
Stereoselective synthesis of Δ(1)-pyrroline sulfonamides via a chiral aldehyde mediated cascade reaction†
Wei
Wen‡
 *,
Zhao-Pin
Ai‡
,
Zhu-Lian
Wu
,
Tian
Cai
and
Qi-Xiang
Guo
*
*,
Zhao-Pin
Ai‡
,
Zhu-Lian
Wu
,
Tian
Cai
and
Qi-Xiang
Guo
*
Chongqing Key Laboratory of Soft-Matter Material Chemistry and Function Manufacturing, School of Chemistry and Chemical Engineering, Southwest University, Chongqing 400715, China. E-mail: wenwei1989@swu.edu.cn; qxguo@swu.edu.cn
First published on 15th November 2023
Abstract
Functionalized chiral heterocycles play important roles in drug discovery and development. Cascade reactions are powerful strategies for the construction of structurally complex chiral heterocycles. Herein, a catalytic asymmetric Michael addition/spiroannulation/ring cleavage cascade reaction between NH2-free glycinate and cyclic 1-azadienes is established by using a novel chiral aldehyde catalyst, which results in the formation of optically active Δ(1)-pyrroline sulfonamides with high efficiency and high diastereo- and enantioselectivities. Notably, the use of the newly developed chiral aldehyde catalyst led to not only an enhanced reaction rate but also reversed chiral induction compared with its analogue bearing the same configurations.
Introduction
Sulfonamides have extensive biological applications due to their diverse pharmacological activities, namely antibacterial, antifungal, antiviral, antitumor, diuretic and anti-inflammatory activities, among others.1 The discovery and development of the first sulfonamide-based antibacterial drug, Prontosil, opened a new era in medicine. Currently, numerous molecules with functionalized sulfonamide moieties are clinically used to cure different diseases.1 Primary sulfonamides are common and are widely found in marketed drug compounds, pesticides, and bioactive molecules (Fig. 1).1 Meanwhile, unsubstituted primary sulfonamides are prone to chemical modifications of the sulfonamide group that regulate their efficacy, absorption and metabolism. On the other hand, optically active Δ(1)-pyrroline is a type of important molecule that is widely found in bioactive compounds and used as an intermediate for the synthesis of pharmacologically active molecules.2 In this scenario, the combination of these two key motifs (primary sulfonamide and Δ(1)-pyrroline) will provide potential candidates for drug screening and discovery.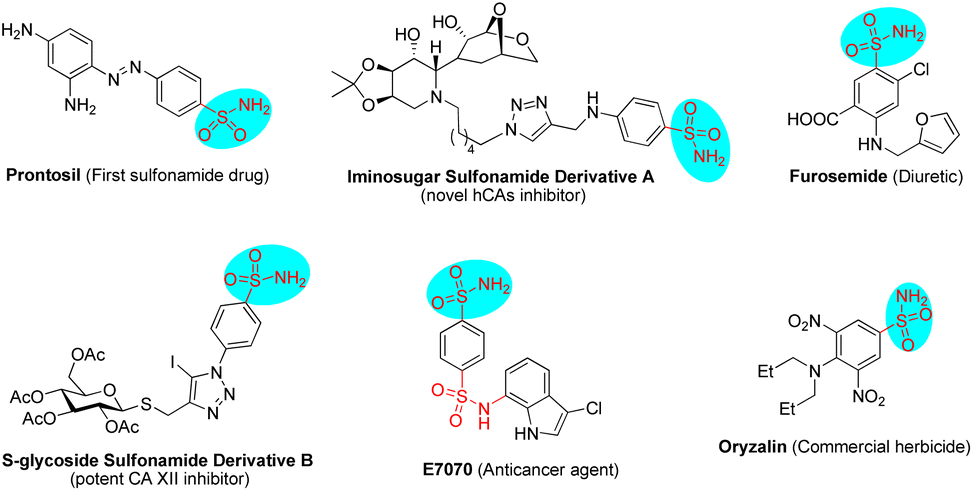 | ||
| Fig. 1 Representative structures of primary sulfonamide-based pharmacological agents. | ||
Chiral aldehyde catalysis has been proven to be a brilliant approach for the asymmetric α-functionalization of NH2-unprotected amino acid esters.3–5 It is vital that the α-functionalized products have free amino groups that can participate as nucleophilic sites in subsequent cascade reactions to construct optically active nitrogen heterocyclic molecules (Scheme 1). In 2018, we disclosed the first chiral aldehyde catalyzed cascade reaction of glycinate, in which the conjugate addition and condensation occurred successively, resulting in optically active Δ(1)-pyrrolines (Scheme 1, eqn (1)).4c Zhao and colleagues reported in 2021 that chiral aldehydes catalyze conjugate addition/lactamization cascade reactions of glycinate to produce chiral pyroglutamic acid esters (Scheme 1, eqn (2)).5a Subsequently, we also reported a similar cascade process to construct fully substituted pyroglutamic acid esters (Scheme 1, eqn (2)).5b
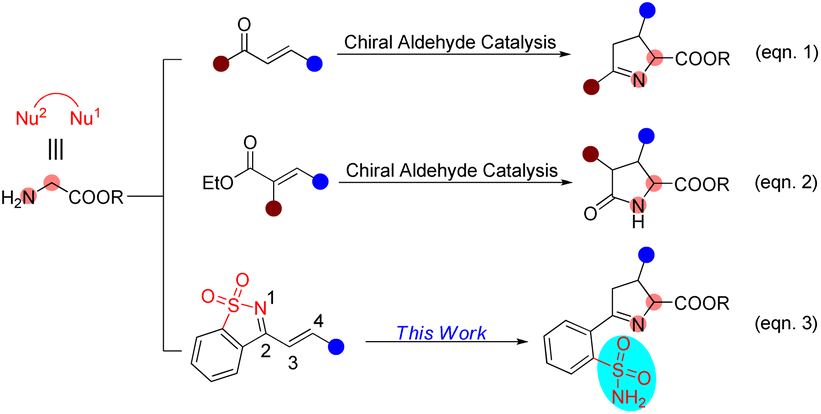 | ||
| Scheme 1 Chiral aldehyde-catalyzed cascade reactions to generate five-membered nitrogen heterocycles. | ||
1-Azadienes have been shown to be utility synthetic building blocks, particularly for building attractive heterocyclic scaffolds in domino reactions.6–10 Herein, we present the results of the asymmetric cascade process of glycine as a C,N-1,2-dinucleophile (Nu1–Nu2) with saccharine-derived cyclic 1-azadienes7–10 catalyzed by a chiral aldehyde, providing an array of optically active Δ(1)-pyrroline derivatives bearing unsubstituted primary sulfonamide moieties in good yields with excellent stereoselectivities (Scheme 1, eqn (3)). It is worth noting that in this work we developed a new chiral aldehyde catalyst to implement our design.
Results and discussion
Reaction condition optimization
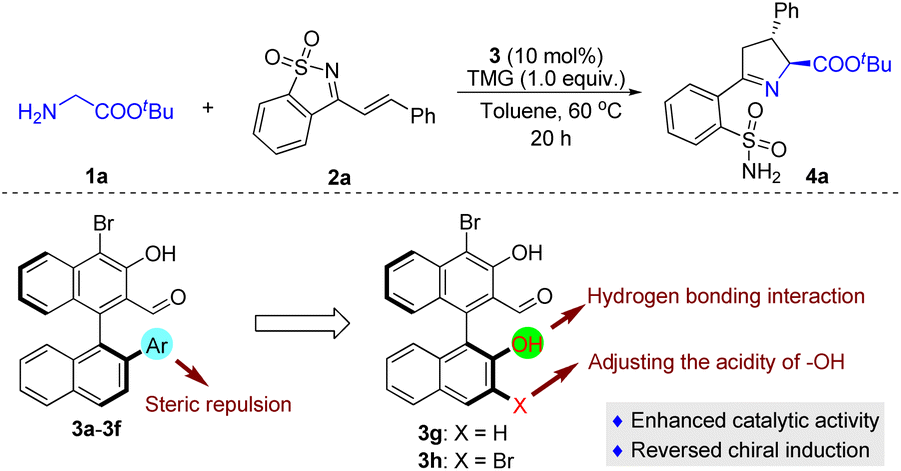
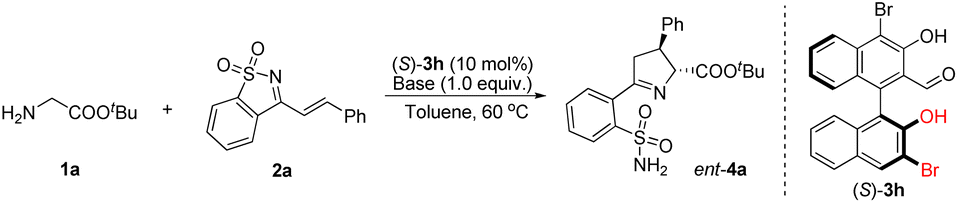
The initial attempt was to examine different aldehyde catalysts 3a–3f; experiments were carried out with tert-butyl glycine ester 1a and saccharine-derived cyclic 1-azadiene 2a (Table 1). Unfortunately, the tested chiral aldehyde catalysts 3a–3d were almost unreactive (Table 1, entries 1–4). Only 3e and 3f smoothly catalyzed the tandem reaction and proceeded to give the desired product 4a with moderate enantioselectivity, but in only low yields (Table 1, entries 5 and 6). We envisage that the introduction of a hydroxyl group in the chiral aldehyde catalysts 3g and 3h, which might form a hydrogen bond with the cyclic 1-azadiene 2a, would be helpful for enantiocontrol, as a synergistic interaction between the organocatalyst and the two reactants could be created. Another important feature is that an electron-withdrawing Br can be introduced at the 3-position, thereby regulating the acidity of the 2′-OH group. Indeed, the reactivity was dramatically improved when the reaction was catalyzed by 3g, while a similar ee value was obtained (Table 1, entry 7), and 3h exhibited higher catalytic activity and enantioselectivity (Table 1, entry 8). In comparison, by replacing the 2′-Ar in the structure of aldehyde catalysts with 2′-OH, we observed an unexpected reversal of chiral induction (Table 1, entries 7 and 8 vs. entries 5 and 6), which also verified that there was a divergence of the stereocontrolling mode and that the 2′-OH group of 3g and 3h played a crucial role in the stereocontrol.|
|
|||
|---|---|---|---|
| Entry | 3, Ar | Yieldb (%) | eec (%) |
| a Reaction conditions: 1a (0.15 mmol), 2a (0.10 mmol), (S)-3 (0.01 mmol) and TMG (0.10 mmol) in toluene (1.0 mL), carried out at 60 °C. b Isolated yield. c Determined by chiral HPLC. d Obtained ent-4a with the opposite configuration. | |||
| 1 | 3a, Ph | Trace | — |
| 2 | 3b, 2-naphthyl | Trace | — |
| 3 | 3c, 4-TMSC6H4 | Trace | — |
| 4 | 3d, 3,5-(tBu)2C6H3 | Trace | — |
| 5 | 3e, 4-PhC6H4 | 15 | 40 |
| 6 | 3f, 3,5-(CF3)2C6H3 | 7 | 69 |
| 7 | 3g | 44 | −54d |
| 8 | 3h | 62 | −62d |
We found that bases had a crucial effect on the reaction conversion (Table 2). When an organic strong base, such as TMG, DBU, or TDMAIP, was used in this reaction, most of the reactant 2a would be consumed within 20 minutes. Some of them would be converted to product 4a with good diastereoselectivity, and moderate yield and enantioselectivity, while the rest would rapidly decompose (Table 2, entries 1–3). When using Et3N or DMAP with weaker alkalinity, the decomposition of 2a slowed down and product 4a could be obtained with lower yields and moderate enantioselectivity (Table 2, entries 4 and 5). The ee values of 4a could be increased to 82–83% when quinuclidine or DABCO was used as a base; however, the yields did not increase (Table 2, entries 6 and 7). Secondary amines have not yet been used as bases in chiral aldehyde catalytic reactions, as they are prone to form hemiaminals or iminiums with aldehydes. Surprisingly, pyrrolidine greatly promoted the reaction, providing the desired Δ(1)-pyrroline product 4a in 92% yield, with 72% ee and 88![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 dr (Table 2, entry 8). Subsequently, various other secondary amines were also tested (Table 2, entries 9–15). Other four-, five- and six-membered cyclic secondary amines, such as azetidine, piperidine, morpholine and indoline, were less efficient than pyrrolidine (Table 2, entries 9–12). Analogously, in the presence of the bulky 2,2-dimethylpyrrolidine and acyclic secondary amines, the conversions observed were lower than those achieved with pyrrolidine (Table 2, entries 13–15). It should be noted that in the presence of a bulky primary amine, the reaction could also proceed smoothly, achieving 46% yield, 90:10 dr and 56% ee (Table 2, entry 16). However, cis-1,2-cyclohexanediamine would cause the reaction to deactivate, which is possibly due to the irreversible formation of condensates with the aldehyde catalyst (Table 2, entry 17). In the absence of a base, it takes longer for 4a to achieve a comparable yield (Table 2, entry 18 vs. entries 1–7). When the reaction temperature drops to 0 °C, the enantioselectivity of 4a could be effectively improved without affecting the activity and diastereoselectivity (Table 2, entry 8 vs. entry 19). Notably, on combination of pyrrolidine and quinuclidine as bases, the stereoselectivity could be further improved to 95:5 dr and 94% ee, with a yield of 92% (Table 2, entry 20).
:12 dr (Table 2, entry 8). Subsequently, various other secondary amines were also tested (Table 2, entries 9–15). Other four-, five- and six-membered cyclic secondary amines, such as azetidine, piperidine, morpholine and indoline, were less efficient than pyrrolidine (Table 2, entries 9–12). Analogously, in the presence of the bulky 2,2-dimethylpyrrolidine and acyclic secondary amines, the conversions observed were lower than those achieved with pyrrolidine (Table 2, entries 13–15). It should be noted that in the presence of a bulky primary amine, the reaction could also proceed smoothly, achieving 46% yield, 90:10 dr and 56% ee (Table 2, entry 16). However, cis-1,2-cyclohexanediamine would cause the reaction to deactivate, which is possibly due to the irreversible formation of condensates with the aldehyde catalyst (Table 2, entry 17). In the absence of a base, it takes longer for 4a to achieve a comparable yield (Table 2, entry 18 vs. entries 1–7). When the reaction temperature drops to 0 °C, the enantioselectivity of 4a could be effectively improved without affecting the activity and diastereoselectivity (Table 2, entry 8 vs. entry 19). Notably, on combination of pyrrolidine and quinuclidine as bases, the stereoselectivity could be further improved to 95:5 dr and 94% ee, with a yield of 92% (Table 2, entry 20).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Base | t (h) | Yieldb (%) | drc | eec (%) |
| n.d. = not determined. TMG = 1,1,3,3-tetramethylguanidine. TDMAIP = tris(dimethylamino)iminophosphorane.a Reaction conditions: 1a (0.15 mmol), 2a (0.10 mmol), (S)-3h (0.01 mmol) and base (0.10 mmol) in toluene (1.0 mL), carried out at 60 °C.b Isolated yield.c Determined by chiral HPLC.d The reaction was performed at 0 °C.e Pyrrolidine (0.05 mmol) and quinuclidine (0.10 mmol) were used. | |||||
| 1 | TMG | 0.3 | 62 | 88:12 |
62 |
| 2 | DBU | 0.3 | 55 | 90:10 |
57 |
| 3 | TDMAIP | 0.3 | 51 | 87:13 |
72 |
| 4 | Et3N | 6 | 39 | 86:14 |
60 |
| 5 | DMAP | 5 | 27 | n.d. | 58 |
| 6 | Quinuclidine | 6 | 51 | 87:13 |
83 |
| 7 | DABCO | 6 | 54 | 85:15 |
82 |
| 8 | Pyrrolidine | 6 | 92 |
88![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :12 :12
|
72 |
| 9 | Azetidine | 6 | 49 | 91:9 |
59 |
| 10 | Piperidine | 6 | 74 | 86:14 |
64 |
| 11 | Morpholine | 6 | 65 | 89:11 |
55 |
| 12 | Indoline | 24 | 41 | n.d. | 54 |
| 13 | 2,2-Dimethylpyrrolidine | 8 | 58 | 88:12 |
62 |
| 14 | Diethylamine | 6 | 54 | 91:9 |
60 |
| 15 | N-Methylaniline | 17 | 40 | n.d. | 55 |
| 16 | t BuNH2 | 12 | 46 | 90:10 |
56 |
| 17 | cis-1,2-Cyclohexanediamine | 12 | Trace | n.d. | n.d. |
| 18 | No base | 24 | 56 | n.d. | 53 |
| 19d | Pyrrolidine | 5 | 89 | 91:9 |
86 |
| 20 , | Pyrrolidine + quinuclidine | 3 | 92 |
95:5
|
94 |
Substrate scope investigation
We studied the substrate range using the optimal reaction conditions shown in Table 2, entry 20, and using (R)-3h as a catalyst (Table 3). Other glycine esters, including glycine methyl, ethyl, and isopropyl ester, were found to be suitable donors in this reaction, resulting in the corresponding products being obtained in high yields with good diastereo- and enantioselectivities (Table 3, 4b–4d). The absolute configuration of compound 4b was unambiguously determined by single crystal X-ray crystallography, and the configurations of the stereogenic centers were 2S,3R.11 The absolute stereochemistry of other compounds 4 was assigned by analogy upon the assumption of a uniform stereochemical pathway.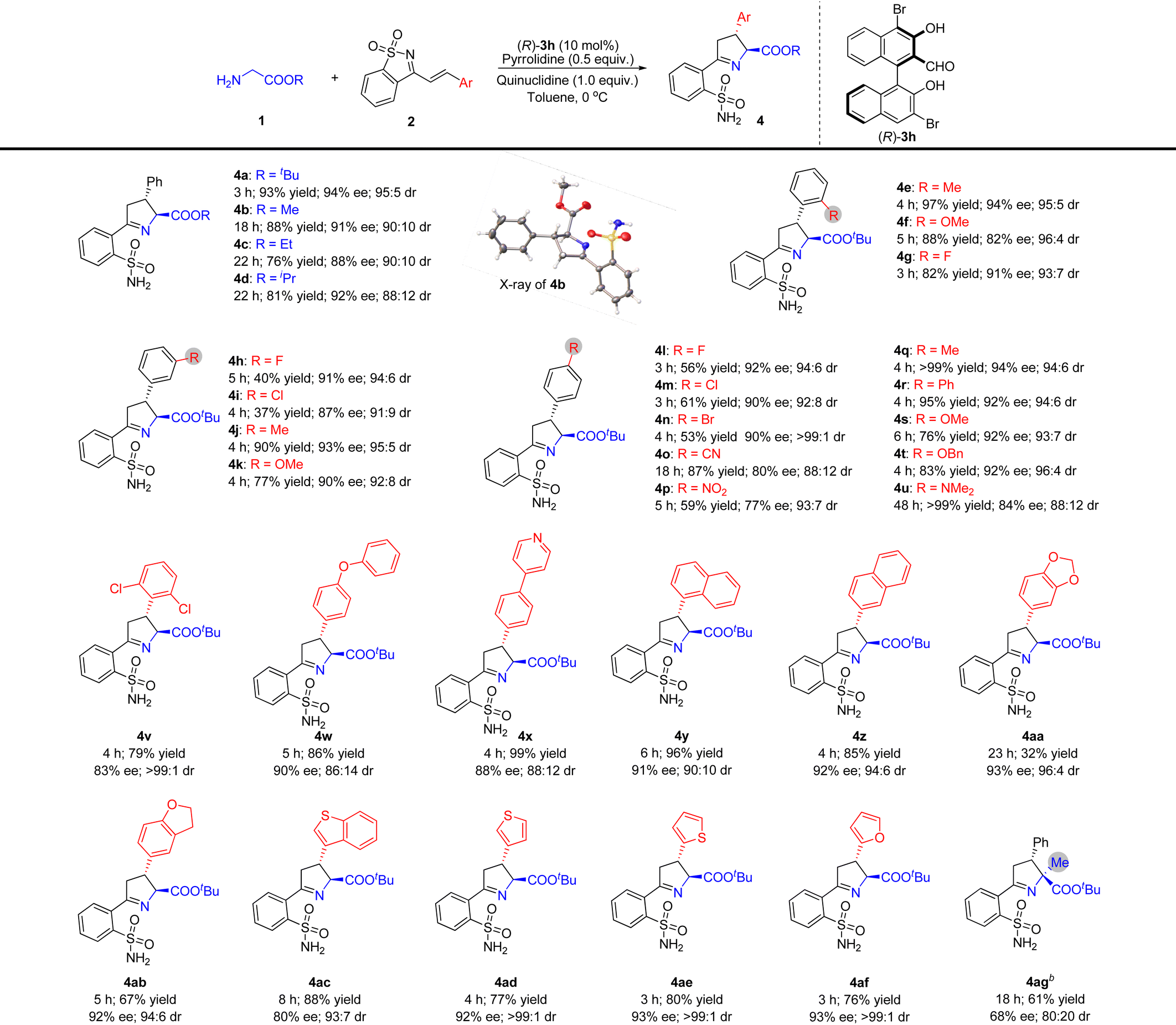
Subsequently, the research for evaluating the tolerance of different substituents toward cyclic 1-azadienes 2 was pushed forward by reactions with tert-butyl glycine ester 1a. The results indicated that cyclic 1-azadienes 2 containing ortho-substituted phenyls were good reaction partners in this reaction, and the yields and stereoselectivities could not be obviously affected by the electronic and steric properties of the corresponding ortho-substituents (Table 3, 4e–4g). Similarly, the reactions of various substituted phenyl cyclic 1-azadienes with different electronic properties at the meta- and para-positions of the benzene ring with glycinate 1a also proceeded smoothly, furnishing the corresponding chiral Δ(1)-pyrrolines 4h–u in moderate to good yields (37 to >99%) with high diastereo- (88:12 to >99:1 dr) and enantioselectivities (77–94% ee). The electronic properties of the meta- and para-substituted phenyl groups seemed to have an impressive impact on the yield. For example, somewhat lower yields were obtained when electron-withdrawing halogens and a nitro group were installed at the meta- or para-position of the phenyl group of 1-azadienes 2 (Table 3, 4h, 4i, 4l–4n and 4p). Cyclic 1-azadiene 2v with 2,6-dichlorodisubstituted phenyl was also used as a substrate, and its steric hindrance did not affect the activity, but the stereoselectivity of 4v was slightly reduced. In addition, the reaction conditions were also tolerant to the structure of diaryl ether (Table 3, 4w). Remarkably, the reaction is also compatible with naphthyl and heteroaryl decorated substrates 2 to achieve the desired products 4x–4af in 32–99% yields with outstanding stereoselectivities (80–93% ee and 88:12 to >99:1 dr). For tert-butyl alaninate with an α-substituent, reaction at 20 °C is required, likely due to steric hindrance, resulting in 61% yield, 68% ee and 80:20 dr, affording product 4ag with a quaternary carbon stereocentre.
Mechanism studies
Next, we compared the catalytic performance of the novel catalyst 3f and catalyst 3h under the optimal reaction conditions (Scheme 2a). As expected, both the catalytic activation and stereocontrol ability of catalyst 3f were better. It should be noted that when (R)-3f and (R)-3h were used as catalysts, respectively, a switch in the enantioselectivity was observed, indicating that 3f and 3h had distinct stereocontrolling modes during the catalytic process. On the basis of our experiments as well as previous studies,4c transition states TS1 and TS2 were proposed to account for the observed stereoselectivities (Scheme 2a). In the process of catalyzation by 3f, the formation of the C–C bond would follow an Re–Si facial attack mode, attributed to the steric effect of 3f's 2′-aryl group (Scheme 2a, TS1). During the catalytic process of 3h, the 2′-hydroxyl group of 3h forms a hydrogen bond with 1-azadiene, which not only improved the electrophilicity of 2a but also adjusted the spatial arrangement of 2a in the transition state, leading to an Si–Re facial attack mode (Scheme 2a, TS2).12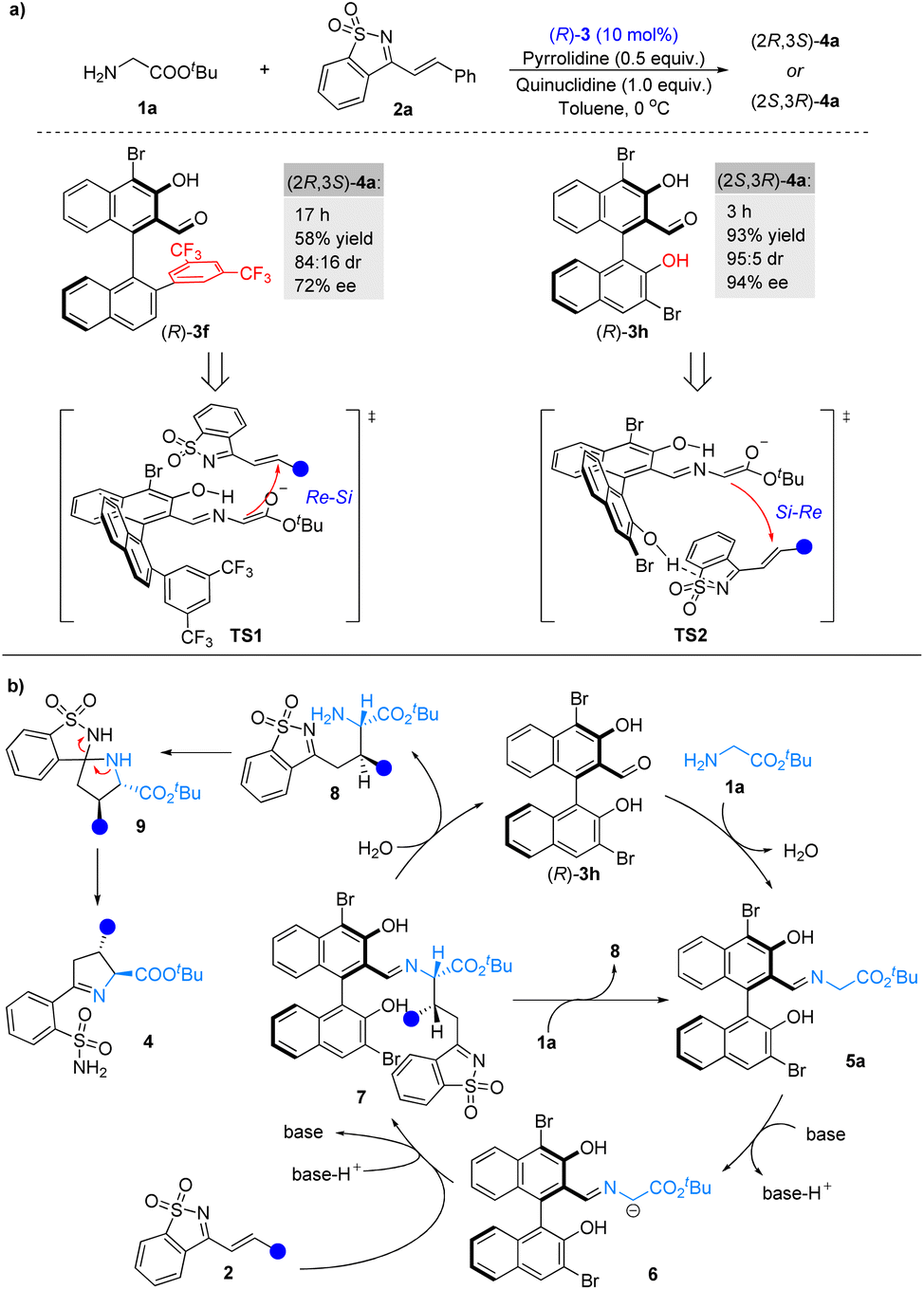 | ||
| Scheme 2 (a) Control experiments of modified chiral aldehyde catalysts and proposed transition states. (b) Proposed catalytic cycle. | ||
Combining the control experiments (see the ESI†), a plausible pathway has been proposed for the reaction (Scheme 2b). Chiral aldehyde catalyst 3h condenses with glycinate 1a to form imine 5a, which is deprotonated by the base to generate α-imine anion 6. Intermediate 6 undergoes asymmetric 1,4-conjugate addition to 1-azadiene 2avia a possible transition state TS2, followed by hydrolysis or amine exchange to produce 8 and complete the chiral aldehyde catalytic cycle.13 Compound 8 is in situ converted into Δ(1)-pyrroline sulfonamides 4via sequential spiroannulation and ring cleavage.
Pyrrolidine was crucial for improving the reactivity. To gain insight into the role of pyrrolidine, a series of control experiments were conducted (Scheme 3). When N-methylpyrrolidine was used instead of pyrrolidine under the standard conditions, the reactivity to generate 4a was significantly reduced (Scheme 3a). Nevertheless, the stereoselectivity was not affected, which implies that the pyrrolidine likely does not participate in the transition state of stereoselective addition.
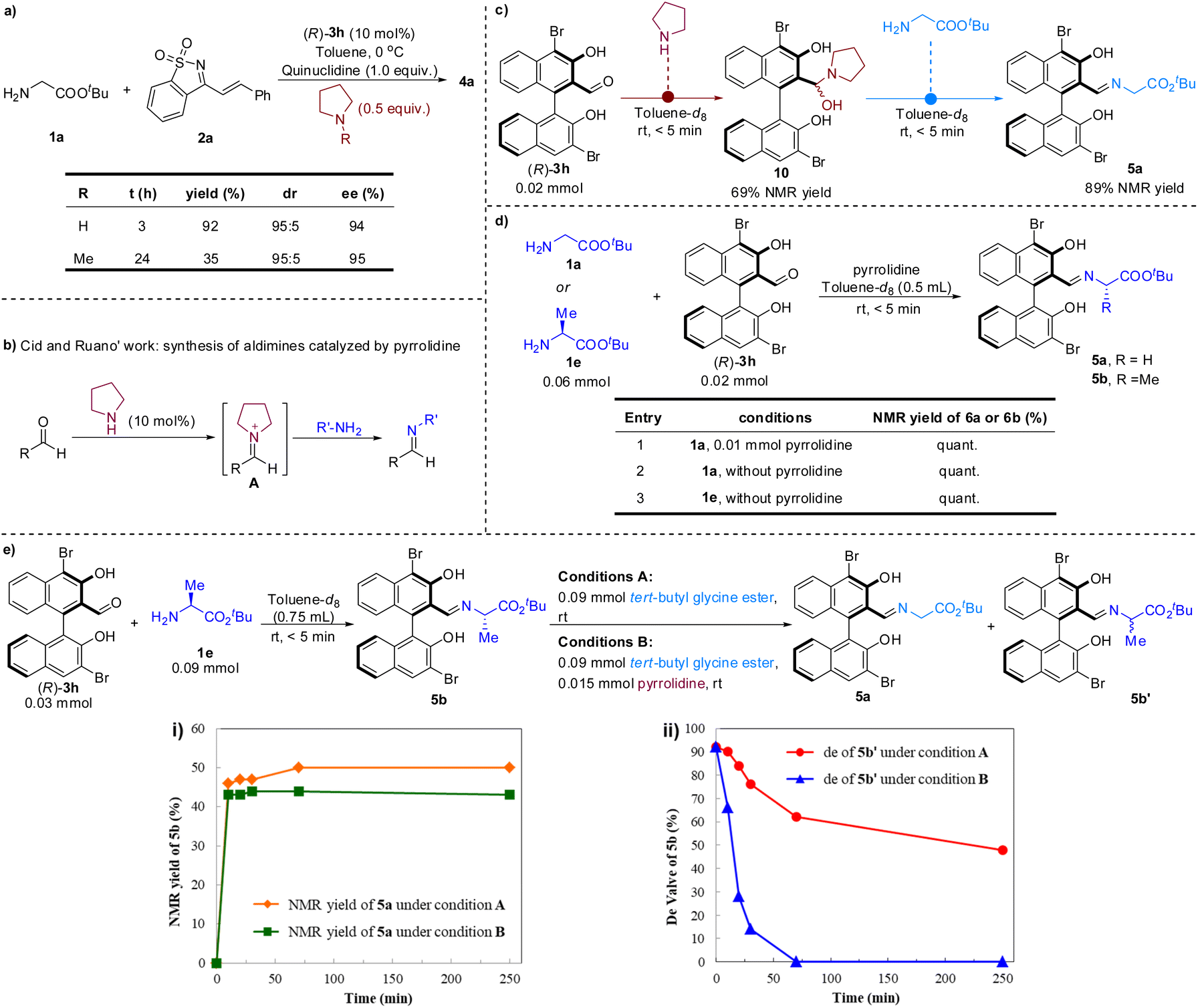 | ||
| Scheme 3 Study on the action of pyrrolidine. | ||
It is a consensus that secondary amines and aldehydes can reversibly form iminium ions. Iminium catalysis based on this mechanism was the canonical organocatalytic activation mode for asymmetric organic synthesis.14 Recently, Cid and colleagues also described a method for preparing aldimines using pyrrolidine as a nucleophilic catalyst (Scheme 3b).15 In this process, the pyrrolidine first reacts with aldehyde to form iminium ion A, which then undergoes a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N+/CN transimination to afford aldimines. To gain insight into whether the aldehyde catalyzed cascade procedure has proceeded through the aforementioned nucleophilic catalysis to accelerate the formation of the imine between the chiral aldehyde and glycinate, several control experiments were carried out (Scheme 3c and d). It was found that chiral aldehyde (R)-3h and pyrrolidine could rapidly form hemiaminal intermediate 10, which can quickly undergo an exchange reaction with 1a to form imine 5a (Scheme 3c). However, it was further found that the condensation of chiral aldehyde 3h with amino acid ester 1a or 1e was very prompt, regardless of the presence of pyrrolidine (Scheme 3d). Therefore, we speculate that the formation of imine 6a does not require the promotion of pyrrolidine.
N+/CN transimination to afford aldimines. To gain insight into whether the aldehyde catalyzed cascade procedure has proceeded through the aforementioned nucleophilic catalysis to accelerate the formation of the imine between the chiral aldehyde and glycinate, several control experiments were carried out (Scheme 3c and d). It was found that chiral aldehyde (R)-3h and pyrrolidine could rapidly form hemiaminal intermediate 10, which can quickly undergo an exchange reaction with 1a to form imine 5a (Scheme 3c). However, it was further found that the condensation of chiral aldehyde 3h with amino acid ester 1a or 1e was very prompt, regardless of the presence of pyrrolidine (Scheme 3d). Therefore, we speculate that the formation of imine 6a does not require the promotion of pyrrolidine.
Pyrrolidine may also act as a nucleophilic catalyst to form a new iminium with intermediate 7, thus releasing compound 8 from 7, and then hydrolysis or amine exchange occurs to complete the catalytic cycle of the aldehyde.16 To test this possibility, imine 5b produced in situ in toluene-d8 was treated with tert-butyl glycine ester 1a at room temperature and the yield of the new imine 5a was monitored over time using 1H-NMR spectroscopy (Scheme 3e). A dynamic equilibrium between 5a and 5b was quickly reached within the time required to record the spectrum (<5 min), and 46% NMR yield of 5a was attained. Analogous results were obtained with the addition of pyrrolidine, with only a slightly lower NMR yield for 5a (Scheme 3e(i)). Unexpectedly, at the same time as imine metathesis occurred, the epimerization of imine 5b also occurred. Moreover, pyrrolidine showed a significant acceleration effect on the process of the epimerization (Scheme 3e(ii)). Taken together, these results indicate that pyrrolidine did not play the role of a nucleophilic catalyst in this reaction, but may act as an unusual base to promote the deprotonation of imine 5a to generate α-imine anion 6 in the catalytic cycle (Scheme 2b).
Conclusions
In summary, we have developed a new type of bifunctional chiral aldehyde catalyst 3h and have proven that it is an efficient catalyst for the asymmetric cascade reaction of NH2-free glycinate with cyclic 1-azadienes, producing various chiral Δ(1)-pyrroline sulfonamides with high efficiency and excellent diastereo- and enantioselectivities under mild conditions. The aldehyde catalyst 3h not only led to a much enhanced reaction rate, but also reversed chiral induction compared with 3a–f bearing the same configurations. Mechanistic investigations have revealed working mode of pyrrolidine in tuning the reactivity of this reaction. These studies shed new light on the design of efficient chiral aldehyde catalysts and their applications in asymmetric catalysis.Author contributions
W. W. and Q.-X. G. conceived and directed the project. W. W. and Z.-P. A. carried out the experiments. Z.-L. W. and T. C. performed the HRMS analysis. W. W. wrote the paper. All authors discussed the results.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge the National Natural Science Foundation of China (22201235, 22071199 and 22371233), the Natural Science Foundation of Chongqing (cstc2021jcyj-msxmX0278), and the Innovation Research 2035 Pilot Plan of Southwest University (SWU-XDZD22011) for financial support.Notes and references
- (a) J. E. Lesch, The first miracle drugs: how the sulfa drugs transformed medicine, Oxford University Press, 2006 CrossRef; (b) F. A. Khan, S. Mushtaq, S. Naz, U. Farooq, A. Zaidi, S. M. Bukhari, A. Rauf and M. S. Mubarak, Sulfonamides as potential bioactive scaffolds, Curr. Org. Chem., 2018, 22, 818–830 CrossRef CAS; (c) S. Mondal and S. Malakar, Synthesis of sulfonamide and their synthetic and therapeutic applications: Recent advances, Tetrahedron, 2020, 131662 CrossRef CAS; (d) P. Devendar and G.-F. Yang, Sulfur-containing agrochemicals, Top. Curr. Chem., 2017, 375, 82 CrossRef PubMed; (e) A. Scozzafava, T. Owa, A. Mastrolorenzo and C. T. Supuran, Anticancer and antiviral sulfonamides, Curr. Med. Chem., 2003, 10, 925–953 CrossRef CAS PubMed; (f) C. T. Supuran, A. Innocenti, A. Mastrolorenzo and A. Scozzafava, Antiviral sulfonamide derivatives, Mini-Rev. Med. Chem., 2004, 4, 189–200 CrossRef CAS PubMed.
- (a) M. Birouk, S. Harraga, J. Panouse-Perrin, J. F. Robert, M. Damelincourt, F. Theobald, R. Mercier and J. Panouse, Dérivés arylés et éthoxycarbonylés de 3,4-dihydro 2H pyrrole, 2H pyrrole et pyrrole immunoactifs sur le lymphocyte T humainAryl and ethoxycarbonyl derivatives of pyrroles, 2H pyrroles and 3,4-dihydropyrroles and their immunoactivity of human T lymphocytes, Eur. J. Med. Chem., 1991, 26, 91–99 CrossRef CAS; (b) S. Rosset, J. P. Célérier and G. Lhommet, Enantioselective syntheses of Monomorium minutum ant venom alkaloids: (5R)-2-(5-Hexenyl)-5-nonyl-3,4-dihydro-2H-pyrrole and (2R,5R)-2-(5-hexenyl)-5-nonylpyrrolidine from (s)-pyroglutamic acid, Tetrahedron Lett., 1991, 32, 7521–7524 CrossRef CAS; (c) G. Dannhardt and W. Kiefer, 1-Pyrrolines (3,4-dihydro-2H-pyrroles) as a template for new drugs, Arch. Pharm., 2001, 334, 183–188 CrossRef CAS; (d) A. Stapon, R. Li and C. A. Townsend, Carbapenem biosynthesis: confirmation of stereochemical assignments and the role of CarC in the ring stereoinversion process from L-proline, J. Am. Chem. Soc., 2003, 125, 8486–8493 CrossRef CAS PubMed.
- (a) S. Li, X.-Y. Chen and D. Enders, Aldehyde catalysis: new options for asymmetric organocatalytic reactions, Chem, 2018, 4, 2026–2028 CrossRef CAS; (b) Q. Wang, Q. Gu and S.-L. You, Enantioselective carbonyl catalysis enabled by chiral aldehydes, Angew. Chem., Int. Ed., 2019, 58, 6818–6825 CrossRef CAS PubMed; (c) W. Wen and Q.-X. Guo, Recent advances in chiral aldehyde catalysis for asymmetric functionalization of amines, Synthesis, 2023, 719–732 CrossRef CAS; (d) X. Xiao and B. Zhao, Vitamin B6-based biomimetic asymmetric catalysis, Acc. Chem. Res., 2023, 56, 1097–1117 CrossRef CAS PubMed.
- (a) B. Xu, L.-L. Shi, Y.-Z. Zhang, Z.-J. Wu, L.-N. Fu, C.-Q. Luo, L.-X. Zhang, Y.-G. Peng and Q.-X. Guo, Catalytic asymmetric direct α-alkylation of amino esters by aldehydes via imine activation, Chem. Sci., 2014, 5, 1988–1991 RSC; (b) J. Chen, X. Gong, J. Li, Y. Li, J. Ma, C. Hou, G. Zhao, W. Yuan and B. Zhao, Carbonyl catalysis enables a biomimetic asymmetric Mannich reaction, Science, 2018, 360, 1438–1442 CrossRef CAS PubMed; (c) W. Wen, L. Chen, M.-J. Luo, Y. Zhang, Y.-C. Chen, Q. Ouyang and Q.-X. Guo, Chiral aldehyde catalysis for the catalytic asymmetric activation of glycine esters, J. Am. Chem. Soc., 2018, 140, 9774–9780 CrossRef CAS PubMed.
- (a) J. Ma, Q. Zhou, G. Song, Y. Song, G. Zhao, K. Ding and B. Zhao, Enantioselective synthesis of pyroglutamic acid esters from glycinate via carbonyl catalysis, Angew. Chem., Int. Ed., 2021, 60, 10588–10592 CrossRef CAS PubMed; (b) W.-Z. Wang, H.-R. Shen, J. Liao, W. Wen and Q.-X. Guo, A chiral aldehyde-induced tandem conjugated addition-lactamization reaction for constructing fully substituted pyroglutamic acids, Org. Chem. Front., 2022, 9, 1422–1426 RSC.
- (a) B. Groenendaal, E. Ruijter and R. V. A. Orru, 1-Azadienes in cycloaddition and multicomponent reactions towards N-heterocycles, Chem. Commun., 2008, 5474–5489 RSC; (b) J.-C. M. Monbaliu, K. G. R. Masschelein and C. V. Stevens, Electron-deficient 1- and 2-azabuta-1,3-dienes: a comprehensive survey of their synthesis and reactivity, Chem. Soc. Rev., 2011, 40, 4708–4739 RSC.
- For selected examples of 1,4-cyclization of saccharine-derived cyclic 1-azadienes, see:
(a) X. Feng, Z. Zhou, C. Ma, X. Yin, R. Li, L. Dong and Y.-C. Chen, Trienamines derived from Interrupted cyclic 2,5-dienones: remote δ,ε-CC bond activation for asymmetric inverse-electron-demand aza-Diels-Alder reaction, Angew. Chem., Int. Ed., 2013, 52, 14173–14176 CrossRef CAS PubMed;
(b) J. Gu, C. Ma, Q.-Z. Li, W. Du and Y.-C. Chen, β,γ-Regioselective inverse-electron-demand aza-Diels-Alder reactions with α,β-unsaturated aldehydes via dienamine catalysis, Org. Lett., 2014, 16, 3986–3989 CrossRef CAS PubMed;
(c) Z. Zhou, Z.-X. Wang, Q. Ouyang, W. Xiao, W. Du and Y.-C. Chen, Cross-conjugated trienamine catalysis with α′-alkylidene 2-cyclohexenones: application in β,γ-regioselective aza-Diels-Alder reaction, Chem. – Eur. J., 2017, 23, 2945–2949 CrossRef CAS PubMed;
(d) X.-X. Yang, R.-J. Yan, G.-Y. Ran, C. Chen, J.-F. Yue, X. Yan, Q. Ouyang, W. Du and Y.-C. Chen, π-Lewis-base-catalyzed asymmetric vinylogous umpolung reactions of cyclopentadienones and tropone, Angew. Chem., Int. Ed., 2021, 60, 26762–26768 CrossRef CAS PubMed;
(e) C. M. Young, D. G. Stark, T. H. West, J. E. Taylor and A. D. Smith, Exploiting the imidazolium effect in base-free ammonium enolate generation: synthetic and mechanistic studies, Angew. Chem., Int. Ed., 2016, 55, 14394–14399 CrossRef CAS PubMed;
(f) J. Izquierdo and M. A. Pericàs, Immobilized analogue of benzotetramisole for catalytic enantioselective domino Michael addition/cyclization reactions in batch and flow, ACS Catal., 2016, 6, 348–356 CrossRef CAS;
(g) Z. Wang, H. Xu, Q. Su, P. Hu, P.-L. Shao, Y. He and Y. Lu, Enantioselective synthesis of tetrahydropyridines/piperidines via stepwise [4 + 2]/[2 + 2] cyclizations, Org. Lett., 2017, 19, 3111–3114 CrossRef CAS PubMed;
(h) Y. Chen, B. Shi, H. Yin, Y. Liu, C. Yu, K. Zhang, T. Li and C. Yao, Stereoselective synthesis of chiral sultam-fused dihydropyridinones via photopromoted NHC catalyzed [4 + 2] annulation, Org. Chem. Front., 2022, 9, 5191–5196 RSC;
(i) X.-R. Ren, J.-B. Lin, X.-Q. Hua and P.-F. Xu, Bifunctional Brønsted base catalyzed inverse-electron-demand aza-Diels-Alder reactions of saccharin-derived 1-azadienes with azlactones, Org. Chem. Front., 2019, 6, 2280–2283 RSC;
(j) Q. An, J. Shen, N. Butt, D. Liu, Y. Liu and W. Zhang, The construction of 3-methyl-4-arylpiperidines via a trans-perhydroindolic acid-catalyzed asymmetric aza-Diels-Alder reaction, Adv. Synth. Catal., 2015, 357, 3627–3638 CrossRef CAS;
(k) Y. Song, J. Wang, S. Deng, G. Liu and T. Cheng, Quinidine-catalyzed enantioselective domino Michael addition/cyclization process: Synthesis of chiral 1,4-dihydro-pyridine containing benzosultams, Mol. Catal., 2022, 520, 112165 CrossRef CAS;
(l) Z.-Q. Liang, D.-L. Wang, C.-L. Zhang and S. Ye, Enantioselective N-heterocyclic carbenecatalyzed synthesis of saccharine-derived dihydropyridinones with cis-selectivity, Org. Biomol. Chem., 2016, 14, 6422–6425 RSC;
(m) C. Li, K. Jiang and Y.-C. Chen, Diastereodivergent and enantioselective [4 + 2] annulations of γ-butenolides with cyclic 1-azadienes, Molecules, 2015, 20, 13642–13658 CrossRef CAS PubMed;
(n) H. Jin, E. Li and Y. Huang, Divergent synthesis of hydropyridine derivatives via nitrogen-containing Lewis base mediated regioselective [4 + 2] cyclizations, Org. Chem. Front., 2017, 4, 2216–2220 RSC;
(o) D. G. Stark, C. M. Young, T. J. C. O'Riordan, A. M. Z. Slawin and A. D. Smith, Enantioselective isothiourea-catalysed trans-dihydropyridinone synthesis using saccharin-derived ketimines: scope and limitations, Org. Biomol. Chem., 2016, 14, 8068–8073 RSC.
- For selected examples of 2,4-cyclization of saccharine-derived cyclic 1-azadienes, see: (a) X. Yin, Y. Zheng, X. Feng, K. Jiang, X.-Z. Wei, N. Gao and Y.-C. Chen, Asymmetric [5 + 3] formal cycloadditions with cyclic enones through cascade dienamine-dienamine catalysis, Angew. Chem., Int. Ed., 2014, 53, 6245–6248 CrossRef CAS PubMed; (b) X.-L. He, Y.-C. Xiao, W. Du and Y.-C. Chen, Enantioselective formal [3 + 3] cycloadditions of ketones and cyclic 1-azadienes by cascade enamine-enamine catalysis, Chem. – Eur. J., 2015, 21, 3443–3448 CrossRef CAS PubMed; (c) X. Chen, J.-Q. Zhang, S.-J. Yin, H.-Y. Li, W.-Q. Zhou and X.-W. Wang, Asymmetric construction of spiro[thiopyranoindolebenzoisothiazole] scaffold via a formal [3 + 3] spiroannulation, Org. Lett., 2015, 17, 4188–4191 CrossRef CAS PubMed; (d) S.-H. Chen, Y.-H. Miao, G.-J. Mei, Y.-Z. Hua, S.-K. Jia and M.-C. Wang, Dinuclear zinc catalyzed asymmetric [3 + 2] spiroannulation for the synthesis of diverse bispirocyclic saccharines, Org. Chem. Front., 2022, 9, 5010–5015 RSC.
- For selected examples of 3,4-cyclization of saccharine-derived cyclic 1-azadienes, see: (a) K.-K. Wang, T. Jin, X. Huang, Q. Ouyang, W. Du and Y.-C. Chen, α-Regioselective asymmetric [3 + 2] annulations of Morita-Baylis-Hillman carbonates with cyclic 1-azadienes and mechanism elucidation, Org. Lett., 2016, 18, 872–875 CrossRef CAS PubMed; (b) L. Yu, Y. Cheng, F. Qi, R. Li and P. Li, Diastereoselective, and enantioselective annulation of cyclic 1-azadienes with γ-nitro ketones via 3,4-cyclization, Org. Chem. Front., 2017, 4, 1336–1340 RSC; (c) H.-P. Lv, X.-P. Yang, B.-L. Wang, H.-D. Yang, X.-W. Wang and Z. Wang, Chiral bidentate phosphoramidite-Pd catalyzed asymmetric decarboxylative dipolar cycloaddition for multistereogenic tetrahydrofurans with cyclic N-sulfonyl ketimine moieties, Org. Lett., 2021, 23, 4715–4720 CrossRef CAS PubMed; (d) L. Chang, G.-Y. Zhu, T. Yang, X.-L. Zhao, M. Shi and M.-X. Zhao, Organocatalytic asymmetric formal [3 + 2] cycloaddition reaction of isocyanoacetates with saccharin-derived 1-azadienes, Org. Biomol. Chem., 2021, 19, 3687–3697 RSC; (e) M. Ke, B. Qiao, Y. Yu, X. Li, X. Xiao, S.-J. Li, Y. Lan and F. Chen, Palladium-catalyzed asymmetric [3 + 2] annulation of vinylethylene carbonates with alkenes installed on cyclic N-sulfonyl imines: highly enantio- and diastereoselective construction of chiral tetrahydrofuran scaffolds bearing three vicinal and quaternary stereocenters, J. Org. Chem., 2022, 87, 5166–5177 CrossRef CAS PubMed; (f) Y.-H. Chen, D.-H. Li and Y.-K. Liu, Diversified synthesis of chiral chromane-containing polyheterocyclic compounds via asymmetric organocatalytic cascade reactions, ACS Omega, 2018, 3, 16615–16625 CrossRef CAS PubMed.
- (a) E. Li, H. Jin, P. Jia, X. Dong and Y. Huang, Bifunctional-phosphine-catalyzed sequential annulations of allenoates and ketimines: construction of functionalized polyheterocycle rings, Angew. Chem., Int. Ed., 2016, 55, 11591–11594 CrossRef CAS PubMed; (b) C. Ma, J. Gu, B. Teng, Q.-Q. Zhou, R. Li and Y.-C. Chen, 1-Azadienes as regio- and chemoselective dienophiles in aminocatalytic asymmetric Diels-Alder reaction, Org. Lett., 2013, 15, 6206–6209 CrossRef CAS PubMed; (c) P. Jia and Y. Huang, Sequential annulation domino reaction of sulfur ylides and α,β-unsaturated cyclic ketimines: synthesis of cyclic 2-alkenyl aziridines, Org. Lett., 2016, 18, 2475–2478 CrossRef CAS PubMed.
- CCDC 2283443† (4b) contains the supplementary crystallographic data for this paper.
- (a) L.-L. Han, Q.-Y. Zhang, X. Li, Y. Qiao, Y. Lan and D. Wei, The chiral pyridoxal-catalyzed biomimetic Mannich reaction: the mechanism and origin of stereoselectivity, Org. Chem. Front., 2022, 9, 4401–4410 RSC; (b) L. Zhu and D. Wang, Deciphering the cooperative effect of base and N-substituents on the origin of enantioselectivity switching for Mannich reactions of glycinate by carbonyl catalysts, J. Catal., 2022, 415, 1–11 CrossRef CAS; (c) X. Cui, Q. Li, L. Yao, Y. Ma, L. Zhang, C. Zhang and L. Zhao, Mechanistic study of the N-quaternized pyridoxal-catalyzed biomimetic asymmetric Mannich reaction: insights into the origins of enantioselectivity and diastereoselectivity, J. Org. Chem., 2021, 86, 6592–6599 CrossRef CAS PubMed; (d) A. Cheng, L. Zhang, Q. Zhou, T. Liu, J. Cao, G. Zhao, K. Zhang, G. Song and B. Zhao, Efficient asymmetric biomimetic Aldol reaction of glycinates and trifluoromethyl ketones by carbonyl catalysis, Angew. Chem., Int. Ed., 2021, 60, 20166–20172 CrossRef CAS PubMed; (e) P. Ji, X. Liu, J. Xu, X. Zhang, J. Guo, W.-W. Chen and B. Zhao, Direct asymmetric α-C-H addition of N-unprotected propargylic amines to trifluoromethyl ketones by carbonyl catalysis, Angew. Chem., Int. Ed., 2022, 61, e202206111 CrossRef CAS PubMed.
- (a) M. Ciaccia and S. D. Stefano, Mechanisms of imine exchange reactions in organic solvents, Org. Biomol. Chem., 2015, 13, 646–654 RSC; (b) M. Ciaccia, R. Cacciapaglia, P. Mencarelli, L. Mandolini and S. D. Stefano, Fast transimination in organic solvents in the absence of proton and metal catalysts. A key to imine metathesis catalyzed by primary amines under mild conditions, Chem. Sci., 2013, 4, 2253–2261 RSC; (c) M. Ciaccia, S. Pilati, R. Cacciapaglia, L. Mandolini and S. D. Stefano, Effective catalysis of imine metathesis by means of fast transiminations between aromatic–aromatic or aromatic-aliphatic amines, Org. Biomol. Chem., 2014, 12, 3282–3287 RSC.
- (a) K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, New strategies for organic catalysis: the first highly enantioselective organocatalytic Diels-Alder reaction, J. Am. Chem. Soc., 2000, 122, 4243–4244 CrossRef CAS; (b) A. Erkkilä, I. Majander and P. M. Pihko, Iminium catalysis, Chem. Rev., 2007, 107, 5416–5470 CrossRef PubMed; (c) Science of Synthesis: Asymmetric Organocatalysis, ed. B. List and K. Maruoka, Thieme Chemistry, Stuttgart, Germany, 2012 Search PubMed.
- S. Morales, F. G. Guijarro, J. L. G. Ruano and M. B. Cid, A General aminocatalytic method for the synthesis of aldimines, J. Am. Chem. Soc., 2014, 136, 1082–1089 CrossRef CAS PubMed.
- N. Wilhelms, S. Kulchat and J.-M. Lehn, Organocatalysis of CN/CN and CC/CN exchange in dynamic covalent chemistry, Helv. Chim. Acta, 2012, 95, 2635–2651 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2283443. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qo01675g |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2024 |