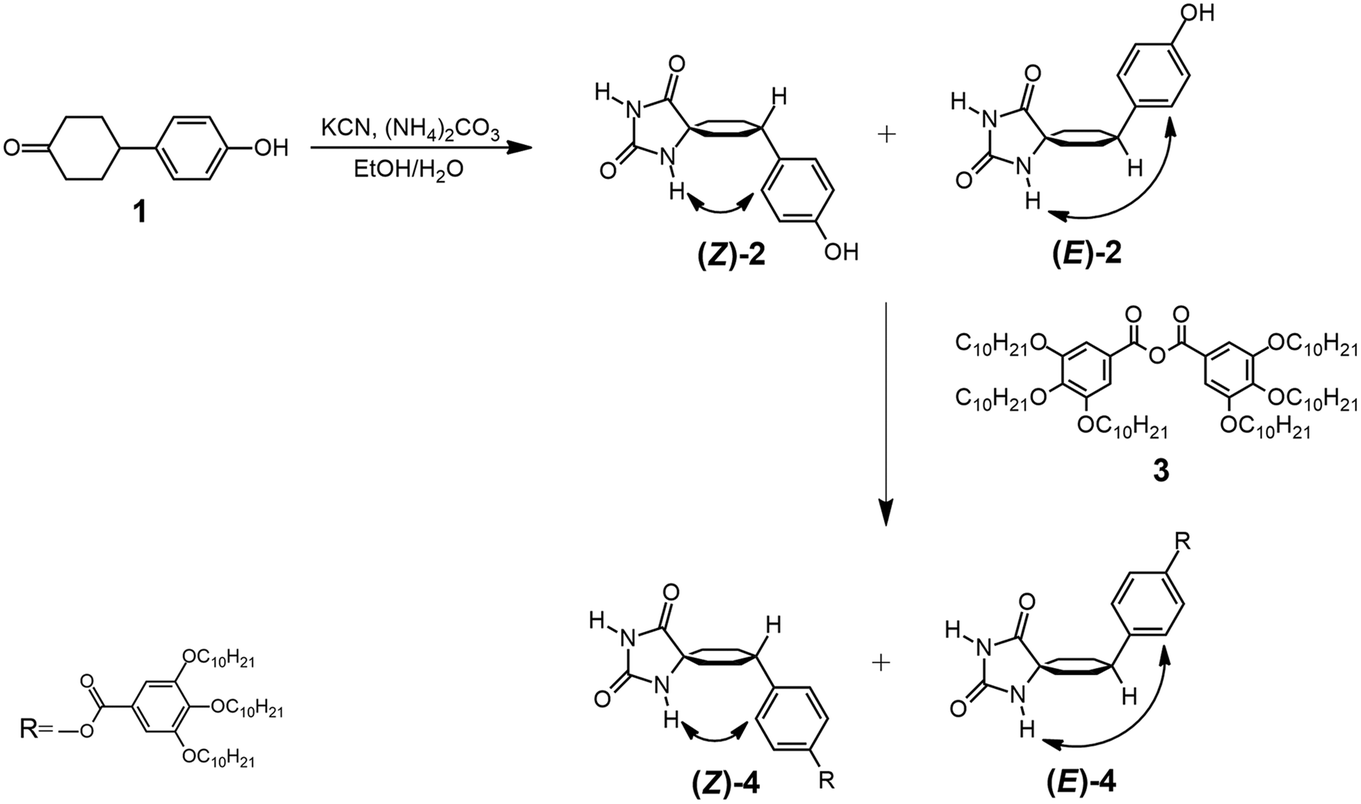
The 1H-NMR spectra obtained at room temperature confirmed that, in both diastereomers, only the conformer that has the bulky aromatic group at the equatorial position appears. The configuration of H4 can be elucidated by studying the coupling constants between H4 and H3ax. The proton signal at approximately 2.6 ppm, corresponding to H4, exhibits a substantial coupling with H3ax (J ≈ 12 Hz), indicating that both H4 and H3ax are at the axial position. It is well-known that axial–axial couplings exhibit larger coupling constants (3Jax–ax = 10–12 Hz) compared to those of equatorial–axial (3Jax–eq = 2–6 Hz) or equatorial–equatorial (3Jeq–eq = 2–5 Hz) couplings. To further elucidate the relative configuration of the two hydantoins within the substituted cyclohexanes, ROESY experiments were conducted (Fig. 3). The 1H–1H ROESY experiment of diastereomer (Z)-4 reveals cross peaks between the hydantoin's NH and H3ax and H2ex, whereas the 1H–1H ROESY spectrum of (E)-4 indicates that hydantoin's NH exhibits cross peaks only with H2ax. Consequently, the NMR studies allow the assignment of (Z)-4 as the major diastereomer (75%) and (E)-4 as the minor one (25%) (Fig. S1–S10 in the ESI†). This observation aligns with the fact that the Bucherer–Bergs reaction of cyclohexanone derivatives predominantly yields hydantoins with the NH group at the axial position.51 In addition, a search of the Cambridge Structural Database (November CSD 2022 release) with both unsubstituted hydantoin NH groups and carbocyclic spiro fusion at the C5 position yielded 54 hits (excluding duplicate structure determinations), 19 of them being cyclohexanespiro 5 hydantoin derivatives.52 Importantly, all of these cyclohexane-5-spirohydantoin derivatives feature hydantoin's NHα, located close to cyclohexane, at the axial position. Besides this, in the ROESY studies, the NOE effect between the H6 proton of the benzene ring and the H3ax protons of the cyclohexane ring is observed in both diastereomers, confirming the substitution of the aromatic ring at the equatorial position. From these data, it is deduced that for the (Z)-4 diastereomer only the NHax–Φeq conformer appears, while for the (E)-4 diastereomer only the NHeq–Φeq conformer appears.
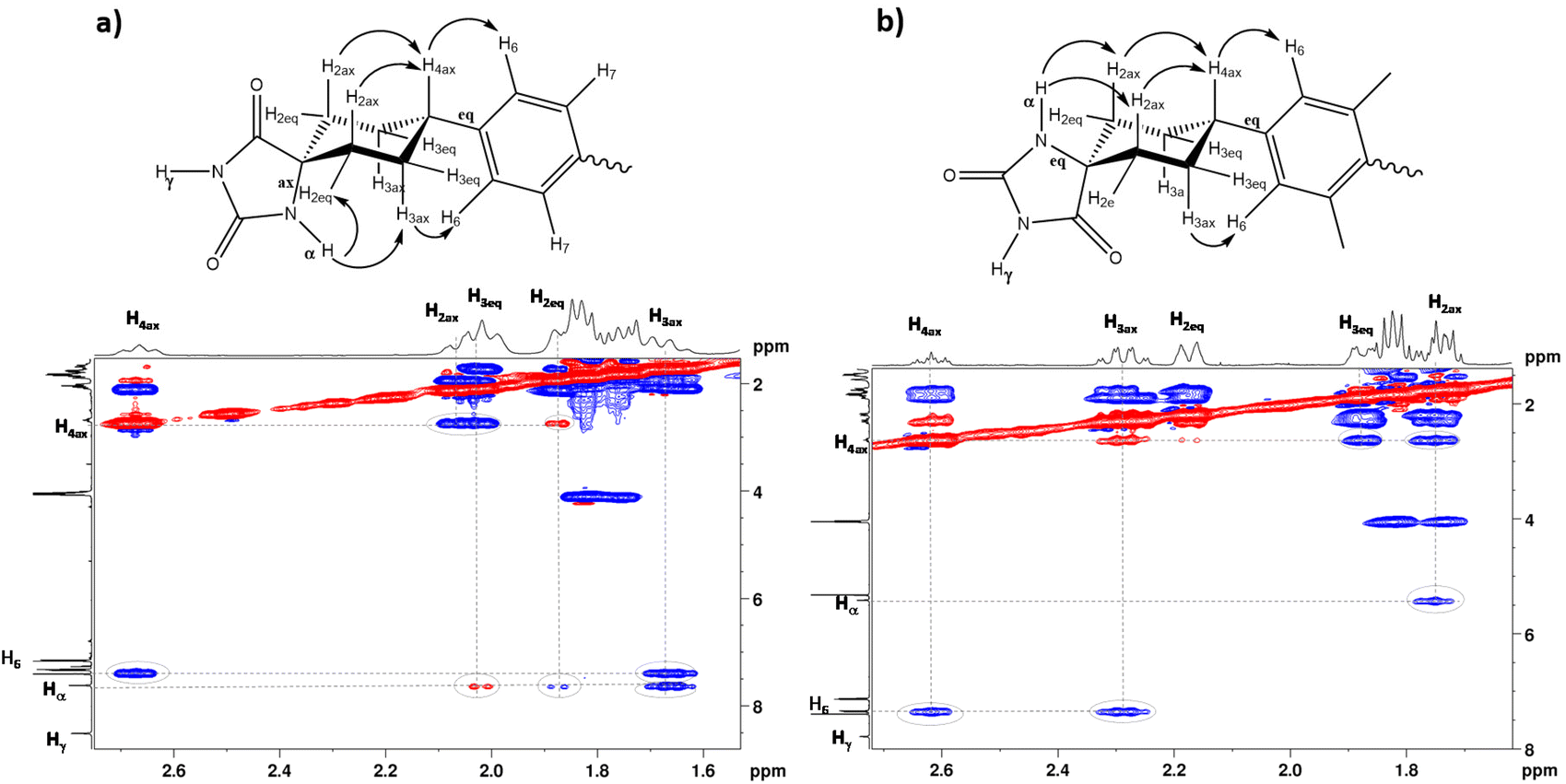 | ||
| Fig. 3 Partial 1H–1H ROESY spectra of (a) (Z)-4 and (b) (E)-4. (Red portions are due to interchange). The letters ax and eq. indicate the axial or equatorial position of the bond in the corresponding diastereomer; α and γ indicate the position of NH in relation of the spirocarbon. | ||
Self-assembly in solution and gelation properties
The self-assembly of (Z)-4 and (E)-4via H-bonding interactions in solution was investigated using 1H-NMR in solvents including CDCl3, CD2Cl2 and C6D12. Our observations revealed that the type of self-assembly exhibited depends on the chosen solvent for the 1H-NMR study. Moreover, when examining the 1H-NMR spectrum of (Z)-4 in a non-polar solvent, such as cyclohexane-d12, all signals, including the NH signals, were observed to broaden and showed no significant shifts with changes in the concentration. This phenomenon strongly suggests the occurrence of H-bonding in a remarkably stable manner and rules out the involvement of π–π interactions in driving the self-assembly process (Fig. S11 in ESI†). Furthermore, we assessed the size of the aggregates in solution using 1H-DOSY experiments (Fig. 4), which has proven to be a highly effective approach for estimating the sizes of supramolecular assemblies.53,54 Therefore, we calculated the reduced diffusion coefficients in relation to TMS (tetramethylsilane) for (Z)-4 in CDCl3, a polar solvent where the supramolecular association can be attributed to the formation of dimeric species in equilibrium with the monomeric species (temperature and concentration dependent experiments in relation to this point are shown in Fig. S12 and S13 in the ESI†). In contrast, in cyclohexane d12, where the association is stronger and remains independent of concentration, we also determined the diffusion coefficients. By comparing these two diffusion coefficients, we could estimate the number of molecules constituting the aggregates in solution. Specifically, (Z)-4 aggregates were found to form through the association of approximately six molecules (see section 3.2 in the ESI†).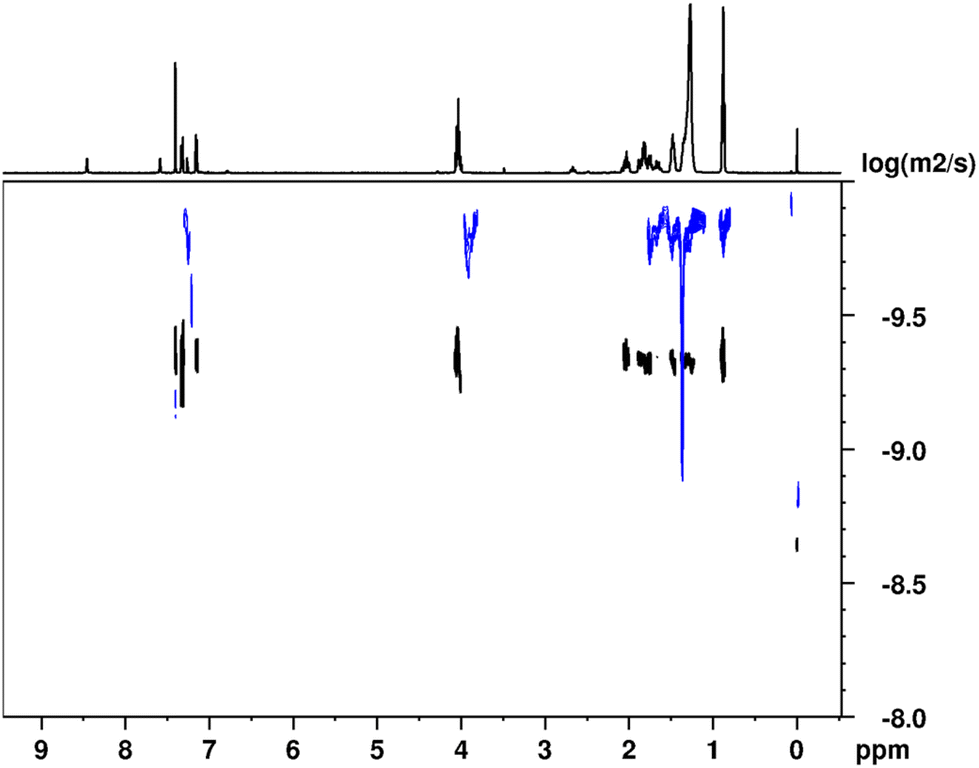 | ||
| Fig. 4 1H DOSY NMR spectra at 298 K of (Z)-4 in CDCl3 (bottom trace in black) and in C6D12 (top trace in blue). The projection corresponds to the 1H-NMR spectrum in CDCl3. | ||
Diastereomer (E)-4 is not soluble in pure cyclohexane but it is soluble when adding 10% (v/v) chloroform (cyclohexane–chloroform, 9:1). The aggregation in these two solvents is different since in the latter case it tends to form dimers. There may be a competition between the formation of dimers in chloroform and rosettes in cyclohexane, so only a qualitative approximation of the aggregation was performed (see Fig. S14 in the ESI†).
Interestingly, during some NMR studies of both diastereomers in cyclohexane, we observed the formation of gels. Consequently, we conducted 1H–1H NOESY experiments using a HRMAS probe to investigate the supramolecular arrangement within these gels. A comparison of 1H–1H NOESY spectra in solution and in the gel phase confirmed that the organizational structure observed in solution is maintained in the gel phase (Fig. S15–17 in the ESI†).
This intriguing observation prompted us to examine the gelation capabilities of (Z)-4 and (E)-4 through H-bonding in various nonpolar or apolar solvents, as well as in aprotic or protic solvents. The solvents included dodecane, heptane, cyclohexane, toluene, dioxane, and octan-1-ol. In the gelation test, the solution was heated until the compound was completely dissolved, and the resulting clear solution was then cooled to room temperature. Gel formation was determined using the tube inversion method, which involves verifying whether the solution flows when the tube is turned upside down (Fig. 5a). Hydantoins 4 demonstrated the ability to form gels in a range of nonpolar solvents, such as dodecane, heptane, and cyclohexane, while they remained soluble in toluene, dioxane, and octan-1-ol. In all the cases, gel formation was immediate, resulting in transparent gels that remained stable at room temperature. To obtain information on the structures of these gels, the xerogels obtained in the three solvents at concentrations of 1 and 2% were studied by transmission electron microscopy (TEM) and scanning electron microscopy (SEM) (see Fig. 5b and c and Tables S2 and S3 in section 3.5 in the ESI†). Notably, the lowest critical gelling concentration was observed with cyclohexane, at just 0.5 wt%, indicating that it acted as a supergelator (Fig. 5b and c). TEM images revealed that the xerogels of (Z)-4 and (E)-4 consisted of a network of entangled fibers, of several tens of nanometers, capable of encapsulating the solvent. SEM observations yielded similar findings, confirming the presence of a fibrillar network, consistent with that observed in TEM (see Table S3 in the ESI†). It is worth noting that in both cases, the xerogel tended to collapse into a thin film due to solvent removal during the sample preparation.
 | ||
| Fig. 5 (a) Gels obtained for (Z)-4 (1.0 wt%) in: (i) dodecane, (ii) heptane, and (iii) cyclohexane. (b) SEM image of the xerogel of (Z)-4 in cyclohexane at 0.5 wt% concentration. (c) TEM image of the xerogel of (Z)-4 in cyclohexane at 0.5 wt% concentration. | ||
To gain a more profound understanding of the molecular architecture within the fibers, we conducted X-ray diffraction (XRD) experiments (Fig. S18 in the ESI†). The XRD patterns obtained from the organogels displayed two sharp maxima in the small-angle region with d-spacings in the ratio of 1:1√3 arising from the (1 0 0) and (1 1 0) reflections of a two-dimensional hexagonal lattice in a columnar hexagonal arrangement. The calculated lattice parameter (a) of the organogels prepared in cyclohexane is 48.1 Å. These materials also displayed diffuse scattering in the high-angle region (around 4.4 Å) that corresponds to the aliphatic chains with conformational disorder and the solvent. Remarkably, these XRD patterns closely resemble those of the liquid crystal phase (vide infra), suggesting that the supramolecular arrangement along the fibers of the organogels corresponds to a columnar mesomorphic-like organization.
Liquid crystal properties
The liquid crystal properties of the two diastereomers, (Z)-4 and (E)-4, were investigated using polarized light optical microscopy (POM), differential scanning calorimetry (DSC), and X-ray diffraction (XRD). Both compounds exhibited liquid crystal behavior, with distinct thermal characteristics depending on the diastereomer, as summarized in Table 1. In the DSC experiments, after the first scan, three heating–cooling cycles were conducted, and it is noteworthy that the results were perfectly reproducible from the second heating–cooling cycle onwards (Fig. S20 and S21 in the ESI†).|
T
2%
(°C) |
Thermal transitionsb | XRD parametersc | |
|---|---|---|---|
| a Temperature at which 2% mass loss is detected in the thermogravimetric curve. b DSC data of the second heating and cooling scans at a rate of 10 °C min−1. Temperatures (°C) are read at the maximum of the corresponding peaks, and enthalpies (kJ mol−1) are given in brackets. I: isotropic liquid, Colh: hexagonal columnar mesophase, Cr: crystal. c a: lattice constant of the Colh phase (Å), h: estimated mean stacking distance (Å), Z: molecules per disc. | |||
| (Z)-4 | 312 | Colh 112 (4.3) I | a = 48.4 Å |
| I 77 (5.0) Colh | h = 4.1 Å | ||
| Z = 6 | |||
| (E)-4 | 285 | Cr1 51 (2.8) Cr2 122 (38.5) Colh 146 (0.9) I | a = 49.5 Å |
| I 142 (1.0) Colh 74 (33.2) Cr2 15 (3.4) Cr1 | h = 3.9 Å | ||
| Z = 6 | |||
The DSC traces of (Z)-4 exhibited a single peak corresponding to the isotropization temperature (Fig. 6a). This transition was represented by as a complex and broad peak, which appeared to result from the overlap of two distinct transitions and was likely associated with a mesophase-to-mesophase transformation. It is worth noting that this transition was not observed through POM analysis (further details below), and as a result, it was treated and integrated as a single transition. In contrast, (E)-4 displayed distinct thermal characteristics. It exhibited a notably higher isotropization temperature compared to (Z)-4. Furthermore, (E)-4 demonstrated a higher propensity to crystallize and exhibited a crystal-to-crystal transition at lower temperatures. The thermal properties were also studied by POM (Fig. 6b). (Z)-4 appeared as a waxy solid at room temperature. Following the first heating–cooling cycle, it exhibited a grained texture, characterized by a mosaic texture with small domains. On the other hand, (E)-4 initially appeared as a waxy substance containing microcrystals and displayed higher birefringence than (Z)-4. After the first heating–cooling cycle, it exhibited a well-defined schlieren mosaic texture.
 | ||
| Fig. 6 (a) DSC traces corresponding to the second heating (red) and cooling (blue) scan of hydantoin diastereomers (10 °C min−1). (b) POM microphotographs observed in the cooling process. (c) Room temperature XRD pattern of (E)-4 in the Colh mesophase. | ||
The absolute assignment of the liquid crystal phases was performed by powder X ray diffraction (XRD). Each sample was heated up to the isotropic liquid and then cooled down to room temperature. The two compounds showed similar X ray patterns with a broad, diffuse halo in the high angle region that corresponds to an average distance of 4.5 Å (Fig. 6c). This halo is usually found in liquid crystal phases and is assigned to the short range correlations between the conformationally disordered hydrocarbon chains. In the low angle region, the patterns exhibited a set of four sharp maxima in a reciprocal ratio of 1:√3:√4:√7. This ratio is characteristic of a well-organized hexagonal columnar phase (Colh) and corresponds to the (1 0), (1 1), (2 0) and (2 1) reflections from the two dimensional hexagonal lattice. The distances measured by XRD are listed in Table S1 of the ESI,† and the calculated lattice constants (a) are displayed in Table 1.
These liquid crystal hydantoins do not possess the traditional discotic shape, typically required for the formation of columnar liquid crystal phases. However, they can engage in the formation of H-bonded aggregates that adopt a discotic shape, enabling further self-assembly into columns. To gain a deeper understanding of the molecular packing within these columns, we conducted density calculations.55–58
Taking into consideration the XRD parameters and assuming a density of approximately 1 g cm−3, a typical value for liquid crystals, we estimated that the number of molecules occupying the cross-sectional area of the column (denoted as Z) was six, giving mean stacking distances ranging from 3.9 to 4.1 Å (see Table 1). Based on these findings, we propose a model for the molecular arrangement within a columnar stratum, wherein six hydantoin molecules self-assemble through H-bonding to form a rosette-like supramolecular disc. This particular arrangement, favored by intermolecular H-bonding, facilitates efficient space filling within the column cross-section. Despite the notable differences in the thermal behavior between the two diastereomers, it is worth highlighting that they exhibited similar lattice parameters, indicative of a similar supramolecular organization.
Theoretical calculations
The experimental results observed in 1H-NMR studies both in the gel and solution states and in the liquid crystal state confirm the supramolecular organization of hydantoin derivatives through H bonds. On the other hand, in the liquid crystal state both diastereomers show a hexagonal columnar organization and X ray studies allow us to estimate an aggregation of six molecules per disk. Despite this, the thermal and thermodynamic properties of both isomers are clearly different, thus the (Z)-4 isomer appears as a waxy material while the (E)-4 isomer shows crystalline polymorphism, and important differences in the clearing points have been observed. To explain this different behavior in relation to the supramolecular organization, quantum chemical calculations were carried out.In order to facilitate the theoretical calculation, the geometry (only of the most stable conformers NHa φe and NHe φe) of diastereomers (Z)-4 and (E)-4 was simplified by replacing the R group (Scheme 1) with a hydrogen atom. The resulting model compounds were named (Z)-4 M and (E)-4 M (Fig. 7). These models were optimized at the B3LYP D3/6 31G** level (see computational details in the ESI†). Hydrogen bonded dimers of optimized (Z)-4 M and (E)-4 M were optimized at the same level, and the calculated structures are shown in Fig. 7. The hydantoin rings of the optimized (E)-4 M dimer are coplanar, but, unexpectedly, those of (Z)-4 M show a dihedral angle of 28 degrees. The distances of hydrogen bonds in the coplanar dimer of (E)-4 M are 1.803 and 1.798 Å, while in the (Z)-4 M dimer, the H-bond distances are 1.881 and 1.782 Å (Fig. S22 in the ESI†). Despite the distance of hydrogen interactions being smaller on average in the (E)-4 M dimer, the interaction energy of the non-planar (Z)-4 M dimer is −77.0 kJ mol−1 and that of the coplanar (E)-4 M dimer is −66.3 kJ mol−1 (Table S4 in the ESI†). These results seem to suggest that the (Z)-4 M dimer is supported by two hydrogen bonds and, probably, by other non-covalent interactions that enhance the stability of this non-planar dimer.
 | ||
| Fig. 7 B3LYP-D3/6-31G** optimized structures calculated for (E)-4-M and (Z)-4-M and their dimers (top). Three-dimensional NCI isosurfaces of the intra- and intermolecular interactions of the calculated structures (bottom). Colored surfaces have sign(λ2)r values of [−0.025, 0.025] a.u. (isosurface s = 0.5). | ||
To gain a deeper understanding of these results, the NCI analysis was performed.59Fig. 7 displays the three-dimensional image of this analysis for (E)-4 M, (Z)-4 M and their corresponding dimers (for NCI plots, see Fig. S23 in the ESI†). The first noteworthy result of the NCI analysis of the monomers is that the dispersive interactions (green isosurface in Fig. 7) are established between the hydrogen atoms of the cyclohexane ring and the hydantoin moiety. In fact, these interactions are significantly stronger in the (E)-4 M isomer, where they involved the oxygen atom. The NCI analysis also revealed dispersive interactions between the hydrogen atoms of the cyclohexane ring and the aromatic hydrogens, which are similar in both isomers. As expected, the NCI analysis of the dimers shows the hydrogen bonds as blue disc-shaped isosurfaces, characteristic of highly localized attractive interactions. The analysis of dispersive interactions in the dimers reveals the second remarkable result of the NCI analysis: in the coplanar dimer, (E)-4 M, the hydrogen bonds are the only interactions established between the two molecules, while in the non-planar dimer, (Z)-4 M, in addition to the hydrogen bonds, extended dispersive interactions are established throughout the length of the molecules. In summary, the (Z)-4 M dimer achieves stabilization by adopting a non-planar conformation that, while maintaining the hydrogen bonds, allows for the extension of dispersive interactions. However, the (E)-4 M dimer prefers to maintain the intramolecular dispersive interactions and relies on the hydrogen bonds alone to support its structure. These results justify the differences in the interaction energy between both dimers.
From the XRD data, it has been established that the liquid crystal phases of (Z)-4 and (E)-4 are hexagonal columnar. The columns of these phases are formed by the stacking of complexes consisting of six hydantoin molecules supported, mainly, by intermolecular H bonding. With this information and considering the results of the theoretical calculations, we built up hexamers of both diastereomers, (Z)-4 M and (E)-4 M, combining three optimized dimers. These complexes were optimized at the B3LYP D3/6 31G** level. Fig. 8 shows the structure of the optimized hexamers and Table S4 in the ESI† shows their interaction energies. The optimized (Z)-4 M hexamer exhibits a bowl-like geometry with C6 symmetry, while the optimized (E)-4 M hexamer is a disc-like structure with C3 symmetry. The NCI analysis of the hexamers confirmed that a bowl-like structure allows, in addition to hydrogen bonding, the dispersive interactions between the molecules of (Z)-4 M, while only hydrogen bonds support the coplanar disc-like structure of the (E)-4 M hexamer (Fig. 8). Table S4 in the ESI† shows the total interaction energies of optimized dimers and hexamers. As expected, the interaction energy of the (Z)-4-M hexamer (−444.9 kJ mol−1) is lower than that of the (E)-4-M hexamer (−317.5 kJ mol−1), which confirms more intense intermolecular interactions in the (Z)-4-M hexamer.
 | ||
| Fig. 8 B3LYP-D3/6-31G** optimized structures calculated for (E)-4-M and (Z)-4-M hexamers (top). Three-dimensional NCI plots of the intra- and intermolecular interactions of the calculated structures (down). Colored surfaces have sign(λ2)r values of [−0.025, 0.025] a.u. (isosurface s = 0.5). | ||
The application of the results from the theoretical studies of the (Z)-4-M and (E)-4-M models to the (Z)-4 and (E)-4 isomers provide very valuable information to explain the different properties of both diastereomers. These studies confirm a different structure of the rosette formed in the supramolecular organization of the two isomers. Thus, the bowl-like structure of the (Z)-4 hexamer, although favors the columnar stacking, seems to hinder the non-covalent interactions between the staked bowls. Consequently, a vitrification process is observed in the cooling process, affording a non-crystalline waxy material. In addition to this, after passing into an isotropic liquid this compound exhibits an important hysteresis in the appearance of the mesophase in the cooling process, probably due to the important reorganization of the molecules necessary to form the supramolecular structure. On the other hand, a disk-like planar structure appears as the most stable in the hexameric structure of the minority (E)-4 isomer. This structure favors the interaction between the discs, increasing the π–π interactions and favoring the formation of the crystalline state in the cooling process. Also, in this compound the transition temperatures among different phases are clearly defined in both, the heating and the cooling process, and do not present hysteresis in this last process. Finally, the bowl-like structure of the (Z)-4 M hexamer as opposed to the disc-like geometry of the (E)-4 M hexamer might well be the reason for the slightly-larger stacking distance estimated for the mesophase of (Z)-4 (h = 4.1 Å, Table 1), compared to the value estimated for the disc-like (E)-4 M hexamer (h = 3.9 Å, Table 1).
Conclusions
In conclusion, the successful preparation of a cyclohexane-5-spirohydantoin derivative is reported. This derivative bears a phenyl 3,4,5-tris(dodecyloxy) benzoate terminal group, synthesized through a modification of the Bucherer–Bergs method. During this process, a mixture of two geometrical diastereomers is obtained in a 3:1 ratio, which is separated using silica gel flash chromatography. Both diastereomers have the remarkable capability of forming hexameric supramolecular rosettes through hydrogen bonding interactions. These rosettes further self-organize into columns, exhibiting both liquid crystal and gelation properties. It is noteworthy that both compounds display significantly distinct liquid crystal behaviors, underscoring the profound impact of minor structural variations on the supramolecular organization and, consequently, the bulk properties of the materials. The major isomer behaves as a glassy liquid crystalline material, while the minor one exhibits liquid crystalline behavior with a high propensity to crystallize. The experimental results combined with theoretical studies suggest that the major isomer forms a bowl-like hexameric rosette, facilitating columnar stacking but impeding strong π–π interactions among neighboring rosettes. Conversely, the most favorable hexameric rosette in the minor diastereoisomer exhibits a planar structure that promotes π–π interactions among neighboring rosettes, thereby favoring crystallization of the hexagonal columnar liquid crystal phase.
Author contributions
L. González, I. Marín: investigation, validation. R. Tejedor: data curation, formal analysis. P. Romero, J. Barberá: formal analysis, validation. A. Concellón: formal analysis, writing – original draft, writing – review & editing. S. Uriel: supervision, writing – original draft. J.L. Serrano: supervision, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the MINECO–FEDER funds PGC2018-097583-B-I00, PID2021-122882NB-I00 and PID2021-126132-NB-I00 (MCIN/AEI/10.13039/501100011033/and by “ERDF A way of making Europe”) and Gobierno de Aragón–FEDER (Research Group E47_23R). A. C. acknowledges grant RYC2021-031154-I funded by the MICINN/AEI/FEDER/EU–NextGenerationEU. The authors would like to acknowledge the use of the SAI (UZ), CEQMA (UZ–CSIC) Services and the Laboratorio de Microscopias Avanzadas-LMA (INMA-UZ).References
- K. Y. Kim, J. Noh, K. Nayani and L. N. Abbott, Soft matter from liquid crystals, Soft Matter, 2019, 15(35), 6913–6929 RSC.
- P. Van der Asdonk and P. H. J. Kouwer, Liquid crystal templating as an approach to spatially and temporally organise soft matter, Chem. Soc. Rev., 2017, 46(19), 5935–5949 RSC.
- A. Concellón, D. Fong and T. M. Swager, Complex Liquid Crystal Emulsions for Biosensing, J. Am. Chem. Soc., 2021, 143(24), 9177–9182 CrossRef.
- K. Nayani, Y. Yang, H. Yu, P. Jani, M. Mavrikakis and N. Abbott, Areas of opportunity related to design of chemical and biological sensors based on liquid crystals, Liq. Cryst. Today, 2020, 29(2), 24–35 CrossRef CAS.
- M. Pilz da Cunha, M. G. Debije and A. P. H. J. Schenning, Bioinspired light driven soft robots based on liquid crystal polymers, Chem. Soc. Rev., 2020, 49(18), 6568–6578 RSC.
- T. Kato, J. Uchida, T. Ichikawa and T. Sakamoto, Functional Liquid Crystals towards the Next Generation of Materials, Angew. Chem., Int. Ed., 2018, 57(16), 4355–4371 CrossRef CAS.
- A. Concellón, M. Marcos, P. Romero, J. L. Serrano, R. Termine and A. Golemme, Not Only Columns: High Hole Mobility in a Discotic Nematic Mesophase Formed by Metal Containing Porphyrin Core Dendrimers, Angew. Chem., Int. Ed., 2017, 56(5), 1259–1263 CrossRef PubMed.
- T. Kato, M. Yoshio, T. Ichikawa, B. Soberats, H. Ohno and M. Funahashi, Transport of ions and electrons in nanostructured liquid crystals, Nat. Rev. Mater., 2017, 2, 17001 CrossRef.
- J. Zheng, A. Suwardi, C. J. E. Wong, X. J. Loh and Z. Li, Halogen Bonding Regulated Functional Nanomaterials, Nanoscale Adv., 2021, 3(22), 6342–6357 RSC.
- H. L. Sun, S. Zhang and V. Percec, From structure to function via complex supramolecular dendrimer systems, Chem. Soc. Rev., 2015, 44(12), 3900–3923 RSC.
- T. Kato, N. Mizoshita and K. Kishimoto, Functional Liquid Crystalline Assemblies: Self Organized Soft Materials, Angew. Chem., Int. Ed., 2006, 45(1), 38–68 CrossRef CAS.
- U. Kumar, T. Kato and J. M. J. Fréchet, Use of intermolecular hydrogen bonding for the induction of liquid crystallinity in the side chain of polysiloxanes, J. Am. Chem. Soc., 1992, 114(17), 6630–6639 CrossRef CAS.
- T. Kato and J. M. J. Fréchet, A new approach to mesophase stabilization through hydrogen bonding molecular interactions in binary mixtures, J. Am. Chem. Soc., 1989, 111(22), 8533–8534 CrossRef CAS.
- C. Fouquey, J. M. Lehn and A. M. Levelut, Molecular recognition directed self assembly of supramolecular liquid crystalline polymers from complementary chiral components, Adv. Mater., 1990, 2(5), 254–257 CrossRef CAS.
- J. Monti, A. Concellón, R. Dong, M. Simmler, A. Münchinger, C. Huck, P. Tegeder, H. Nirschl, M. Wegener, C. O. Osuji and E. Blasco, Two Photon Laser Microprinting of Highly Ordered Nanoporous Materials Based on Hexagonal Columnar Liquid Crystals, ACS Appl. Mater. Interfaces, 2022, 14(29), 33746–33755 CrossRef CAS.
- J. Kloos, N. Joosten, A. Schenning and K. Nijmeijer, Self assembling liquid crystals as building blocks to design nanoporous membranes suitable for molecular separations, J. Membr. Sci., 2021, 620, 118849 CrossRef CAS.
- A. Concellón, T. Liang, A. P. H. J. Schenning, J. L. Serrano, P. Romero and M. Marcos, Proton conductive materials formed by coumarin photocrosslinked ionic liquid crystal dendrimers, J. Mater. Chem. C, 2018, 6(5), 1000–1007 RSC.
- D. J. Broer, C. M. W. Bastiaansen, M. G. Debije and A. P. H. J. Schenning, Functional Organic Materials Based on Polymerized Liquid Crystal Monomers: Supramolecular Hydrogen Bonded Systems, Angew. Chem., Int. Ed., 2012, 51(29), 7102–7109 CrossRef CAS.
- B. Feringán, P. Romero, J. L. Serrano, C. L. Folcia, J. Etxebarria, J. Ortega, R. Termine, A. Golemme, R. Giménez and T. Sierra, H Bonded Donor–Acceptor Units Segregated in Coaxial Columnar Assemblies: Toward High Mobility Ambipolar Organic Semiconductors, J. Am. Chem. Soc., 2016, 138(38), 12511–12518 CrossRef PubMed.
- Y. Yu Wang, H. Wu and J. Fraser Stoddart, Molecular Triangles: A New Class of Macrocycles, Acc. Chem. Res., 2021, 54(8), 2027–2039 CrossRef CAS PubMed.
- M. J. Mayoral, N. Bilbao and D. González-Rodríguez, Hydrogen Bonded Macrocyclic Supramolecular Systems in Solution and on Surfaces, ChemistryOpen, 2016, 5(1), 10 CrossRef CAS.
- A. S. Tayi, A. Kaeser, M. Matsumoto, T. Aida and S. I. Stupp, Supramolecular ferroelectrics, Nat. Chem., 2015, 7(4), 281–294 CrossRef CAS.
- S. Yagai, Y. Goto, X. Lin, T. Karatsu, A. Kitamura, D. Kuzuhara, H. Yamada, Y. Kikkawa, A. Saeki and S. Seki, Self Organization of Hydrogen Bonding Naphthalene Chromophores into J type Nanorings and H type Nanorods: Impact of Regioisomerism, Angew. Chem., Int. Ed., 2012, 51(27), 6643–6647 CrossRef CAS.
- D. Miyajima, F. Araoka, H. Takezoe, J. Kim, K. Kato, M. Takata and T. Aida, Ferroelectric Columnar Liquid Crystal Featuring Confined Polar Groups Within Core–Shell Architecture, Science, 2012, 336(6078), 209–213 CrossRef CAS PubMed.
- D. Miyajima, F. Araoka, H. Takezoe, J. Kim, K. Kato, M. Takata and T. Aida, Columnar Liquid Crystal with a Spontaneous Polarization along the Columnar Axis, J. Am. Chem. Soc., 2010, 132(25), 8530–8531 CrossRef CAS PubMed.
- S. Yagai, S. Kubota, H. Saito, K. Unoike, T. Karatsu, A. Kitamura, A. Ajayaghosh, M. Kanesato and Y. Kikkawa, Reversible Transformation between Rings and Coils in a Dynamic Hydrogen Bonded Self Assembly, J. Am. Chem. Soc., 2009, 131(15), 5408–5410 CrossRef CAS.
- K. E. Maly, D. Dauphin and J. D. Wuest, Self assembly of columnar mesophases from diaminotriazines, J. Mater. Chem., 2006, 16(48), 4695–4700 RSC.
- K. Kanie, M. Nishii, T. Yasuda, T. Taki, S. Ujiie and T. Kato, Self assembly of thermotropic liquid crystalline folic acid derivatives: hydrogen bonded complexes forming layers and columns, J. Mater. Chem., 2001, 11(11), 2875–2886 RSC.
- C. T. Seto and G. M. Whitesides, Molecular self assembly through hydrogen bonding: supramolecular aggregates based on the cyanuric acid melamine lattice, J. Am. Chem. Soc., 1993, 115(3), 905–916 CrossRef CAS.
- S. Takahashi and S. Yagai, Harmonizing Topological Features of Self-Assembled Fibers by Rosette-Mediated Random Supramolecular Copolymerization and Self-Sorting of Monomers by Photo-Cross-Linking, J. Am. Chem. Soc., 2022, 144, 13374–13383 CrossRef CAS PubMed.
- K. Aratsu, R. Takeya, B. R. Pauw, M. J. Hollamby, Y. Kitamoto, N. Shimizu, H. Takagi, R. Haruk, S.-i. Adachi and S. Yagai, Supramolecular copolymerization driven by integrative self-sorting of hydrogen-bonded rosettes, Nat. Commun., 2020, 11, 1623 CrossRef CAS PubMed.
- B. Adhikari, X. Lin, M. Yamauchi, H. Ouchi, K. Aratsu and S. Yagai, Hydrogen bonded rosettes comprising π conjugated systems as building blocks for functional one dimensional assemblies, Chem. Commun., 2017, 53(70), 9663–9683 RSC.
- B. Adhikari, X. Lin, M. Yamauchi, H. Ouchi, K. Aratsua and S. Yagai, Hydrogen-bonded rosettes comprising p-cnjugated systems as building blocks for functional one-dimensional assemblies, Chem. Commun., 2017, 53, 9663–9683 RSC.
- M. Crego-Calama, D. N. Reinhoudt, J. J. García López and J. M. C. A. Kerckhoffs, Nanostructured Hydrogen Bonded Rosette Assemblies Self Assembly and Self Organization, in Nanoscale Assembly: Chemical Techniques, ed. W. T. S. Huck, Springer US, Boston, MA, 2005, pp. 65–78 Search PubMed.
- B. Feringán, C. L. Folcia, R. Termine, A. Golemme, J. M. Granadino-Roldán, A. Navarro, J. L. Serrano, R. Giménez and T. Sierra, Inspecting the Electronic Architecture and Semiconducting Properties of a Rosette Like Supramolecular Columnar Liquid Crystal, Chem. – Eur. J., 2018, 24(66), 17459–17463 CrossRef PubMed.
- F. Vera, J. Barberá, P. Romero, J. L. Serrano, M. B. Ros and T. Sierra, Orthogonal Action of Noncovalent Interactions for Photoresponsive Chiral Columnar Assemblies, Angew. Chem., Int. Ed., 2010, 49(29), 4910–4914 CrossRef CAS PubMed.
- S. Cho, S. H. Kim and D. Shin, Recent applications of hydantoin and thiohydantoin in medicinal chemistry, Eur. J. Med. Chem., 2019, 164, 517–545 CrossRef CAS.
- L. Konnert, F. Lamaty, J. Martinez and E. Colacino, Recent Advances in the Synthesis of Hydantoins: The State of the Art of a Valuable Scaffold, Chem. Rev., 2017, 117(23), 13757–13809 CrossRef CAS.
- L. Vatannavaz, S. J. Sabounchei, A. Sedghi, R. Karamian, S. H. M. Farida and N. Rahmani, New Nickel, Palladium and Platinum Complexes of Hydantoin Derivative: Synthesis, Characterization, Theoretical Study and Biological Activity, Polyhedron, 2020, 181, 114478 CrossRef CAS.
- S. J. Sabounchei, P. Shahriary, S. Salehzadeh, Y. Gholiee and H. R. Khavasi, Mercury(II) Complexes with 5 Methyl 5 (4 Pyridyl) 2,4 Imidazolidenedione: Synthesis, Structural Characterization, and Theoretical Studies, J. Mol. Struct., 2013, 1051, 15–22 CrossRef CAS.
- M. Puszyńska-Tuszkanow, T. Grabowski, M. Daszkiewicz, J. Wietrzyk, B. Filip, G. MacIejewska and M. Cieślak-Golonka, Silver(I) Complexes with Hydantoins and Allantoin: Synthesis, Crystal and Molecular Structure, Cytotoxicity and Pharmacokinetics, J. Inorg. Biochem., 2011, 105(1), 17–22 CrossRef.
- K. Oyaizu, Y. Ohtani, A. Shiozawa, K. Sugawara, T. Saito and M. Yuasa, Highly Stable Gold(III) Complex with a Hydantoin Ligand in Alkaline Media, Inorg. Chem., 2005, 44(20), 6915–6917 CrossRef CAS PubMed.
- D. Maddileti and A. Nangia, Polymorphism in Anti Hyperammonemic Agent N Carbamoyl l Glutamic Acid, CrystEngComm, 2015, 17(28), 5252–5265 RSC.
- V. Gerhardt, M. Tutughamiarso and M. Bolte, Conformational studies of hydantoin 5 acetic acid and orotic acid, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2012, 68(2), o92–o98 CrossRef CAS PubMed.
- B. Chattopadhyay, A. K. Mukherjee, N. Narendra, H. P. Hemantha, V. V. Sureshbabu, M. Helliwell and M. Mukherjee, Supramolecular Architectures in 5,5′ Substituted Hydantoins: Crystal Structures and Hirshfeld Surface Analyses, Cryst. Growth Des., 2010, 10, 4476–4484 CrossRef CAS.
- F. L. Yu, C. H. Schwalbe and D. J. Watkin, Hydantoin and hydrogen bonding patterns in hydantoin derivatives, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60(10), o714–o717 CrossRef.
- S. Graus, S. Uriel and J. L. Serrano, Supramolecular Hydrogen Bonding Patterns in 4′ Substituted Cyclohexane 5 Spirohydantoin, CrystEngComm, 2012, 14(10), 3759–3766 RSC.
- S. Graus, D. Casabona, S. Uriel, C. Cativiela and J. L. Serrano, Supramolecular Arrangements Based on Cyclohexane 5 Spirohydantoin Derivatives, CrystEngComm, 2010, 12, 3132–3137 RSC.
- H. T. Bucherer and H. Fischbeck, Hexahydrodiphenylamine and its derivatives, J. Prakt. Chem., 1934, 140, 69–89 CAS.
- M. Kalník, P. Gabko, M. Bella and M. Koóš, The Bucherer–Bergs Multicomponent Synthesis of Hydantoins—Excellence in Simplicity, Molecules, 2021, 26(13), 4024 CrossRef PubMed.
- J. T. Edward and C. Jitrangsri, Stereochemistry of the Bucherer–Bergs and Strecker Reactions of 4 Tert Butylcyclohexanone, Can. J. Chem., 1975, 53(22), 3339–3350 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, The Cambridge Structural Database, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72(2), 171–179 CrossRef CAS PubMed.
- Y. Cohen, L. Avram and L. Frish, Diffusion NMR Spectroscopy in Supramolecular and Combinatorial Chemistry: An Old Parameter—New Insights, Angew. Chem., Int. Ed., 2005, 44(4), 520–554 CrossRef CAS PubMed.
- A. Pérez, D. de Saá, A. Ballesteros, J. L. Serrano, T. Sierra and P. Romero, NMR Spectroscopic Study of the Self Aggregation of 3 Hexen 1,5 diyne Derivatives, Chem. – Eur. J., 2013, 19(31), 10271–10279 CrossRef.
- V. Iguarbe, A. Concellón, R. Termine, A. Golemme, J. Barberá and J. L. Serrano, Making Coaxial Wires Out of Janus Dendrimers for Efficient Charge Transport, ACS Macro Lett., 2018, 1138–1143 CrossRef CAS.
- B. Feringán, P. Romero, J. L. Serrano, R. Giménez and T. Sierra, Supramolecular Columnar Liquid Crystals Formed by Hydrogen Bonding between a Clicked Star Shaped s Triazine and Benzoic Acids, Chem. – Eur. J., 2015, 21(24), 8859–8866 CrossRef PubMed.
- A. Pérez, J. L. Serrano, T. Sierra, A. Ballesteros, D. de Saá and J. Barluenga, Control of Self Assembly of a 3 Hexen 1,5 diyne Derivative: Toward Soft Materials with an Aggregation Induced Enhancement in Emission, J. Am. Chem. Soc., 2011, 133(21), 8110–8113 CrossRef PubMed.
- Y. Li, A. Concellón, C. J. Lin, N. A. Romero, S. Lin and T. M. Swager, Thiophene fused polyaromatics: synthesis, columnar liquid crystal, fluorescence and electrochemical properties, Chem. Sci., 2020, 11(18), 4695–4701 RSC.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, Revealing Noncovalent Interactions, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3qo01832f |
| This journal is © the Partner Organisations 2024 |