
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclododecene isomeric separation by (supported) rhodium(I)-catalysed selective dehydrogenative borylation reaction†
Amravati S.
Singh
and
Antonio
Leyva-Pérez
 *
*
Instituto de Tecnología Química (UPV-CSIC), Universidad Politècnica de València-Consejo Superior de Investigaciones Científicas, Avda. de los Naranjos s/n, 46022 Valencia, Spain. E-mail: anleyva@itq.upv.es
First published on 18th December 2023
Abstract
Cyclododecene, commercially available as a cis/trans mixture, reacts with dipinacolborane (BPin)2 only through the cis isomer, during the Rh(I)–XantPhos-catalyzed dehydrogenative monoborylation reaction, leaving the starting trans-cyclododecene untouched. In this way, both diastereoisomers can now be easily separated. The catalytic Rh(I)-phosphine complex progressively degrades under the reaction conditions; thus we also present here a cheap, ligandless Rh(I)-supported zeolite NaY catalyst, equally selective for the dehydrogenative monoborylation reaction. These results provide a new methodology for macrocyclic alkene diastereoisomeric separation.
1 Introduction
Simple alkene macrocycles, with application in the fragrance and pharma industries, for example,1 are usually obtained as cis/trans mixtures due to the similar size and energetic values of the isomers, and their separation becomes extremely sluggish. Dozens of examples can be found in the literature, particularly after a Wittig reaction,2 after elimination,3 and in particular after ring-closing alkene metathesis reactions4 (Fig. 1A). Indeed, some commercially available alkene macrocycles are directly sold as cis/trans mixtures, and a prominent example is one of the simpler alkene macrocycles, i.e. cyclododecene 1.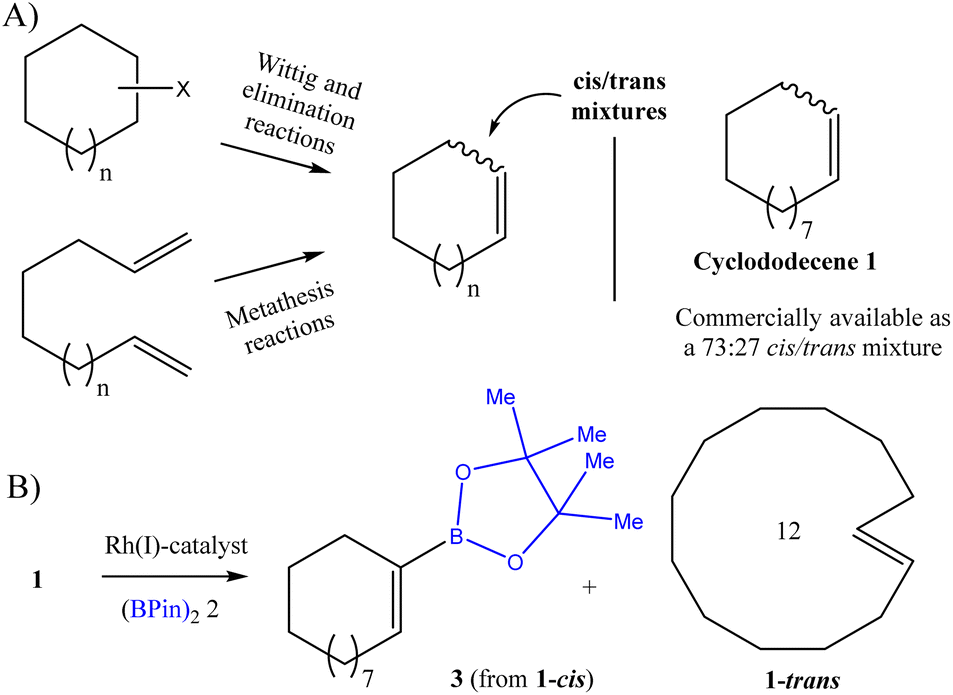 | ||
| Fig. 1 (A) Synthesis of alkene macrocycles to generate cis/trans mixtures, and cyclododecene 1 as an example. (B) Rh(I)-catalyzed dehydrogenative monoborylation reaction of 1, to give 3 from 1-cis, and recover 1-trans. | ||
We have carried out molecular mechanics calculations (MM2) for the cis and trans isomers of cyclododecene (1-cis and 1-trans, respectively) in vacuum (Fig. S1, see ESI†), and the results show that the free energy [19.1(9) and 18.8(4) kcal mol−1, respectively] and the kinetic radii (3.1 Å in both cases) for both alkene diastereoisomers are very similar, which explains their commercialization as an isomeric mixture. However, the sterics around the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond are significantly different, which suggests that a particular synthetic methodology may allow one of the alkene diastereoisomers to react in the presence of the other. This selective reaction would be of interest not only for the inherent value of the new diastereoselective transformation but also, in practical terms, for the ultimate separation of both diastereomers.
C double bond are significantly different, which suggests that a particular synthetic methodology may allow one of the alkene diastereoisomers to react in the presence of the other. This selective reaction would be of interest not only for the inherent value of the new diastereoselective transformation but also, in practical terms, for the ultimate separation of both diastereomers.
The dehydrogenative monoborylation reaction is a catalytic synthetic methodology based on C–H activation of the alkene group, which allows the CC double bond to be preserved after replacing one of the alkene H atoms by a boron-substituted group (Fig. 1B).5 Thus this reaction is very sensitive to the CC double bond configuration, so it may differentiate the cis/trans alkene diastereoisomers during reaction. Among the different catalysts employed for this reaction, which include the metals Ru,6 Rh,6b,7 Co,8 Pd9 and Pt,10 and recently non-metal catalysts as well,11 only Rh(I) seems to have the capacity to perform alkene functionalization without associated diborylation and hydrogenation reactions, without losing any starting alkene.7b While other alkenes could be used as a sacrificial hydrogenable agent here,6b a reaction system based on the Rh(I)–XantPhos complex as a catalyst and dipinacolborane (BPin)22 as a borylating agent was chosen for our purposes, since no sacrificial alkene is required, which simplifies the purification protocol.7b The Rh(I)–XantPhos catalytic system has, to our knowledge, been tested in all-cis (smaller) cycloalkenes,7b but not yet in cis/trans mixtures. Considering the higher steric hindrance around the CC double bond in 1-trans compared to 1-cis, we envisioned that the latter will react preferentially, to give monoborylated vinyl borane product 3 as the main product, which can then be further converted if required.12 In contrast, 1-trans will remain as unreacted material, ready to be separated from the reaction mixture (Fig. 1B),13 when given low amounts of the corresponding monoborylated product 4.
2 Experimental section
2.1 General preparation of the Rh-supported zeolites
Y zeolites, in Na+ form, are commercially available. First, 500 mg of this zeolite was dried in vacuum at 300 °C for 3 h. Then, impregnation of 0.5 and 5 wt% of Rh precursor in 0.5 mL of DCM was carried out, drop by drop at room temperature. Then this material was dried in vacuum at room temperature for 2 h. These catalysts were named 0.5%RhL@NaY and 5%RhL@NaY, respectively.2.2 Reaction procedure for the dehydrogenative monoborylation reaction of 1 with Rh(I) XantPhos complex catalyst
Reagent 1 (cis/trans 73![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :27 cyclododecene mixture, 0.3 mmol), (BPin)2 (2 equiv.) in toluene (0.5 M) and [Rh(COD)Cl]2/XantPhos (3/6 mol%) were introduced into a sealed vial with a magnetic stirrer. The mixture was allowed to react for 7 h at 100 °C. GC samples were prepared by taking out 50 μL of the reaction mixture and introducing them into a vial with 0.5 mL of toluene and N-dodecane (11 μL, 0.05 mmol) as an external standard. For the isolation of 3, a 1-gram synthesis was performed, and the product was isolated by column chromatography. The same procedure was followed for 4.15 The characterization of both vinyl boronates can be found in the ESI (Fig. S3 and S4,† respectively).
:27 cyclododecene mixture, 0.3 mmol), (BPin)2 (2 equiv.) in toluene (0.5 M) and [Rh(COD)Cl]2/XantPhos (3/6 mol%) were introduced into a sealed vial with a magnetic stirrer. The mixture was allowed to react for 7 h at 100 °C. GC samples were prepared by taking out 50 μL of the reaction mixture and introducing them into a vial with 0.5 mL of toluene and N-dodecane (11 μL, 0.05 mmol) as an external standard. For the isolation of 3, a 1-gram synthesis was performed, and the product was isolated by column chromatography. The same procedure was followed for 4.15 The characterization of both vinyl boronates can be found in the ESI (Fig. S3 and S4,† respectively).
2.3 Reaction procedure for the dehydrogenative monoborylation reaction of 1 with the Rh(I)-supported zeolite
Reagent 1 (cis/trans 73:27 cyclododecene mixture, 0.3 mmol), (BPin)2 (2 equiv.) in toluene (0.5 M) and the solid catalyst (90 mg, unless otherwise indicated) were introduced into a sealed vial with a magnetic stirrer. The mixture was allowed to react and analyzed as above, taking out the samples for the GC analysis from the supernatant.
3 Results and discussion
3.1 Catalytic results with a soluble Rh complex
Table 1 shows the catalytic results obtained for the dehydrogenative borylation reaction of commercial cyclododecene 1 (cis/trans 73:27, Fig. S2†) with 2 equivalents of (BPin)22 in toluene (0.5 M) at 100 °C for 7 h. Reactions were followed by gas chromatography (GC), and products were characterized by a combination of mass spectrometry (GC-MS) and 1H, 13C, 11B, distortionless enhancement by polarization transfer (DEPT) and bidimensional 1H–1H nuclear magnetic resonance (NMR), and also compared with the available literature.14 Gratifyingly, the catalytic combination of [Rh(COD)Cl]2 and XantPhos, to generate in situ the corresponding organometallic complex, selectively produces the monoborylated vinyl borane product 3 from 1-cis (Fig. S3†) in the presence of 1-trans, and the latter barely reacts with vinyl borane 4 (entries 1–3, Fig. S4†). When 3 mol% of Rh and 6 mol% of XantPhos are used, complete conversion of 1-cis is achieved, to give 3 in 38% isolated yield with respect to all starting 1 (76% respect to 1-cis). The use of the ligand in excess with respect to metal has been previously proven to be necessary for the selective reaction7b and further optimization was not undertaken here. Indeed, diborylated product 6 was not observed with the 1:2 metal-to-ligand molar ratio employed. It should be noted here that the amount of 1-trans after reaction is higher than in the starting 1 mixture, since the isomerization reaction of 1-cis to 1-trans also occurs to some extent (see below).
:27) cyclododecene mixture 1 with (BPin)22 in toluene (0.5 M) at 100 °C for 7 h. COD: 1,5-cyclooctadiene
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Rh/XantPhos (mol%) | 1-cis (%) | 1-trans (%) |
3a (%) |
4a (%) |
6a (%) |
| a GC yields, double-checked by 1H-NMR, and referred to the starting 1 mixture. Within brackets, isolated yields in 1-gram experiments. b 1 equivalent (BPin)22. c [Rh(COD)2]NTf2 instead of [Rh(COD)Cl]2. d 1.5 equivalents of norbornene were also added. e 3 mol% Rh; RhCl3, Rh2O3, Rh nanoparticles on carbon or on alumina did not give any conversion. | ||||||
| 1 | 1/2 | 46 | 37 | 15 | 2 | 0 |
| 2 | 2/3 | 31 | 43 | 23 | 3 | 0 |
| 3 | 3/6 | <1 | 50 | 42 [38] | 8 | 0 |
| 4b | 3/6 | 56 | 33 | 10 | 1 | 0 |
| 5 | 3/0 | <1 | 41 | 31 | 5 | 22 |
| 6c | 3 (NTf2)/3 | 32 | 50 | 16 | 2 | 0 |
| 7c | 3 (NTf2)/0 | 28 | 50 | 19 | 3 | 0 |
| 8d | 3/6 | 27 | 48 | 20 | 3 | 0 |
| 9e | 0.5%Rh@NaY | 42 | 31 | 1 | 4 | 22 |
| 10e | 5%Rh@NaY | 15 | 40 | 5 | 10 | 30 |

With the above results in hand, we could now isolate 1-trans from the reaction mixture (i.e. with column chromatography or distillation). Not only that, 3 could be converted back to pure 1-cis by a simple acid treatment.15 In this way, the cyclododecene mixture 1 could be conveniently separated into both diastereoisomers 1-cis and 1-trans. In order to further facilitate the isolation procedure and decrease amounts of by-product, the use of 1 equivalent of (BPin)22 was attempted, but it led to much lower conversion and yield (entry 4), which suggests that HBPin 5 formed as a by-product during the reaction does not react further to produce borylated products. Indeed, the independent use of HBPin 5 as a reactant did not give product 3 under our reaction conditions, and nor did the use of other monoborylated reagents (Fig. S5†).
The lack of ligand XantPhos in the reaction does not preclude the formation of 3, but the prevalence of 1-trans produces diborylated product 6 in significant yield (entry 5).7a Indeed, the reaction rate doubles in the absence of the XantPhos ligand, according to kinetic experiments (Fig. S6 and S7† left), and the reaction order calculated for Rh under these reaction conditions is one, regardless of the presence or not of the phosphine (Fig. S7,† right). In both cases, rapid isomerization of 1-cis to 1-trans takes place, to give a nearly isomolecular starting alkene mixture, which is in good agreement with the ability of group VIII metals to isomerize alkenes to the more thermodynamically stable composition.16 A kinetic study on the isomerization of the starting cyclododecene 1 mixture (73% cis, 27% trans), without reagent 2 in the medium, was then performed, with the homogeneous Rh catalyst in the presence or not of the XantPhos ligand. The results (Fig. S8†) showed that the complex with XantPhos does not modify the starting composition of 1 but, in striking contrast, complex [Rh(COD)Cl]2, alone, rapidly isomerizes 1 to the opposite composition (∼25% cis and 75% trans), which remains stable over time. This result suggests that XantPhos is not strictly necessary to form the catalytically active species during the borylation reaction, since a cis to trans isomerization reaction is observed. When [Rh(COD)2]NTf2 was used instead of [Rh(COD)Cl]2 as the Rh catalyst, complete selectivity for monoborylated products 3 was observed with and without phosphine (entries 6 and 7). These results together suggest that the XantPhos ligand is acting more as a stabilizer than as a necessary electron modifier, and that the chloride ligands may also be responsible for the diborylation reaction. These assumptions fit well with the reported literature, where chelating diphosphines lead to better selectivity for the dehydrogenative monoborylation reaction, but not necessarily to much higher reactivity.7 In other words, the phosphine could theoretically be suppressed under the right conditions; thus the catalytic system can be simplified.
In order to further check the above hypothesis, the borylation reaction with stoichiometric amounts of [Rh(COD)Cl]2 and XantPhos (1:2 metal-to-ligand molar ratio) was followed by in situ31P, 1H and 11B NMR in toluene-d8. Fig. 2 shows the 31P NMR spectra after sequential addition of the catalyst and reactants. The original 31P NMR signal for XantPhos at −18 ppm transforms in ca. 50% to a new signal centered at 2 ppm, constituted by a doublet with JRh–P = 91 Hz, a coupling constant value in good agreement with the expected value for the tri-coordinated octahedral Rh(I)–XantPhos complex.17 The fact that 50% of free XantPhos ligand remains in solution strongly supports the diphosphine complex not being formed and that an excess of ligand is added to enhance the formation of the monophosphine complex and not to generate a six-coordinated Rh–P complex.18 Accordingly, the corresponding in situ1H NMR spectra show that the COD ligand mostly remains coordinated to Rh after addition of XantPhos and the XantPhos methyl groups appear as a singlet signal, accounting for six equivalent H atoms (Fig. S9†). Thus, we can conclude that the resulting complex must respond to a symmetric structure of formula Rh(COD)(XantPhos)Cl.
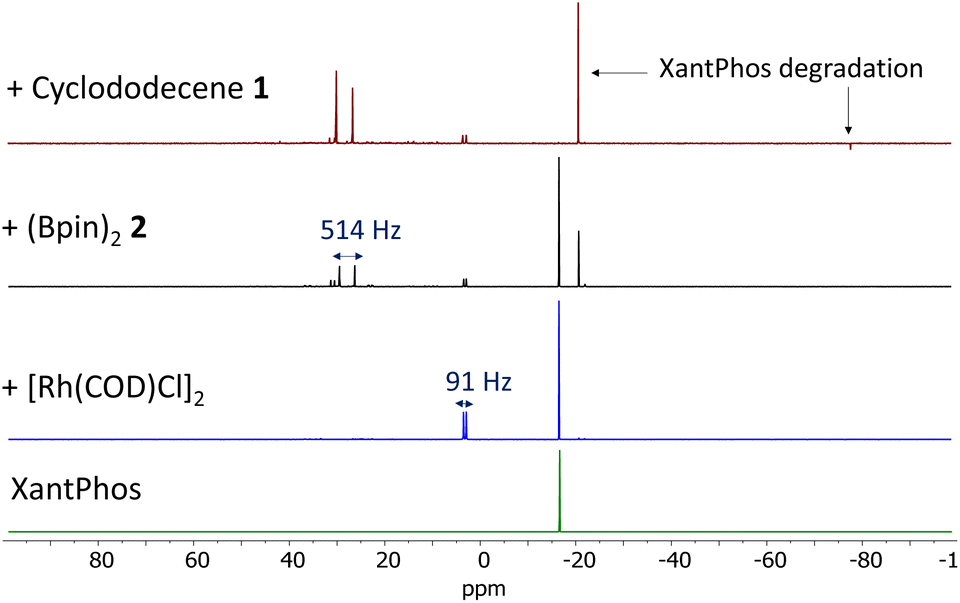 | ||
| Fig. 2
In situ
31P NMR measurements for the dehydrogenative borylation reaction of a cis/trans (73:27) cyclododecene mixture 1 with 2 equivalents of (BPin)22 in toluene-d8 (0.5 M) at room temperature, in the presence of equimolecular amounts of [Rh(COD)Cl]2 and XantPhos (1:2 molar ratio). Reactants were sequentially added from bottom to top. | ||
The addition of (BPin)22 to the Rh(COD)(XantPhos)Cl + free XantPhos mixture (ca. 1:1) results in the formation of two new major 31P NMR signals at −22 and ∼30 ppm. The downshifted signal at ∼30 ppm could be assigned to a doublet with JRh–P = 514 Hz. However, this very high JRh–P constant may only be ascribed to either the coordination of an extremely structural trans ligand, perhaps BPin, the protonation of the phosphine or the formation of direct P–P bonds.19 However, the last two hypotheses must be discarded since additional JH–P or JP–P coupling constants are not observed in either the 31P or the 1H NMR spectra (Fig. 2 and S9†).20 In contrast, the in situ11B NMR study shows the plausible formation of an Rh–B complex by the appearance of a new 11B NMR signal at around 25 ppm, which suggests the formation of the expected Rh(III)–B oxidative addition species.21 Notice here that the JB–P coupling constant could be too small to be observed. Thus, we assign the 31P NMR signals at ∼30 ppm to the Rh(III) complex after oxidative addition of 2, although an alternative assignment of this doublet to independent oxidized species of XantPhos must not be discarded, since the intensity of these signals increases asymmetrically after the addition of alkene 1 (Fig. 2, top red line). In any case, the addition of 1 definitively consumes the original XantPhos 31P NMR signal at −18 ppm, leaving just a residual amount of the Rh(COD)(XantPhos)Cl catalytic species. In other words, the XantPhos ligand is completely degraded under the reaction conditions, according not only to the 31P NMR but also to the corresponding 1H NMR study (Fig. S9†). Besides, the borylation reaction of the COD ligand is also observed by 1H NMR (Fig. S9 and S10†); thus none of the organic ligands in the original complex, i.e. XantPhos and COD, resists the dehydrogenative borylation reaction conditions. A competitive reaction test using norbornene as an additional alkene substrate6b confirms the higher reactivity of tensioned secondary cyclic alkenes such as COD in comparison with 1 (entry 8 in Table 1), since the conversion of 1 decreases significantly.
The reaction mixture was also studied with ultra-performance liquid chromatography-high-resolution mass spectrometry after direct infusion (UPLC-HRMS, Fig. S11†). A signal was found corresponding exactly (just 1 ppm of difference) to the mass of the protonated Rh(cyclooctene)(XantPhos) (3 or 4) complex (but may also be assigned to a mixture of 1 and BPin instead of the product), but not the peaks corresponding to the Rh complex with chloride. Indeed, a new NMR study with NTf2 as counteranion instead of Cl shows that the reaction also progresses, in accordance with the results in Table 1. These results confirm that Cl is not absolutely necessary for the catalytic activity of the Rh complex.
Fig. 3 shows a plausible reaction pathway for the Rh complex, which illustrates that the role of the tri-coordinated XantPhos would be to isolate the Rh site and avoid diborylation reactions, probably by steric hindrance. The reaction starts with the oxidation addition of 2 to the Rh(I) site. COD and XantPhos can both be partially removed at this stage; thus cis/trans isomerization may occur without the participation of the XantPhos ligand. Then, the incorporation of 1 into the complex occurs, after ligand removal (probably Cl), and then the catalyst is ready to form product 3. The combined catalytic, kinetic, NMR and UPLC-HRMS studies show that neither the XantPhos, COD nor Cl ligands are strictly necessary to accomplish the selective synthesis of 3.
 | ||
| Fig. 3 Reaction pathway for the Rh(I)–XantPhos catalyst in solution during the dehydrogenative borylation reaction of 1 with 2, highlighting the degradation pathways for the ligands. | ||
3.2 Catalytic results with Rh-supported zeolites
With the above data in hand, we envisioned that the incorporation of [Rh(COD)Cl]2 in a solid catalyst, particularly a solid able to isolate, stabilize and generate steric hindrance around the Rh catalytic site, would provide an efficient and selective solid catalyst for the dehydrogenative monoborylation reaction of cyclododecene 1. In the first approach we chose commercially available zeolite NaY, since it is a crystalline tridimensional microporous aluminosilicate which easily coordinates metal cations within channels and cavities of 7.4 Å and 12 Å diameter size, respectively, large enough to impart size selectivity while letting reactants and products diffuse (see Fig. S1†).22 Besides, zeolite NaY contains a ∼5 wt% Na+ cations within its framework, able to react with the Cl− anions of the complex and remove the unnecessary and unselective halide.23The incorporation of [Rh(COD)Cl]2 in zeolite NaY was carried out in two different loadings (0.5 and 5 wt%) by the wetness impregnation method [0.5%RhL@NaY and 5%RhL@NaY respectively, see Experimental section], to avoid any loss of precious metal, and the resulting solid was characterized by inductively coupled plasma-atomic emission spectroscopy (ICP-AES), powder X-ray diffraction (PXRD), Brunauer–Emmett–Teller surface area (BET), diffuse-reflectance ultraviolet visible spectrophotometry (DR-UVvis), Fourier transformed infrared spectroscopy (FT-IR), X-ray energy dispersive spectroscopy (EDS) coupled to high-angle-annular-dark field scanning transmission electron microscopy (HAADF-STEM), X-ray photoelectron spectrometry (XPS) and field emission scanning electron microscopy (FE-SEM), and also by 29Si and 27Al solid-state magic angle spinning nuclear magnetic resonance (29Si and 27Al SS-MAS NMR). ICP-AES analyses confirmed the incorporation of Rh in the solid (5%RhL@NaY: 2.9 wt% of pure Rh) and the PXRD technique confirmed the integrity of the faujasite zeolite crystallinity after Rh incorporation (Fig. S12†). Moreover, the latter also showed that Rh nanoparticles are not formed during the incorporation process, since no peak corresponding to nanocrystals could be found. Accordingly, the BET surface area calculated for the higher loaded solid, i.e. 5%RhL@NaY, is 559 m2 g−1, in the range expected for an atomically dispersed zeolite NaY (Fig. S13†), and the corresponding DR-UVvis spectrum does not show any absorbing Rh cluster or nanoparticles (Fig. S14†). FT-IR measurements show the signals corresponding to the COD ligand of the Rh complex after the impregnation process (Fig. S15†).
EDS-HAADF-STEM images of the outer surface of the 5%RhL@NaY material (Fig. S16†) show that Rh is homogeneously distributed across the structured microporous zeolite, particularly visible in the elemental mappings. However, some nanoparticles are observable, and the corresponding zeolite EDX spectrum gives an elemental Rh composition of about 3.9 wt%, higher than in the whole zeolite framework (2.9 wt%, Fig. S17†). These results indicate that a higher proportion of the Rh complex is deposited on the outer surface of the zeolite during the wet impregnation procedure, and that the high voltage microscopy electron beam incudes Rh agglomeration during measurement. It is noteworthy here that the outer surface is about one order of magnitude (10 times) lower that the inner surface of the zeolite; thus the Rh distribution is good and it must be better inside the zeolite. The corresponding field emission scanning electron microscopy (FESEM) images of 5%RhL@NaY display the presence of crystals with a slightly hexagonal shape, and a porous structure suitable for the diffusion of reactants/products during heterogeneous catalysis (Fig. S18†).
The oxidation states of Al, O, Si, and Rh in 5%RhL@NaY were thoroughly investigated using X-ray photoelectron spectroscopy (XPS, Fig. S19†). Survey spectra of NaY, 0.5%RhL@NaY and 5%RhL@NaY confirm the incorporation of Rh on the zeolite support with a new peak appearing at 310 eV. The high-resolution, deconvoluted Si 2p signals, depicted in Fig. 4A, show three peaks located at 101.0 eV for Si–O–Al, and 102.9 and 103.6 eV assigned to SiO2.24 The high-resolution spectrum for O 1s is shown in Fig. 4B, and three peaks located at 529.2, 531 and 534.6 eV were obtained after deconvolution, which were assigned to the lattice oxygen (OLat), Si-OH and absorbed water, respectively.25 There are two Rh 3d peaks in the 5%RhL@NaY sample, shown in Fig. 4C, at 309.4 and 314.1 eV, corresponding to Rh(I) and Rh oxide in the Rh3+ oxidation state.26,27 The high-resolution Rh 3d peak of the 0.5%RhL@NaY sample (Fig. S20†) shows a major presence of Rh(I) in this Rh-diluted zeolite. To complete the XPS analysis, Fig. 4D shows the characteristic XPS peaks for Al 2p at 74.8 eV, attributable to Al2O3 species.24
 | ||
| Fig. 4 X-ray photoelectron spectroscopy (XPS) for the 5%RhL@NaY sample: (A) Si 2p, (B) O 1s, (C) Rh 3d and (D)Al 2p. | ||
27Al SS-MAS NMR spectroscopy was then used to determine the coordination of aluminum atoms in the zeolite. Fig. 5 shows a clear 1.8 ppm shift in the tetrahedrally coordinated Al signal when the Rh complex is incorporated into the NaY zeolite, which indicates that cationic Rh is interacting with the inner and outer surface Al–O groups.28
 | ||
| Fig. 5 27Al solid-state magic angle spinning nuclear magnetic resonance (27Al SS-MAS NMR) of 5%RhL@NaY and zeolite NaY. | ||
The corresponding 29Si SS-MAS NMR results (Fig. S21†) show the characteristic four peaks of the zeolite, with two main peaks denoted Q3 = HOSi(OSi)3 (94.5 ppm) and Q4 = Si(OSi)4 (100.3 ppm) downshifted about 10 ppm from typical values, due to Al3+ substitution.29,30 This large Q3/Q4 ratio is directly proportional to the number of surface hydroxyl groups, which implies better cationic Rh incorporation and interaction with the zeolite framework.
The new solids 0.5%RhL@NaY and 5%RhL@NaY were tested as catalysts for the dehydrogenative borylation reaction of cyclododecene 1, under the reaction conditions shown in Table 1. The results (entries 9 and 10) show that the 5%RhL@NaY zeolite is active and selective for the borylation of 1-cis, but with diborylation product 6 as the major product. Neither RhCl3, Rh2O3, or Rh nanoparticles on carbon or on alumina gave any conversion of 1, which strongly supports the idea that the active species in the zeolite must be isolated Rh sites. To compare the catalytic activity of the different Rh sites in both zeolites, we performed kinetic experiments with the same wt% of solid catalyst and we compared the initial reaction rate with the % of each Rh species in the zeolite according to XPS, as shown in Fig. 6. The 0.5%RhL@NaY catalyst gave a 6 times slower rate than 5%RhL@NaY, despite having 10 times less total Rh (Fig. S22†). Thus, the turnover frequency (TOF) per Rh atom is higher for 0.5%RhL@NaY. If we represent the initial rate vs. the amount of Rh(I) and not the total amount of Rh, the TOFs for both catalysts are the same, since the 0.5%RhL@NaY catalyst contains a significant amount, >20%, of Rh(I) than the 5% counterpart. In other words, the TOF value perfectly matches the amount of Rh(I) in both catalysts; thus Rh(I) seems to be the catalytically active species for the reaction, as occurred for the complex in solution.
 | ||
| Fig. 6 Comparison of the catalytic activity of 0.5%RhL@NaY and 5%RhL@NaY with respect to the amount of supported Rh species, according to the XPS data. TOF: turnover frequency (h−1). | ||
The reusability of 5%RhL@NaY was studied for the selective dehydrogenative monoborylation of cyclododecene 1 under optimized reaction conditions. First, a hot filtration test was performed, in order to evaluate whether the catalytically active species remain in the solid during reaction. The solid was filtered at intermediate conversion and the reaction was followed in the presence or not of the solid catalyst. The results (Fig. S23†) show that no catalytic species is present in solution; thus the whole catalytic activity comes from the solid, which can therefore be defined as a truly heterogenous catalyst. Then, the solid catalyst was employed for subsequent use. After the completion of the reaction, the solid catalyst was centrifuged and washed at least three times with dichloromethane. The recovered catalyst was dried under vacuum and used for the next reaction. The results (Fig. S24†) show that a significant deterioration in the conversion was observed after the first use. To determine the factor decreasing the catalytic activity, we characterized the recycled solid catalyst by XRD, XPS and DR-UVvis. A new broad diffraction peak is observed at 41.2° in XRD, consistent with the formation of Rh nanoparticles (Fig. S25†).31 The XPS data of the spent solid catalyst show the formation of Rh(0) as the major oxidation state, with peaks at 307.6 and 312.3 eV (Fig. S26†). In addition, a new broad absorption band between 300 and 400 nm arises in the spent catalyst, typical of plasmonic nanoparticles (Fig. S27†). Thus, we must conclude that the formation of Rh nanoparticles (NPs) during the reaction explains the decrease in catalytic activity. To further check this, the spent catalyst was impregnated again with the Rh complex, and the resulting zeolite recovered the original catalytic activity (Fig. S24†). Besides, a kinetic experiment with XantPhos added externally together with the zeolite (Fig. S28†) showed that neither the catalytic activity nor the selectivity of the zeolite is modified, confirming that Rh is not in solution (XantPhos is too big to enter into the zeolite). A hot filtration test for this experiment confirms that there is no Rh leaching; thus XantPhos cannot undertake any catalytic modification. Although the loss of catalytic activity observed for the solid may indicate a failure in the catalyst design, this approach opens the way for employing other recyclable solid catalysts for the dehydrogenative borylation reaction after regenerative treatments of the catalyst or, at least, to in situ recover the amount of noble metal, which in any case is better than using a non-recoverable, deteriorating Rh(I) phosphine complex in solution.
4 Conclusions
We have shown here that the Rh(I)-catalyzed dehydrogenative monoborylation reaction of a cis/trans mixture of cyclododecene 1 proceeds with high selectivity towards the cis starting material, leaving the trans isomer unreacted. In this way, both original cis/trans isomers were separated. The benchmark catalyst for this transformation, an Rh(I) complex with XantPhos as a ligand, is progressively degraded under the reaction conditions, and we have developed a solid catalyst based on Rh(I) incorporated in NaY zeolite, without any ligand. This simple solid catalyzes the desired reaction, but at the expense of forming Rh nanoparticles (as the homogeneous system probably does). Despite this loss of catalytic activity, the Rh(I)-supported solid allows the noble metal to be recovered in situ, so we are now able to separate cycloalkene cis/trans mixtures.Author contributions
A. S. S. performed part of the experimental work and analysis, including zeolite synthesis and characterization. A. L.-P. performed the experimental part with homogeneous catalysts and supervised the project. Both authors wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the projects PID2020-115100GB-I00 (funded by Spanish MCIINN, MCIN/AEI/10.13039/501100011033MICIIN) and Severo Ochoa centre of excellence program (CEX2021-001230-S, also from MCIINN) is acknowledged. A. S. S. thanks ITQ for the concession of a contract.References
- (a) A. Sytniczuk, M. Dąbrowski, L. Banach, M. Urban, S. C. Śniadała, M. Milewski, A. Kajetanowicz and K. Grela, At long last: Olefin metathesis macrocyclization at high concentration, J. Am. Chem. Soc., 2018, 140(28), 8895–8901 CrossRef CAS; (b) F. Garnes-Portolés, J. Sanchez-Quesada, E. Espinós-Ferri and A. Leyva-Pérez, Synthesis of dehydromuscone by an alkene metathesis macrocyclization reaction at 0.2 M concentration, Synlett, 2022, 33, 1933–1937 CrossRef.
- K. B. Becker, Cycloalkenes by intramolecular Wittig reaction, Tetrahedron, 1980, 36, 1717–1745 CrossRef CAS.
- T. Ohgiya, N. Kutsumura and S. Nishiyama, DBU-promoted elimination reactions of vicinal dibromoalkanes mediated by adjacent O-functional groups, and applications to the synthesis of biologically active natural products, Synlett, 2008, 3091–3105 CAS.
- (a) A. Fürstner, Olefin metathesis and beyond, Angew. Chem., Int. Ed., 2000, 39, 3012–3043 CrossRef; (b) A. Gradillas and J. Pérez-Castells, Macrocyclization by ring–closing metathesis in the total synthesis of natural products: reaction conditions and limitations, Angew. Chem., Int. Ed., 2006, 45, 6086–6101 CrossRef CAS; (c) A. Fürstner, Lessons from natural product total synthesis: Macrocyclization and postcyclization strategies, Acc. Chem. Res., 2021, 54, 861–874 CrossRef; (d) R. S. Phatake, N. B. Nechmad, O. Reany and N. G. Lemcoff, Highly substrate–selective macrocyclic ring closing metathesis, Adv. Synth. Catal., 2022, 364, 1465–1472 CrossRef CAS.
- S. Li, X. Cui and Y. Wang, Catalyst Development in the dehydrogenative borylation of alkenes for the synthesis vinylboronate esters, Synlett, 2020, 32, 102–108 Search PubMed.
- (a) A. Caballero and S. Sabo-Etienne, Ruthenium-catalyzed hydroboration and dehydrogenative borylation of linear and cyclic alkenes with pinacolborane, Organometallics, 2007, 26, 1191–1195 CrossRef CAS; (b) M. Murata, K. Kawakita, T. Asana, S. Watanabe and Y. Masuda, Rhodium and ruthenium-catalyzed dehydrogenative borylation of vinylarenes with pinacolborane: stereoselective synthesis of vinylboronates, Bull. Chem. Soc. Jpn., 2002, 75, 825–829 CrossRef CAS.
- (a) M. Morimoto, T. Miura and M. Murakami, Rhodium–catalyzed dehydrogenative borylation of aliphatic terminal alkenes with pinacolborane, Angew. Chem., Int. Ed., 2015, 54, 12659–12663 CrossRef CAS; (b) A. Kondoh and T. F. Jamison, Rhodium-catalyzed dehydrogenative borylation of cyclic alkenes, Chem. Commun., 2010, 46, 907–909 RSC; (c) I. A. Mkhalid, R. B. Coapes, S. N. Edes, D. N. Coventry, F. E. Souza, R. L. Thomas, J. J. Hall, S. W. Bi, Z. Lin and T. B. Marder, Rhodium catalysed dehydrogenative borylation of alkenes: Vinylboronates via C–H activation, Dalton Trans., 2008, 8, 1055–1064 RSC; (d) L. Tao, X. Guo, J. Li, R. Li, Z. Lin and W. Zhao, Deoxygenation and borylation of ketones: a combined experimental and theoretical investigation, J. Am. Chem. Soc., 2020, 42, 18118–18127 CrossRef.
- H. Wen, L. Zhang, S. Zhu, G. Liu and Z. Huang, Stereoselective synthesis of trisubstituted alkenes via cobalt-catalyzed double dehydrogenative borylations of 1-alkenes, ACS Catal., 2017, 10, 6419–6425 CrossRef.
- N. Kirai, S. Iguchi, T. Ito, J. Takaya and N. Iwasawa, PSiP-pincer type palladium-catalyzed dehydrogenative borylation of alkenes and 1, 3-dienes, Bull. Chem. Soc. Jpn., 2013, 7, 784–799 CrossRef.
- T. Ohmura, Y. Takasaki, H. Furukawa and M. Suginome, Rhodium-catalyzed selective olefination of arene esters via C–H bond activation, Angew. Chem., Int. Ed., 2009, 13, 2372–2375 CrossRef.
- V. Desrosiers, S. M. Knight and F. G. Fontaine, A Metal-free approach for the C–H activation and transfer borylation of electron-rich alkenes, ACS Catal., 2022, 21, 13609–13618 CrossRef.
- J. Carreras, A. Caballero and P. J. Pérez, Alkenyl boronates: synthesis and applications, Chem. – Asian J., 2019, 3, 329–343 CrossRef.
- J. Prunet, Recent methods for the synthesis of (E)–alkene units in macrocyclic natural products, Angew. Chem., Int. Ed., 2003, 25, 2826–2830 CrossRef PubMed.
- V. Rauniyar, H. Zhai and D. G. Hall, Catalytic enantioselective allyl- and crotylboration of aldehydes using chiral diol·SnCl4 complexes. optimization, substrate scope and mechanistic investigations, J. Am. Chem. Soc., 2008, 26, 8481–8490 CrossRef.
- J. Bhattacharjee, A. Harinath, K. Bano and T. K. Panda, Highly chemoselective hydroboration of alkynes and nitriles catalyzed by group 4 metal amidophosphine–borane complexes, ACS Omega, 2020, 3, 1595–1606 CrossRef.
- S. Sanz-Navarro, M. Mon, A. Doménech-Carbó, R. Greco, J. Sánchez-Quesada, E. Espinós-Ferri and A. Leyva-Pérez, Parts–per–million of ruthenium catalyze the selective chain–walking reaction of terminal alkene, Nat. Commun., 2022, 1, 2831 CrossRef PubMed.
- (a) M. S. Goedheijt, P. C. Kamer and P. W. van Leeuwen, A water-soluble diphosphine ligand with a large natural bite angle for two-phase hydroformylation of alkenes, J. Mol. Catal. A: Chem., 1998, 3, 243–249 CrossRef; (b) P. Ren, S. D. Pike, I. Pernik, A. S. Weller and M. C. Willis, Rh–POP pincer xantphos complexes for C–S and C–H activation. implications for carbothiolation catalysis, Organometallics, 2015, 4, 711–723 CrossRef.
- L. M. Klingensmith, E. R. Strieter, T. E. Barder and S. L. Buchwald, New insights into Xantphos/Pd-Catalyzed C–N bond forming reactions: a structural and kinetic study, Organometallics, 2006, 1, 82–91 CrossRef.
- (a) F. A. Cotton, J. L. Eglin and S. J. Kang, Stereo-and regiospecific ligand substitution reactions of mono-and dinuclear rhodium(III) phosphine complexes, J. Am. Chem. Soc., 1992, 10, 4015–4016 CrossRef; (b) V. Varela-Izquierdo, A. M. Geer de, B. Bruin, J. A. López, M. A. Ciriano and C. Tejel, Rhodium complexes in P–H bond activation reactions, Chem. – Eur. J., 2019, 69, 15915–15928 CrossRef.
- T. H. Brown and P. J. Green, Phosphorus-31 and rhodium-103 nuclear magnetic resonance spectra of some rhodium(I) and rhodium(III) phosphine complexes, J. Am. Chem. Soc., 1970, 8, 2359–2362 CrossRef.
- C. Borner, K. Brandhorst and C. Kleeberg, N–H deprotonation of a diaminodialkoxido diborane(4) – a structural study on bifunctional Lewis acids/bases and their dimerisation to B(sp2)2B(sp3)2N2 six membered rings, Dalton Trans., 2015, 18, 8600–8604 RSC.
- J. R. Cabrero-Antonino, A. Leyva-Pérez and A. Corma, Beyond acid strength in zeolites: soft framework counteranions for stabilization of carbocations on zeolites and its implication in organic synthesis, Angew. Chem., Int. Ed., 2015, 19, 5658–5661 CrossRef.
- P. Mingueza-Verdejo, J. C. Hernández-Garrido, A. Vidal-Moya, J. Oliver-Meseguer and A. Leyva-Pérez, Zeolites catalyze the halogen exchange reaction of alkyl halides, Catal. Sci. Technol., 2023, 8, 2308–2316 RSC.
- S. Senthilkumar, W. Zhong, M. Natarajan, C. Lu, B. Xu and X. Liu, A green approach for aerobic oxidation of benzylic alcohols catalysed by CuI–Y zeolite/TEMPO in ethanol without additional additives, New J. Chem., 2021, 45, 705–713 RSC.
- S. Daniel, C. K. Monguen, A. El Kasmi, M. F. Arshad and Z. Y. Tian, Oxidative dehydrogenation of propane to olefins promoted by Zr modified ZSM-5, Catal. Lett., 2023, 153, 285–299 CrossRef CAS.
- A. S. Singh, S. Jindani, B. Ganguly and A. V. Biradar, Highly regioselective tandem hydroformylation of substituted styrene using Iminophosphine rhodium complex immobilized on carbon, J. Ind. Eng. Chem., 2022, 25, 218–232 CrossRef.
- J. Wang, Z. Yu, L. Wang, B. Wang, F. Liu, X. Liang, P. Sun, X. Yan, X. Chuai and G. Lu, Improvement of NO 2 sensing characteristic for mixed potential type gas sensor based on YSZ and Rh/Co 3 V 2 O 8 sensing electrode, RSC Adv., 2017, 7, 49440–49445 RSC.
- H. V. Bekkum, J. C. Jansen and E. M. Flanigen, Introduction to zeolite science and practice, Elsevier, 1991 Search PubMed.
- L. S. Yuan, J. Efendi, N. S. H. Razali and H. Nur, Fine-tuning the local structure and catalytic activity of titanium-amine functionalized silica in oxidation of limonene by aqueous hydrogen peroxide, Catal. Commun., 2012, 20, 85–88 CrossRef CAS.
- C. P. Grey, Handbook of Zeolite Science and Technology, CRC Press, 2003 Search PubMed.
- W. Alsalahi, W. Tylus and A. M. Trzecia, Green synthesis of rhodium nanoparticles that are catalytically active in benzene hydrogenation and 1-hexene hydroformylation, ChemCatChem, 2018, 10, 2051–2058 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Further experimental details and Fig. S1–S28. See DOI: https://doi.org/10.1039/d3qo01786a |
| This journal is © the Partner Organisations 2024 |