
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective synthesis of chiral BCPs
Irene
Sánchez-Sordo†
 ,
Sergio
Barbeira-Arán†
and
Martín
Fañanás-Mastral
*
,
Sergio
Barbeira-Arán†
and
Martín
Fañanás-Mastral
*
Centro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS), Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain. E-mail: martin.fananas@usc.es
First published on 21st December 2023
Abstract
Bicyclo[1.1.1]pentanes (BCPs) have emerged as an interesting scaffold in drug design. These strained molecules can act as bioisosteres of para-substituted phenyl rings, tert-butyl groups or internal alkynes, leading to drug analogues with enhanced pharmacokinetic and physicochemical properties. Thus, catalytic methodologies for the synthesis of BCPs represent a major goal in modern organic synthesis. In particular, asymmetric transformations that provide chiral BCPs bearing an adjacent stereocenter are particularly valuable to expand the chemical space of this important scaffold. In this article, we discuss the available methodologies for the asymmetric synthesis of α-chiral BCPs, their key mechanistic features and their application in bioisosteric replacements in drug design.
 Irene Sánchez-Sordo | Irene Sánchez-Sordo obtained her PhD from the University of Oviedo (Spain) in 2018 under the supervision of Prof. Pilar Gamasa. During this period, she did a short stay at the group of Prof. Martin Albrecht at the University of Bern (Switzerland). In 2020 she moved to Gakushuin University (Tokyo, Japan) and joined the group of Prof. Takahiko Akiyama as a postdoctoral researcher. In 2021 she moved to the group of Prof. Martín Fañanás-Mastral where she is currently working on the development of methodologies for the stereoselective carboboration of hydrocarbons and the direct functionalization of alkanes. |
 Sergio Barbeira-Arán | Sergio Barbeira-Arán obtained his BSc in chemistry (2020) and MSc (2021) from the University of Santiago de Compostela, where he worked in Cu-catalyzed synthetic transformations under the supervision of Prof. Martín Fañanás-Mastral. Currently, he is a PhD candidate in the Fañanás-Mastral group at CiQUS working on the functionalization of light hydrocarbons. During that time, he did a short stay at the group of Prof. Bill Morandi at ETH Zürich (Switzerland). |
 Martín Fañanás-Mastral | Martín Fañanás-Mastral obtained his PhD at the University of Oviedo (Spain) in 2007. During that time, he did a short stay at the group of Prof. Steven Ley at the University of Cambridge (UK). In 2009 he joined the group of Prof. Ben L. Feringa at the University of Groningen (The Netherlands) as a postdoctoral researcher. In 2014 he moved to CiQUS (University of Santiago de Compostela, Spain), where he leads the “Sustainable Catalysis and Asymmetric Synthesis” group. His current research interests focus on the development of synthetic methods towards the stereoselective carboboration of hydrocarbons and the direct functionalization of alkanes. |
Introduction
In recent years many efforts have been devoted to the design of small-molecule drugs. However, nonspecific interactions with off-target proteins can end up in higher risks of toxicity. In this context, some studies point to the fact that drug development is more likely to be successful for compounds having a small number of aromatic rings and a more-three-dimensional structure.1 In addition, these considerations can enhance the pharmacokinetic and physicochemical properties of drug candidates. In this sense, highly strained carbocycles such as bicyclo[1.1.1]pentanes (BCPs) have attracted much interest since they can act as bioisosteres of para-substituted phenyl rings, tert-butyl groups or internal alkynes (Fig. 1), leading to new drugs with improved aqueous solubility, membrane permeability, and/or metabolic stability.2–7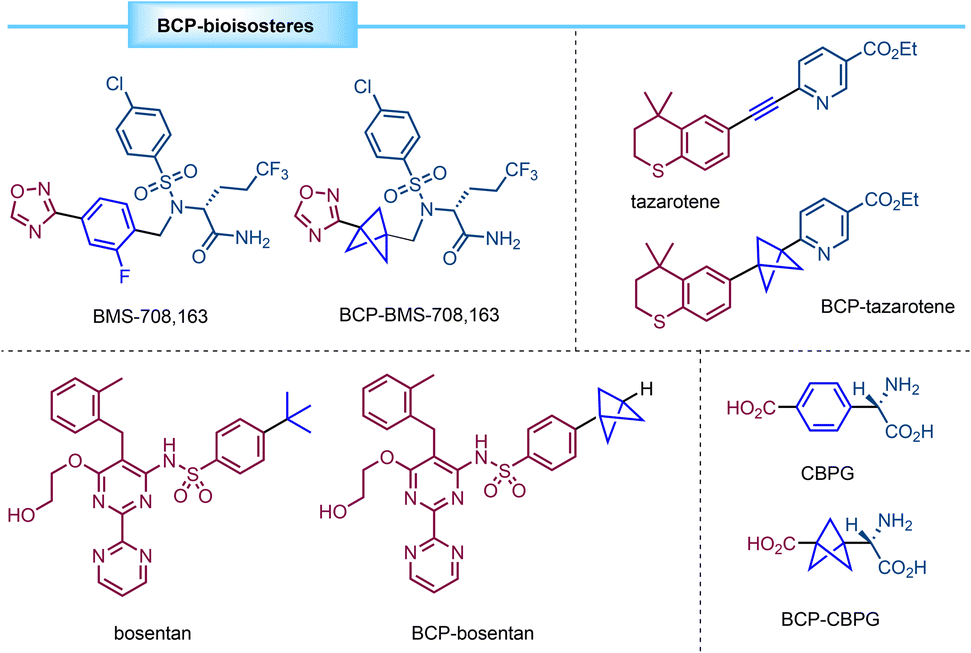 | ||
| Fig. 1 Examples of BCP bioisosteres of different groups. | ||
Bicyclo[1.1.1]pentane (BCP, 1) was first synthesized by Wiberg and co-workers in 1964 by a Na-mediated ring closure of 3-bromocyclobutane-1-methyl bromide (Scheme 1a). At that time, the synthesis of this compound represented a synthetic challenge with the aim of expanding the number of bicyclic small ring compounds.8 However, the chemistry involving this scaffold experienced a major breakthrough when the first synthesis of [1.1.1]propellane (2) was achieved.9 Propellane was first described by Wiberg and Walker in 1982.10 Szeimies and co-workers reported in 1985 a refined methodology which provides [1.1.1]propellane from a readily available precursor (1,1-bis(chloromethyl)-2,2-dibromocyclopropane).11 Moreover, the group of Baran reported that PhLi can promote this transformation with an improved yield and purity.12 In these methodologies, propellane is usually stored in an ethereal solution. Sometimes, due to storage issues and relative instability of [1.1.1]propellane, prefunctionalized disubstituted BCPs such as biacetyl-BCP (3) have acted as precursors for the synthesis of BCP scaffolds (Scheme 1b).13
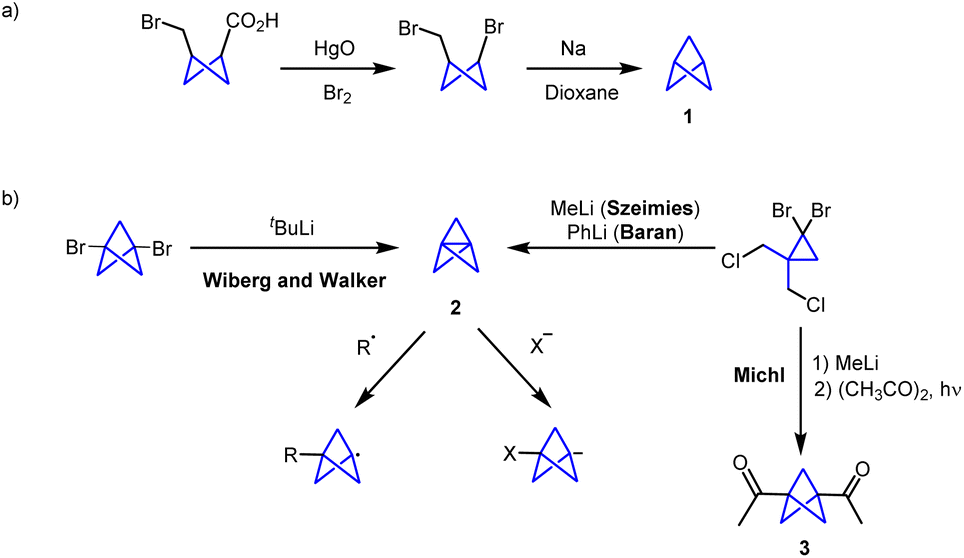 | ||
| Scheme 1 (a) First synthesis of bicyclo[1.1.1]pentane; (b) bicyclo[1.1.1]pentane synthesis from propellane. | ||
The ability to react through its stressed central bond via radical or anionic pathways makes [1.1.1]propellane the most general precursor for the synthesis of BCPs.14–16
Despite the great advances in the development of synthetic methodologies to access BCP derivatives,14 the synthesis of chiral BCPs bearing an adjacent stereocenter has been much less developed. Since chiral centers are a common motif in bioactive molecules, the development of methodologies capable of functionalizing the stressed bond of [1.1.1]propellane in an enantioselective manner affording BCPs with adjacent stereocenters is important to obtain chiral drug analogues. Additionally, these chiral BCPs could be employed as surrogates for benzylic, propargylic or neopentylic stereocenters.
This article covers an overview of the state-of-the-art methodologies for the synthesis of α-chiral BCPs highlighting the major achievements and limitations in order to identify future challenges in the field. We have divided the review considering whether the transformation involves an enantioselective post-functionalization of already prepared BCP derivatives or a direct asymmetric functionalization of the [1.1.1]propellane ring. Furthermore, we have also reviewed other stereoselective methodologies that allow access to other types of chiral BCPs.
Enantioselective post-functionalization of BCP derivatives
Synthesis of chiral BCP-α-amino acids
The first strategy for the synthesis of α-chiral bicyclo[1.1.1]pentanes was described by Pellicciari and co-workers and was based on a chiral resolution strategy for the synthesis of enantioenriched BCP-α-amino acids.7 The synthetic route starts with a Strecker reaction in which the aldehyde (4), previously synthesized in seven steps from propellane, is reacted with (R)-2-phenylglycinol, which acts as a chiral auxiliary. The reaction affords a Schiff base that allows the attack of trimethyl silyl cyanide (TMSCN) resulting in the formation of a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomers 5 and 6 (Scheme 2).5 Both diastereomers were separated by medium pressure liquid chromatography (MPLC). Finally, oxidative cleavage with Pb(OAc)2 followed by acid hydrolysis and ion exchange chromatography on Dowex 50 × 2–200 resin provided, respectively, α-chiral BCPs 7 and 8. An important feature of this work is that 7 is the first example reported in the literature of a BCP analogue of a bioactive molecule. In particular, 7 showed enhanced selectivity and activity compared to its analogue (S)-(4-carboxyphenyl)glycine (CBPG) which is an antagonist of the metabolic glutamate receptor mGluR1.
:1 mixture of diastereomers 5 and 6 (Scheme 2).5 Both diastereomers were separated by medium pressure liquid chromatography (MPLC). Finally, oxidative cleavage with Pb(OAc)2 followed by acid hydrolysis and ion exchange chromatography on Dowex 50 × 2–200 resin provided, respectively, α-chiral BCPs 7 and 8. An important feature of this work is that 7 is the first example reported in the literature of a BCP analogue of a bioactive molecule. In particular, 7 showed enhanced selectivity and activity compared to its analogue (S)-(4-carboxyphenyl)glycine (CBPG) which is an antagonist of the metabolic glutamate receptor mGluR1.
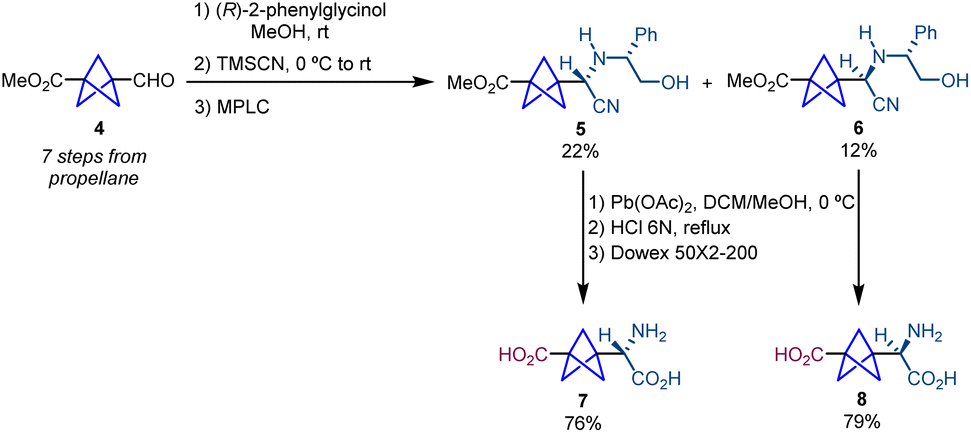 | ||
| Scheme 2 Synthesis of BCP-CBGP analogues 7 and 8via chemical resolution. | ||
A similar methodology based on the use of (R)-phenylglycinol as a chiral auxiliary in a Strecker reaction was employed by Pritz and Pätzel in the synthesis of amino acid 14, a BCP analogue of (S)-(4-tert-butylphenyl)glycine (Scheme 3).17 The synthesis starts with a 1,3-functionalization of propellane to afford BCP 9. Reduction using LiAlH4 followed by a Dess–Martin oxidation of the resulting alcohol gives rise to 11 which is involved in Strecker-type amino acid synthesis providing 12 and 13. Chromatographic separation of diastereomers, removal of the chiral auxiliary and subsequent hydrolysis give rise to the target product 14. It is important to note that the Fmoc-protected amino acid 14 was used for the preparation of cyclic and linear peptides as replacements of tryptophan residues. The antimicrobial activity of these BCP-containing peptides was determined, which showed an enhancement compared to that of the unmodified molecules.
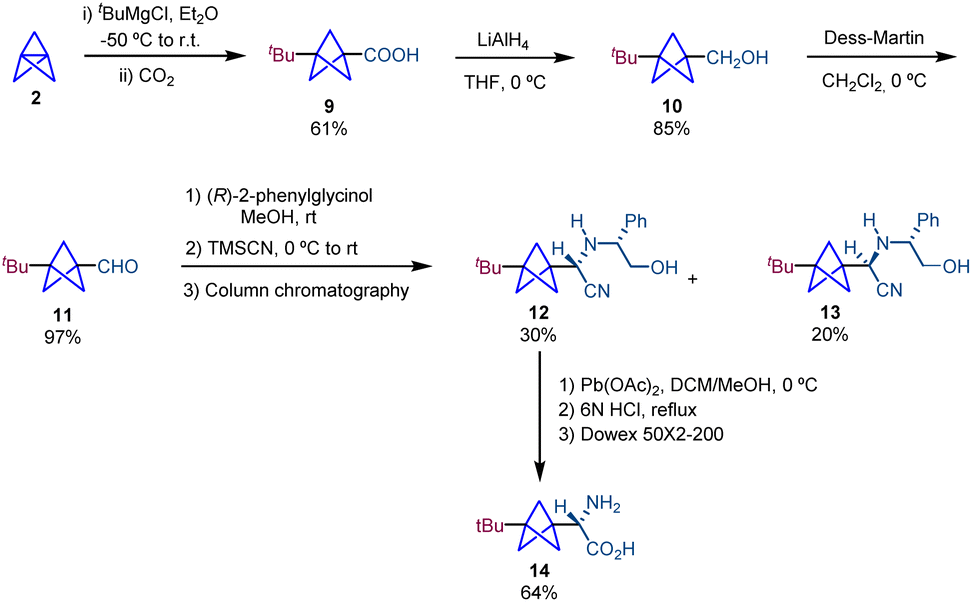 | ||
| Scheme 3 Synthesis of (S)-((4-tBu-phenyl)glycine) BCP analogue 14via resolution. | ||
Following on the use of this Strecker/chiral resolution strategy, Pellicciari and co-workers published in 2009 the synthesis of both enantiomers of BCP-homoCBGP and BCP-homoPBGP (Schemes 4 and 5).4 In this case, the preparation of BCP-homoCBGP analogues (23 and 24) requires the insertion of one methylene unit between the carboxylate moiety and the bicyclopentane core in substrate 15. For this purpose, BCP 15 is homologated by a modified Arndt–Eistert reaction18 which involves the formation of an α-diazomethyl ketone (16) followed by a Wolff rearrangement. This sequence gives rise to product 17 which is converted into diester 18 which subsequently undergoes alkaline hydrolysis giving rise to product 19. Reduction of the carboxylic acid 19 to the corresponding primary alcohol and subsequent reoxidation afford aldehyde 20. As reported previously, this aldehyde is involved in a Strecker-type resolution which after acid hydrolysis provides final products (S)-23 and (R)-24 (Scheme 4).
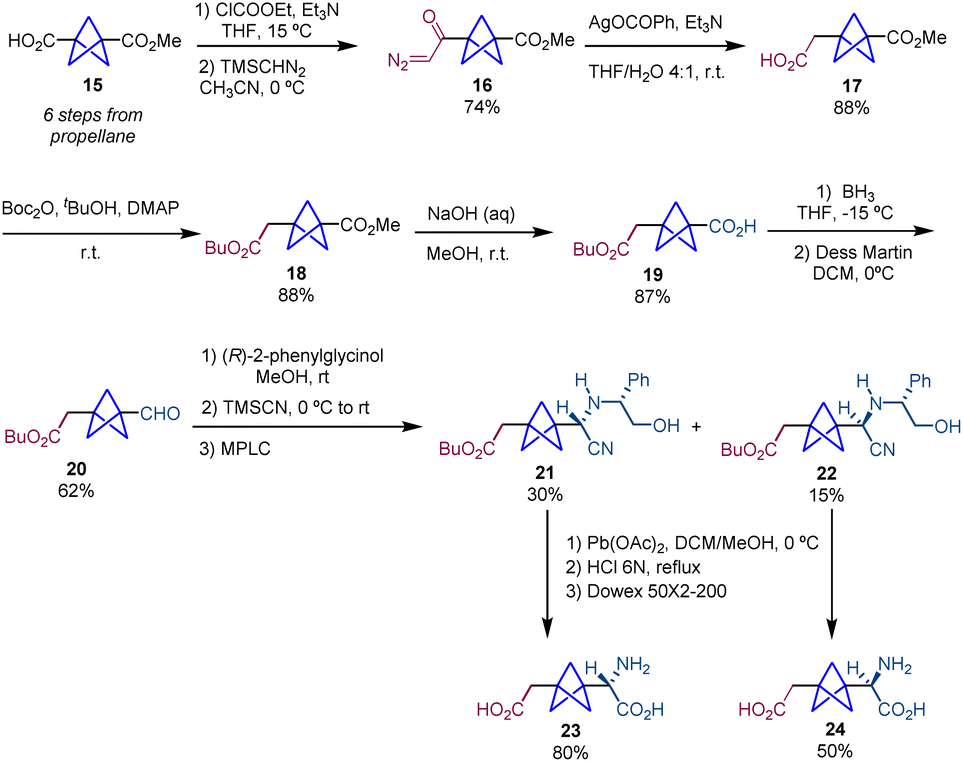 | ||
| Scheme 4 Synthesis of BCP-homoCBGP analogues 23 and 24via chemical resolution. | ||
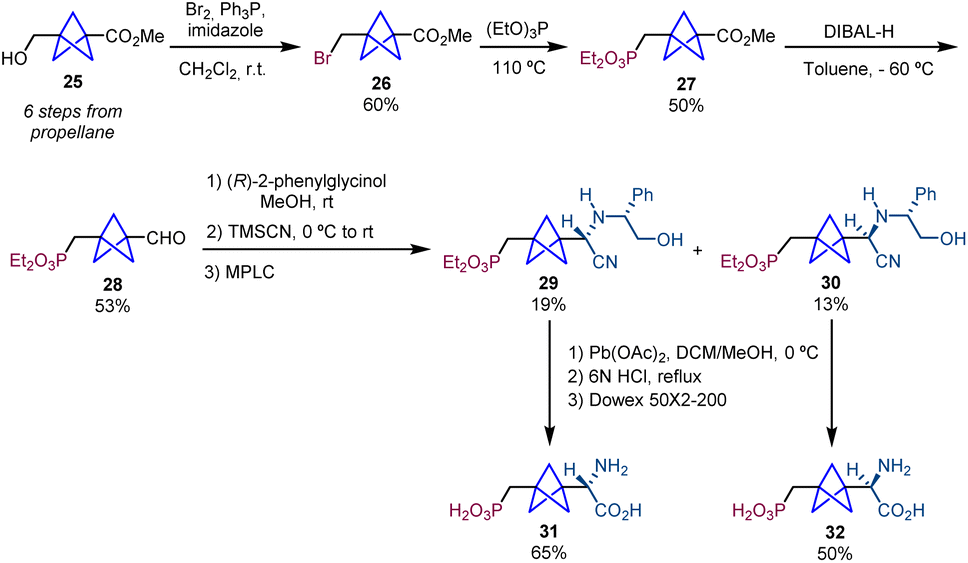 | ||
| Scheme 5 Synthesis of BCP-homoPBGP analogues 31 and 32via chemical resolution. | ||
The synthesis of the phosphonic acid BCP-homoPBGP, (S)-homo-PBPG (31) and (R)-homoPBPG (32), was achieved using the same strategy starting from methyl 3-(hydroxymethyl)bicyclo[1.1.1]pentane-1-carboxylate (25).7 The sequence involves the formation of bromide 26 by treatment of 25 with Br2 and triphenylphosphine in the presence of imidazole, a subsequent Arbuzov reaction affording product 27 and final reduction that provides aldehyde 28 which is then used in the Strecker/chiral resolution strategy (Scheme 5).
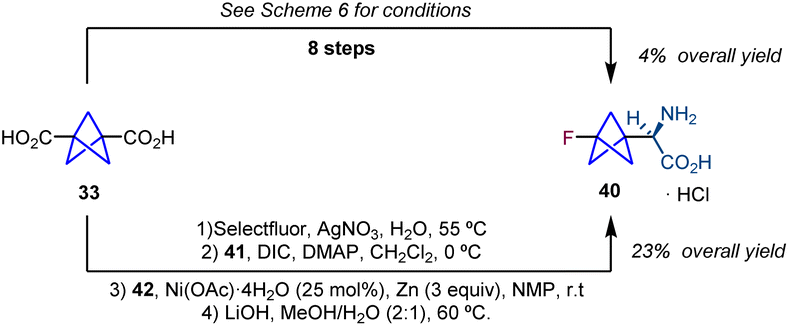
The synthesis of chiral BCP α-amino acids was further pursued by the groups of Ulrich and Mykhailiuk. They reported the synthesis and application of a monofluorinated aliphatic amino acid (3-fluorobicyclo[1.1.1]pentyl)glycine (F-Bpg, 40) with the aim of using it in the determination of interatomic distances in membrane-active peptides by solid-state 19F NMR spectroscopy.19 Their synthetic sequence starts with diacid 33,20via the transformation of one of the carboxylic acid groups into a fluorine atom using Selectfluor and AgNO3.21 Carboxylic acid 34 is then converted into ester 35, and a subsequent reduction provides aldehyde 36 which is used in a Strecker reaction affording diastereomers 37 and 38. Oxidative cleavage of the chiral auxiliary and the following hydrolysis and Boc protection lead to protected amino acid 39. Cleavage of the N-Boc group provides the target product 40 (Scheme 6).
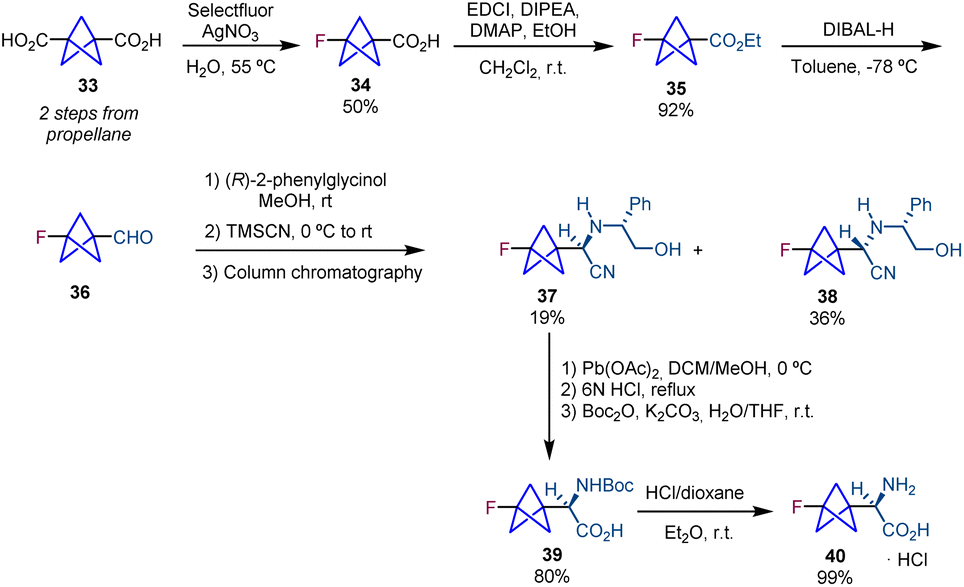 | ||
| Scheme 6 Synthesis of monofluorinated aliphatic BCP α-amino acid 40. | ||
In 2018, Baran and co-workers reported a new strategy for the synthesis of chiral BCP α-amino acids that significantly shortens the number of steps required by the Strecker-based methodologies. The protocol involves a nickel-catalyzed radical cross-coupling between a redox active ester (41) and a chiral imine radical acceptor (42) (Scheme 7).22
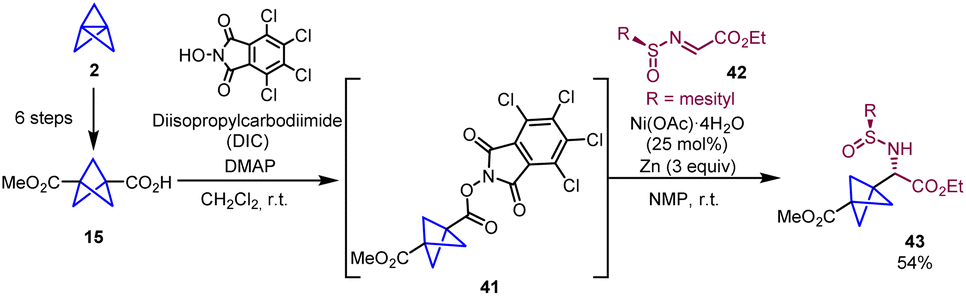 | ||
| Scheme 7 Synthesis of chiral BCP α-amino acid 43. | ||
This reduction in steps was illustrated by the synthesis of amino acid 40 which was previously synthesized from diacid 33 in 8 steps improving the overall yield of the synthetic sequence (Scheme 8).
 | ||
| Scheme 8 Improved synthetic sequence for chiral BCP α-amino acid 40. | ||
Synthesis of α-chiral BCPs using an oxazolidinone chiral auxiliary
In 2019, Anderson and co-workers reported a protocol for obtaining chiral BCPs with an adjacent stereocenter based on the use of a chiral oxazolidinone as a chiral auxiliary (Scheme 9).23 The synthesis of the required chiral intermediate 47 was inspired by previous reports of the same research group in which highly functionalized disubstituted BCPs were obtained via atom transfer radical addition to [1.1.1]propellane.24 Using triethylborane as an initiator, radicals derived from alkyl halides were able to perform the ring opening of [1.1.1]propellane giving rise to 1-halo-3-substituted BCPs. This methodology, followed by a deiodination step using (Me3Si)3SiH (TTMSS) afforded BCP 47. The authors showed that this compound can be alternatively accessed on a multigram scale by the acylation of oxazolidinone 46 with acid chloride 45. Formation of the corresponding enolate and trapping with several electrophiles provided the corresponding chiral α-BCPs in good yields and excellent diastereoselectivity. In this process, the substitution pattern of the chiral auxiliary 46 proved to be crucial for the efficiency of the alkylation. The use of 4-substituted oxazolidin-2-one derivatives furnished the products in modest yields and/or with low diastereomeric ratios, while SuperQuat oxazolidinones (4-substituted 5,5-dimethyloxazolidine-2-one) provided the optimal conditions.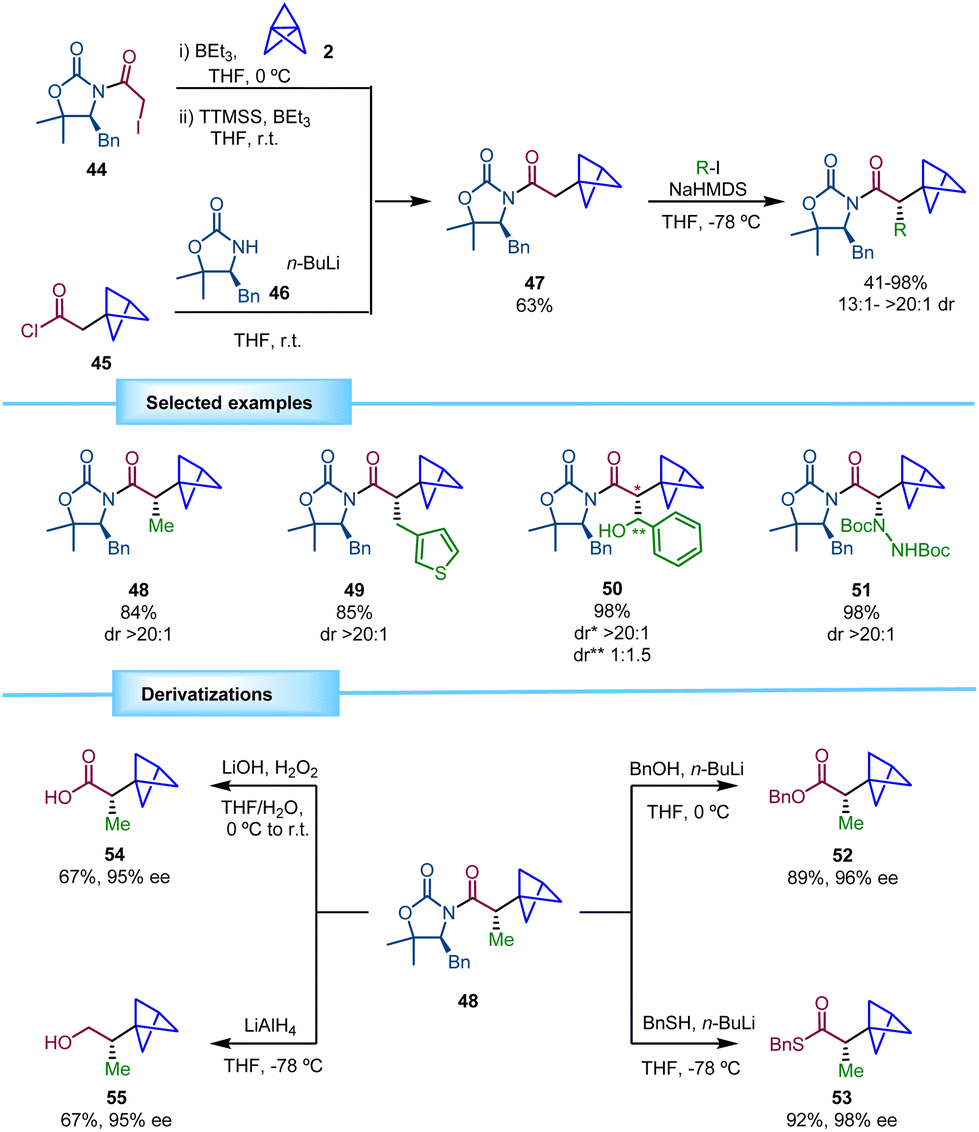 | ||
| Scheme 9 Synthesis of α-chiral BCPs by using a chiral oxazolidinone auxiliary and synthetic transformations performed with 48. | ||
The scope of the reaction was evaluated using a wide range of electrophiles leading to products with high yields and excellent diastereoselectivities. Alkyl halides bearing saturated and unsaturated chains performed well in the reaction. Heteroaromatic substituents in the alkyl halide were also tolerated. Direct introduction of heteroatoms using different electrophiles was also possible. Additionally, the employment of benzaldehyde led to an aliphatic alcohol (50) with a new stereocenter albeit with low diastereoselectivity.
In addition, the cleavage of the chiral auxiliary by transforming it into a variety of functional groups was demonstrated. Transesterification (52, 53), hydrolysis (54) and reduction (55) provided excellent results in terms of yields and enantioselectivities (Scheme 9).
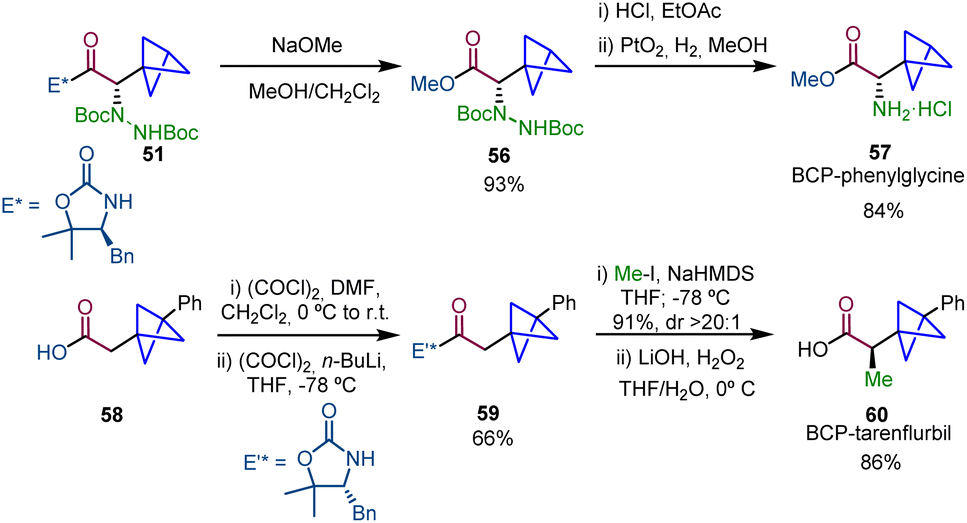
Interestingly, this strategy was applied to the synthesis of α-chiral BCP analogues of bioactive compounds (Scheme 10). Methanolysis of 51 afforded ester 56 and the subsequent Boc deprotection and Pt-catalyzed hydrogenation25,26 gave rise to the BCP analogue of L-(+)-α-phenylglycine methyl ester hydrochloride (57). In addition, the synthesis of drug analogue BCP-tarenflurbil 60 was achieved from carboxylic acid 58 by formation of acyl oxazolidinone 59, subsequent diastereoselective alkylation and final hydrolysis.
 | ||
| Scheme 10 Synthesis of BCP-analogues 57 and 60. | ||
Asymmetric transformation of boron containing BCPs
In 2020, Aggarwal and co-workers described how BCP-Grignard intermediates (61), generated from the reaction of [1.1.1]propellane (2) and an aryl Grignard reagent using conditions reported by Knochel,3 can be successfully employed in a Zweifel olefination to produce alkenyl-substituted BCPs via reaction with an alkenyl boronate and subsequent treatment with iodine and sodium hydroxide (Scheme 11).27 This alkenyl BCP was further functionalized through a copper-catalyzed enantioselective hydroboration,28 giving rise to α-chiral BCP 64 with excellent yield and enantioselectivity.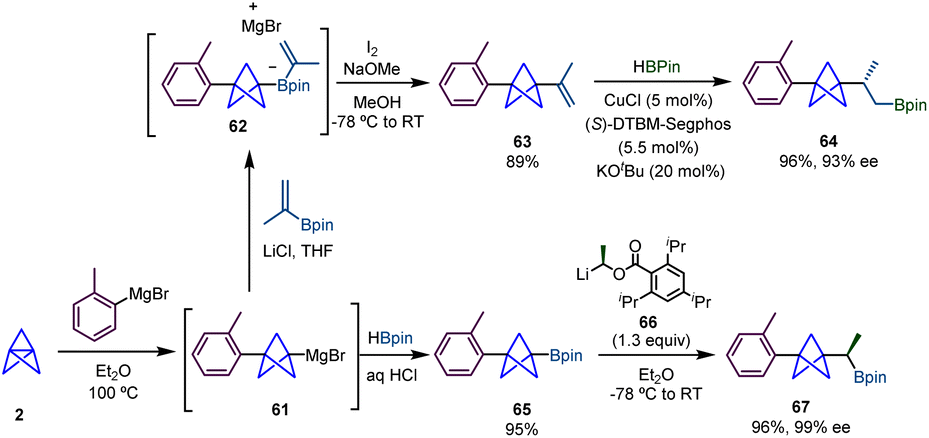 | ||
| Scheme 11 Synthesis of α-chiral BCPS via 1,2-metallate rearrangements of BCP boronate complexes. | ||
In the same work, the authors extended the range of BCP derivatives accessible by 1,2-metallate rearrangements by the preparation of BCP-boronic ester 65 from a reaction between BCP Grignard and pinacol borane. Among different types of transformations, BCP boronic ester 65 proved to be efficient to undergo homologation via a lithiation–borylation reaction with enantioenriched carbenoid 66,29 to provide boronic ester 67 which was obtained in very good yield and with high enantioselectivity.
Uchiyama and co-workers reported an alternative method for the difunctionalization of [1.1.1]propellane via an additive-free silaboration reaction, which allows concomitant introduction of boron and silicon groups into the BCP scaffold.30 The reaction of [1.1.1]propellane (2) towards Me2PhSi-Bpin gives rise to silaborate-BCP 68 (Scheme 12). This reaction is enhanced by UV-light irradiation suggesting a radical chain pathway initiated by oxygen in the triplet state which was further confirmed by radical trapping experiments and DFT calculations. Moreover, 68 was obtained on a gram scale in a single step without column chromatography purification. Additionally, it is easy to store and handle, making it a versatile synthetic platform for BCP scaffolds. The synthetic utility of the silaboration adduct was evaluated by different C–B bond transformations. Among them, the stereospecific installation of a chiral C(sp3) unit with high enantiospecificity was accomplished via a lithiation–borylation homologation strategy using chiral reagent 69.31 The obtention of 70 demonstrates the ability of the silaboration adduct to facilitate selective and controlled chemical transformations of BCPs (Scheme 12).
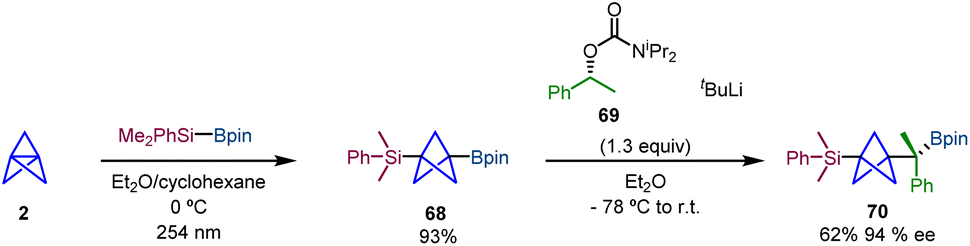 | ||
| Scheme 12 Silaboration of [1.1.1]propellane and subsequent enantioselective homologation via a lithiation–borylation reaction. | ||
Synthesis of α-chiral BCPs by asymmetric transfer hydrogenation of BCP ketones
Another strategy for the enantioselective synthesis of α-chiral BCPs is the asymmetric transfer hydrogenation (ATH) of BCP-ketones, as reported by Wills in 2021.32 The process relies on the formation of the BCP ketone from trapping of the BCP Grignard with an aldehyde and subsequent oxidation with MnO2. Attempts to directly generate the ketone by trapping with PhCOCl were not productive due to the formation of significant amounts of benzophenone, impossible to separate from the ketone product.The obtained ketones were reduced in an asymmetric fashion to their corresponding alcohols by means of an ATH using chiral ruthenium catalyst 71 (Scheme 13). This methodology was successfully extended to several substrates, including aromatic rings and heterocycles, demonstrating remarkable tolerance towards different substitution patterns and functional groups. In addition, ketones flanked by BCP and alkyne moieties were reduced with good yields and excellent enantioselectivities.
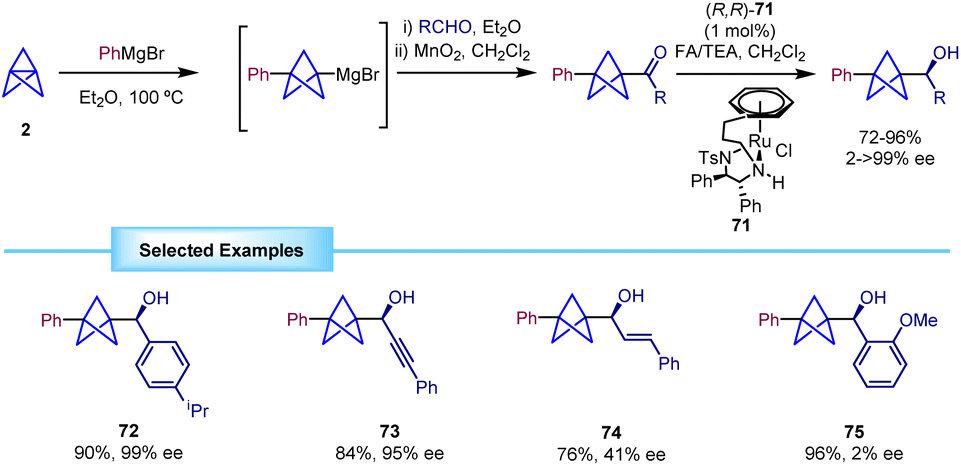 | ||
| Scheme 13 Synthesis of α-chiral BCPs by ATH. | ||
In general, this strategy provided the products with high enantioselectivity. However, a low enantiomeric excess was observed when substrates bearing an alkenyl substituent (74) or an ortho-substituted aryl ring (75) were employed.
The synthetic utility of the ATH methodology was demonstrated with the synthesis of the BCP analogue of (S)-neobenodine in which the BCP moiety acts as a bioisostere of the phenyl ring (Scheme 14). In this case, ketone 77 was generated from BCP-iodide (76) by generation of an organolithium intermediate via lithium–halogen exchange, trapping with benzaldehyde and subsequent oxidation. ATH was performed using the (S,S)-enantiomer of catalyst 71 to obtain the BCP with an analogous configuration of neobenodine.
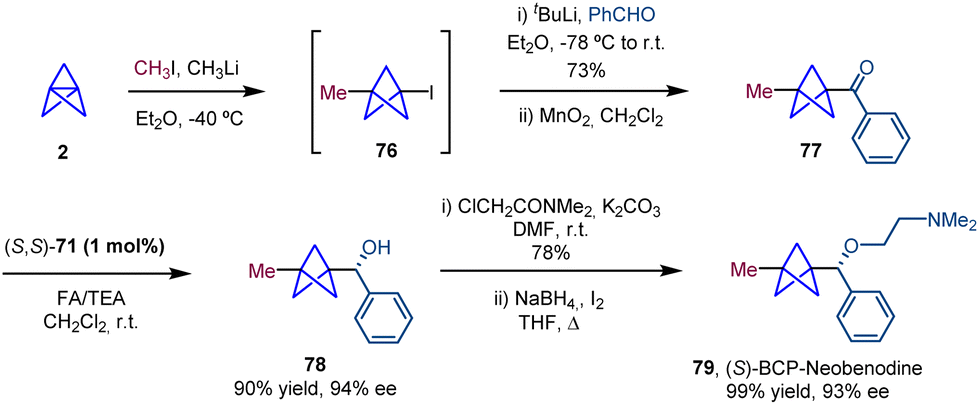 | ||
| Scheme 14 Synthesis of (S)-BCP-neobenodine. | ||
Synthesis of chiral BCPs by enantioselective rhodium-catalyzed carbene C–H insertion
Intermolecular metal carbene insertion into C–H bonds is a powerful tool in the field of C–H functionalization.33 Davies and co-workers successfully achieved the site-selective and stereoselective activation of tertiary C–H bonds using this strategy by means of a chiral dirhodium catalyst.34 Based on this background, they reported in 2020 a catalytic methodology that allows the transformation of monosubstituted BCPs into 1,3-disubstituted BCPs bearing an α-stereocenter via an intermolecular insertion of a stabilized diazo compound facilitated by a dirhodium catalyst (Scheme 15).35 The methodology showed broad tolerance towards diazo compounds bearing aromatic rings featuring different substitution patterns and electronic properties. In addition, different prefunctionalized BCPs containing aryl, benzyl and functionalized alkyl groups were used, which led to the corresponding chiral products with moderate to good enantioselectivity.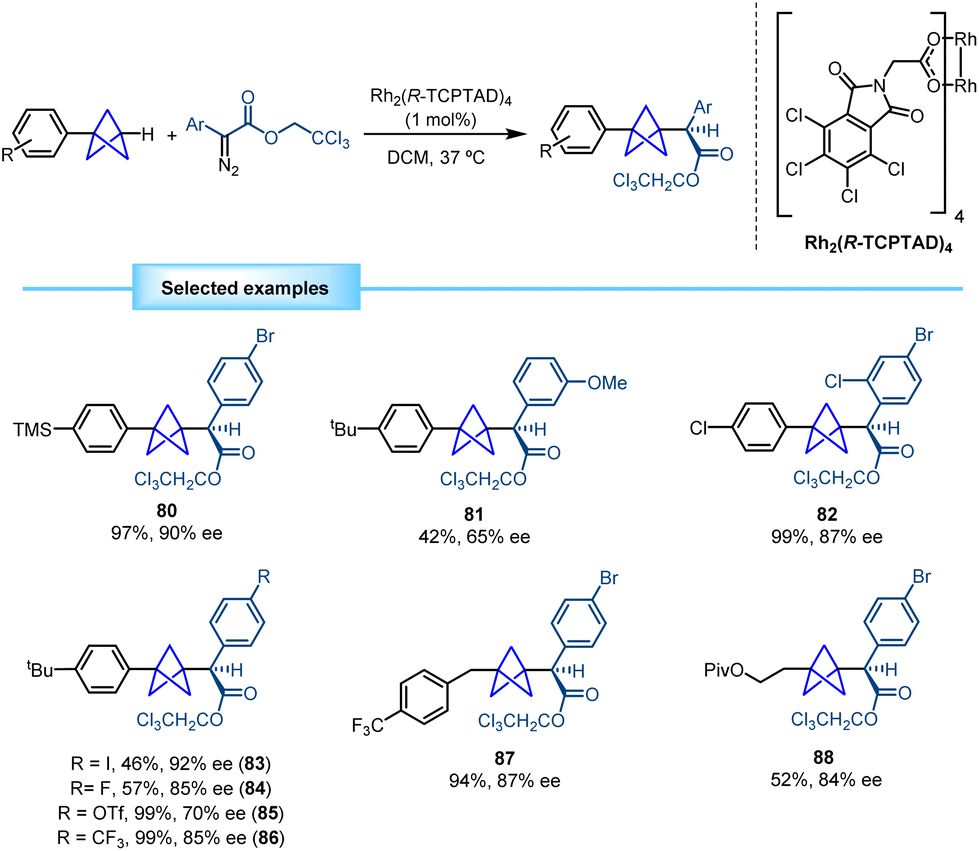 | ||
| Scheme 15 Synthesis of α-chiral BCPs by enantioselective rhodium catalyzed C–H activation. | ||
Moreover, this methodology was applied to the synthesis of compounds 92 and 93, which are BCP analogues of bioactive compounds 94 (anticancer screen) and 95 (antagonistic activity on human α4β2 nAChR) (Scheme 16). Pharmacokinetic properties of these new products such as solubility (measured by the decrease in lipophilicity) and microsomal clearance were determined, concluding that increased three-dimensionality can improve absorption, distribution, metabolism and excretion properties.
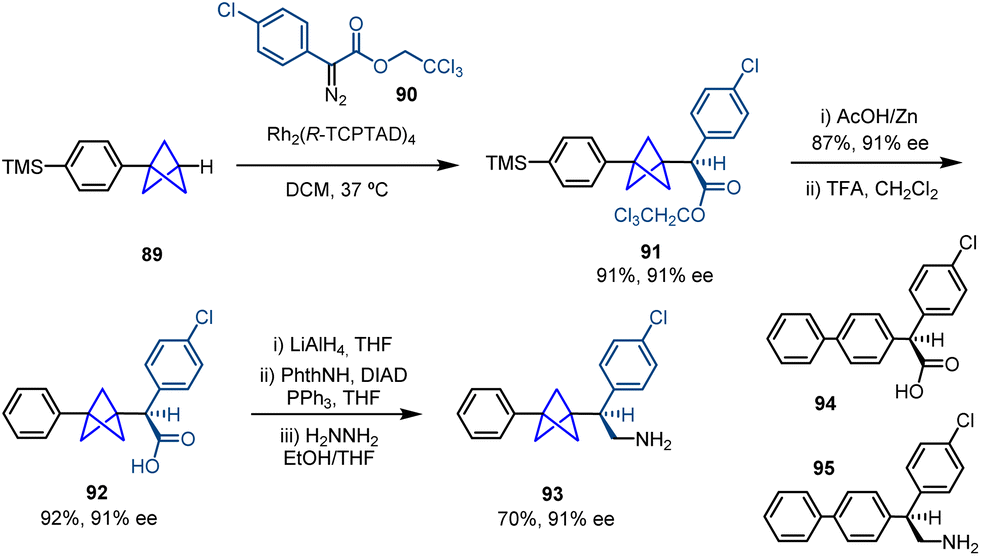 | ||
| Scheme 16 Synthesis of BCP analogues 92 and 93via Rh-catalyzed enantioselective carbene C–H insertion. | ||
Synthesis of BCPs bearing α-quaternary centers via synergistic organophotoredox and HAT catalysis
In 2021, the research team led by Anderson identified an underexplored area in the preparation of α,α-dicarbonyl tertiary radicals and envisioned an opportunity to develop an innovative new methodology that tackles the creation of quaternary centers adjacent to the BCP skeleton.36To address this challenge, they proposed a tricomponent strategy based on photoredox catalysis to obtain tertiary α,α-dicarbonyl radicals via one-electron oxidation of β-ketoesters. The addition of these radicals to [1.1.1]propellane (2) resulted in the formation of a radical species containing two contiguous quaternary centers that engaged in a thiol-mediated hydrogen atom transfer (HAT) process to provide a BCP bearing an α-quaternary center (Scheme 17).37
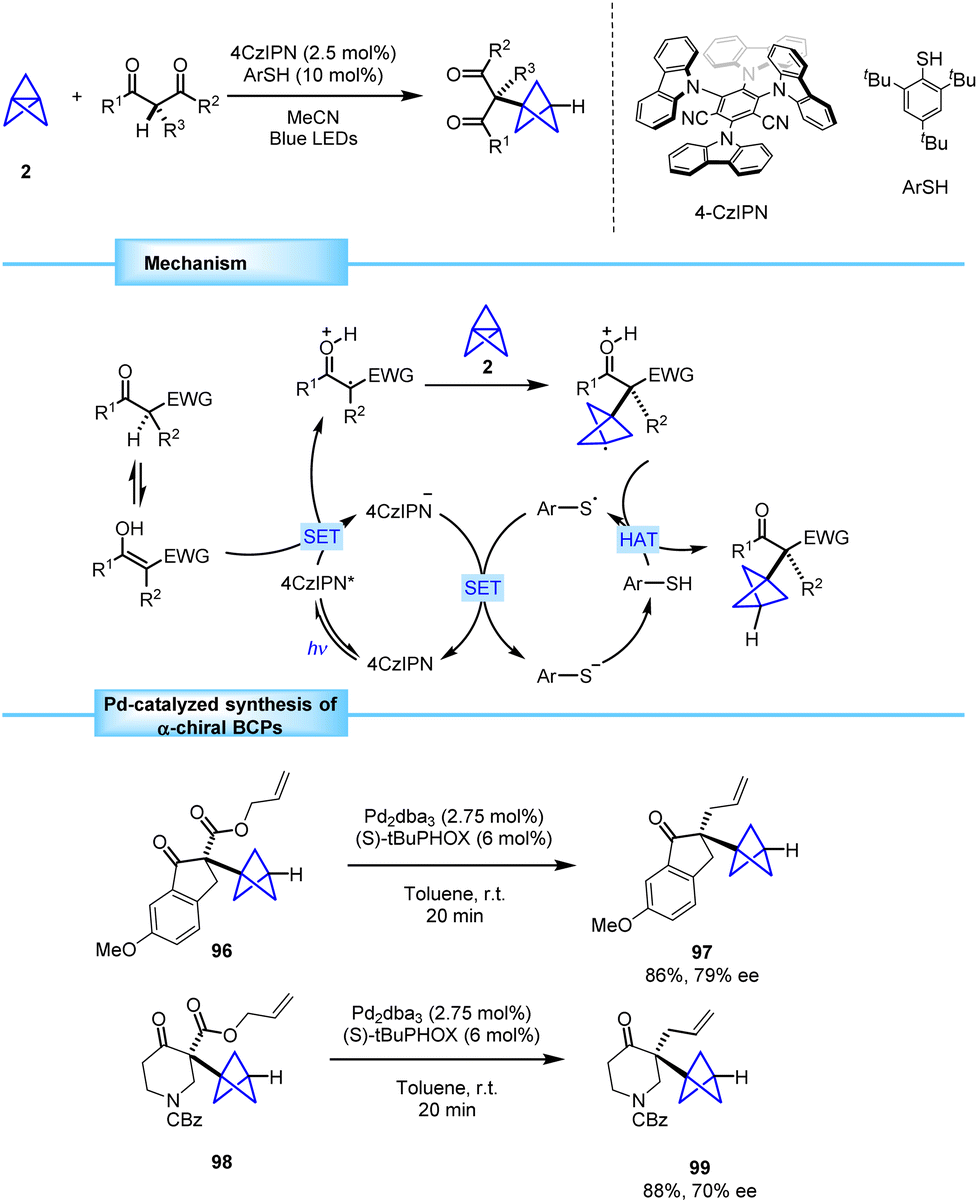 | ||
| Scheme 17 Synthesis of α-chiral BCPs bearing quaternary centers via photoredox/HAT catalysis and subsequent Pd-catalyzed asymmetric decarboxylative allylation. | ||
The resulting products 96 and 98 were demonstrated to efficiently participate in a Pd-catalyzed decarboxylative allylation that provided enantioenriched BCPs 97 and 99 in good yields and with moderate enantioselectivity. Noteworthily, this methodology represents the first example of asymmetric synthesis of α-chiral BCPs bearing quaternary centers.
Direct catalytic asymmetric functionalization of [1.1.1]propellane
Synthesis of α-chiral BCPs by diastereoselective radical functionalization of [1.1.1]propellane
In 2018 Shenvi and co-workers reported a catalytic radical hydrofunctionalization of olefins based on a metal-hydride hydrogen atom transfer (MHAT) process that couples the alkene with a radicalophilic electrophile.38 In a single example, Shenvi showed how the central bond of [1.1.1]propellane behaves as an alkene in this process and the catalytically generated BCP radical can be coupled with chiral sulfinimine 100 with excellent diastereoselectivity (Scheme 18).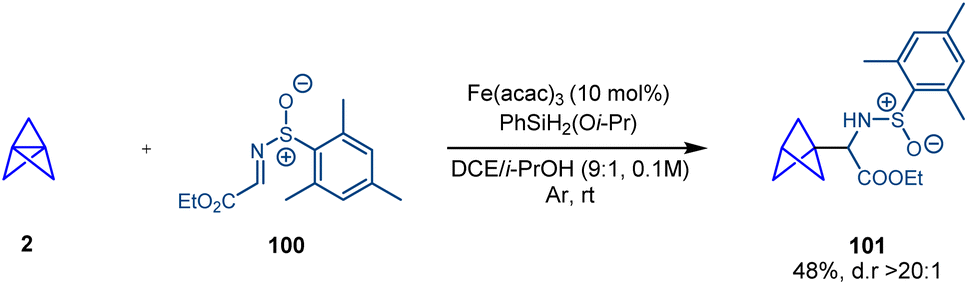 | ||
| Scheme 18 Radical hydrofunctionalization of [1.1.1]propellane using metal-hydride hydrogen atom transfer (MHAT) catalysis. | ||
In 2022, Walsh and co-workers devised a novel methodology for the synthesis of bicyclo[1.1.1]pentane benzylamine derivatives using a strategy similar to the one previously described by Shenvi. Conditions for the MHAT process diverged, with manganese serving as the metallic source instead of iron in this case. The reaction proved to be efficient with different chiral aryl sulfinimines, furnishing the corresponding monosubstituted BCP benzylamines in good yields with excellent diastereoselectivity. Furthermore, the methodology demonstrated tolerance towards heterocyclic compounds (Scheme 19).39 The reaction is proposed to start with the formation of a manganese hydride through reaction between the manganese catalyst and the hydrosilane. The metal hydride engages [1.1.1]propellane in a hydrogen atom transfer (HAT), leading to the reduction of the catalyst and the formation of the BCP radical. This radical couples with the sulfinimine, generating a nitrogen-centered radical which is subsequently reduced via a single electron transfer (SET) that regenerates the catalyst and forms an anion that yields the final BCP product by protonation.
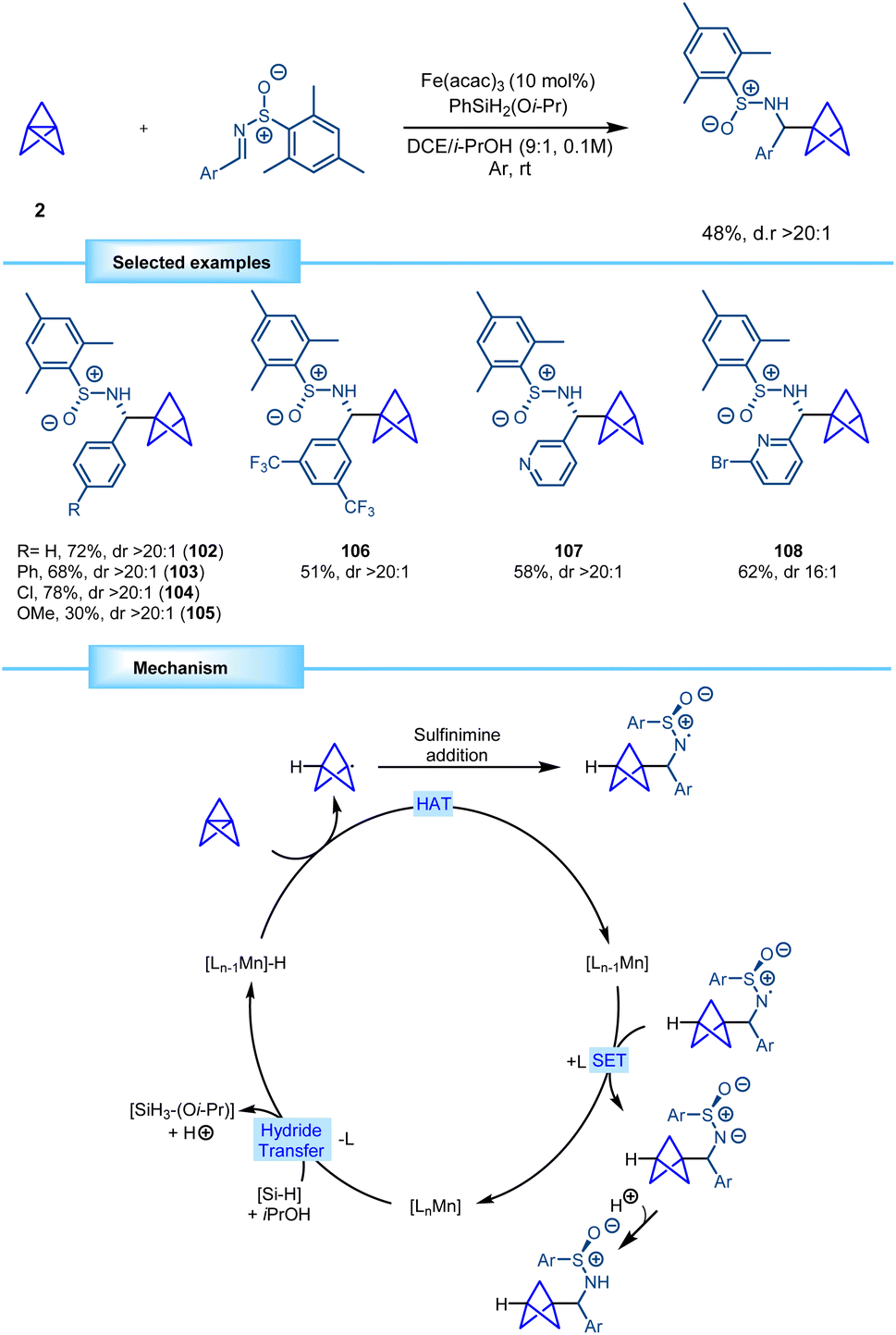 | ||
| Scheme 19 Diastereoselective synthesis of chiral BCP benzylamines via MHAT catalysis. | ||
Walsh and co-workers showed how the chiral BCP sulfinamides can be easily deprotected by hydrolysis to yield the corresponding enantioenriched BCP benzylamine derivatives, as exemplified by the synthesis of 109 (95% ee). Additionally, 4-chloro substituted substrate 110 was further functionalized to access the core structure of various analogues of important pharmaceuticals, such as levocetirizine, meclizine, and hydroxyzine (Scheme 20).
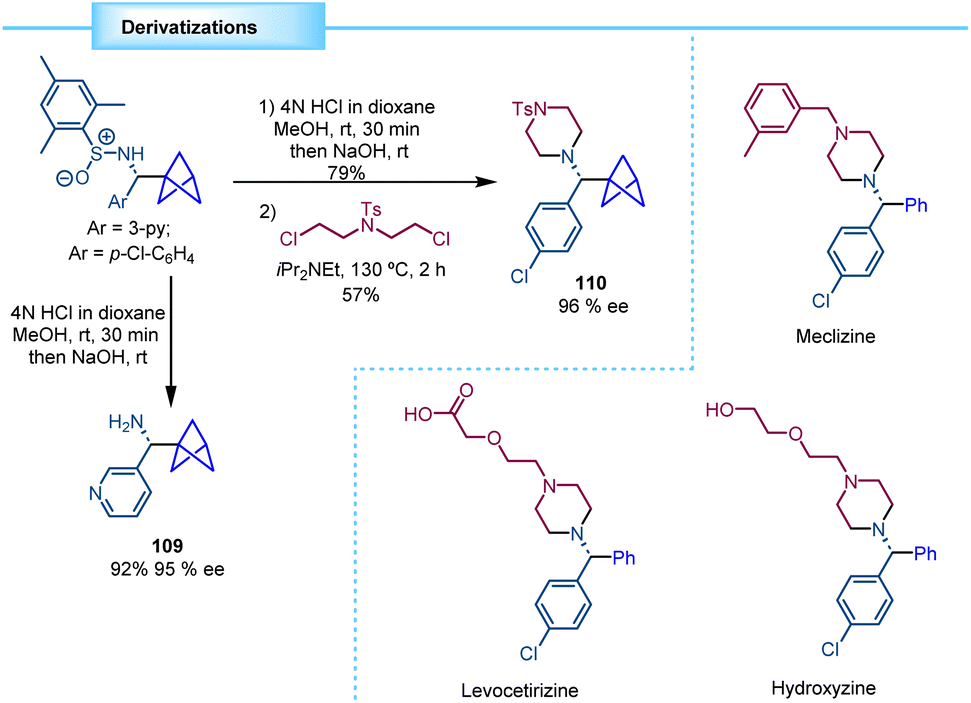 | ||
| Scheme 20 Diastereoselective synthesis of chiral BCP benzylamines via MHAT catalysis. | ||
In 2023, Molander reported a photocatalytic methodology for the synthesis of BCP alkyl amines through a multicomponent functionalization of [1.1.1]propellane (Scheme 21).40 This method, when compared to previous approaches, demonstrates the ability to achieve disubstitution on the BCP scaffold. The strategy employed in this study involved the generation of nucleophilic alkyl radicals by oxidation of trifluoroborate and fluoroalkylsulfinate salts with an excited photocatalyst. These alkyl radicals readily engage in addition reactions at the central bond of the propellane leading to a BCP radical that can be efficiently trapped by imines, sulfinimines, and sulfonimines. Notably, the use of enantiopure N-mesitylsulfinimines produced the corresponding chiral BCPs with excellent diastereoselectivity, thus resulting in a new method for the synthesis of enantioenriched α-chiral BCP amines.
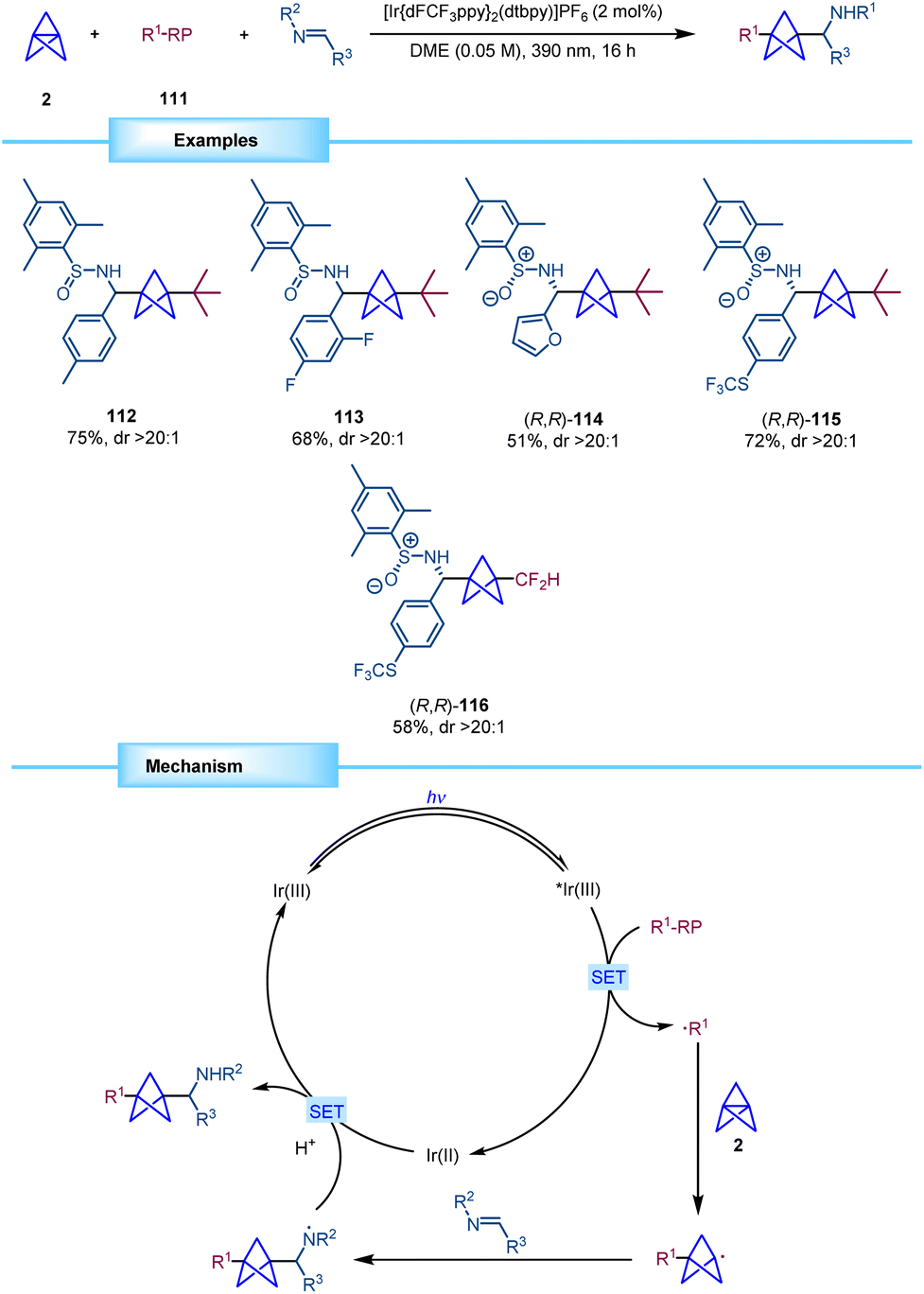 | ||
| Scheme 21 Diastereoselective multicomponent radical difunctionalization of [1.1.1]propellane. | ||
Synthesis of α-chiral BCPs by transition-metal free multi-component difunctionalization of [1.1.1]propellane
Molander and co-workers reported a novel strategy for the synthesis of synthetically versatile BCP boronates based on a transition metal free three-component reaction (Scheme 22).41 In this process, radicals derived from either redox active esters or organic halides add to [1.1.1]propellane leading to a BCP radical that further engages in a polarity-matched borylation step. Notably, the reaction proved to be stereospecific with chiral secondary radical precursors leading to the corresponding α-chiral BCPs with excellent diastereoselectivity. Mechanistically, in the reaction with redox active esters, B2pin2 serves as a twofold reagent, being involved in the radical formation through a SET process between the excited redox active ester and a phthalimide-B2pin2 adduct and acting as the BCP radical acceptor. For organohalides, photoexcitation promotes the C–X bond homolysis leading to an alkyl radical that gets trapped by [1.1.1]propellane providing the BCP radical that adds to Suginome's reagent (Me2PhSi-Bpin). This borylation step generates a silyl radical that propagates the chain reaction by a halogen atom transfer (XAT) event.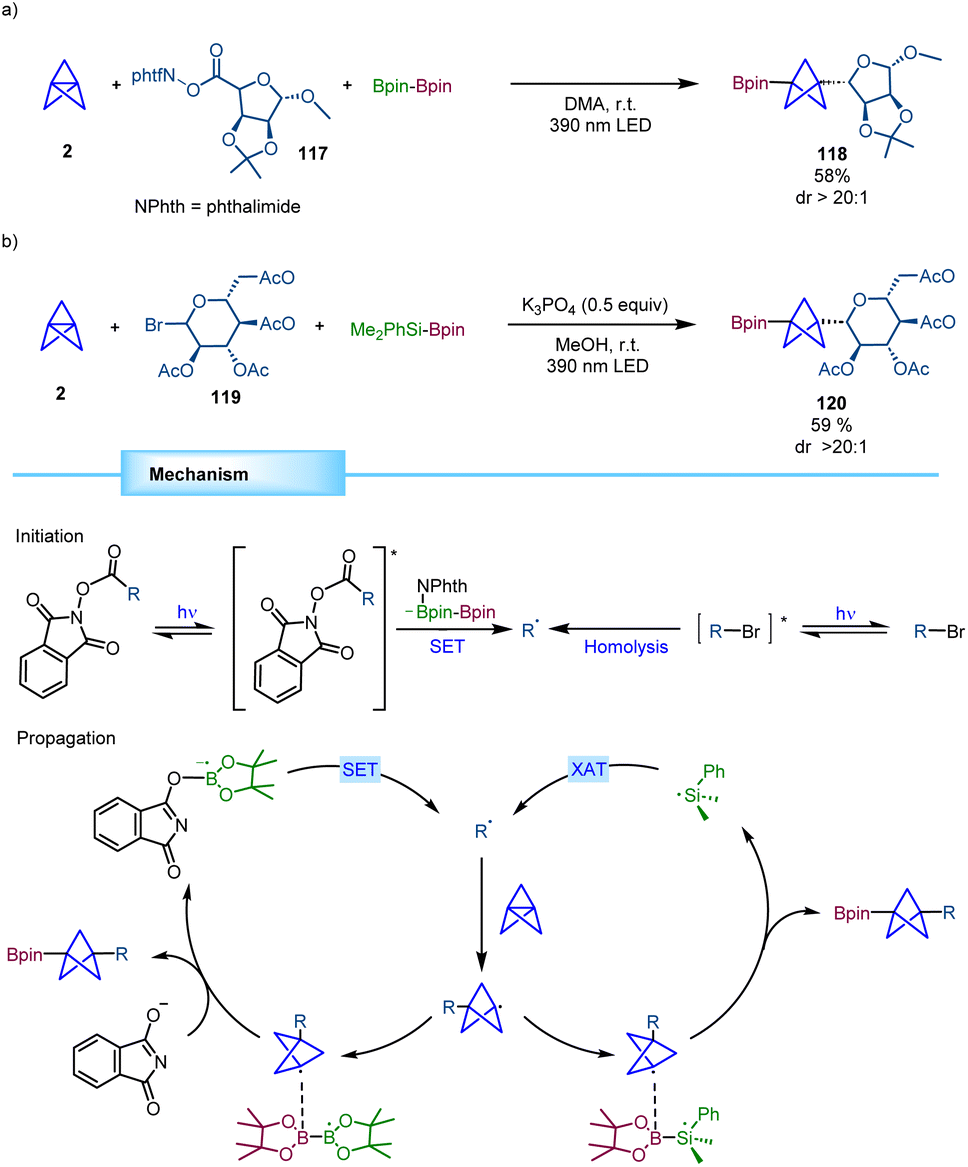 | ||
| Scheme 22 Stereospecific three-component transition metal-free radical difunctionalization of [1.1.1]propellane. | ||
Synthesis of α-chiral BCPs by iridium-catalyzed asymmetric allylic substitution
Aggarwal and co-workers developed in 2020 a direct methodology for the synthesis of BCPs with an adjacent chiral center based on a multicomponent iridium-catalyzed asymmetric allylic substitution (Scheme 23).42 The reaction of [1.1.1]propellane with the corresponding Grignard reagent and subsequent transmetallation employing ZnCl2 afford a BCP–zinc complex which is reacted with a racemic allylic carbonate in the presence of a chiral iridium/phosphoramidite catalyst. Of note, the direct use of the BCP-Grignard led to significantly diminished regio- and enantioselectivity. Despite the high steric hindrance of the BCP nucleophile, it was found to be compatible with the bulky iridium(π-allyl) intermediate providing branched allylic products with high regio- and enantioselectivity. This methodology tolerates a wide variety of cinnamyl carbonates with different substituents such as halide, trifluoromethyl phenylsulfonyl, ester and alkoxy groups. Substrates derived from nitrogen heterocycles, including quinolines and pyridines, were also suitable for this transformation. A 1,4-enyne (124) and a polysubstituted 1,4-diene derived from the natural product (−)-perillaldehyde (125) also provided the products in high yields and stereoselectivities. However, a more sterically hindered diene derived from (1R)-(−)-myrtenal provided a linear regioisomer (126) as the major product in low yield. Moreover, BCP intermediates generated from alkyl Grignard reagents proved also efficient in this transformation. Of note, the use of simple alkyl-substituted allylic carbonates was not reported.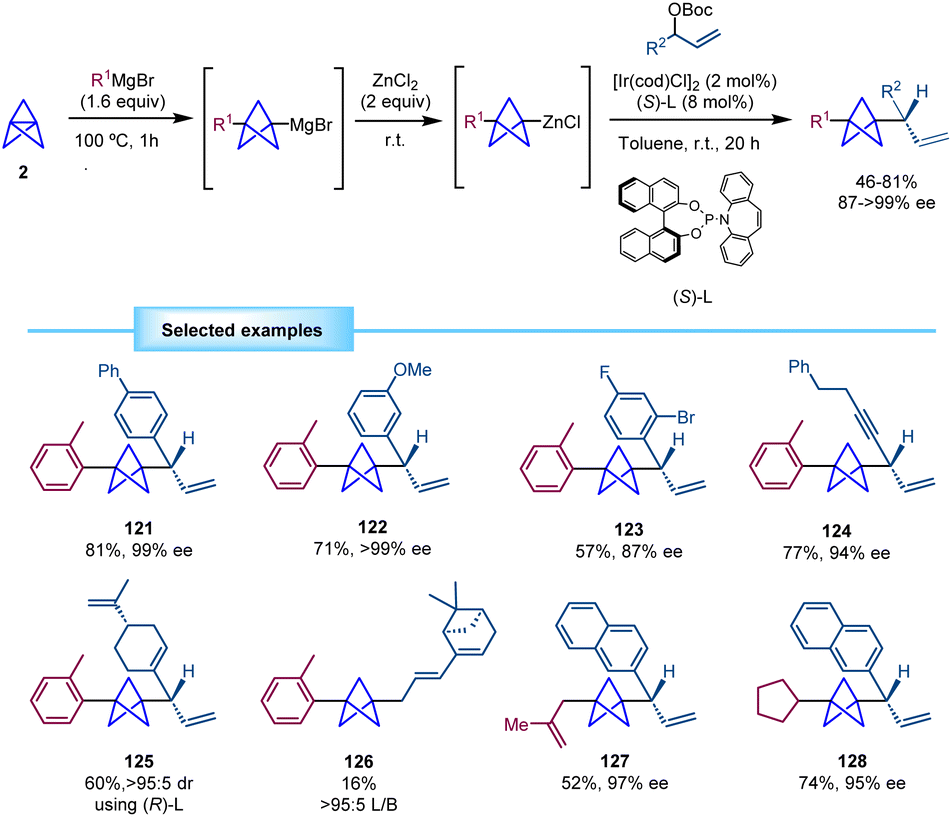 | ||
| Scheme 23 Synthesis of α-chiral BCPs by iridium-catalyzed asymmetric allylic substitution. | ||
Synthesis of α-chiral BCPs by merging asymmetric organocatalysis and photoredox/HAT catalysis
One year later, the group of Anderson reported a direct methodology for the synthesis of α-chiral BCPs based on the merging of asymmetric organocatalysis and photoredox catalysis (Scheme 24).43 The proposed strategy involves the generation of an electron-rich enamine obtained by the condensation between an aldehyde and chiral organocatalyst 129. The subsequent generation of an α-iminyl radical cation by means of single electron oxidation by an excited-state iridium photocatalyst and its addition to the central bond of [1.1.1]propellane provide a chiral BCP radical that undergoes a reductive hydrogen atom transfer (HAT) with HAT catalyst 130 to afford the final product after hydrolysis of the iminium ion. The resulting sulfur radical oxidizes the reduced iridium photocatalyst closing the photocatalytic cycle and leading to a thiolate anion that gets protonated to restart the HAT catalytic cycle. The final product was reduced to the corresponding alcohol to prevent potential epimerization and facilitate product isolation.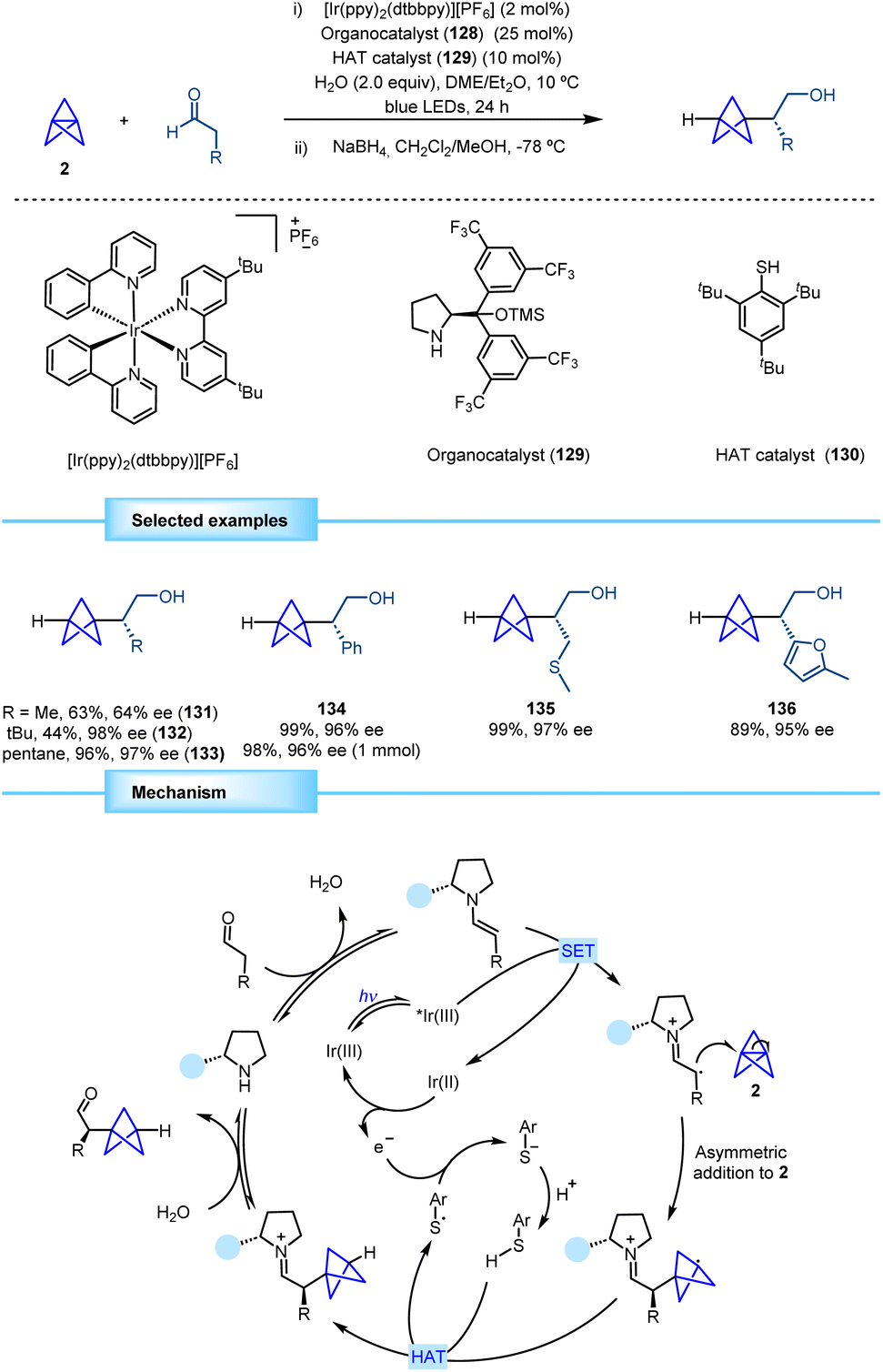 | ||
| Scheme 24 Synthesis of α-chiral BCPs by merging asymmetric organocatalysis and photoredox/HAT catalysis. | ||
The optimized reaction enables the synthesis of previously inaccessible enantioenriched BCP products. This transformation exhibits broad scope and affords excellent levels of enantioselectivity. Aldehydes bearing simple alkyl moieties performed well in the reaction. Aryl, alkenyl and alkynyl aldehydes afforded products with high yields and enantioselectivities. Aldehydes bearing heteroatoms such as thioethers, carbamates, halides, esters, and ethers also gave rise to products with high efficiency and enantioselectivity. Although heteroaromatic aldehydes thiophenyl and pyridynil derivatives proved to be less efficient in some cases, furanyl BCP product 136 was obtained with excellent yield and enantioselectivity. As a limitation, this methodology only provides monofunctionalized BCPs.
Enantioselective synthesis of other chiral BCPs
Besides the methodologies for the stereoselective synthesis of α-chiral BCPs described above, in this section we highlight some strategies for the enantioselective synthesis of other types of chiral [1.1.1]bicyclopentanes.In analogy with the synthesis of chiral BCP-α-amino acids, Pellicciari and co-workers extended their methodology based on a Strecker reaction followed by a chiral resolution to the preparation of chiral BCP β-amino acids S-CBPA (140) and R-CBPA (141) (Scheme 25).4
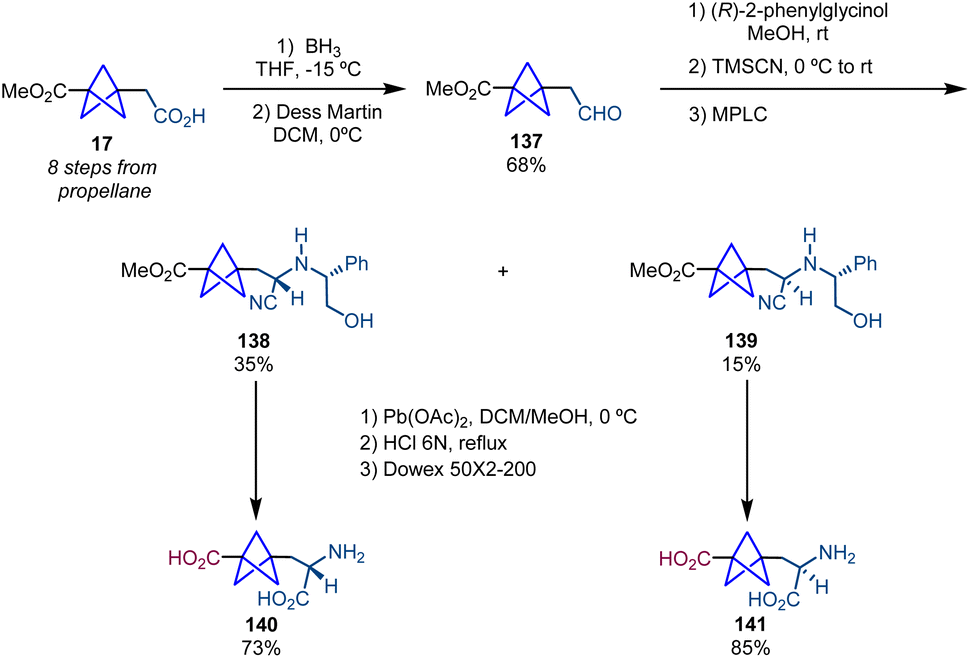 | ||
| Scheme 25 Synthesis of β-chiral amino acids 140 and 141via chemical resolution. | ||
The group of Guiry reported the asymmetric synthesis of a series of BCP-containing synthetic lipoxin A4 mimetics (Scheme 26).44 Lipoxins possess important anti-inflammatory properties, although their chemical and metabolic instability decrease their therapeutic exploitation. Consequently, structural modifications of the molecule, including a BCP moiety, can provide a potential solution to this instability. The synthetic strategy to access these lipoxin analogues relied on the preparation of an α-iodoketone (145) in which the BCP moiety was installed through a triethylborane-initiated atom transfer radical addition (ATRA) to [1.1.1]propellane,24 followed by a tris(trimethylsilyl)silane (TTMSS)-mediated deiodination reaction. The resulting BCP-ketone 146 was then reduced via asymmetric hydrogenation using Noyori's catalyst, RuCl2[DM-BINAP][DAIPEN], under 20 bar of hydrogen gas. Both enantiomers (147 and 148) were prepared in high enantiomeric excess by choosing the (R,R)- or the (S,S)-Ru catalyst, respectively. Final Suzuki coupling and zirconium-mediated acetonide deprotection led to the β-chiral BCP analogues of lipoxin A4 (149 and 150). The biological studies performed by Guiry and co-workers revealed that compound 149 has an enhanced inflammatory response compared to lipoxin A4 (151).
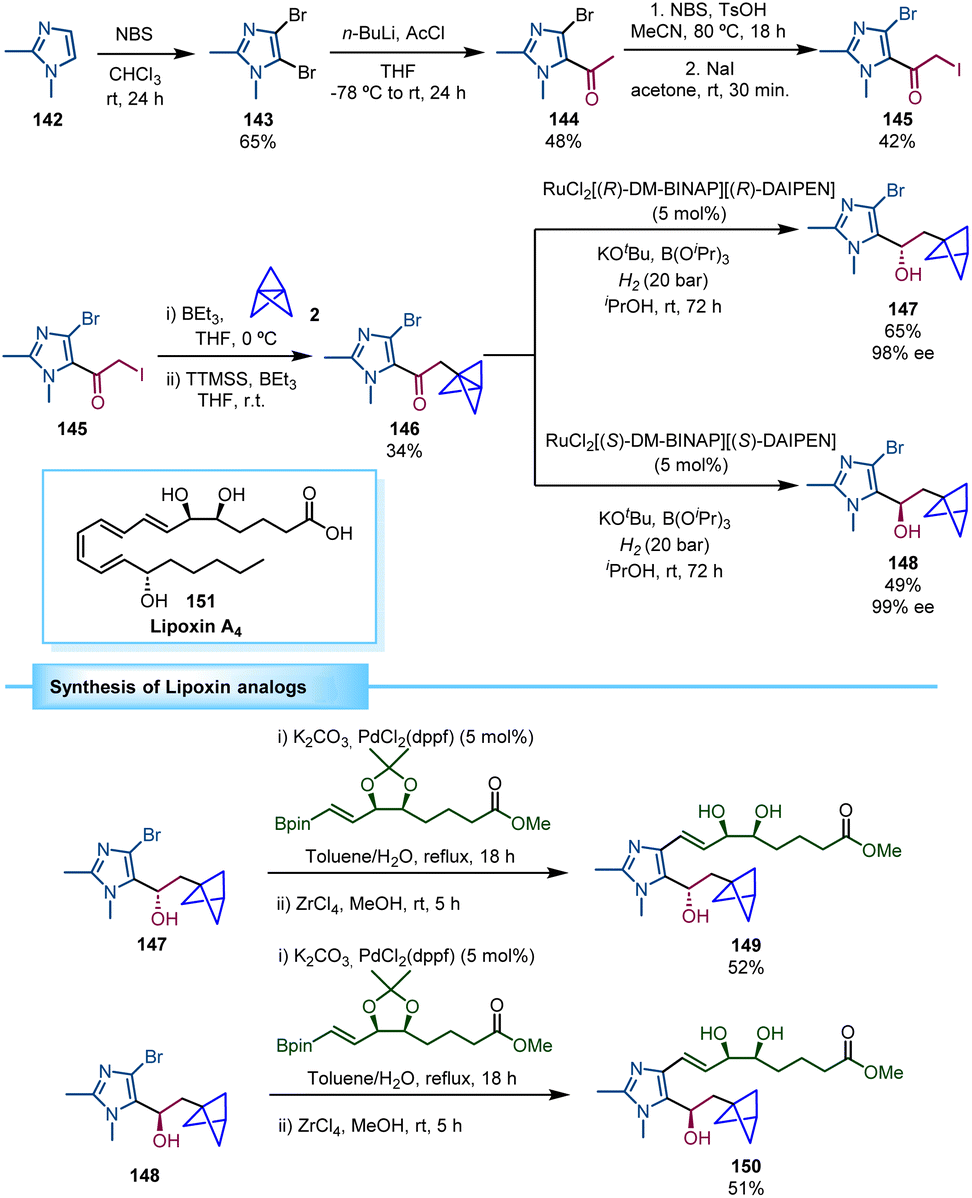 | ||
| Scheme 26 Synthesis of β-chiral BCPs 149 and 150 as analogues of lipoxin A4. | ||
A method for the construction of chiral multisubstituted BCPs bearing substituents at the C1, C2 and C3 positions has been described by Qin and co-workers through intramolecular cyclization of boronic ester-tethered cyclobutanones (Scheme 27).45 This approach relies on a two-step process involving condensation with a sulfonyl hydrazide to provide a sulfonylhydrazone and its subsequent base-mediated conversion into a diazo intermediate,46 which intramolecularly reacts with the boronate group to generate the bicyclic [2.1.1] zwitterionic intermediate 155 which then undergoes 1,2-metallate rearrangement to generate the BCP through the extrusion of N2. A range of di-, tri-, and tetra-substituted BCPs could be obtained by this methodology. In cases where the reaction was performed with an enantioenriched substrate (152), a multisubstituted BCP product was obtained with high enantiospecificity (156).
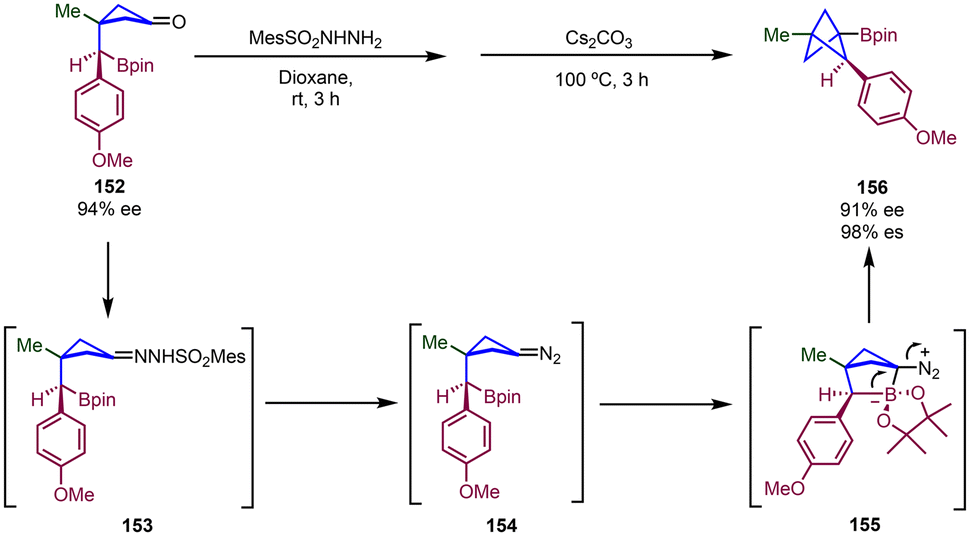 | ||
| Scheme 27 Synthesis of chiral multisubstituted BCPs via intramolecular cyclization of boronic ester-tethered cyclobutanone derivatives. | ||
Conclusions
The area of BCP functionalization has grown significantly in recent years. However, enantioselective synthesis of α-chiral BCPs has been much less developed. As we have outlined here, several groups have reported different strategies for the asymmetric synthesis of these scaffolds. Most of these methodologies rely on the use of stoichiometric chiral auxiliaries involved in long synthetic sequences. Other processes take advantage of an asymmetric catalytic reaction that affords chiral products from an already functionalized BCP derivative.The field of direct enantioselective functionalization of propellane has been less explored and represents an almost uncharted territory. As far as we know, only two reported methodologies that are able to directly functionalize [1.1.1]propellane in an enantioselective manner have been described to date. Despite the versatility of these methodologies, structural limitations are mainly associated with the lack of methods to directly access fully aliphatic chiral difunctionalized BCPs in an enantioselective manner, and BCPs bearing two α-stereocenters or quaternary centers still exist.
Thus, future efforts should be directed towards the development of novel catalytic asymmetric transformations that provide access to a new chemical space in BCP chemistry.
Author contributions
All the authors conceptualized and discussed the concept of this article. I. S.-S. and S. B.-A. performed the literature research and wrote the article (equal contributions). The manuscript was co-written and corrected by M. F.-M.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the European Research Council (863914), AEI (PID2020-118237RB-I00), Xunta de Galicia (ED431C 2022/27; Centro singular de investigación de Galicia accreditation 2019–2022, ED431G 2019/03) and the European Regional Development Fund (ERDF) is gratefully acknowledged.References
- J. Kanazawa and M. Uchiyama, Recent Advances in the Synthetic Chemistry of Bicyclo[1.1.1]pentane, Synlett, 2019, 30, 1–11 CrossRef CAS.
- A. F. Stepan, C. Subramanyam, I. V. Efremov, J. K. Dutra, T. J. O'Sullivan, K. J. DiRico, W. S. McDonald, A. Won, P. H. Dorff, C. E. Nolan, S. L. Becker, L. R. Pustilnik, D. R. Riddell, G. W. Kauffman, B. L. Kormos, L. Zhang, Y. Lu, S. H. Capetta, M. E. Green, K. Karki, E. Sibley, K. P. Atchison, A. J. Hallgren, C. E. Oborski, A. E. Robshaw, B. Sneed and C. J. O'Donnell, Application of the Bicyclo[1.1.1]pentane Motif as a Nonclassical Phenyl Ring Bioisostere in the Design of a Potent and Orally Active γ-Secretase Inhibitor, J. Med. Chem., 2012, 55, 3414–3424 CrossRef CAS PubMed.
- I. S. Makarov, C. E. Brocklehurst, K. Karaghiosoff, G. Koch and P. Knochel, Synthesis of Bicyclo[1.1.1]pentane Bioisosteres of Internal Alkynes and para–Disubstituted Benzenes from [1.1.1]Propellane, Angew. Chem., Int. Ed., 2017, 56, 12774–12777 CrossRef CAS PubMed.
- R. Filosa, M. C. Fulco, M. Marinozzi, N. Giacchè, A. Macchiarulo, A. Peduto, A. Massa, P. De Caprariis, C. Thomsen, C. T. Christoffersen and R. Pellicciari, Design, synthesis and biological evaluation of novel bicyclo[1.1.1]pentane-based ω-acidic amino acids as glutamate receptors ligands, Bioorg. Med. Chem., 2009, 17, 242–250 CrossRef CAS PubMed.
- M. V. Westphal, B. T. Wolfstädter, J.-M. Plancher, J. Gatfield and E. M. Carreira, Evaluation of tert-Butyl Isosteres: Case Studies of Physicochemical and Pharmacokinetic Properties, Efficacies, and Activities, ChemMedChem, 2015, 10, 461–469 CrossRef CAS PubMed.
- N. D. Measom, K. D. Down, D. J. Hirst, C. Jamieson, E. S. Manas, V. K. Patel and D. O. Somers, Investigation of a Bicyclo[1.1.1]pentane as a Phenyl Replacement within an LpPLA2 Inhibitor, ACS Med. Chem. Lett., 2017, 8, 43–48 CrossRef CAS PubMed.
- R. Pellicciari, M. Raimondo, M. Marinozzi, B. Natalini, G. Costantino and C. Thomsen, (S)-(+)-2-(3′-Carboxybicyclo[1.1.1]pentyl)- glycine, a Structurally New Group I Metabotropic Glutamate Receptor Antagonist, J. Med. Chem., 1996, 39, 2874–2876 CrossRef CAS PubMed.
- K. B. Wiberg, D. S. Connor and G. M. Lampman, The reaction of 3-bromocyclobutane-1-methyl bromide with sodium: bicyclo[1. 1. 1]pentane, Tetrahedron Lett., 1964, 5, 531–534 CrossRef.
- K. B. Wiberg and S. T. Waddell, Reactions of [1.1.1]propellane, J. Am. Chem. Soc., 1990, 112, 2194–2216 CrossRef CAS.
- K. B. Wiberg and F. H. Walker, [1.1.1]Propellane, J. Am. Chem. Soc., 1982, 104, 5239–5240 CrossRef CAS.
- K. Semmler, G. Szeimies and J. Belzner, Tetracyclo[5.1.0.01,6.02,7]octane, a [1.1.1]propellane derivative, and a new route to the parent hydrocarbon, J. Am. Chem. Soc., 1985, 107, 6410–6411 CrossRef CAS.
- R. Gianatassio, J. M. Lopchuk, J. Wang, C.-M. Pan, L. R. Malins, L. Prieto, T. A. Brandt, M. R. Collins, G. M. Gallego, N. W. Sach, J. E. Spangler, H. Zhu, J. Zhu and P. S. Baran, Strain-release amination, Science, 2016, 351, 241–246 CrossRef CAS PubMed.
- P. Kaszynski and J. Michl, A practical photochemical synthesis of bicyclo[1.1.1]pentane-1,3-dicarboxylic acid, J. Org. Chem., 1988, 53, 4593–4594 CrossRef CAS.
- (a) B. R. Shire and E. A. Anderson, Conquering the Synthesis and Functionalization of Bicyclo[1.1.1]pentanes, JACS Au, 2023, 3, 1539–1553 CrossRef CAS PubMed; (b) T. Yasukawa, K. S. Håheim and J. Cossy, Synthesis of 1,3-disubstituted bicyclo[1.1.1]pentanes by cross-coupling induced by transition metals - formation of C-C bonds, Org. Biomol. Chem., 2023, 21, 7666–7680 RSC; (c) J. Turkowska, J. Durka and D. Gryko, Strain release – an old tool for new transformations, Chem. Commun., 2020, 56, 5718–5734 RSC; (d) J. H. Kim, A. Ruffoni, Y. S. S. Al-Faiyz, N. S. Sheikh and D. Leonori, Divergent Strain-Release Amino-Functionalization of [1.1.1]Propellane with Electrophilic Nitrogen-Radicals, Angew. Chem., Int. Ed., 2020, 59, 8225–8231 CrossRef CAS PubMed; (e) J. M. Anderson, N. D. Measom, J. A. Murphy and D. L. Poole, Bridge Functionalisation of Bicyclo[1.1.1]pentane Derivatives, Angew. Chem., Int. Ed., 2021, 60, 24754–24769 CrossRef CAS PubMed.
- A. M. Dilmaç, E. Spuling, A. de Meijere and S. Bräse, Propellanes-From a Chemical Curiosity to “Explosive” Materials and Natural Products, Angew. Chem., Int. Ed., 2017, 56, 5684–5718 CrossRef PubMed.
- M. D. Levin, P. Kaszynski and J. Michl, Bicyclo[1.1.1]pentanes, [n]Staffanes, [1.1.1]Propellanes, and Tricyclo[2.1.0.02,5]pentanes, Chem. Rev., 2000, 100, 169–234 CrossRef CAS PubMed.
- S. Pritz, M. Pätzel, G. Szeimies, M. Dathe and M. Bienert, Synthesis of a chiral amino acid with bicyclo[1.1.1]pentane moiety and its incorporation into linear and cyclic antimicrobial peptides, Org. Biomol. Chem., 2007, 5, 1789–1794 RSC.
- R. Pellicciari, B. Natalini and M. Marinozzi, L-α-Aminoadipic Acid from L-Glutamic Acid, Synth. Commun., 1988, 18, 1707–1714 CrossRef CAS.
- S. O. Kokhan, A. V. Tymtsunik, S. L. Grage, S. Afonin, O. Babii, M. Berditsch, A. V. Strizhak, D. Bandak, M. O. Platonov, I. V. Komarov, A. S. Ulrich and P. K. Mykhailiuk, Design, Synthesis, and Application of an Optimized Monofluorinated Aliphatic Label for Peptide Studies by Solid–State 19F NMR Spectroscopy, Angew. Chem., Int. Ed., 2016, 55, 14788–14792 CrossRef CAS PubMed.
- M. D. Levin, P. Kaszynski and J. Michl, Photochemical synthesis of bicyclo[1.1.1]pentane-1,3-dicarboxylic acid, Org. Synth., 2000, 77, 249 CrossRef CAS.
- F. Yin, Z. Wang, Z. Li and C. Li, Silver-Catalyzed Decarboxylative Fluorination of Aliphatic Carboxylic Acids in Aqueous Solution, J. Am. Chem. Soc., 2012, 134, 10401–10404 CrossRef CAS PubMed.
- (a) S. Ni, A. F. Garrido-Castro, R. R. Merchant, J. N. de Gruyter, D. C. Schmitt, J. J. Mousseau, G. M. Gallego, S. Yang, M. R. Collins, J. X. Qiao, K.-S. Yeung, D. R. Langley, M. A. Poss, P. M. Scola, T. Qin and P. S. Baran, A General Amino Acid Synthesis Enabled by Innate Radical Cross-Coupling, Angew. Chem., Int. Ed., 2018, 57, 14560–14565 CrossRef CAS PubMed; (b) S. Ni, N. M. Padial, C. Kingston, J. C. Vantourout, D. C. Schmitt, J. T. Edwards, M. M. Kruszyk, R. R. Merchant, P. K. Mykhailiuk, B. B. Sanchez, S. Yang, M. A. Perry, G. M. Gallego, J. J. Mousseau, M. R. Collins, R. J. Cherney, P. S. Lebed, J. S. Chen, T. Qin and P. S. Baran, A Radical Approach to Anionic Chemistry: Synthesis of Ketones, Alcohols, and Amines, J. Am. Chem. Soc., 2019, 141, 6726–6739 CrossRef CAS.
- M. L. J. Wong, J. J. Mousseau, S. J. Mansfield and E. A. Anderson, Synthesis of Enantioenriched α-Chiral Bicyclo[1.1.1]pentanes, Org. Lett., 2019, 21, 2408–2411 CrossRef CAS.
- D. F. J. Caputo, C. Arroniz, A. B. Dürr, J. J. Mousseau, A. F. Stepan, S. J. Mansfield and E. A. Anderson, Synthesis and applications of highly functionalized 1-halo-3-substituted bicyclo[1.1.1]pentanes, Chem. Sci., 2018, 9, 5295–5300 RSC.
- J. Kanazawa, K. Maeda and M. Uchiyama, Radical Multicomponent Carboamination of [1.1.1]Propellane, J. Am. Chem. Soc., 2017, 139, 17791–17794 CrossRef CAS PubMed.
- K. D. Bunker, N. W. Sach, Q. Huang and P. F. Richardson, Scalable Synthesis of 1-Bicyclo[1.1.1]pentylamine via a Hydrohydrazination Reaction, Org. Lett., 2011, 13, 4746–4748 CrossRef CAS PubMed.
- S. Yu, C. Jing, A. Noble and V. K. Aggarwal, 1,3−Difunctionalizations of [1.1.1]Propellane via 1,2−Metallate Rearrangements of Boronate Complexes, Angew. Chem., Int. Ed., 2020, 59, 3917–3921 CrossRef CAS PubMed.
- W. J. Jang, S. M. Song, J. H. Moon, J. Y. Lee and J. Yun, Copper-Catalyzed Enantioselective Hydroboration of Unactivated 1,1-Disubstituted Alkenes, J. Am. Chem. Soc., 2017, 139, 13660–13663 CrossRef CAS PubMed.
- M. Burns, S. Essafi, J. R. Bame, S. P. Bull, M. P. Webster, S. Balieu, J. W. Dale, C. P. Butts, J. N. Harvey and V. K. Aggarwal, Assembly-line synthesis of organic molecules with tailored shapes, Nature, 2014, 513, 183–188 CrossRef CAS PubMed.
- M. Kondo, J. Kanazawa, T. Ichikawa, T. Shimokawa, Y. Nagashima, K. Miyamoto and M. Uchiyama, Silaboration of [1.1.1]Propellane: A Storable Feedstock for Bicyclo[1.1.1]pentane Derivatives, Angew. Chem., Int. Ed., 2020, 59, 1970–1974 CrossRef CAS PubMed.
- J. L. Stymiest, V. Bagutski, R. M. French and V. K. Aggarwal, Enantiodivergent conversion of chiral secondary alcohols into tertiary alcohols, Nature, 2008, 456, 778–782 CrossRef CAS PubMed.
- V. K. Vyas, G. J. Clarkson and M. Wills, Enantioselective Synthesis of Bicyclopentane-Containing Alcohols via Asymmetric Transfer Hydrogenation, Org. Lett., 2021, 23, 3179–3183 CrossRef CAS.
- H. M. L. Davies and D. Morton, Guiding principles for site selective and stereoselective intermolecular C–H functionalization by donor/acceptor rhodium carbenes, Chem. Soc. Rev., 2011, 40, 1857 RSC.
- K. Liao, T. C. Pickel, V. Boyarskikh, J. Bacsa, D. G. Musaev and H. M. L. Davies, Site-selective and stereoselective functionalization of unactivated C–H bonds, Nature, 2017, 551, 609–613 CrossRef CAS PubMed.
- Z. J. Garlets, J. N. Sanders, H. Malik, C. Gampe, K. N. Houk and H. M. L. Davies, Enantioselective C–H functionalization of bicyclo[1.1.1]pentanes, Nat. Catal., 2020, 3, 351–357 CrossRef CAS.
- J. Nugent, A. J. Sterling, N. Frank, J. J. Mousseau and E. A. Anderson, Synthesis of α-Quaternary Bicyclo[1.1.1]pentanes through Synergistic Organophotoredox and Hydrogen Atom Transfer Catalysis, Org. Lett., 2021, 23, 8628–8633 CrossRef CAS PubMed.
- F. Zhou, L. Zhu, B.-W. Pan, Y. Shi, Y.-L. Liu and J. Zhou, Catalytic enantioselective construction of vicinal quaternary carbon stereocenters, Chem. Sci., 2020, 11, 9341–9365 RSC.
- J. L. M. Matos, S. Vásquez-Céspedes, J. Gu, T. Oguma and R. A. Shenvi, Branch-Selective Addition of Unactivated Olefins into Imines and Aldehydes, J. Am. Chem. Soc., 2018, 140, 16976–16981 CrossRef CAS PubMed.
- R. Shelp, R. R. Merchant, J. M. E. Hughes and P. J. Walsh, Enantioenriched BCP Benzylamine Synthesis via Metal Hydride Hydrogen Atom Transfer/Sulfinimine Addition to [1.1.1]Propellane, Org. Lett., 2022, 24, 110–114 CrossRef CAS PubMed.
- W. Huang, Y. Zheng, S. Keess and G. A. Molander, A General and Modular Approach to BCP Alkylamines via Multicomponent Difunctionalization of [1.1.1]Propellane, J. Am. Chem. Soc., 2023, 145, 5363–5369 CrossRef CAS PubMed.
- W. Dong, E. Yen-Pon, L. Li, A. Bhattacharjee, A. Jolit and G. A. Molander, Exploiting the sp2 character of bicyclo[1.1.1]pentyl radicals in the transition-metal-free multi-component difunctionalization of [1.1.1]propellane, Nat. Chem., 2022, 14, 1068–1077 CrossRef CAS PubMed.
- S. Yu, C. Jing, A. Noble and V. K. Aggarwal, Iridium-Catalyzed Enantioselective Synthesis of α-Chiral Bicyclo[1.1.1]pentanes by 1,3-Difunctionalization of [1.1.1]Propellane, Org. Lett., 2020, 22, 5650–5655 CrossRef CAS PubMed.
- M. L. J. Wong, A. J. Sterling, J. J. Mousseau, F. Duarte and E. A. Anderson, Direct catalytic asymmetric synthesis of α-chiral bicyclo[1.1.1]pentanes, Nat. Commun., 2021, 12, 1644 CrossRef CAS PubMed.
- B. Owen, M. De Gaetano, A. Gaffney, C. Godson and P. J. Guiry, Synthesis and Biological Evaluation of Bicyclo[1.1.1]pentane-Containing Aromatic Lipoxin A4 Analogues, Org. Lett., 2022, 24, 6049–6053 CrossRef CAS PubMed.
- Y. Yang, J. Tsien, J. M. E. Hughes, B. K. Peters, R. R. Merchant and T. Qin, An intramolecular coupling approach to alkyl bioisosteres for the synthesis of multisubstituted bicycloalkyl boronates, Nat. Chem., 2021, 13, 950–955 CrossRef CAS PubMed.
- J. Barluenga, M. Tomás-Gamasa, F. Aznar and C. Valdés, Metal-free carbon–carbon bond-forming reductive coupling between boronic acids and tosylhydrazones, Nat. Chem., 2009, 1, 494–499 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © the Partner Organisations 2024 |