DOI:
10.1039/D3QO01615C
(Research Article)
Org. Chem. Front., 2024,
11, 211-216
Chiral phosphoric acid catalyzed enantioselective inverse-electron-demand oxa-Diels–Alder reactions to synthesize chiral tricyclic tetrahydropyran derivatives†
Received
2nd October 2023
, Accepted 22nd November 2023
First published on 23rd November 2023
Abstract
A series of enantioenriched tricyclic tetrahydropyran derivatives were achieved through chiral phosphoric acid catalyzed intramolecular inverse electron demand oxa-Diels–Alder (IEDODA) reactions, with good yields (up to 95%) and excellent stereocontrol (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, up to 99% ee). The achievement of such high enantioselectivity was ascribed to the rationally designed remote dual hydrogen bonding interaction, by which the conformation of the flexible substrate was well arranged by the catalyst in a reactive and stereoselective manner to participate in the transformation.
:1 dr, up to 99% ee). The achievement of such high enantioselectivity was ascribed to the rationally designed remote dual hydrogen bonding interaction, by which the conformation of the flexible substrate was well arranged by the catalyst in a reactive and stereoselective manner to participate in the transformation.
Introduction
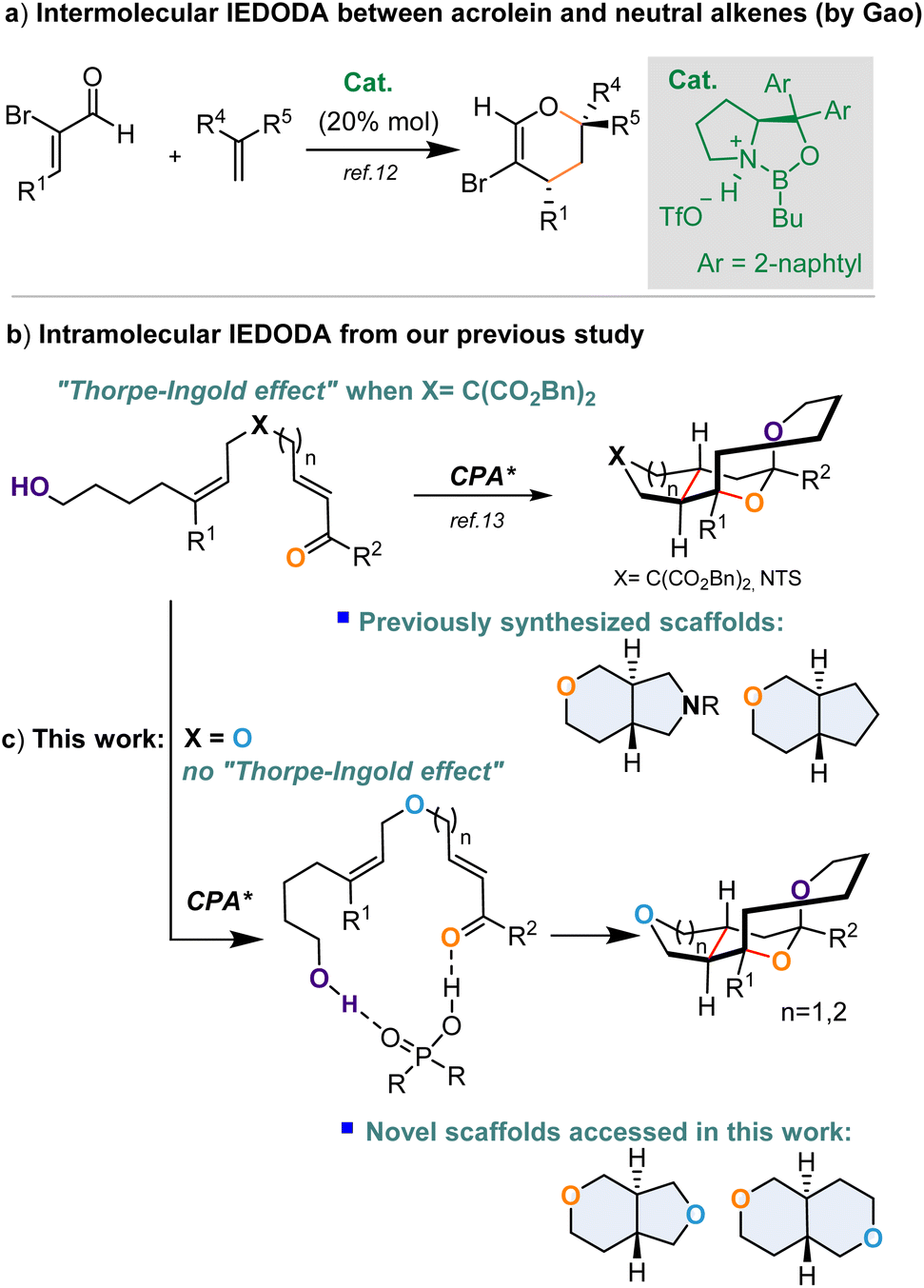
The asymmetric inverse-electron demand hetero Diels–Alder (IEDHDA) reaction is widely utilized as a powerful protocol to construct chiral six-membered heterocyclic molecules, among which tetrahydropyran-containing derivatives are found in numerous naturally occurring products and bioactive compounds.1–4 Thus, the synthesis of tetrahydropyran scaffolds by inverse-electron demand oxa-Diels–Alder (IEDODA) reactions intrigued considerable interest from the synthetic community over the past decades.1–4 Generally, either electron rich substrates, i.e., enol ether, enamine or thioenol ether, serving as nucleophilic components or “chelatable” electrophilic partners, such as β,γ-unsaturated ketoester, α,β-unsaturated oxazolidinone etc. able to coordinate to transition metal catalysts, were mainly employed in the reported asymmetric IEDODA reactions to enhance the reactivity or enantiocontrol.1–4 In this context, neutral alkenes as nucleophilic partners participating in the IEDODA reactions were much less explored, due to their low reactivity and also the difficulty in controlling the stereoselectivity. A few examples were documented by Cheng, Zhang and Ishihara's groups respectively, in which chiral transition metal complexes promoted the IEDODA reactions involving neutral alkenes and β,γ-unsaturated ketoesters to deliver an array of enantioenriched didydropryans.5–7 Furthermore, highly reactive ortho-quinone methides were able to react with neutral alkenes in the presence of chiral Brønsted acids to form chiral chromanes, independently disclosed by Rueping and Shi groups.8,9 In contrast to the intermolecular version of the IEDODA reaction using non-activated alkenes as nucleophilic components, intramolecular versions of this reaction were reported by Kumaran and Campagne groups, respectively. They independently demonstrated the formation of enantioenriched trans-fused bicyclic dihydropyran by a metal catalyzed IEDODA reaction between β,γ-unsaturated ketoester, α,β-unsaturated acyl imidazole and a non-polarized alkene motif.10,11 Although the enantioselectivities of these two reactions were high, the generality of substrate scope was quite narrow.
Except for the above described “chelatable” unsaturated carbonyl substrates as the electrophilic partners frequently applied in IEDODA reactions to react with neutral alkenes, employing “simple” and “unchelatable” α,β-unsaturated ketones or aldehydes to cyclize with no-polarized alkenes was undoubtedly a challenging task. Very recently, two successful cases were revealed on tackling this issue. One was from Gao and co-workers, who disclosed that an oxazaborolidinium cation catalyst could promote the intermolecular IEDODA reaction between α-bromo acroleins and non-activated alkenes to deliver enantioenriched dihydropyran derivatives with broad substrate scope (Fig. 1a).12 Alternatively, our group has also successfully developed an intramolecular IEDODA reaction involving α,β-unsaturated ketones or aldehydes to react with neutral alkenes on the basis of a remote dual hydrogen bonding interaction, in which tetrahydropyran-fused-pyrrolidine and -cyclopentane derivatives were formed with broad substrate scope and excellent enantioselectivity (Fig. 1b).13
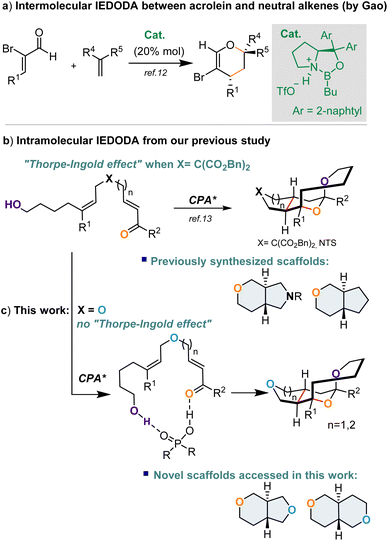 |
| | Fig. 1 (a) Reported intermolecular IDEODA reactions between α,β-unsaturated aldehydes and neutral alkenes; (b) an intramolecular IDEODA reaction between an α,β-unsaturated ketone, aldehyde and neutral alkene motif reported in our previous study; (c) synthesis of new tetrahydropyran-containing scaffolds by the IDEODA reaction in this work. CPA = chiral phosphoric acid. | |
It would be interesting to know whether the IEDODA reaction would still proceed efficiently if an oxygen atom (X![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) is incorporated into a linear substrate instead of NTs or C(CO2Bn) as the “linker”, of which the latter more likely exerted a Thorpe–Ingold effect on this cyclization (Fig. 1b).14,15 Furthermore, the bicyclic trans-fused octahydropyranopyrrole and tetrahydro-pyranocyclopentane motifs as shown in Fig. 1b are privileged structures present in numerous natural products and bioactive molecules;16–19 nevertheless, their analogues trans-fused octahydropyranofuran and octahydropyranopyran are hardly found in natural resources, and thus less explored in terms of synthesis and biological properties. Therefore, development of synthetic approaches to access those unprecedented skeletons will be highly desirable in terms of expanding the chemical space and structural diversity of trans-fused bicyclic ring systems and exploring their bioactivity. In this context, herein, we explore the enantioselective intramolecular IEDODA–acetalization cascade reaction involving α,β-unsaturated ketone and non-activated alkene catalyzed by chiral phosphoric acid to generate a series of novel trans-fused octahydropyranofuran or octahydropyranopyran scaffolds with excellent enantio- and diastereocontrol on the basis of a remote dual hydrogen bonding activation model between the catalyst and substrate. (Fig. 1c).
O) is incorporated into a linear substrate instead of NTs or C(CO2Bn) as the “linker”, of which the latter more likely exerted a Thorpe–Ingold effect on this cyclization (Fig. 1b).14,15 Furthermore, the bicyclic trans-fused octahydropyranopyrrole and tetrahydro-pyranocyclopentane motifs as shown in Fig. 1b are privileged structures present in numerous natural products and bioactive molecules;16–19 nevertheless, their analogues trans-fused octahydropyranofuran and octahydropyranopyran are hardly found in natural resources, and thus less explored in terms of synthesis and biological properties. Therefore, development of synthetic approaches to access those unprecedented skeletons will be highly desirable in terms of expanding the chemical space and structural diversity of trans-fused bicyclic ring systems and exploring their bioactivity. In this context, herein, we explore the enantioselective intramolecular IEDODA–acetalization cascade reaction involving α,β-unsaturated ketone and non-activated alkene catalyzed by chiral phosphoric acid to generate a series of novel trans-fused octahydropyranofuran or octahydropyranopyran scaffolds with excellent enantio- and diastereocontrol on the basis of a remote dual hydrogen bonding activation model between the catalyst and substrate. (Fig. 1c).
Results and discussion
Reaction condition optimization
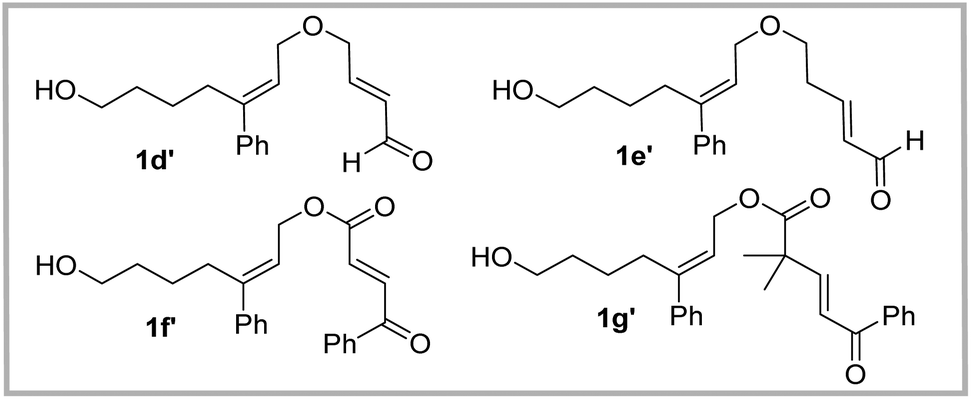
At the outset, to validate the efficacy of an oxygen-containing linear substrate undergoing the IDEODA reaction, α,β-unsaturated ketone 1a with two methyl substituents next to the oxygen atom was prepared and subjected to BINOL-derived chiral phosphoric acid (CPA) 3a.20,21 Gratifyingly, the tricyclic product 2a was formed as a sole stereoisomer albeit with only 17% yield and 43% ee (entry 1, Table 1). Encouraged by this primary result, we investigated other chiral Brønsted acids including N-triflylphosphoramide 3b and SPINOL-CPA 3c, and found that the latter resulted in a superior yield and enantioselectivity (47% yield, 72% ee, entry 3, Table 1). Then, a series of SPINOL CPAs 3d–j were evaluated, and the highest enantioselectivity of 2a was achieved in the presence of catalyst 3j (87% ee, entry 10, Table 1); meanwhile, when catalyst 3i was applied, excellent yield was obtained in spite of a slightly lower ee value (85% yield, 83% ee, entry 9, Table 1). Then the other reaction parameter solvent was surveyed; however, no better satisfactory results were obtained compared with those for DCM (entries 11–18 vs. entry 10, Table 1). Although the enantiocontrol of this cyclization reaction enhanced in toluene in the presence of catalyst 3j, only a low yield was obtained (39%, 91% ee, entry 13, Table 1), which was likely ascribed to the poor reactivity and low conversion of substrate 1a. After slight modification of methyl-ketone to phenyl ketone 1b, surprisingly, an excellent ee value was observed (94% ee, entry 20, Table 1), indicating that the aromatic substituent on the enone moiety was beneficial for the enantiocontrol of the IEDODA reaction; however, the low conversion of the starting material still limited the improvement of the yield of product 2b (19% yield, entry 20, Table 1). We anticipated that the steric hindrance from the gem-methyl substituents on 2a or 2b was likely responsible for its low conversion. With this idea in mind, substrate 1c without the gem-methyl substituent was then prepared and subjected to catalyst 3j in DCM, and to our delight, excellent stereocontrol was obtained as well as with improved yield compared to the result of 1b (44% yield, 97% ee, entry 21, Table 1). Furthermore, when the solvent 1,2-dichloroethane was applied, the cyclization reaction of substrate 1c underwent smoothly and delivered the product 2c as a single stereoisomer with the optimal yield and excellent enantioselectivity so far obtained (64% yield, 98% ee, dr > 20:1, entry 22, Table 1). These results showed that the Thorpe–Ingold effect induced by the gem-methyl was supposed to be unessential to trigger the occurrence of the IEDODA reaction. Nevertheless, when we subjected α,β-unsaturated aldehyde 1d′ to the optimal reaction conditions, unexpectedly, it was found ineffective to undergo the cyclization reaction, in contrast to the outcome of its counterpart substrate in our previous study when the “linker” was NTs or C(CO2Bn) (Fig. 2).12 Analogously, other substrates such as esters 1e′–g′ either provided no products or afforded products only in trace amounts (Fig. 2).
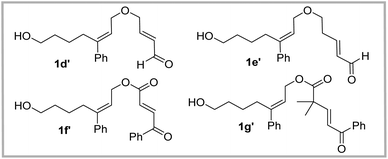 |
| | Fig. 2 Other ineffective substrates in the IEDODA reaction. | |
Table 1 Optimization of the reaction conditionsa
Investigation of substrate scope
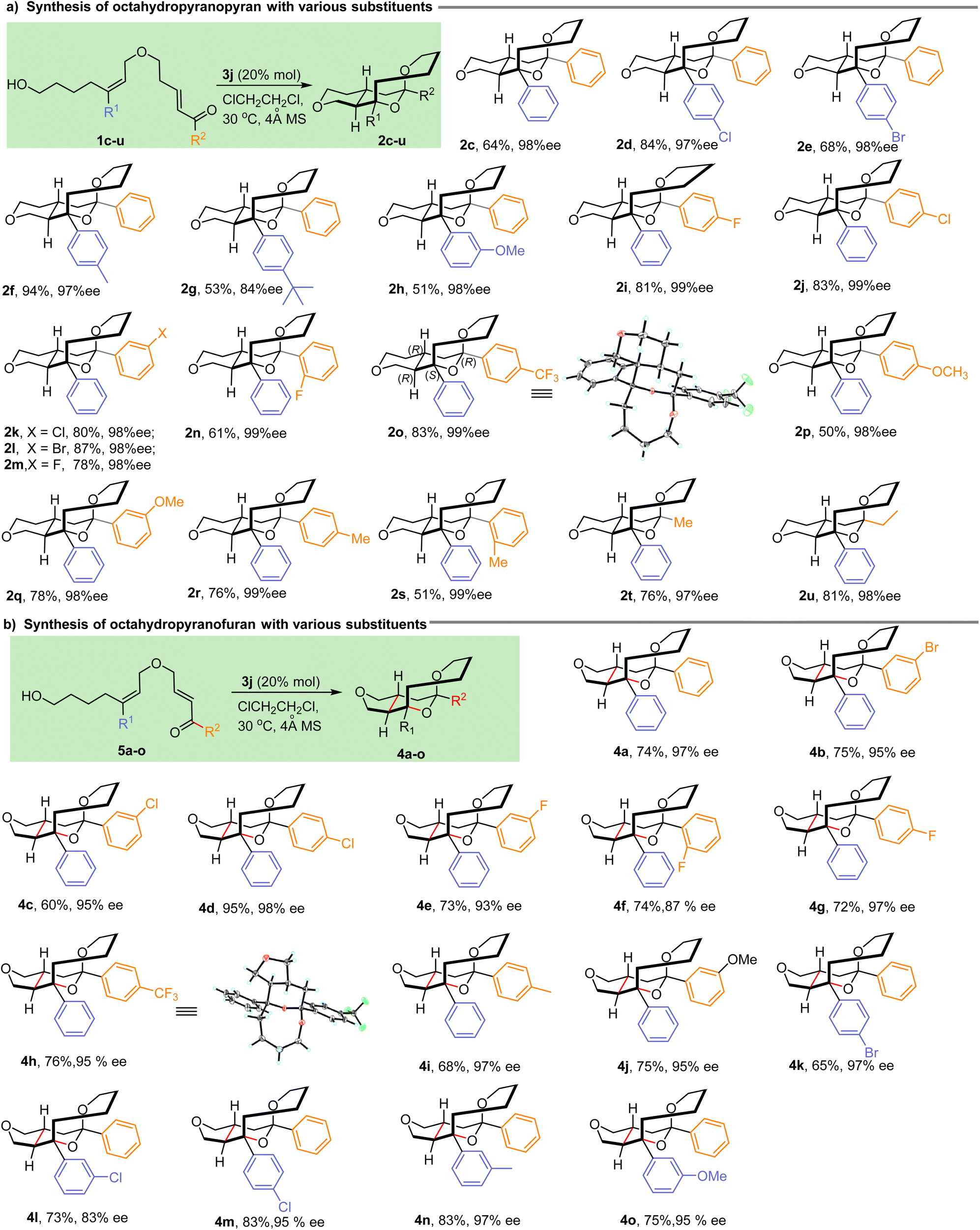
With the optimal reaction parameters established, we then explored the substrate scope of the IEDODA reaction, and the results are summarized in Scheme 1. In the cyclization reactions to access [6,6]-trans-fused octahydropyranopyran scaffolds, various aromatic substituents on the trisubstituted alkenes were tolerated regardless of the electron-deficient or electron-rich group on the phenyl, affording the desired products 2d and 2e or 2f–h, respectively, in excellent diastereoselectivities, enantioselectivities and good yields (dr > 20:1, 84–98% ee, 51–94% yields, Scheme 1a). Then, the substituents on the enone motif also were examined. Almost no influences by the nature of substituents on the phenyl were observed, and uniformly, the products 2i–s were generated with excellent enantiocontrol (98–99% ee). Besides the various aromatic groups on the enone, aliphatic substituents such as Me and Et could also be well compatible with this reaction, and the products 2t and 2u were formed with good yields and enantioselectivities (76%–81% yields, 97–98% ee). Unambiguously, the absolute configuration of all products was confirmed as (R,S,R,R) based on the X-ray analysis result of 2o (CCDC 2286029†). Besides the [6,6]-trans-fused octahydropyranopyran successfully constructed by the IEDODA reaction, an array of [5,6]-trans-fused octahydropyranofuran derivatives could also be effectively generated under the optimal conditions as shown in Scheme 1b. Similarly, substituents on both the trisubstituted alkene and enone motifs were investigated, and in most cases, the tricyclic products 4b–o were obtained with good yields (60–95%) and excellent enantioselectivities (83–98% ee). Furthermore, the X-ray analysis of product 4h (CCDC 2286031†) confirmed the absolute configuration of the formed tricyclic tetrahydropyran analogues as (R,S,R,R). In a word, the present method represents a versatile tool to access the unprecedented tetrahydropyran-containing tricyclic skeletons with high enantiopurity and broad substrate scope.
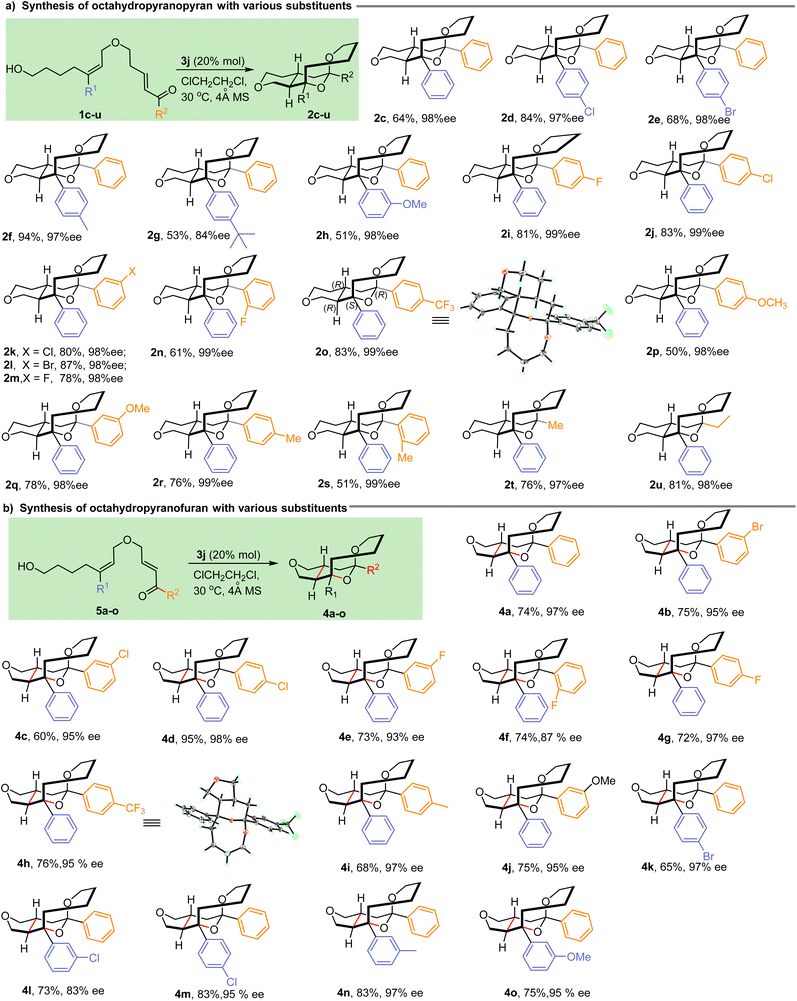 |
| | Scheme 1 Substrate scope for the IEDODA reactions. | |
Derivation of products
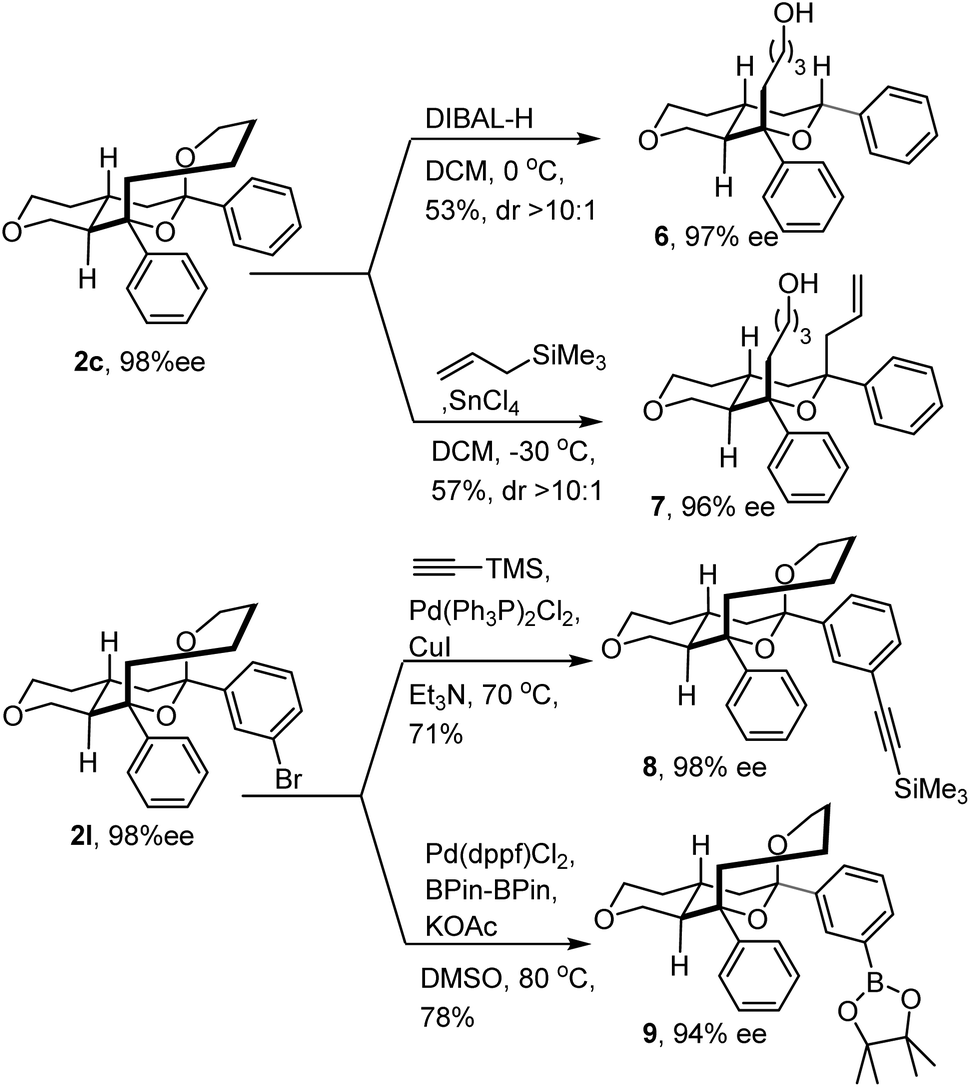
With the tricyclic IEDODA products in hand, a variety of derivations were conducted (Fig. 3). The ketal motif on product 2c could be readily reduced by DIBAL-H to generate the trans-fused octahydropyranopyran 6. Alternatively, stereoselective allylation of the same product could afford the bicyclic 7 bearing four contiguous stereogenic centers with excellent enantiopurity. Furthermore, at elevated temperature, 2l was smoothly coupled with trimethylsilylacetylene and provided the alkyne 8. Attached with an alkyne motif, this functionalized compound 8 was supposed to be able to click with biotin-azide to form a molecular probe and thus lay the foundation for exploring its bioactivity in the future.22 Meanwhile, the Miyaura borylation reaction could also readily occur at 80 °C and afford boronate 9 with good yield and enantioselectivity.23 These examples demonstrated the promising thermostability of the tricyclic scaffold, and the potential for conducting diverse post-transformations of IEDODA products.
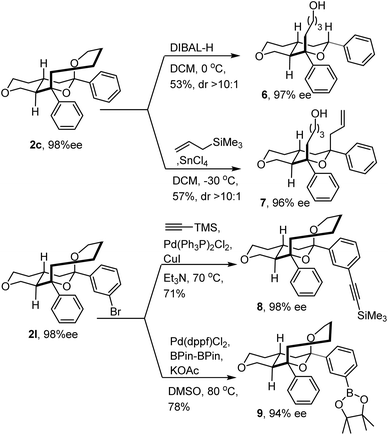 |
| | Fig. 3 Derivation of IEDODA products. | |
Conclusions
We have developed a chiral phosphoric acid catalyzed intramolecular IEDODA reaction between α,β-unsaturated ketone and a neutral alkene motif, which furnished an array of [6,6] or [5,6]-trans-fused tricyclic tetrahydropyran derivatives with excellent enantioselectivities, diastereoselectivities and good yields. This research combined with our previous study presents a powerful strategy for stereocontrol, namely by introducing a remote dual hydrogen bonding interaction between the catalyst and linear, flexible substrate. We believe that this concept will be applicable to other asymmetric transformations. With these enantioenriched tetrahydropyran derivatives in hand, we will be interested in probing their biological activities in the future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the High-Level Talent Program of Yunnan Province, the Regional Development Program for Young Scholars, the Chinese Academy of Sciences (CAS), the Youth Innovation Promotion Association, the Chinese Academy of Sciences (CAS), the Key Laboratory for Asymmetric Synthesis and Chirotechnology of Sichuan Province, the Chengdu Institute of Organic Chemistry (2021KFKT01), and the Yunan Province Science and Technology Department (202305AH34005).
References
- For selected reviews on asymmetric IEDHDA reactions: G. Desimoni, G. Faita and P. Quadrelli, Forty Years after “Heterodiene Syntheses with α,β-Unsaturated Carbonyl Compounds”: Enantioselective Syntheses of 3,4-Dihydropyran Derivatives, Chem. Rev., 2018, 118, 2080–2248 CrossRef CAS PubMed.
- M. Xie, L. Lin and X. Feng, Catalytic Asymmetric Inverse-Electron-Demand Hetero-Diels-Alder Reactions, Chem. Rec., 2017, 17, 1184–1202 CrossRef CAS PubMed.
- A. Pałasz, Recent Advances in Inverse-Electron-Demand Hetero-Diels-Alder Reactions of 1-Oxa-1,3-Butadienes, Top. Curr. Chem., 2016, 374, 24 CrossRef PubMed.
- X. Jiang and R. Wang, Recent Developments in Catalytic Asymmetric Inverse-Electron-Demand Diels-Alder Reaction, Chem. Rev., 2013, 113, 5515–5546 CrossRef CAS PubMed.
- J. Lv, L. Zhang, S. Luo and J.-P. Cheng, Switchable Diastereoselectivity in Enantioselective [4+2] Cycloadditions with Simple Olefins by Asymmetric Binary Acid catalysis, Angew. Chem., Int. Ed., 2013, 52, 9786–9790 CrossRef CAS PubMed.
- Y. Matsumura, T. Suzuki, A. Sakakura and K. Ishihara, Catalytic Enantioselective Inverse Electron Demand Hetero-Diels-Alder Reaction with Allylsilanes, Angew. Chem., Int. Ed., 2014, 53, 6131–6134 CrossRef CAS PubMed.
- X.-K. Guan, G.-F. Liu, D. An, H. Zhang and S.-Q. Zhang, Chiral Imidodiphosphoric Acid-Catalyzed Highly Diastereo- and Enantioselective Synthesis of Poly-substituted 3, 4-Dihydro-2H-Pyrans: [4+2] Cycloadditions of β, γ-Unsaturated α-Ketoesters and 3-Vinylindoles, Org. Lett., 2019, 21, 5438–5442 CrossRef CAS PubMed.
- C.-C. Hsiao, S. Raja, H.-H. Liao, I. Atodiresei and M. Rueping, Ortho-Quinone Methides as Reactive Intermediates in Asymmetric Brønsted Acid Catalyzed Cycloadditions with Unactivated Alkenes by Exclusive Activation of the Electrophile, Angew. Chem., Int. Ed., 2015, 54, 5762–5765 CrossRef CAS PubMed.
- J.-J. Zhao, S.-B. Sun, S.-H. He, Q. Wu and F. Shi, Catalytic Asymmetric Inverse-Electron-Demand Oxa-Diels–Alder Reaction of in situ Generated Ortho-Quinone Methides with 3-Methyl-2-Vinylindoles, Angew. Chem., Int. Ed., 2015, 54, 5460–5464 CrossRef CAS PubMed.
- E. Wada, H. Koga and G. Kumaran, A Novel Catalytic Enantioselective Tandem Transetherification−Intramolecular Hetero Diels−Alder Reaction of Methyl (E,)-4-Methoxy-2-Oxo-3-Butenoate with δ, ε-Unsaturated Alcohols, Tetrahedron Lett., 2002, 43, 9397–9400 CrossRef CAS.
- J. Lauberteaux, A. Lebrun, A. V. D. Lee, M. Mauduit, R. M. D. Figueiredo and J.-M. Campagne, Iron-Catalyzed Enantioselective Intramolecular Inverse Electron-Demand Hetero Diels−Alder Reactions: an access to bicyclic dihydropyran derivatives, Org. Lett., 2019, 21, 10007–10012 CrossRef CAS PubMed.
- L. Zeng, S. Liu, Y. Lan and L. Gao, Catalytic Asymmetric Oxa-Diels–Alder Reaction of Acroleins with Simple Alkenes, Nat. Commun., 2023, 14, 3511 CrossRef CAS PubMed.
- M. Jin, C. Tang, Y. Li, S. Yang, Y.-T. Yang, L. Peng, X.-N. Li, W. Zhang, Z. Zuo, F. Gagosz and L.-L. Wang, Enantioselective Access to Tricyclic Tetrahydropyran Derivatives by a Remote Hydrogen Bonding Mediated Intramolecular IEDHDA Reaction, Nat. Commun., 2021, 12, 7188 CrossRef CAS PubMed.
- R. M. Beesley, C. K. Ingold and J. F. Thorpe, CXIX-The Formation and Stability of Spiro-Compounds. Part I. Spiro-Compounds from Cyclohexane, J. Chem. Soc., Trans., 1915, 107, 1080–1106 RSC.
- M. E. Jung and G. Piizzi,
Gem-disubstituent Effect: Theoretical Basis and Synthetic Applications., Chem. Rev., 2005, 105, 1735–1766 CrossRef CAS PubMed.
-
R. J. Devita, J. Bao and S. G. Mills, Octahydropyra-no[3,4-C]pyrrole Tachykinin Receptor Antagonists, 2007, WO2007075528A2 Search PubMed.
-
M. D. Meyer, et al., Benzopyanopyrrole and Benzopy-ranopyridine alpha-1 Adrenergic Compounds, 1998, WO98/24791 Search PubMed.
- S.-B. Mou, W. Xiao, H.-Q. Wang, S.-J. Wang and Z. Xiang, Syntheses of Epoxyguaiane Sesquiterpenes (−)-Englerin A, (−)-Oxyphyllol, (+)-Orientalol E, and (+)-Orientalol F: a Synthetic Biology Approach, Org. Lett., 2020, 22, 1976–1979 CrossRef CAS PubMed.
- D. J. Paymode and C. V. Ramana, Studies toward the Total Synthesis of Parvifolals A/B: an Intramolecular o-Quinone Methide [4+2]-Cycloaddition to Construct the Central Tetra-cyclic Core, ACS Omega, 2019, 4, 810–818 CrossRef CAS PubMed.
- For selected examples of CPA catalyzed cyclization reactions from our lab:
(a) L.-L. Wang, T. Jiang, P.-H. Li, R.-J. Sun and Z. Zuo, Asymmetric Syntheses of Spirooxindoledihydro-quinazolinones by Cyclization Reactions between N-substituted Anthranilamides and Isatins, Adv. Synth. Catal., 2018, 360, 4832–4836 CrossRef CAS;
(b) L.-L. Wang, Q. Yu, W. Zhang, S. Yang, L. Peng, L. Zhang, X.-N. Li, F. Gagosz and A. Kirschning, Asymmetric Total Synthesis of Antibiotic Elansolid A, J. Am. Chem. Soc., 2022, 144, 6871–6881 CrossRef CAS PubMed.
- For reviews on chiral phosphoric acid catalyzed reactions:
(a) T. Akiyama, Stronger Brønsted Acids, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS PubMed;
(b) M. Terada, Chiral Phosphoric Acids as Versatile Catalysts for Enantioselective Transformations, Synthesis, 2010, 1929–1982 CrossRef CAS;
(c) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brønsted Acid and Metal Catalysis: History and Classification by Mode of Activation; Brønsted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed;
(d) T. Akiyama and K. Mori, Stronger Brønsted Acids: Recent Progress, Chem. Rev., 2015, 115, 9277–9306 CrossRef CAS PubMed;
(e) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Addition and Correction to Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brønsted Acid and Metal Catalysis: History and Classification by Mode of Activation; Brønsted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates, Chem. Rev., 2017, 117, 10608–10620 CrossRef CAS PubMed.
- For selected reviews on click reactions applied in biosystems: T. Cañeque, S. Müller and R. Rodriguez, Visualizing Biologically Active Small Molecules in Cells Using Click Chemistry, Nat. Rev. Chem., 2018, 2, 202–215 CrossRef.
- T. Ishiyama, M. Murata and N. Miyaura, Palladium(0)-Catalyzed Cross-Coupling Reaction of Alkoxydiboron with Haloarenes: A Direct Procedure for Arylboronic Esters, J. Org. Chem., 1995, 60, 7508–7510 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2286029 and 2286031. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qo01615c |
| ‡ Equal contribution by these authors. |
|
| This journal is © the Partner Organisations 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *c
*c