
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Halogen bonding and mechanochemistry combined: synthesis, characterization, and application of N-iodosaccharin pyridine complexes†
Christian
Schumacher‡§
 ab,
Khai-Nghi
Truong‡¶
a,
Jas S.
Ward‡
a,
Rakesh
Puttreddy
a,
Anssi
Rajala
a,
Elias
Lassila
a,
Carsten
Bolm
b and
Kari
Rissanen
*a
ab,
Khai-Nghi
Truong‡¶
a,
Jas S.
Ward‡
a,
Rakesh
Puttreddy
a,
Anssi
Rajala
a,
Elias
Lassila
a,
Carsten
Bolm
b and
Kari
Rissanen
*a
aUniversity of Jyvaskyla, Department of Chemistry, Survontie 9 B, 40014 Jyväskylä, Finland. E-mail: kari.t.rissanen@jyu.fi
bInstitute of Organic Chemistry, RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany
First published on 5th December 2023
Abstract
Halogen-bonded complexes are utilized across a myriad of synthetic chemistry fields, with halogen(I) complexes such as Barluenga's reagent being ubiquitous in halogenation reactions. The preparation of Barluenga's reagent requires the use of heavy metal salts and vast amounts of chlorinated solvents. In line with a more modern, environmentally conscious ethos, halogen-bonded adducts and a halogen(I) complex similar to Barluenga's reagent based on N-iodosaccharin were prepared by mechanochemical processes for the first time. The general absence of solvents or the use of vanishingly small amounts of ethyl acetate in a liquid-assisted grinding approach during mechanochemical preparations enabled the homoleptic [(DMAP)–I–(DMAP)]+ iodine(I) complex to be synthesized. The as prepared mechanochemical materials were used in the iodination of antipyrine, demonstrating their potential use as surrogates for Barluenga's reagent in both solution and solid-state syntheses.
Introduction
Noncovalent interactions are an unambiguously important aspect in nature, as strikingly demonstrated by proteins and the double helix structure of DNA.1–3 Hence, noncovalent interactions are extensively studied in the field of natural and materials sciences.2,4,5 Out of the plethora of these interactions, the concept of σ-hole bonding has attracted vast interest in the last few decades,6–10 halogen bonding being the most prominent ambassador. Discovered in the 19th century, albeit not named so,11 halogen bonding was the main aspect of the Nobel prize awarded to the works of Mulliken on the chemical bond (1966) and fundamental to the conformational studies of Hassel (1969).11–20 Since then, the concept of halogen bonding made its way into various fields,11e.g., supramolecular chemistry,21–26 crystal engineering,27–30 liquid crystals,31–35 polymers,36–39 gels,40–44 biomolecular systems,45–49 or organocatalysis.50–56Nowadays, a halogen bond (XB) is defined as the net attractive interaction between the electrophilic region of a halogen, the halogen bond donor, and a Lewis base, the halogen bond acceptor.57 The nature of this attraction is discussed to be mostly based on dispersion,58–63 charge transfer,64–68 and electrostatic interactions.69–73 Spanning a broad range of interaction energies,11 it ranges from weak but distinctive interactions between quinuclidine and iodobenzene [or (iodoethynyl)benzenes (ca. −1.1 kJ mol−1)]74,75 to strong interactions found in N-halosaccharin pyridine N-oxide complexes (up to −120.3 kJ mol−1).76 Additionally, XBs offer distinct characteristics, such as high directionality (interaction angle of ∼180°), short contacts between donor and acceptor [shorter than the sum of the van der Waals (vdW) radii], and tuneability.11,57 As it presents comparable features, XB is often referred to as the “(long lost) brother” of hydrogen bonding (HB),77 endowing it with the potential to become as widely applicable.11,50–56
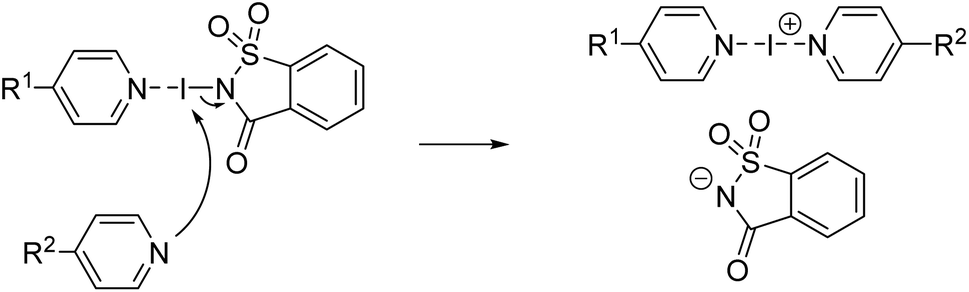
Considering these similarities, Fourmigué, Espinosa, and coworkers posed the question: Are halogen-bonded adducts based on a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry between XB donor and acceptor co-crystals or salts (Fig. 1a)?78 As a matter of fact, so far, salt formation was only observed when dihalogens were reacted with pyridines (and thiones), leading to N-iodopyridinium salts, bonded to the halide anion by a XB.79–83
:1 stoichiometry between XB donor and acceptor co-crystals or salts (Fig. 1a)?78 As a matter of fact, so far, salt formation was only observed when dihalogens were reacted with pyridines (and thiones), leading to N-iodopyridinium salts, bonded to the halide anion by a XB.79–83
 | ||
| Fig. 1 (a) Schematic representation of the co-crystal and “close-to-ionic” coordination mode in NIS/NISac based XB complexes. (b) Molecular structure of Barluenga's reagent (CCDC 202334;† thermal ellipsoids shown at the 50% probability level; distances are given in Å).94–101 (c) Possible asymmetric and symmetric coordination modes of a three-center-four-electron halogen bond. | ||
In Fourmigué's study, it was reasoned that the position of the iodine atom in these systems (D–I⋯A ⇌ [D]−⋯[I–A]+) would define the degree of ionicity. To favor strong halogen bonds, electron-donating substituents were attached to the C4-position of the pyridines increasing the electron density on the sp2 nitrogen making it a stronger XB acceptor.78,84 A “close-to-neutral” co-crystal for halogen-bonded complexes between N-iodosuccinimide and pyridine derivatives was found, whereas a “close-to-ionic” nature was observed when N-iodosaccharin (NISac), a much stronger XB donor,76 was used. The predominant ionic character is well-founded by a much shorter distance between iodine and the sp2 nitrogen atom of the pyridine derivative than the nitrogen atom of the saccharin. Compared to the “close-to-ionic” nature, a genuine ionic form based on charge conservation is found in symmetrical trihalide anions and in bis(pyridine)iodine(I) cations.85–93 One of the most prominent examples is bis(pyridine)iodine(I) tetrafluoroborate, Barluenga's reagent (Fig. 1b).94–101 Since its re-emergence in 1985,94 it (and analogous complexes) has found a myriad of applications in organic synthesis for oxidations, or as a versatile electrophilic halogenation reagent,94–101e.g., with alkenes, alkynes, and aromatics.102–110 Due to the three-center four-electron bond, the [N–I–N]+ iodine(I) complexes differ from the “classical XB” by manifesting a strong simultaneous interaction (≥−100 kJ mol−1)76,111 between two electron donor moieties and the electrophilic iodine atom. This three-center-four-electron halogen bond should not be referred to as a “coordinative halogen bond” anymore,86–91,112 simply because halogen(I) complexes and coordinative metal complexes behave differently. Being a three-center complex, an asymmetric or symmetric coordination mode for iodine(I) is possible (Fig. 1c). Therefore, such complexes have been analyzed in detail by the groups of Rissanen et al. and Erdélyi et al. in the solid state, as well as in solution. In these studies, a highly symmetric coordination mode was found, regardless of the counteranion, substituent effects, or the solvent.85–91,111 Only when two different pyridines, covalently bonded by an aryldiyne linker, viz. a clamp ligand, are used is an asymmetric halogen bond formed with iodine(I) located closer to the more electron-rich pyridine.113 Then, in 2020, the first unrestrained, asymmetric halogen-bonded iodine(I) complexes were reported in the solid-state by Ward et al.114 However, scrambling between asymmetric and symmetric complexes in solution occurred, demonstrating the lability and reactivity of these complexes in solution.115,116
Usually, bis(pyridine)halogen(I) complexes like Barluenga's reagent are prepared in a two-step Ag(I) to I(I) cation exchange reaction.117–122 Initially, the corresponding Ag(I) complex is formed, which then reacts with molecular iodine to form the desired iodine(I) complex in a metathesis reaction. Not only does this approach involve the use of heavy and expensive metal salts (originally, even HgO was applied)94 but also generates a tremendous amount of by-products (i.e., precipitated AgI or AgBr) while using an extensive volume of chlorinated solvent.117 Motivated by the description of a “close-to-ionic” coordination mode in NISac-based XB complexes,78 the preparation of bis(pyridine)iodine(I) complexes by an exchange reaction of the saccharinato anion with another pyridine-based XB acceptor (Scheme 1) was targeted.123 Not only would this approach avoid the use of heavy metals but hopefully lead to a straightforward synthesis of symmetric iodine(I) complexes making use of the entatic iodine atom in the XB complex.
 | ||
| Scheme 1 Schematic representation of the intended exchange reaction in XB complexes with a “close-to-ionic” halogen bond to prepare symmetric and asymmetric iodine(I) complexes. | ||
Results and discussion
Preparation of NISac-based XB complexes in solution
The investigation began via the preparation of four complexes with halogen bonds between NISac (1) and pyridine derivatives 2. 4-Dimethylaminopyrdine (2a, DMAP), 4-pyrrolidinopyridine (2b, PPY), 4-morpholinopyrdine (2c, MPY), and 4-piperidinopyridine (2d, PiPY) were selected for the preparation of the corresponding XB complexes 3a–3d that would offer a “close-to ionic” XB based on the results of Fourmigué's study. After slowly evaporating solutions containing equal amounts of XB donor 1 and the selected pyridine derivative 2, crystals suitable for single crystal X-ray diffraction (SCXRD) analysis were obtained and compared to structures reported in the literature where available.124 The corresponding X-ray crystal structures of desired XB complexes 3a–3d are shown in Fig. 2.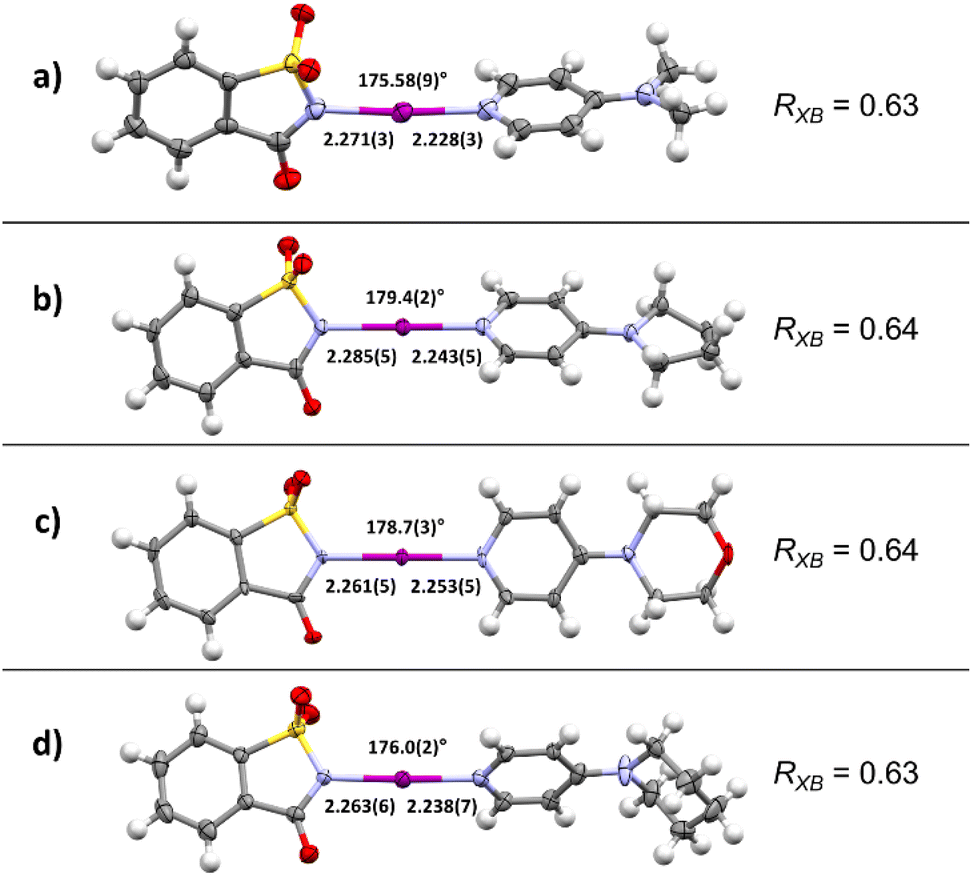 | ||
| Fig. 2 X-ray crystal structures of XB complexes between NISac and (a) DMAP (3a), (b) PPY (3b), (c) MPY (3c), (d) PiPY (3d), and corresponding average reduction ratios, RXB (thermal ellipsoids shown at the 50% probability level; distances are given in Å). | ||
In all cases XB complexes with a 1:1 stoichiometry and close to linear coordination geometry were formed with a very short N–I⋯N′ halogen bond. Even though the distance between donor and acceptor does not linearly correlate with the interaction strength, the normalized contact distance, RXB, of the prepared XB complexes are calculated to compare them with each other as these systems have very similar scaffolds. The RXB is defined as the distance between the donor and the acceptor, dXB [Å], and is divided by the sum of vdW radii [Å] of the donor (X) and the acceptor (B) [RXB = dXB/(XvdW + BvdW)].86,125–128 Using 1.55 Å for N and 1.98 Å for I,125–128 complexes 3a and 3d gave a value of 0.63, and a value of 0.64 for 3b and 3c. A more detailed analysis of the crystal structures revealed possible secondary interactions (see ESI, Fig. S3–S10†). For 3a π-stacking is present, with centroid-to-centroid distances of ca. 3.71 Å (between the aromatic rings of DMAP and the N-iodosaccharin). The secondary interactions are present in each complex and their RXB value variation is negligible, indicating that crystal packing of very strong N-iodosaccharin–pyridine XB complexes does not have an impact on the N–I⋯N′ halogen bond itself. These observations are in accordance with the work of Fourmigué and Espinosa, who observed the same secondary interactions for a different polymorph of XB complex 3a as well.78 Due to iodine transfer from N-iodosaccharin to the base occurring in 3a–3d, the binding constants cannot be determined.78 However, iodine transfer does not occur when the XB acceptor is a less nucleophilic pyridine129 or pyridine N-oxide130 instead of 4-dimethylaminopyrdine (2a, DMAP), 4-pyrrolidinopyridine (2b, PPY), 4-morpholinopyrdine (2c, MPY), or 4-piperidinopyridine (2d, PiPY), making binding constant determination possible. The binding constants for N-iodosaccharin–pyridine129 and N-iodosaccharin–pyridine N-oxide76,130 XB complexes are high, varying from 236 to 1.44 × 105 and 1180 to 108 M−1, respectively. As the XB complexes 3a–3d constituted the foundation of further studies, a robust and scalable synthesis protocol for their preparation was devised. During initial crystallization attempts, colorless precipitates were always found when EtOAc was used as a solvent. Additional analysis by SCXRD (single crystal X-ray diffraction) identified them as the desired XB complexes 3a–3d. Therefore, the synthesis of these XB complexes was attempted by a precipitation procedure (Scheme 2). First, equimolar amounts of XB donor 1 and the chosen acceptor 2 were dissolved separately in EtOAc, then combined and the desired XB complex precipitated immediately. Through use of this protocol, the desired complexes were obtained in high yields ranging from 81% for 3b to 91% for 3c. Being first performed on a scale of a few milligrams, the protocol was effortlessly scaled up to a quantity of 100 mg NISac for complexes 3a, 3c, and 3d. In addition, a 1 mmol batch gave XB complex 3a in 87% yield demonstrating the robustness of the developed protocol. Only in the case of PPY might solubility problems interfere with the standard protocol. Potential water contamination of the pyridine derivative results in protonation and therefore in lower solubility. In this case, pyridine 2b was dissolved in DCM, combined with a solution of 1 in EtOAc, and kept overnight for precipitation. If freshly prepared pyridine 2b was used, the solubility increased, and the standard protocol was applied.
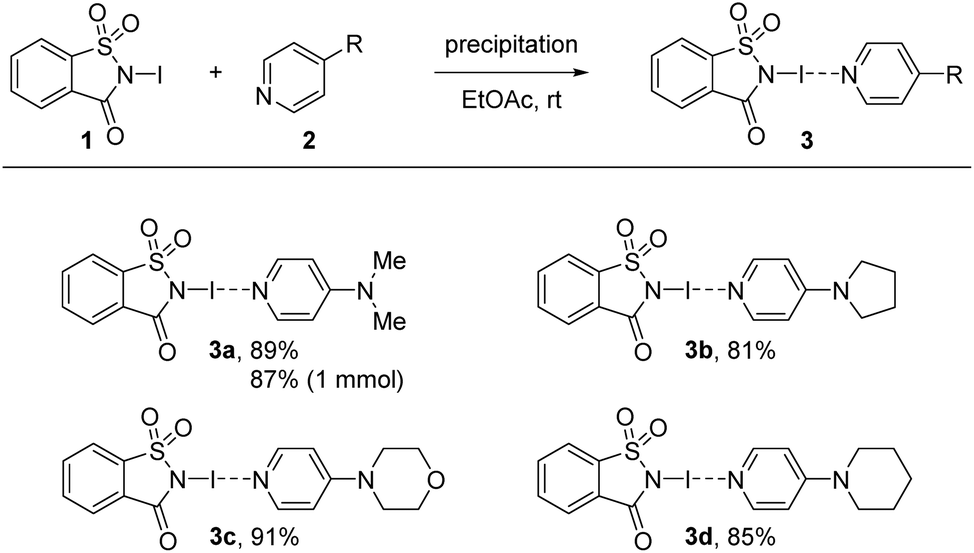 | ||
| Scheme 2 Synthesis of XB complexes 3 in the reaction between NISac (1) and 4-substituted pyridine derivatives 2 by precipitation. All reactions were performed on a 0.32 mmol scale. NISac (1, 100 mg, 0.32 mmol, 1.0 equiv.), pyridine derivative 2 (0.32 mmol, 1.0 equiv.), both dissolved in 1.5 mL EtOAc, then combined. The XB complexes 3a–3d were isolated by filtration. | ||
Preparation of the symmetric and asymmetric iodine(I) complexes by crystallization
After obtaining sufficient quantities of 3a–3d, the hypothesis that a strongly polarized “entatic” iodine atom enabled an exchange reaction to proceed (Scheme 3) was tested. The exchange reaction of the N-saccharinato part of complexes 3a–3d with another XB acceptor 2 would lead to a metal-free synthesis of symmetric or asymmetric iodine(I) complexes 4a–4cd, similar to Barluenga's reagent (Scheme 3).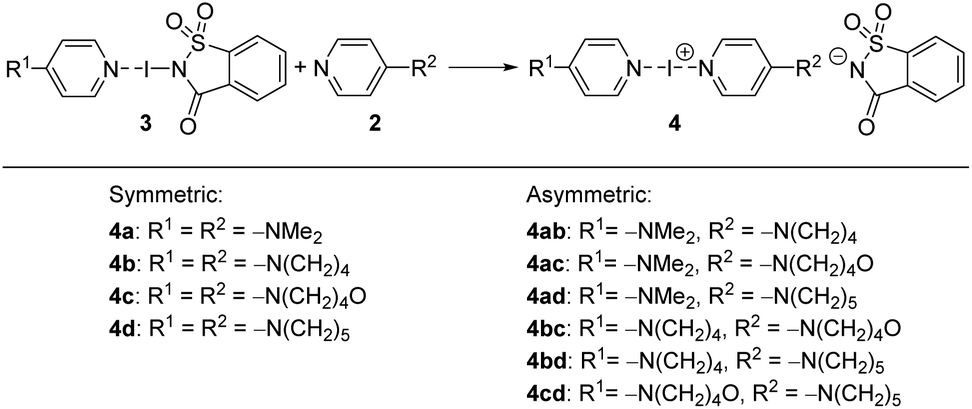 | ||
| Scheme 3 Preparation of symmetric and asymmetric iodine(I) complexes 4a–4cd by an exchange reaction between XB complexes 3 and the corresponding pyridines 2. | ||
First, as a proof-of-concept, the crystallization of bis(4-dimethylaminopyridine)iodine(I) complex 4a was performed. Equal amounts of DMAP and 3a were dissolved in different solvents and kept at ambient temperature for co-crystallization by slow evaporation. After a few days, crystals suitable for SCXRD analysis were formed in acetonitrile. The crystal structure confirmed the desired iodine(I) complex 4a (4a_1, Fig. 3a) with symmetric N–I bond lengths, 2.25(1) and 2.25(1) Å, of the [N–I–N]+ moiety and a N–I–N angle of 178.0(5)°, very similar to those reported for Barluenga's reagent (Fig. 1b). The N-saccharinato anion does not manifest any interactions with the iodine(I) cation. When the same reaction was done in DCM, a slow evaporation of the DCM solution resulted in a different polymorph of 4a, namely, 4a_2. The geometry of the three-center-four-electron halogen bond in 4a_2 is very similar, with a N–I–N angle of 177.67(11)° and N–I bond lengths of 2.248(3) and 2.250(3) Å. Despite the success in the crystallization of the desired complex 4a, it was realized that co-crystallization under even slightly altered conditions can lead to a different composition.115,116 For instance, using DCM as solvent, the desired bis(2a)iodine(I) cation was formed. However, the counter anion is not the expected NSac−. Instead, the linear triiodide was found resulting from a decomposition of the in situ formed 4a (ESI, Fig. S30–S31†). Besides the desired complexes, the used starting materials 2a (in protonated form) or 3a tend to crystallize out from the solution, yielding a complex mixture (ESI, Fig. S23–S29†).
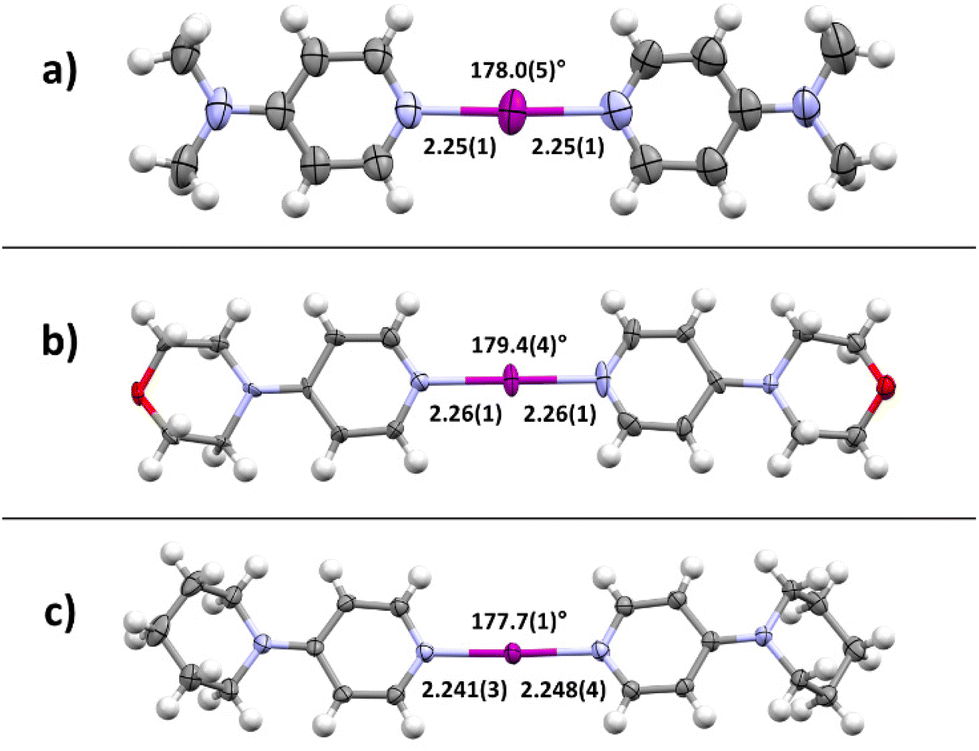 | ||
| Fig. 3 X-ray crystal structures of symmetric iodine(I) complexes 4: (a) [I(DMAP)2]+ (4a_1), (b) [I(MPY)2]+ (4c_1), (c) [I(PiPY)2]+ (4d) (thermal ellipsoids shown at the 50% probability level; anions, minor disordered positions, and solvates omitted for clarity; distances are given in Å). | ||
Motivated by the presence of crystals of symmetric iodine(I) complex 4a through the co-crystallization method, the crystallization of the symmetric iodine(I) complexes 4b–d was attempted. Unfortunately, the crystallization of complex 4b from 3b proved to be difficult. In all attempts, only decomposition products, the 2bH-saccharinato salt, or the XB complex 3b was found (ESI, Fig. S27–S29†). Therefore, the crystal structure of 4b remains unknown. The desired symmetric [2c–I–2c]+ complex 4c was successfully crystallized at −20 °C (Fig. 3b) using pyridine 2c and XB complex 3c as the starting materials in acetone. The N–I bond lengths of 2.26(1) and 2.26(1) Å, and the N–I–N angle of 179.4(4)° were found for 4c_1 (acetone solvate, ESI Fig. S17†). Two additional structures for 4c were obtained, 4c_2 (non-solvated, ESI Fig. S18†) and 4c_3 (chloroform solvate, ESI Fig. S19†), with similar XB geometries to 4c_1, with N–I bond lengths of 2.26(1) Å (4c_2) and 2.256(7)/2.251(7) Å (4c_3), and N–I–N angles of 180° (4c_2) and 179.4(3)° (4c_3). Similar behavior was found for complex 4d (Fig. 3c) which crystallized from DCM at 4 °C using pyridine 2d and XB adduct 3d as starting materials. It shows N–I bond lengths of 2.241(3) and 2.248(4) Å, with a N–I–N angle of 177.7(1)° and crystallizes as a DCM solvate (ESI, Fig. S21†). In the three prepared symmetric complexes of 4c no secondary interaction on the [N–I–N]+ cation was observed. The slight asymmetry in the XB bond length for complexes 4c and 4d is very likely caused by crystal packing, especially as two of these compounds crystallize as solvates. The preparation of asymmetric iodine(I) complexes 4ab–4cd was then attempted, knowing about the existence of similar complexes.114–116 As the preparation of the symmetric iodine(I) complexes 4a, 4c, and 4d was possible by the exchange reaction between the N-saccharinato anion and pyridine derivative 2, the same procedure for the preparation of the asymmetric complexes was used. Equal amounts of the chosen XB complex 3 and pyridine derivative 2 were used in the crystallization. Out of the six possible asymmetric iodine(I) complexes 4ab–4cd, the presence of four complexes (Fig. 4) was demonstrated by the analysis of the corresponding crystalline materials that were formed by co-crystallization. Slow cooling at 4 °C of a DCM solution over 15 hours resulted in single crystals of 4ab (Fig. 4a, ESI, Fig. S14†). The asymmetric iodine(I) complex 4ab exhibits a N–I–N angle of 178.6(3)° and asymmetric N–I bond lengths. The distance for N(2a)–I is 2.258(9) Å and for I–N(2b) 2.240(9) Å. This result was expected as the pyrrolidino group was slightly more electron-donating than the dimethylamino group. As a result, the electron density on the sp2 nitrogen atom of 2b is higher, making it a stronger XB acceptor. Starting from XB complex 3c and 2a or vice versa (XB complex 3a and pyridine 2c), the asymmetric complex 4ac was crystallized from acetone, or MeCN respectively, at 4 °C (Fig. 4b). Consequently, successful crystallization cannot be attributed solely to a difference in the relative (already alike) Lewis base characteristic of the pyridines used, since otherwise only the symmetrical complex of the stronger Lewis basic pyridine is to be expected. Therefore, it is reasoned that the association constants play an important role, too. In a similar way, complex 4bd was prepared from 3d and 2b using DCM as a solvent at 4 °C (Fig. 4c). For both iodine(I) complexes, N–I–N angles of 178.4(3)° (4ac) and 177.4(2)° (4bd) were found. On the other hand, an unexpected symmetric [N–I–N]+ moiety was found with N–I bond lengths of 2.234(5) Å and 2.254(5) Å for 4ac and 4bd, respectively. It should be noted that both 4ac and 4bd demonstrated symmetry-based disorder of 2a/2c (in 4ac) and 2b/2d (in 4bd), and therefore their individual N–I bond lengths were indistinguishable from one another. The fourth asymmetric iodine(I) complex 4cd was prepared by crystallization at −20 °C from 3c and 2d in chloroform. It showed the expected asymmetric [N–I–N]+ moiety with a N–I–N angle of 178.9(6)° and N–I lengths of 2.25(1) Å towards 2c and 2.28(2) Å for 2d. The implementation of oxygen in the six-membered ring in the morpholino group makes it a slightly stronger electron-donating group than the piperidino group resulting in the distinct difference in bond length towards the iodine. Complexes 4ad and 4bc were not obtained upon crystallization in any of the methods attempted, as the starting materials or decomposition products crystallized preferentially. Other products, such as [2bH]I and the [3cH]saccharinato salts crystallized out in attempts to prepare complex 4bc, and the [2dH]I3 salt for 4ad. As an undesired product, the symmetric iodine(I) complex 4c was observed in attempts to prepare the asymmetric complex 4cd from 2c and 3d. Similar results were obtained in an attempt to crystallize complex 4bc from 2b and 3c. As asymmetric iodine(I) complexes are also highly reactive and only a few examples have been crystallized,114–116 the presence of undesired side products was expected (ESI, Fig. S23–S32†). This is underlined by the fact that all obtained crystal structures of complexes 4ab, 4ac, 4db and 4cd were obtained at lowered temperatures and [2H]saccharinato salts were detected as side products. The combination of their structural instability in solution and the described co-occurrence with side products made further characterization, e.g., by NMR, difficult, and SCXRD remained the only characterization method for the asymmetric complexes.
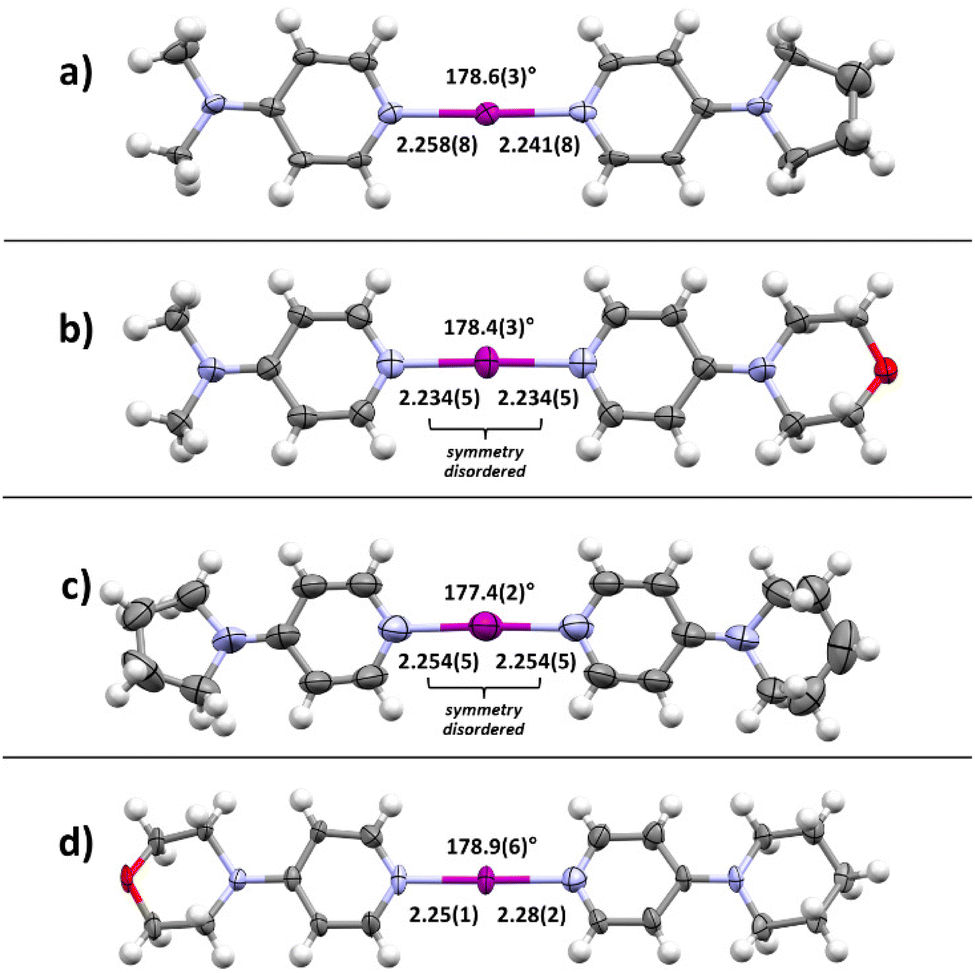 | ||
| Fig. 4 X-ray crystal structures of asymmetric iodine(I) complexes 4: (a) [I(DMAP)(PPY)]+ (4ab), (b) [I(DMAP)(MPY)]+ (4ac), (c) [I(PPY)(PiPY)]+ (4bd), (d) [I(MPY)(PiPY)]+ (4cd) (thermal ellipsoids shown at the 50% probability level; anions, minor disordered positions, and solvates omitted for clarity; distances are given in Å). | ||
Mechanochemical synthesis of the NISac XB complexes
The crystallization of iodine(I) complexes 4 in solution was successful but proved to be difficult due to the observation of numerous side products. Therefore, a solid-state synthesis approach was considered to be a promising alternative. Due to the absence of solvent, the solid-state properties of NISac complexes 3 are unaltered and would enable the direct synthesis of Barluenga-type iodine(I) complexes 4. When successful, the solid-state synthesis could be used for the preparation of larger quantities of the desired complexes and promote their application as electrophilic iodinating agents, whilst also promoting an environmentally friendly methodology. For this purpose, a mechanochemical approach using a mortar and pestle for triturating the starting materials was adapted.131–133 The concept of mechanochemistry is defined as the induction of a chemical reaction by the direct absorption of mechanical energy.134 Due to the absence of solvent, shorter reaction times, cleaner reactions,131–133,135–141 and access to products that are inaccessible by other means,142–149 mechanochemistry enjoys increasing popularity in numerous fields,131,136,137,139,141e.g., organic synthesis,132,135,140,142,147 supramolecular chemistry,150 materials science,143,144,146,148 catalysis,138,151 crystal engineering,152–158 or coordination chemistry,145,159 just to name a few. Due to the high popularity and numerous advantages, IUPAC has nominated it as one of ten innovations with the potential to change the world,160 and it has already been successfully applied in the contexts of co-crystallization and halogen bonding.150,152–158,161,162Co-crystal formation between XB donor 1 and acceptor 2a as a model reaction was selected (Scheme 4). To avoid any influence of the solvent, a focus was made on ex situ analysis using IR spectroscopy and PXRD (powder X-ray diffraction) for optimization and a comparison of spectra to the complexes obtained from solution. As a first attempt, equal amounts of NISac and DMAP were mixed in a vial using a spatula. Just by this gentle mixing, the desired XB complex, 3a, was not formed according to IR analysis (ESI, Fig. S44†). As a next try, equal amounts of DMAP and NISac were ground in a mortar. After 1 minute of grinding, the formation of complex 3a could already be detected, together with unreacted starting materials, by IR (ESI, Fig. S45†). A total grinding time of 15 min was necessary to quantitively convert the starting materials to the desired product according to IR (Fig. 5), however, the sample was amorphous and could not be analyzed by PXRD.
 | ||
| Scheme 4 Mechanochemical synthesis of XB complexes 3 from NISac (1) and pyridine derivatives 2 using EtOAc for a liquid-assisted grinding (LAG) approach. | ||
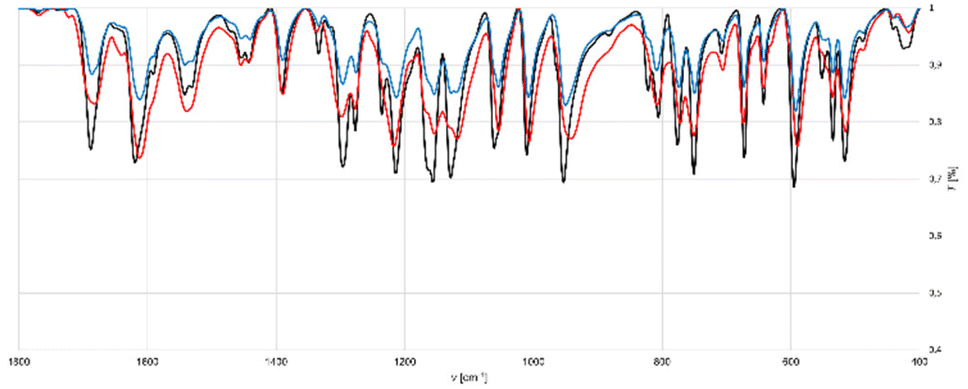 | ||
| Fig. 5 Comparison of IR spectra around 1800–400 cm−1 obtained in the mechanochemical synthesis of XB complex 3a. Black: XB complex 3a from solution. Red: Neat grinding of equal amounts of DMAP and NISac for 15 min. Blue: LAG (EtOAc) of equal amounts of DMAP and NISac for 5 min. | ||
Similar behavior was found for the formation of complexes 3b–3d, of which 3d required a longer grinding time due to the slower conversion reaction (ESI, Fig. S46–S50†). The slower conversion to 3d compared to the fast reaction in solution was probed by analyzing the XB donor NISac more deeply. The IR spectrum of NISac (ESI, Fig. S33†) showed broad bands in the region from 3600 to 3200 cm−1 which could be attributed to OH vibrations. This observation agrees well with an NISac hydrate, which was indeed confirmed by elemental analysis and a comparison to the self-prepared sample of 1. Moreover, a search of the CSD revealed that the reported crystal structures of NISac occurred either as a halogen bonded hydrate or a solvate (THF).100 In addition, attempts to prepare non-hydrated or non-solvated NISac crystals failed. Based on these findings, it was assumed that the hydrate was hampering the reactivity of NISac in the solid state. Therefore, an investigation of the positive influence of small quantities of solvent with the hope of enhancing the reactivity of NISac by liquid-assisted grinding (LAG) was done.151,159,163–166 By adding a single drop of EtOAc prior to grinding of NISac and DMAP for 1 min, conversion to complex 3a was observed, and IR (Fig. 5) indicated its quantitative formation after 5 cycles of adding 1 drop of EtOAc and grinding the mixture for 1 min (Scheme 4). Moreover, freshly prepared XB complex 3a was analyzed by PXRD. Even though mechanochemically prepared 3a had an amorphous character, the PXRD pattern was in good agreement with a simulated pattern obtained from the SCXRD analysis of a solution-prepared pure sample of 3a (ESI, Fig. S74†). Altering the procedure by adding 3 drops at the beginning and grinding the mixture for 3 min or reducing the amount of solvent by just adding one drop of solvent did not prove to be as efficient (ESI, Fig. S51–S53†). Subsequently, the protocol was extended to the preparation of 3b–3d and successfully confirmed by IR and PXRD analysis (ESI, Fig. S54–S57 and S75–S77†). The milling time for 3c and 3d had to be extended to 6 min as traces of starting materials could still be detected in the IR spectra after shorter grinding times. PXRD analysis of the mechanochemically prepared complexes 3a–3d confirmed them to be pure as they perfectly fit with the simulated PXRD pattern based on SCXRD structures of solution prepared pure 3a–3d (ESI, Fig.S75–S77†). The successful mechanochemical preparation of NISac complexes 3 was further verified by their successful subsequent crystallization and confirmation of their identities from SCXRD studies. In addition to EtOAc as the LAG solvent, MeCN was also tested as an alternative. However, the decomposition reaction was visually observed for 2d after only 2 min, as the reaction mixture turned from the usual colorless powder to a brown slurry. Decomposition could also be seen by IR analysis (ESI, Fig. S57†).
As a proof-of-concept, the developed protocol was transferred to a planetary ball mill, which would simplify scaling up of the synthesis, making it more reproducible as it could be performed under well-controlled conditions. The experiment was executed for 3a on a 0.2 mmol scale. An agate vessel, 3 agate balls (10 mm in Ø), and 400 rpm frequency were used. After ball-milling of equal amounts of 1 and 2a in an agate vessel for 5 cycles of 1 min and with the addition of one drop of EtOAc prior to each cycle, 3a was quantitatively obtained according to IR. Even though the solution approach is quite straightforward to scale up, the benefits of the mechanochemical protocol are substantial as only minute amounts of the LAG solvent is used.
Mechanochemical preparation of the iodine(I) complexes
Having demonstrated the successful utilization of a mechanochemical protocol for the synthesis of NISac complexes 3, we attempted to prepare symmetric and asymmetric iodine(I) complexes 4. The preparation of symmetric iodine(I) complex 4a was selected as the model target. Equal amounts of DMAP and pure, solution-prepared solid 3a were ground under neat conditions using an agate mortar. IR analysis of the product indicated the formation of 4a after 10 min grinding. The desired complex 4a could already be detected after 2 min grinding (ESI, Fig. S59 and S60†). LAG using EtOAc was then applied to probe the possibility of reducing the reaction time. The LAG experiment consisted of 5 cycles by adding a drop of solvent and grinding equal amounts of starting materials 2a and 3a in an agate mortar (Scheme 5, route A). Not only did the reaction quantitatively produce desired 4a after 5 min grinding, but it also allowed us to use other solvents such as DCM, CHCl3, acetone, or MeCN (ESI, Fig. S61 and S62†). To further utilize the mechanochemistry in two subsequent reactions, the above LAG protocol was first applied to the synthesis of 3a, and then continued by adding an equivalent amount of DMAP to as-obtained 3a to convert it into symmetric iodine(I) complex 4a. IR and PXRD analysis confirmed (ESI, Fig. S63–S65 and S78–S81†) the quantitative conversion (Scheme 5, route B).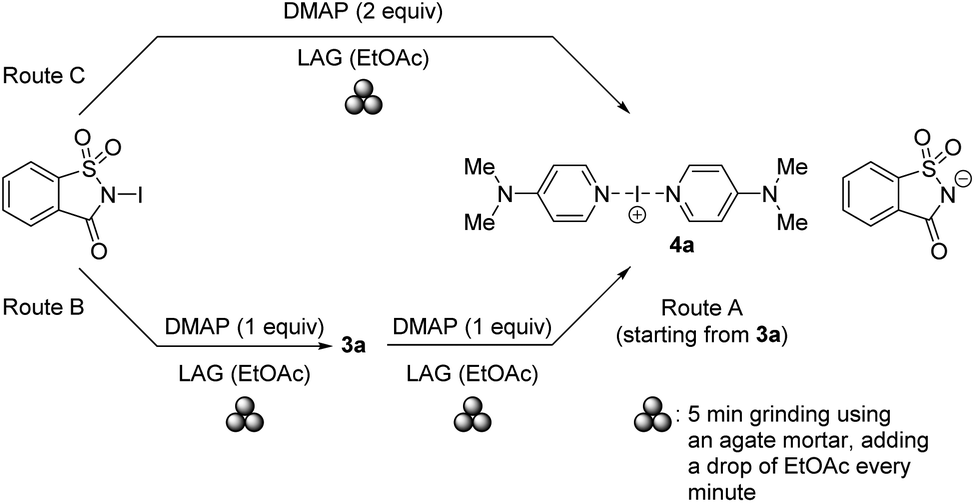 | ||
| Scheme 5 Investigated mechanochemical routes towards iodine(I) complex 4a. | ||
This result suggested that 4a could be obtained mechanochemically through a one-step (concerted or domino-type) reaction, by simply grinding 2 equivalents of DMAP and 1 equivalent of NISac using a LAG approach with EtOAc in a mortar (Scheme 5, route C). IR and PXRD results (Fig. 6) were very satisfying by revealing the quantitative formation of 4a in just 5 min. Dissolving 4a, from the above reaction, in DCM and leaving it to slowly evaporate led to single crystals suitable for SCXRD analysis, which confirmed the identity of 4a.
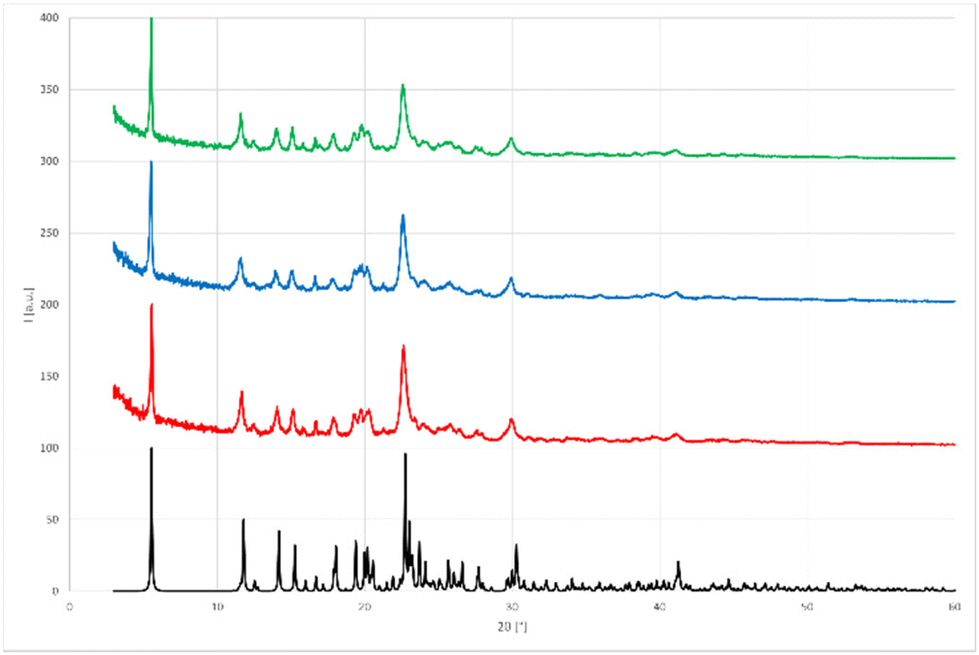 | ||
| Fig. 6 Comparison of PXRD patterns obtained in the mechanochemical preparation of symmetric iodine(I) complex 4a.33 Black: Simulated PXRD pattern of symmetric iodine(I) complex 4a. Red: route A (3a and DMAP for 5 min under LAG (EtOAc) conditions). Blue: route B (two step synthesis under LAG (EtOAc) conditions). Green: route C (grinding one equivalent of NISac and two equivalents of DMAP for 5 min under LAG (EtOAc) conditions). | ||
After establishing the above, fast and robust, one-step protocol, the preparation of other symmetric iodine(I) complexes 4c–d was carried out using the two above-mentioned LAG protocols. The first is based on grinding equal amounts of solution-prepared 3c and 3d and matching pyridines 2c and 2d (Scheme 5, route A), whereas the second protocol uses the one-pot synthesis method by grinding NISac and 2 equivalents of the chosen pyridines 2c and 2d (Scheme 5, route C). For the reactions of both 2c–3c and 2d–4d, IR and PXRD analysis revealed the presence of desired products 4c and 4d. As expected, the products were partially amorphous. Nonetheless a good match for the simulated PXRD patterns and those from as-prepared 4c and 4d was found (ESI, Fig. S66–S70 and S82–S83†). Moreover, the products prepared by two-step or one-step LAG methods showed almost identical spectra. The same observations were made for the attempted synthesis of complex 4b. However, as any crystallization attempt was not successful at this stage and the molecular structure remains unknown so far, the extension of the protocols to the synthesis of product 4b is still to be confirmed. Only the comparison between the IR spectra of used starting materials and the prepared sample differs, indicating the presence of a new species (ESI, Fig. S71–S72 and S84†).
As with the mortar-based mechanochemical preparation of XB complex 3a, subsequent transfer of the protocol to a planetary ball mill, with a focus on the preparation of symmetric iodine(I) complex 4a as a proof-of-concept target, took place. The same mechanochemical conditions to those implemented for the proof-of-concept preparation of 3a in the planetary ball mill were utilized. Only the molar ratios were changed, with 1 equivalent of NISac and 2 equivalents of DMAP being milled under the aforementioned LAG conditions using EtOAc. The effective preparation was shown by IR analysis, and it could effortlessly be scaled up to a 1 mmol approach.
Attempts were made to extend the mechanochemical methodology to the synthesis of asymmetric iodine(I) complexes 4ab–4cd by grinding equal amounts of XB complex 3 and another pyridine 2, however, the systems seemed to undergo scrambling, which made their analysis difficult. Thus, the synthesis of asymmetric iodine(I) complexes under mechanochemical conditions was not investigated further.
Iodination of antipyrine
Being able to prepare sufficiently large quantities of the materials 3a and 4a, an attempt was made to use them as surrogates for Barluenga's reagent in a proof-of-concept electrophilic iodination reaction. A report in the literature indicates that the iodination of tertiary enaminones using Barluenga's reagent is feasible,167 whereas other methods are described as being difficult.167–171 Motivated by these reports, the α-iodination of antipyrine (5) to 4-iodoantipyrine (6) was selected as the model reaction (Scheme 6).167,172 Antipyrine derivatives are known to be biologically active as they exhibit antipyretic, analgesic and anti-inflammatory properties167–171 and are used for medicinal screening processes as well.173–180 Therefore, iodo-derivative 6 could be a useful building block for other biologically active antipyrine derivatives since iodination opens the door for further structural diversity.167,180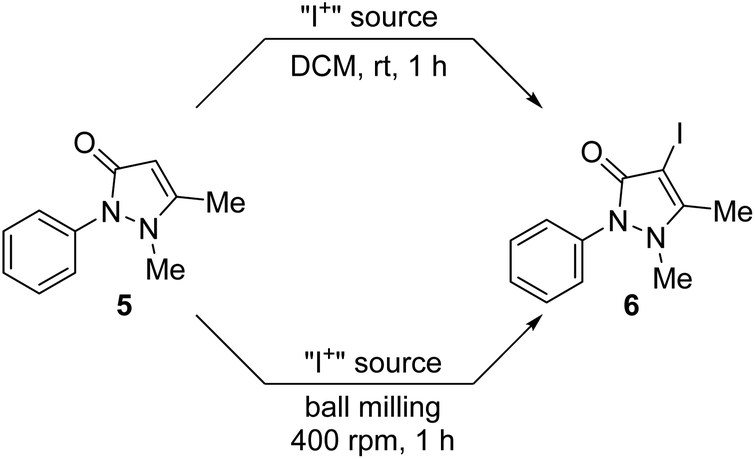 | ||
| Scheme 6 Iodination of antipyrine. | ||
Initially, the reported solution iodination protocol was followed as a baseline reaction.167,172 Equimolar amounts of antipyrine and the chosen iodination reagent were stirred in DCM for 1 h at ambient temperature. A parallel mechanochemical protocol was devised, which used a planetary ball mill operating at 400 rpm. Freshly prepared Barluenga reagent bis(pyridine)iodine(I) tetrafluoroborate was used as a benchmark solution reaction (Table 1, entry 1), which gave 6 in an excellent 91% yield. Using 10 agate balls (5 mm in Ø) the same yield (Table 1, entry 1) was obtained for the mechanochemical protocol; instead, when using 3 agate balls of 10 mm in diameter, the yield was increased to 96% (Table 1, entry 1). Using NISac-based 3a, a very good yield of 87% for both the solution and mechanochemical protocols (Table 1, entry 2) was achieved. Symmetric iodine(I) complex 4a resulted in a good yield of 80% in solution and 76% from ball milling (Table 1, entry 3). However, when the mechanochemical reaction was performed on a 1 mmol scale, product 6 was obtained in a very good yield of 89% (Table 1, entry 3). The SCXRD determined unit cell confirmed the product to be 6.172 The above results confirm that both the charge-neutral NISac and cationic iodine(I) complexes can be used for iodination reactions and achieve yields similar to those with Barluenga's reagent. Even though the yield is slightly lower when made mechanochemically, the advantage of using 3a and 4a is clearly based on their very simple and metal-free synthesis compared to preparations that utilize Barluenga's reagent. In addition, 59% of DMAP and 11% of saccharin could be recovered under unoptimized conditions in a 1 mmol approach. This enables saccharin to be recovered and converted back into NISac, which would provide an avenue for the continuous recovery and usage of the starting materials. Consequently, the as-prepared mechanochemical materials 3a and 4a represent suitable surrogates for Barluenga's reagent. The influence of ligand modulation of the Barluenga reagent was studied by using the DMAP derivative of Barluenga's reagent. Good yields of 85% and 83% for both protocols (Table 1, entry 4) were achieved for the solution and mechanochemical strategies, respectively. As these results are similar to the ones using reagents 3a and 4a, it was reasoned that the influence and nature of the ligand of the cationic iodine(I) complex was negligible. Finally, NISac and elemental iodine were tested (Table 1, entries 5 and 6). In contrast to results reported in the literature (vide supra), we found that iodination did take place. However, as this puts the use of Barluenga's reagent for the iodination of antipyrine into question, it does not interfere with the fact that the mechanochemically NISac based materials 3a and 4a are potential substitutes for Barluenga's reagent. Not only is the performance similar but they also evade the disadvantages of preparing Barluenga's reagent such as chlorinated solvents and heavy metals.
| Entry | Reagent | Solutionb | Ball millingc |
|---|---|---|---|
| a All reactions were performed on a 0.2 mmol scale. 5 (37.6 mg, 0.2 mmol), “I+” source (0.2 mmol). Yields after column chromatography. b DCM (3 mL), 1 h at RT. c Agate vessel, 12 mL, 10 agate balls (5 mm in Ø), 400 rpm, 4 × 15 min, reversing rotation every cycle. d 3 balls (10 mm in Ø). e Performed on a 1 mmol scale. | |||
| 1 | Barluenga's reagent | 91 | 91 (96)d |
| 2 | 3a | 87 | 87 |
| 3 | 4a | 80 | 76 (89)e |
| 4 | [(DMAP)2I]BF4 | 85 | 83 |
| 5 | NISac | 85 | 93 |
| 6 | I2 | 89 | 49 |
Conclusions
In summary, the current study reports the synthesis of NISac-based XB complexes 3a–3d and the presence of symmetric and asymmetric cationic iodine(I) complexes 4a–4d, both in solution and under mechanochemical conditions. The studied systems were based on NISac (1) as a strong halogen bond donor, and electron-rich pyridine derivatives 2 as halogen bond acceptors. The molecular structures of XB complexes 3 manifested very strong halogen bonds. An innovative solution protocol enabled us to prepare symmetric iodine(I) complexes 4 by a substitution of the saccharinato part of the XB complex for a stronger neutral XB acceptor. Seven complexes were successfully identified by exchanging the NSac anion for another pyridine derivative 2, and were analyzed by SCXRD. However, the preparation proved to be difficult due to the reactivity of the produced Barluenga-type complexes, as manifested by the observation of several decomposition products and ligand scrambling events. As mechanochemistry had not previously been applied in the synthesis of these types of XB complexes, mechanochemical protocols for the synthesis of XB complexes 3 and 4 were developed, with the liquid assisted grinding (LAG) method proving to be highly successful. In the preparation of symmetric iodine(I) complexes 4, the LAG method proved to be superior in the synthesis of large quantities of the desired complex 4a, endearing itself to future commercial uses. Access to large quantities allowed us to survey 3a and 4a as electrophilic iodination reagents for comparison with Barluenga's reagent. Comparable good yields to those found with Barluenga's reagent were obtained both in solution and under mechanochemical conditions, such that materials 3a and 4a proved to be suitable alternatives to Barluenga's reagent. The developed mechanochemical protocols do not involve heavy metal salts and use only minute amounts of environmentally benign solvents, allowing recovery and reuse of the starting materials. Overall, this makes the mechanochemical methodology developed herein a highly attractive novel strategy for the large-scale synthesis and implementation of iodinating reagents.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. S. thanks the Verband der Chemischen Industrie for providing a Kekulé scholarship and the German Academic Exchange Service for a DAAD Scholarship. K. R. and J. S. W. gratefully acknowledge the Academy of Finland (grant numbers: 351121 (K. R.) and 356187 (J. S. W.)) for funding.References
- R. R. Knowles and E. N. Jacobsen, Attractive Noncovalent Interactions in Asymmetric Catalysis: Links between Enzymes and Small Molecule Catalysts, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20678–20685 CrossRef CAS PubMed.
- D. Herschlag and M. M. Pinney, Hydrogen Bonds: Simple after All?, Biochemistry, 2018, 57, 3338–3352 CrossRef CAS PubMed.
- J. D. Watson and F. H. C. Crick, Molecular Structure of Nucleic Acids: A Structure for Deoxyribose Nucleic Acid, Nature, 1953, 171, 737–738 CrossRef CAS PubMed.
- P. Hobza and J. Řezác, Introduction: Noncovalent Interactions, Chem. Rev., 2016, 116, 4911–4912 CrossRef CAS PubMed.
- I. Alkorta, J. Elguero and A. Frontera, Not Only Hydrogen Bonds: Other Noncovalent Interactions, Crystals, 2020, 180 CrossRef CAS.
- P. Politzer, Book Review of Halogen Bonding: Fundamentals and Applications. Structure and Bonding, 126, J. Am. Chem. Soc., 2008, 130, 10446–10447 CrossRef CAS.
- H. Wang, W. Wang and W. J. Jin, σ-Hole Bond vs π-Hole Bond: A Comparison Based on Halogen Bond, Chem. Rev., 2016, 116, 5072–5104 CrossRef CAS PubMed.
- L. Vogel, P. Wonner and S. M. Huber, Chalcogen Bonding: An Overview, Angew. Chem., Int. Ed., 2019, 58, 1880–1891 CrossRef CAS PubMed.
- P. Politzer, J. S. Murray and T. Clark, Halogen Bonding and Other σ-Hole Interactions: A Perspective, Phys. Chem. Chem. Phys., 2013, 15, 11178–11189 RSC.
- A. Docker, C. H. Guthrie, H. Kuhn and P. D. Beer, Modulating Chalcogen Bonding and Halogen Bonding Sigma-Hole Donor Atom Potency and Selectivity for Halide Anion Recognition, Angew. Chem., Int. Ed., 2021, 60, 21973–21978 CrossRef CAS PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, The Halogen Bond, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- R. S. Mulliken, Structures of Complexes Formed by Halogen Molecules with Aromatic and with Oxygenated Solvents1, J. Am. Chem. Soc., 1950, 72, 600–608 CrossRef CAS.
- R. S. Mulliken, Molecular Compounds and Their Spectra. II, J. Am. Chem. Soc., 1952, 74, 811–824 CrossRef CAS.
- R. S. Mulliken, Molecular Compounds and Their Spectra. III. The Interaction of Electron Donors and Acceptors, J. Phys. Chem., 1952, 56, 801–822 CrossRef CAS.
- J. R. Platt, 1966 Nobel Laureate in Chemistry: Robert S. Mulliken, Science, 1966, 154, 745–747 CrossRef CAS PubMed.
- O. Hassel, Structural Aspects of Interatomic Charge-Transfer Bonding, Science, 1970, 170, 497–502 CrossRef CAS PubMed.
- O. Hassel and J. Hvoslef, The Structure of Bromine 1,4-Dioxanate, Acta Chem. Scand., 1954, 8, 873–873 CrossRef CAS.
- O. Hassel and C. Rømming, Direct Structural Evidence for Weak Charge-Transfer Bonds in Solids Containing Chemically Saturated Molecules, Q. Rev., Chem. Soc., 1962, 16, 1–18 RSC.
- O. Hassel and K. O. Strømme, Structure of the Crystalline Compound Benzene-Bromine (1:1), Acta Chem. Scand., 1958, 12, 1146 CrossRef CAS.
- O. Hassel and K. O. Strømme, Crystal Structure of the Addition Compound Benzene-Chlorine (1:1), Acta Chem. Scand., 1959, 13, 1781–1786 CrossRef CAS.
- A. De Santis, A. Forni, R. Liantonio, P. Metrangolo, T. Pilati and G. Resnati, N⋯Br Halogen Bonding: One-Dimensional Infinite Chains through the Self-Assembly of Dibromotetrafluorobenzenes with Dipyridyl Derivatives, Chem. – Eur. J., 2003, 9, 3974–3983 CrossRef CAS PubMed.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Halogen Bonding in Supramolecular Chemistry, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- L. C. Gilday and P. D. Beer, Halogen- and Hydrogen-Bonding Catenanes for Halide-Anion Recognition, Chem. – Eur. J., 2014, 20, 8379–8385 CrossRef CAS PubMed.
- O. Dumele, N. Trapp and F. Diederich, Halogen Bonding Molecular Capsules, Angew. Chem., Int. Ed., 2015, 54, 12339–12344 CrossRef CAS PubMed.
- N. K. Beyeh, F. Pan and K. Rissanen, A Halogen-Bonded Dimeric Resorcinarene Capsule, Angew. Chem., Int. Ed., 2015, 54, 7303–7307 CrossRef CAS PubMed.
- C.-Z. Liu, S. Koppireddi, H. Wang, D.-W. Zhang and Z.-T. Li, Halogen Bonding-Driven Formation of Supramolecular Macrocycles and Double Helix, Chin. Chem. Lett., 2019, 30, 953–956 CrossRef CAS.
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Halogen Bonds in Crystal Engineering: Like Hydrogen Bonds yet Different, Acc. Chem. Res., 2014, 47, 2514–2524 CrossRef CAS PubMed.
- J. Teyssandier, K. S. Mali and S. De Feyter, Halogen Bonding in Two-Dimensional Crystal Engineering, ChemistryOpen, 2020, 9, 225–241 CrossRef CAS PubMed.
- R. B. Walsh, C. W. Padgett, P. Metrangolo, G. Resnati, T. W. Hanks and W. T. Pennington, Crystal Engineering through Halogen Bonding: Complexes of Nitrogen Heterocycles with Organic Iodides, Cryst. Growth Des., 2001, 1, 165–175 CrossRef CAS.
- G. Mínguez Espallargas, F. Zordan, L. Arroyo Marín, H. Adams, K. Shankland, J. van de Streek and L. Brammer, Rational Modification of the Hierarchy of Intermolecular Interactions in Molecular Crystal Structures by Using Tunable Halogen Bonds, Chem. – Eur. J., 2009, 15, 7554–7568 CrossRef PubMed.
- H. L. Nguyen, P. N. Horton, M. B. Hursthouse, A. C. Legon and D. W. Bruce, Halogen Bonding: A New Interaction for Liquid Crystal Formation, J. Am. Chem. Soc., 2004, 126, 16–17 CrossRef CAS PubMed.
- M. Saccone, M. Spengler, M. Pfletscher, K. Kuntze, M. Virkki, C. Wölper, R. Gehrke, G. Jansen, P. Metrangolo, A. Priimagi and M. Giese, Photoresponsive Halogen-Bonded Liquid Crystals: The Role of Aromatic Fluorine Substitution, Chem. Mater., 2019, 31, 462–470 CrossRef CAS.
- D. Devadiga and T. N. Ahipa, An Up-to-Date Review on Halogen-Bonded Liquid Crystals, J. Mol. Liq., 2021, 333, 115961 CrossRef CAS.
- F. Fernandez-Palacio, M. Poutanen, M. Saccone, A. Siiskonen, G. Terraneo, G. Resnati, O. Ikkala, P. Metrangolo and A. Priimagi, Efficient Light-Induced Phase Transitions in Halogen-Bonded Liquid Crystals, Chem. Mater., 2016, 28, 8314–8321 CrossRef CAS PubMed.
- G. Cavallo, G. Terraneo, A. Monfredini, M. Saccone, A. Priimagi, T. Pilati, G. Resnati, P. Metrangolo and D. W. Bruce, Superfluorinated Ionic Liquid Crystals Based on Supramolecular, Halogen-Bonded Anions, Angew. Chem., Int. Ed., 2016, 55, 6300–6304 CrossRef CAS PubMed.
- R. Kampes, S. Zechel, M. D. Hager and U. S. Schubert, Halogen Bonding in Polymer Science: Towards New Smart Materials, Chem. Sci., 2021, 12, 9275–9286 RSC.
- A. Priimagi, G. Cavallo, A. Forni, M. Gorynsztejn-Leben, M. Kaivola, P. Metrangolo, R. Milani, A. Shishido, T. Pilati, G. Resnati and G. Terraneo, Halogen Bonding versus Hydrogen Bonding in Driving Self-Assembly and Performance of Light-Responsive Supramolecular Polymers, Adv. Funct. Mater., 2012, 22, 2572–2579 CrossRef CAS.
- A. Vanderkooy and M. S. Taylor, Solution-Phase Self-Assembly of Complementary Halogen Bonding Polymers, J. Am. Chem. Soc., 2015, 137, 5080–5086 CrossRef CAS PubMed.
- G. Berger, J. Soubhye and F. Meyer, Halogen Bonding in Polymer Science: From Crystal Engineering to Functional Supramolecular Polymers and Materials, Polym. Chem., 2015, 6, 3559–3580 RSC.
- L. Meazza, J. A. Foster, K. Fucke, P. Metrangolo, G. Resnati and J. W. Steed, Halogen-Bonding-Triggered Supramolecular Gel Formation, Nat. Chem., 2013, 5, 42–47 CrossRef CAS PubMed.
- G. Berger, P. Frangville and F. Meyer, Halogen Bonding for Molecular Recognition: New Developments in Materials and Biological Sciences, Chem. Commun., 2020, 56, 4970–4981 RSC.
- A. Pizzi, L. Lascialfari, N. Demitri, A. Bertolani, D. Maiolo, E. Carretti and P. Metrangolo, Halogen Bonding Modulates Hydrogel Formation from Fmoc Amino Acids, CrystEngComm, 2017, 19, 1870–1874 RSC.
- S. Bhattacharjee and S. Bhattacharya, Remarkable Role of C–I⋯N Halogen Bonding in Thixotropic ‘Halo'Gel Formation, Langmuir, 2016, 32, 4270–4277 CrossRef CAS PubMed.
- X. Tong, X. Zhao, Y. Qiu, H. Wang, Y. Liao and X. Xie, Intrinsically Visible Light-Responsive Liquid Crystalline Physical Gels Driven by a Halogen Bond, Langmuir, 2020, 36, 11873–11879 CrossRef CAS PubMed.
- R. Wilcken, X. Liu, M. O. Zimmermann, T. J. Rutherford, A. R. Fersht, A. C. Joerger and F. M. Boeckler, Halogen-Enriched Fragment Libraries as Leads for Drug Rescue of Mutant P53, J. Am. Chem. Soc., 2012, 134, 6810–6818 CrossRef CAS PubMed.
- M. R. Scholfield, C. M. Vander Zanden, M. Carter and P. S. Ho, Halogen Bonding (X-Bonding): A Biological Perspective, Protein Sci., 2013, 22, 139–152 CrossRef CAS PubMed.
- R. Wilcken, M. O. Zimmermann, A. Lange, A. C. Joerger and F. M. Boeckler, Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology, J. Med. Chem., 2013, 56, 1363–1388 CrossRef CAS PubMed.
- L. A. Hardegger, B. Kuhn, B. Spinnler, L. Anselm, R. Ecabert, M. Stihle, B. Gsell, R. Thoma, J. Diez, J. Benz, J.-M. Plancher, G. Hartmann, D. W. Banner, W. Haap and F. Diederich, Systematic Investigation of Halogen Bonding in Protein–Ligand Interactions, Angew. Chem., Int. Ed., 2011, 50, 314–318 CrossRef CAS PubMed.
- J. Heidrich, L. E. Sperl and F. M. Boeckler, Embracing the Diversity of Halogen Bonding Motifs in Fragment-Based Drug Discovery—Construction of a Diversity-Optimized Halogen-Enriched Fragment Library, Front. Chem., 2019, 1–9, DOI:10.3389/fchem.2019.00009.
- A. Bruckmann, M. A. Pena and C. Bolm, Organocatalysis through Halogen-Bond Activation, Synlett, 2008, 900–902 CAS.
- R. L. Sutar and S. M. Huber, Catalysis of Organic Reactions through Halogen Bonding, ACS Catal., 2019, 9, 9622–9639 CrossRef CAS.
- A. Dreger, P. Wonner, E. Engelage, S. M. Walter, R. Stoll and S. M. Huber, A Halogen-Bonding-Catalysed Nazarov Cyclisation Reaction, Chem. Commun., 2019, 55, 8262–8265 RSC.
- Y. Kobayashi and Y. Takemoto, Reactions Catalyzed by 2-Halogenated Azolium Salts: From Halogen-Bond Donors to Brønsted-Acidic Salts, Synlett, 2020, 772–783 CrossRef.
- D. Bulfield and S. M. Huber, Halogen Bonding in Organic Synthesis and Organocatalysis, Chem. – Eur. J., 2016, 22, 14434–14450 CrossRef CAS PubMed.
- S. Kuwano, T. Suzuki, Y. Hosaka and T. Arai, A Chiral Organic Base Catalyst with Halogen-Bonding-Donor Functionality: Asymmetric Mannich Reactions of Malononitrile with N-Boc Aldimines and Ketimines, Chem. Commun., 2018, 54, 3847–3850 RSC.
- S. H. Jungbauer and S. M. Huber, Cationic Multidentate Halogen-Bond Donors in Halide Abstraction Organocatalysis: Catalyst Optimization by Preorganization, J. Am. Chem. Soc., 2015, 137, 12110–12120 CrossRef CAS PubMed.
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Definition of the Halogen Bond (IUPAC Recommendations 2013), Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS.
- P. Politzer and J. S. Murray, Halogen Bonding: An Interim Discussion, ChemPhysChem, 2013, 14, 278–294 CrossRef CAS PubMed.
- F. F. Awwadi, R. D. Willett, K. A. Peterson and B. Twamley, The Nature of Halogen⋯Halogen Synthons: Crystallographic and Theoretical Studies, Chem. – Eur. J., 2006, 12, 8952–8960 CrossRef CAS PubMed.
- K. E. Riley, J. S. Murray, P. Politzer, M. C. Concha and P. Hobza, Br⋯O Complexes as Probes of Factors Affecting Halogen Bonding: Interactions of Bromobenzenes and Bromopyrimidines with Acetone, J. Chem. Theory Comput., 2009, 5, 155–163 CrossRef CAS PubMed.
- T. Clark, σ-Holes, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 13–20 CAS.
- K. E. Riley and P. Hobza, Investigations into the Nature of Halogen Bonding Including Symmetry Adapted Perturbation Theory Analyses, J. Chem. Theory Comput., 2008, 4, 232–242 CrossRef CAS PubMed.
- K. E. Riley, J. S. Murray, J. Fanfrlík, J. Řezáč, R. J. Solá, M. C. Concha, F. M. Ramos and P. Politzer, Halogen Bond Tunability II: The Varying Roles of Electrostatic and Dispersion Contributions to Attraction in Halogen Bonds, J. Mol. Model., 2013, 19, 4651–4659 CrossRef CAS PubMed.
- H. A. Bent, Structural Chemistry of Donor-Acceptor Interactions, Chem. Rev., 1968, 68, 587–648 CrossRef CAS.
- A. E. Reed, F. Weinhold, R. Weiss and J. Macheleid, Nature of the Contact Ion Pair Trichloromethyl-Chloride (CCl3+Cl-). A Theoretical Study, J. Phys. Chem., 1985, 89, 2688–2694 CrossRef CAS.
- J. Řezáč and A. de la Lande, On the Role of Charge Transfer in Halogen Bonding, Phys. Chem. Chem. Phys., 2017, 19, 791–803 RSC.
- B. Inscoe, H. Rathnayake and Y. Mo, Role of Charge Transfer in Halogen Bonding, J. Phys. Chem. A, 2021, 125, 2944–2953 CrossRef CAS PubMed.
- C. Wang, D. Danovich, Y. Mo and S. Shaik, On The Nature of the Halogen Bond, J. Chem. Theory Comput., 2014, 10, 3726–3737 CrossRef CAS PubMed.
- P. Politzer, J. S. Murray and M. C. Concha, Halogen Bonding and the Design of New Materials: Organic Bromides, Chlorides and Perhaps Even Fluorides as Donors, J. Mol. Model., 2007, 13, 643–650 CrossRef CAS PubMed.
- P. Politzer, P. Lane, M. C. Concha, Y. Ma and J. S. Murray, An Overview of Halogen Bonding, J. Mol. Model., 2007, 13, 305–311 CrossRef CAS PubMed.
- P. Politzer, J. S. Murray and T. Clark, Halogen Bonding: An Electrostatically-Driven Highly Directional Noncovalent Interaction, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 RSC.
- J. M. Holthoff, E. Engelage, R. Weiss and S. M. Huber, “Anti-Electrostatic” Halogen Bonding, Angew. Chem., Int. Ed., 2020, 59, 11150–11157 CrossRef CAS PubMed.
- T. Brinck and A. N. Borrfors, Electrostatics and Polarization Determine the Strength of the Halogen Bond: A Red Card for Charge Transfer, J. Mol. Model., 2019, 25, 125 CrossRef PubMed.
- F. Otte, J. Kleinheider, W. Hiller, R. Wang, U. Englert and C. Strohmann, Weak yet Decisive: Molecular Halogen Bond and Competing Weak Interactions of Iodobenzene and Quinuclidine, J. Am. Chem. Soc., 2021, 143, 4133–4137 CrossRef CAS PubMed.
- O. Dumele, D. Wu, N. Trapp, N. Goroff and F. Diederich, Halogen Bonding of (Iodoethynyl)Benzene Derivatives in Solution, Org. Lett., 2014, 16, 4722–4725 CrossRef CAS PubMed.
- R. Puttreddy, J. M. Rautiainen, T. Mäkelä and K. Rissanen, Strong N−X⋯O−N Halogen Bonds: A Comprehensive Study on N-Halosaccharin Pyridine N-Oxide Complexes, Angew. Chem., Int. Ed., 2019, 58, 18610–18618 CrossRef CAS PubMed.
- K. Rissanen, Halogen Bonded Supramolecular Complexes and Networks, CrystEngComm, 2008, 10, 1107–1113 RSC.
- O. Makhotkina, J. Lieffrig, O. Jeannin, M. Fourmigué, E. Aubert and E. Espinosa, Cocrystal or Salt: Solid State-Controlled Iodine Shift in Crystalline Halogen-Bonded Systems, Cryst. Growth Des., 2015, 15, 3464–3473 CrossRef CAS.
- C. Ouvrard, J.-Y. Le Questel, M. Berthelot and C. Laurence, Halogen-Bond Geometry: A Crystallographic Database Investigation of Dihalogen Complexes, Acta Crystallogr., Sect. B: Struct. Sci., 2003, 59, 512–526 CrossRef PubMed.
- M. C. Aragoni, M. Arca, F. A. Devillanova, F. Isaia, V. Lippolis, A. Mancini, L. Pala, A. M. Z. Slawin and J. D. Woollins, First Example of an Infinite Polybromide 2D-Network, Chem. Commun., 2003, 17, 2226–2227 RSC.
- A. S. Batsanov, J. A. K. Howard, A. P. Lightfoot, S. J. R. Twiddle and A. Whiting, Stereoselective Chloro-Deboronation Reactions Induced by Substituted Pyridine–Iodine Chloride Complexes, Eur. J. Org. Chem., 2005, 1876–1883 CrossRef CAS.
- C. Rømming, Refinement of the Crystal Structure of the Charge Transfer Compound Pyridine-Iodomonochloride, Acta Chem. Scand., 1972, 26, 1555–1560 CrossRef.
- M. C. Aragoni, M. Arca, S. J. Coles, F. A. Devillanova, M. B. Hursthouse, S. L. Coles (née Huth), F. Isaia, V. Lippolis and A. Mancini, CT-Adduct vs. Pyridinium Polyhalide Salt Formation in the Reactions between Polypyridyl Donors and Dihalogens: Reactivity of 1,4-Di-(3′-Pyridylethynyl)Benzene towards Br2 and I2, CrystEngComm, 2011, 13, 6319–6322 RSC.
- J.-Y. Le Questel, C. Laurence and J. Graton, Halogen-Bond Interactions: A Crystallographic Basicity Scale towards Iodoorganic Compounds, CrystEngComm, 2013, 15, 3212–3221 RSC.
- G. C. Pimentel, The Bonding of Trihalide and Bifluoride Ions by the Molecular Orbital Method, J. Chem. Phys., 1951, 19, 446–448 CrossRef CAS.
- L. Koskinen, P. Hirva, E. Kalenius, S. Jääskeläinen, K. Rissanen and M. Haukka, Halogen Bonds with Coordinative Nature: Halogen Bonding in a S–I + –S Iodonium Complex, CrystEngComm, 2015, 17, 1231–1236 RSC.
- M. Bedin, A. Karim, M. Reitti, A.-C. C. Carlsson, F. Topić, M. Cetina, F. Pan, V. Havel, F. Al-Ameri, V. Sindelar, K. Rissanen, J. Gräfenstein and M. Erdélyi, Counterion Influence on the N–I–N Halogen Bond, Chem. Sci., 2015, 6, 3746–3756 RSC.
- S. B. Hakkert and M. Erdélyi, Halogen Bond Symmetry: The N–X–N Bond, J. Phys. Org. Chem., 2015, 28, 226–233 CrossRef CAS.
- L. Turunen and M. Erdélyi, Halogen Bonds of Halonium Ions, Chem. Soc. Rev., 2020, 49, 2688–2700 RSC.
- J. S. Ward, K.-N. Truong, M. Erdélyi and K. Rissanen, 1.12 - Halogen-Bonded Halogen(I) Ion Complexes, in Comprehensive Inorganic Chemistry III, ed. J. Reedijk and K. R. B. T. Poeppelmeier, Elsevier, Oxford, 2023, pp. 586–601 Search PubMed.
- A.-C. C. Carlsson, K. Mehmeti, M. Uhrbom, A. Karim, M. Bedin, R. Puttreddy, R. Kleinmaier, A. A. Neverov, B. Nekoueishahraki, J. Gräfenstein, K. Rissanen and M. Erdélyi, Substituent Effects on the [N–I–N] + Halogen Bond, J. Am. Chem. Soc., 2016, 138, 9853–9863 CrossRef CAS PubMed.
- L. P. Wolters and F. M. Bickelhaupt, Halogen Bonding versus Hydrogen Bonding: A Molecular Orbital Perspective, ChemistryOpen, 2012, 1, 96–105 CrossRef CAS PubMed.
- F. Jiménez-Grávalos, M. Gallegos, Á. Martín Pendás and A. S. Novikov, Challenging the Electrostatic σ-Hole Picture of Halogen Bonding Using Minimal Models and the Interacting Quantum Atoms Approach, J. Comput. Chem., 2021, 42, 676–687 CrossRef PubMed.
- J. Barluenga, J. M. González, P. J. Campos and G. Asensio, I(Py) 2 BF 4, a New Reagent in Organic Synthesis: General Method for the 1,2-Iodofunctionalization of Olefins, Angew. Chem., Int. Ed. Engl., 1985, 24, 319–320 CrossRef.
- J. Barluenga, H. Vázquez-Villa, A. Ballesteros and J. M. González, Cyclization of Carbonyl Groups onto Alkynes upon Reaction with IPy2BF4 and Their Trapping with Nucleophiles: A Versatile Trigger for Assembling Oxygen Heterocycles, J. Am. Chem. Soc., 2003, 125, 9028–9029 CrossRef CAS PubMed.
- J. Barluenga, F. González-Bobes, M. C. Murguía, S. R. Ananthoju and J. M. González, Bis(Pyridine)Iodonium Tetrafluoroborate (IPy2BF4): A Versatile Oxidizing Reagent, Chem. – Eur. J., 2004, 10, 4206–4213 CrossRef CAS PubMed.
- J. Barluenga, Transferring Iodine: More than a Simple Functional Group Exchange in Organic Synthesis, Pure Appl. Chem., 1999, 71, 431–436 CrossRef CAS.
- Y. Kim, E. J. Mckinley, K. E. Christensen, N. H. Rees and A. L. Thompson, Toward the Understanding of Modulation in Molecular Materials: Barluenga's Reagent and Its Analogues., Cryst. Growth Des., 2014, 14, 6294–6301 CrossRef CAS.
- C. Alvarez-Rua, S. Garcia-Granda, A. Ballesteros, F. Gonzalez-Bobes and J. M. Gonzalez, Bis(Pyridine)Iodonium(I) Tetrafluoroborate, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2002, 58, o1381–o1383 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, The Cambridge Structural Database, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, Mercury CSD 2.0 - New Features for the Visualization and Investigation of Crystal Structures, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- T. Okitsu, S. Yumitate, K. Sato, Y. In and A. Wada, Substituent Effect of Bis(Pyridines)Iodonium Complexes as Iodinating Reagents: Control of the Iodocyclization/Oxidation Process, Chem. – Eur. J., 2013, 19, 4992–4996 CrossRef CAS PubMed.
- R. D. Evans and J. H. Schauble, Syntheses of α-Iodocarbonyl Compounds Using Bis(Sym-Collidine)Iodine(I) Tetrafluoroborate/Dimethyl Sulfoxide, Synthesis, 1986, 1986, 727–730 CrossRef.
- J. C. López, P. Bernal-Albert, C. Uriel, S. Valverde and A. M. Gómez, IPy2BF4/HF-Pyridine: A New Combination of Reagents for the Transformation of Partially Unprotected Thioglycosides and n-Pentenyl Glycosides to Glycosyl Fluorides, J. Org. Chem., 2007, 72, 10268–10271 CrossRef PubMed.
- J. C. López, C. Uriel, A. Guillamón-Martín, S. Valverde and A. M. Gómez, IPy2BF4-Mediated Transformation of n-Pentenyl Glycosides to Glycosyl Fluorides: A New Pair of Semiorthogonal Glycosyl Donors, Org. Lett., 2007, 9, 2759–2762 CrossRef PubMed.
- J. W. Lown and A. V. Joshua, Stereochemistry and Regiochemistry of the Addition of Lodonium Nitrate to Alkenes, J. Chem. Soc., Perkin Trans. 1, 1973, 2680–2687 RSC.
- J. W. Lown and A. V. Joshua, Electrophilic Additions of Iodonium Nitrate to Unsaturated Substrates, Can. J. Chem., 1977, 55, 122–130 CrossRef CAS.
- J. W. Lown and A. V. Joshua, Eiectrophilic Additions of Bromonium Nitrate to Unsaturated Substrates, Can. J. Chem., 1977, 55, 508–521 CrossRef CAS.
- M. Bigioni, P. Ganis, A. Panunzi, F. Ruffo, C. Salvatore and A. Vito, Electrophilic Attack of [I(Py)2]+(NO3-) on Three-Coordinate Pt0 Precursors: Synthesis and in Vitro Antitumor Activity of Water-Soluble, Five- Coordinate Pt(II) Complexes, Eur. J. Inorg. Chem., 2000, 2000, 1717–1721 CrossRef.
- M. Vilaró, J. Nieto, J. R. La Parra, M. R. Almeida, A. Ballesteros, A. Planas, G. Arsequell and G. Valencia, Tuning Transthyretin Amyloidosis Inhibition Properties of Iododiflunisal by Combinatorial Engineering of the Nonsalicylic Ring Substitutions, ACS Comb. Sci., 2015, 17, 32–38 CrossRef PubMed.
- A.-C. C. Carlsson, J. Gräfenstein, A. Budnjo, J. L. Laurila, J. Bergquist, A. Karim, R. Kleinmaier, U. Brath and M. Erdélyi, Symmetric Halogen Bonding Is Preferred in Solution, J. Am. Chem. Soc., 2012, 134, 5706–5715 CrossRef CAS PubMed.
- D. C. Georgiou, P. Butler, E. C. Browne, D. J. D. Wilson and J. L. Dutton, On the Bonding in Bis-Pyridine Iodonium Cations*, Aust. J. Chem., 2013, 66, 1179–1188 CrossRef CAS.
- S. Lindblad, K. Mehmeti, A. X. Veiga, B. Nekoueishahraki, J. Gräfenstein and M. Erdélyi, Halogen Bond Asymmetry in Solution, J. Am. Chem. Soc., 2018, 140, 13503–13513 CrossRef CAS PubMed.
- J. S. Ward, G. Fiorini, A. Frontera and K. Rissanen, Asymmetric [N–I–N]+ Halonium Complexes, Chem. Commun., 2020, 56, 8428–8431 RSC.
- D. von der Heiden, K. Rissanen and M. Erdélyi, Asymmetric [N–I–N]+ Halonium Complexes in Solution?, Chem. Commun., 2020, 56, 14431–14434 RSC.
- S. Yu and J. S. Ward, Ligand Exchange among Iodine(I) Complexes, Dalton Trans., 2022, 51, 4668–4674 RSC.
- J. M. Chalker, A. L. Thompson, B. G. Davis, N. Zaware and P. Wipf, Safe and Scalable Preparation of Barluenga's Reagent: (Bis(Pyridine) Iodonium(I) Tetrafluoroborate), Org. Synth., 2010, 288–298 CAS.
- J. S. Ward, A. Frontera and K. Rissanen, Iodonium Complexes of the Tertiary Amines Quinuclidine and 1-Ethylpiperidine, Dalton Trans., 2021, 50, 8297–8301 RSC.
- J. S. Ward, R. M. Gomila, A. Frontera and K. Rissanen, Iodine(I) Complexes Incorporating Sterically Bulky 2-Substituted Pyridines, RSC Adv., 2022, 12, 8674–8682 RSC.
- J. S. Ward, The Solid-State Hierarchy and Iodination Potential of [Bis(3-Acetaminopyridine)Iodine(I)]PF6, CrystEngComm, 2022, 24, 7029–7033 RSC.
- K. Rissanen and J. S. Ward, Iodine(I) and Silver(I) Complexes Incorporating 3-Substituted Pyridines, ACS Omega, 2023, 8, 24064–24071 CrossRef CAS PubMed.
- J. S. Ward, E. I. Sievänen and K. Rissanen, Solid-State NMR Spectroscopy of Iodine(I) Complexes, Chem. – Asian J., 2023, 18, e202201203 CrossRef CAS PubMed.
- C. Wang, D. Danovich, H. Chen and S. Shaik, Oriented External Electric Fields: Tweezers and Catalysts for Reactivity in Halogen-Bond Complexes, J. Am. Chem. Soc., 2019, 141, 7122–7136 CrossRef CAS PubMed.
- M. Mattila, K. Rissanen and J. S. Ward, Chiral Carbonyl Hypoiodites, Chem. Commun., 2023, 59, 4648–4651 RSC.
- A. Bondi, Van Der Waals Volumes and Radii, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- J. P. M. Lommerse, A. J. Stone, R. Taylor and F. H. Allen, The Nature and Geometry of Intermolecular Interactions between Halogens and Oxygen or Nitrogen, J. Am. Chem. Soc., 1996, 118, 3108–3116 CrossRef CAS.
- L. Brammer, E. A. Bruton and P. Sherwood, Understanding the Behavior of Halogens as Hydrogen Bond Acceptors, Cryst. Growth Des., 2001, 1, 277–290 CrossRef CAS.
- F. Zordan, L. Brammer and P. Sherwood, Supramolecular Chemistry of Halogens: Complementary Features of Inorganic (M−X) and Organic (C−X‘) Halogens Applied to M−X⋯X‘−C Halogen Bond Formation, J. Am. Chem. Soc., 2005, 127, 5979–5989 CrossRef CAS PubMed.
- S. Yu, J. M. Rautiainen, P. Kumar, L. Gentiluomo, J. S. Ward, K. Rissanen and R. Puttreddy, Ortho-Substituent Effects on Halogen Bond Geometry for N-Haloimide⋯2-Substituted Pyridine Complexes, Adv. Sci., 2023 DOI:10.1002/advs.202307208.
- R. Puttreddy, J. M. Rautiainen, S. Yu and K. Rissanen, N−X⋯O−N Halogen Bonds in Complexes of N-Haloimides and Pyridine-N-Oxides: A Large Data Set Study, Angew. Chem., Int. Ed., 2023, 62, e202307372 CrossRef CAS PubMed.
- S. L. James and T. Friščić, Mechanochemistry, Chem. Soc. Rev., 2013, 42, 7494–7496 RSC.
- C. Oliver Kappe, J. Mack and C. Bolm, Enabling Techniques for Organic Synthesis, J. Org. Chem., 2021, 86, 14242–14244 CrossRef CAS PubMed.
- C. Bolm and J. G. Hernández, Mechanochemistry of Gaseous Reactants, Angew. Chem., Int. Ed., 2019, 58, 3285–3299 CrossRef CAS PubMed.
- K. Horie, M. Barón, R. B. Fox, J. He, M. Hess, J. Kahovec, T. Kitayama, P. Kubisa, E. Maréchal, W. Mormann, R. F. T. Stepto, D. Tabak, J. Vohlídal, E. S. Wilks and W. J. Work, Definitions of Terms Relating to Reactions of Polymers and to Functional Polymeric Materials (IUPAC Recommendations 2003), Pure Appl. Chem., 2004, 76, 889–906 CrossRef CAS.
- T. Friščić, C. Mottillo and H. M. Titi, Mechanochemistry for Synthesis, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef PubMed.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Mechanochemistry: Opportunities for New and Cleaner Synthesis, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- J. L. Howard, Q. Cao and D. L. Browne, Mechanochemistry as an Emerging Tool for Molecular Synthesis: What Can It Offer?, Chem. Sci., 2018, 9, 3080–3094 RSC.
- W. Pickhardt, S. Grätz and L. Borchardt, Direct Mechanocatalysis: Using Milling Balls as Catalysts, Chem. – Eur. J., 2020, 26, 12903–12911 CrossRef CAS PubMed.
- K. J. Ardila-Fierro and J. G. Hernández, Sustainability Assessment of Mechanochemistry by Using the Twelve Principles of Green Chemistry, ChemSusChem, 2021, 14, 2145–2162 CrossRef CAS PubMed.
- C. G. Avila-Ortiz and E. Juaristi, Novel Methodologies for Chemical Activation in Organic Synthesis under Solvent-Free Reaction Conditions, Molecules, 2020, 1–25 Search PubMed.
- J.-L. Do and T. Friščić, Mechanochemistry: A Force of Synthesis, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed.
- L. Konnert, B. Reneaud, R. M. de Figueiredo, J.-M. Campagne, F. Lamaty, J. Martinez and E. Colacino, Mechanochemical Preparation of Hydantoins from Amino Esters: Application to the Synthesis of the Antiepileptic Drug Phenytoin, J. Org. Chem., 2014, 79, 10132–10142 CrossRef CAS PubMed.
- G.-W. Wang, K. Komatsu, Y. Murata and M. Shiro, Synthesis and X-Ray Structure of Dumb-Bell-Shaped C120, Nature, 1997, 387, 583–586 CrossRef CAS.
- K. Komatsu, G.-W. Wang, Y. Murata, T. Tanaka, K. Fujiwara, K. Yamamoto and M. Saunders, Mechanochemical Synthesis and Characterization of the Fullerene Dimer C120, J. Org. Chem., 1998, 63, 9358–9366 CrossRef CAS.
- J. G. Hernández and C. Bolm, Altering Product Selectivity by Mechanochemistry, J. Org. Chem., 2017, 82, 4007–4019 CrossRef PubMed.
- S.-E. Zhu, F. Li and G.-W. Wang, Mechanochemistry of Fullerenes and Related Materials, Chem. Soc. Rev., 2013, 42, 7535–7570 RSC.
- G.-W. Wang, Mechanochemical Organic Synthesis, Chem. Soc. Rev., 2013, 42, 7668–7700 RSC.
- G.-W. Wang, Fullerene Mechanochemistry: Serendipitous Discovery of Dumb-Bell-Shaped C120 and Beyond, Chin. J. Chem., 2021, 39, 1797–1803 CrossRef CAS.
- A. O. Patil, D. Y. Curtin and I. C. Paul, Solid-State Formation of Quinhydrones from Their Components. Use of Solid-Solid Reactions to Prepare Compounds Not Accessible from Solution, J. Am. Chem. Soc., 1984, 106, 348–353 CrossRef CAS.
- T. Friščić, Supramolecular Concepts and New Techniques in Mechanochemistry: Cocrystals, Cages, Rotaxanes, Open Metal–Organic Frameworks, Chem. Soc. Rev., 2012, 41, 3493–3510 RSC.
- P. Ying, J. Yu and W. Su, Liquid-Assisted Grinding Mechanochemistry in the Synthesis of Pharmaceuticals, Adv. Synth. Catal., 2021, 363, 1246–1271 CrossRef CAS.
- Y. Xu, J. Viger-Gravel, I. Korobkov and D. L. Bryce, Mechanochemical Production of Halogen-Bonded Solids Featuring P
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯I–C Motifs and Characterization via X-Ray Diffraction, Solid-State Multinuclear Magnetic Resonance, and Density Functional Theory, J. Phys. Chem. C, 2015, 119, 27104–27117 CrossRef CAS.
O⋯I–C Motifs and Characterization via X-Ray Diffraction, Solid-State Multinuclear Magnetic Resonance, and Density Functional Theory, J. Phys. Chem. C, 2015, 119, 27104–27117 CrossRef CAS. - Y. Xu, L. Champion, B. Gabidullin and D. L. Bryce, A Kinetic Study of Mechanochemical Halogen Bond Formation by in Situ31P Solid-State NMR Spectroscopy, Chem. Commun., 2017, 53, 9930–9933 RSC.
- D. Cinčić, T. Friščić and W. Jones, A Stepwise Mechanism for the Mechanochemical Synthesis of Halogen-Bonded Cocrystal Architectures, J. Am. Chem. Soc., 2008, 130, 7524–7525 CrossRef PubMed.
- M. Karimi-Jafari, L. Padrela, G. M. Walker and D. M. Croker, Creating Cocrystals: A Review of Pharmaceutical Cocrystal Preparation Routes and Applications, Cryst. Growth Des., 2018, 18, 6370–6387 CrossRef CAS.
- D. Braga, L. Maini and F. Grepioni, Mechanochemical Preparation of Co-Crystals, Chem. Soc. Rev., 2013, 42, 7638–7648 RSC.
- T. Carstens, D. A. Haynes and V. J. Smith, Cocrystals: Solution, Mechanochemistry, and Sublimation, Cryst. Growth Des., 2020, 20, 1139–1149 CrossRef CAS.
- D. Hasa, E. Miniussi and W. Jones, Mechanochemical Synthesis of Multicomponent Crystals: One Liquid for One Polymorph? A Myth to Dispel, Cryst. Growth Des., 2016, 16, 4582–4588 CrossRef CAS.
- T. Friščić, D. G. Reid, I. Halasz, R. S. Stein, R. E. Dinnebier and M. J. Duer, Ion- and Liquid-Assisted Grinding: Improved Mechanochemical Synthesis of Metal–Organic Frameworks Reveals Salt Inclusion and Anion Templating, Angew. Chem., Int. Ed., 2010, 49, 712–715 CrossRef PubMed.
- F. Gomollón-Bel, Ten Chemical Innovations That Will Change Our World: IUPAC Identifies Emerging Technologies in Chemistry with Potential to Make Our Planet More Sustainable, Chem. Int., 2019, 41, 12–17 Search PubMed.
- D. Hasa and W. Jones, Screening for New Pharmaceutical Solid Forms Using Mechanochemistry: A Practical Guide, Adv. Drug Delivery Rev., 2017, 117, 147–161 CrossRef CAS PubMed.
- C. Schumacher, H. Fergen, R. Puttreddy, K.-N. Truong, T. Rinesch, K. Rissanen and C. Bolm, N-(2,3,5,6-Tetrafluoropyridyl)Sulfoximines: Synthesis, X-Ray Crystallography, and Halogen Bonding, Org. Chem. Front., 2020, 7, 3896–3906 RSC.
- S. Karki, T. Friščić, W. Jones and W. D. S. Motherwell, Screening for Pharmaceutical Cocrystal Hydrates via Neat and Liquid-Assisted Grinding, Mol. Pharm., 2007, 4, 347–354 CrossRef CAS PubMed.
- J. L. Howard, Y. Sagatov, L. Repusseau, C. Schotten and D. L. Browne, Controlling Reactivity through Liquid Assisted Grinding: The Curious Case of Mechanochemical Fluorination, Green Chem., 2017, 19, 2798–2802 RSC.
- N. Shan, F. Toda and W. Jones, Mechanochemistry and Co-Crystal Formation: Effect of Solvent on Reaction Kinetics, Chem. Commun., 2002, 20, 2372–2373 RSC.
- V. S. Pfennig, R. C. Villella, J. Nikodemus and C. Bolm, Mechanochemical Grignard Reactions with Gaseous CO2 and Sodium Methyl Carbonate**, Angew. Chem., Int. Ed., 2022, 61, e202116514 CrossRef CAS PubMed.
- P. J. Campos, J. Arranz and M. A. Rodríguez, α-Iodination of Enaminones with Bis(Pyridine)Iodonium(I) Tetrafluoroborate, Tetrahedron Lett., 1997, 38, 8397–8400 CrossRef CAS.
- I. Jirkovsky, Studies on Enaminoketones, Can. J. Chem., 1974, 52, 55–65 CrossRef CAS.
- G. G. Habermehl, I. Wippermann, H. C. Krebs and S. tom Dieck, Halogenierung Und Acyloxylierung von N-Cyclohex-2-En-1-on-3-Yl-Aminosäuren/Halogenation and Acyloxylation of N-Cyclohex-2-En-1-on-3-Yl-Amino Acids, Z. Naturforsch., B: J. Chem. Sci., 1991, 46, 1415–1420 CrossRef CAS.
- K. Matsuo, S. Ishida and Y. Takuno, Reaction of Enaminones with Benzyltrimethylammonium Dichloroiodate, Chem. Pharm. Bull., 1994, 42, 1149–1150 CrossRef CAS.
- C. P. Kordik and A. B. Reitz, Unexpected α-Chlorination of Tertiary Enaminones Using Benzyltrimethylammonium Dichloroiodate, J. Org. Chem., 1996, 61, 5644–5645 CrossRef CAS.
- L. M. E. Wilson, K. Rissanen and J. S. Ward, Iodination of Antipyrine with [N–I–N]+ and Carbonyl Hypoiodite Iodine(I) Complexes, New J. Chem., 2023, 47, 2978–2982 RSC.
- A. N. Evstropov, V. E. Yavorovskaya, E. S. Vorob'ev, Z. P. Khudonogova, L. N. Gritsenko, E. V. Shmidt, S. G. Medvedeva, V. D. Filimonov, T. P. Prishchep and A. S. Saratikov, Synthesis and Antiviral Activity of Antipyrine Derivatives, Pharm. Chem. J., 1992, 26, 426–430 CrossRef.
- O. Sakurada, C. Kennedy, J. Jehle, J. D. Brown, G. L. Carbin and L. Sokoloff, Measurement of Local Cerebral Blood Flow with Iodo [14C] Antipyrine, Am. J. Physiol.: Heart Circ. Physiol., 1978, 234, H59–H66 CrossRef CAS PubMed.
- O. Z. Chi, C. Hunter, X. Liu and H. R. Weiss, The Effects of Isoflurane Pretreatment on Cerebral Blood Flow, Capillary Permeability, and Oxygen Consumption in Focal Cerebral Ischemia in Rats, Anesth. Analg., 2010, 110, 1412–1418 CrossRef CAS PubMed.
- D. P. Holschneider, J.-M. I. Maarek, J. Yang, J. Harimoto and O. U. Scremin, Functional Brain Mapping in Freely Moving Rats during Treadmill Walking, J. Cereb. Blood Flow Metab., 2003, 23, 925–932 CrossRef CAS PubMed.
- G. M. Tozer, V. E. Prise and V. J. Cunningham, Quantitative Estimation of Tissue Blood Flow Rate BT - Angiogenesis Protocols: Second Edition, in Methods in Molecular Biology, ed. C. Murray and S. Martin, Humana Press, Totowa, NJ, 2009, vol. 467, pp. 271–286 Search PubMed.
- Z. Tao, S. M. Gomha, M. G. Badrey, T. T. El-Idreesy and T. M. A. Eldebss, Novel 4−Heteroaryl–antipyrines: Synthesis, Molecular Docking, and Evaluation as Potential Anti–breast Cancer Agents, J. Heterocycl. Chem., 2018, 55, 2408–2416 CrossRef CAS.
- F. Dehmel, M. Abarbri and P. Knochel, Preparation of New Magnesiated Functionalized Imidazoles and Antipyrines via an Iodine-Magnesium Exchange, Synlett, 2000, 345–346 CAS.
- E. V. Shchegolkov, Y. V. Burgart, D. A. Matsneva, S. S. Borisevich, R. A. Kadyrova, I. R. Orshanskaya, V. V. Zarubaev and V. I. Saloutin, Polyfluoroalkylated Antipyrines in Pd-Catalyzed Transformations, RSC Adv., 2021, 11, 35174–35181 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data, molecular structures determined by SCXRD, ex situ analysis by IR and PXRD, and NMR spectra. CCDC 2255940–2255965. For ESI and crystallographic data in CIF or other electronic formats see DOI: https://doi.org/10.1039/d3qo01512b |
| ‡ These authors contributed equally. |
| § Present address: InnoSyn B.V. Urmonderbaan 22, 6167 RD Geleen, The Netherlands. |
| ¶ Present address: Rigaku Europe SE, Hugenottenallee 167, 63263 Neu-Isenburg, Germany. |
| This journal is © the Partner Organisations 2024 |