
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intramolecular hydrogen bond activation for kinetic resolution of furanone derivatives by an organocatalyzed [3 + 2] asymmetric cycloaddition†‡
Miguel A.
Valle-Amores
 a,
Claudia
Feberero
ab,
Ana
Martin-Somer
c,
Sergio
Díaz-Tendero
def,
Andrew D.
Smith
g,
Alberto
Fraile
*ae and
José
Alemán
*ae
a,
Claudia
Feberero
ab,
Ana
Martin-Somer
c,
Sergio
Díaz-Tendero
def,
Andrew D.
Smith
g,
Alberto
Fraile
*ae and
José
Alemán
*ae
aOrganic Chemistry Department (Módulo 1), Facultad de Ciencias, Universidad Autónoma de Madrid, 28049 Madrid, Spain. E-mail: alberto.fraile@uam.es; jose.aleman@uam.es; Web: https://www.uam.es/jose.aleman
bOrganic Chemistry Area, Chemistry Department, Universidad de Burgos, Burgos 09001, Spain
cApplied Physical Chemistry Department (Módulo 14), Facultad de Ciencias, Universidad Autónoma de Madrid, 28049 Madrid, Spain
dChemistry Department (Módulo 13), Facultad de Ciencias, Universidad Autónoma de Madrid, 28049 Madrid, Spain
eInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049 Madrid, Spain
fCondensed Matter Physics Center (IFIMAC), Universidad Autónoma de Madrid, 28049 Madrid, Spain
gEaStCHEM, School of Chemistry, University of St Andrews, North Haugh, Fife, Scotland, KY16 9ST, UK
First published on 11th October 2023
Abstract
Herein, a formal highly enantioselective organocatalyzed [3 + 2] cycloaddition of furanone derivatives and azomethine ylides is presented. The success of this reaction resides in intramolecular hydrogen bond activation through an o-hydroxy group at the aromatic ring of the imine, allowing the formation of highly multifunctional bicyclic adducts with five stereogenic centers in a stereocontrolled manner. Furthermore, the reaction is paired with a highly efficient kinetic resolution of butenolides, achieving selectivity factors above 200. Using this methodology, furan-2(5H)-ones and furo[3,4-c]pyrrolidinones were obtained with high enantioselectivities. Quantum chemistry calculations reveal the crucial role of the hydrogen bond formed between the catalyst donor-units and the two reactants, which modifies their arrangement and promotes effective facial discrimination resulting in a highly selective kinetic resolution. In addition, further applicability of the kinetic resolution process is shown.
Introduction
The synthesis of chiral compounds in an efficient way has been and still is one of the principal issues within the organic community in both industrial and academic points of view. Among the numerous methods that exist at present, kinetic resolution (KR) is one of the most powerful tools used since it allows, in a very efficient way, the separation of both enantiomers from racemic mixtures.1Regarding the different processes of kinetic resolution, we can distinguish between the use of chiral auxiliaries1c,2 and catalysts.1c,3 The principle of KR relies on the reaction of a chiral reagent or catalyst with each enantiomer of the racemic mixture that proceeds through the generation of two diastereomeric transition states. The difference in the free energy between these transition states (ΔΔG‡) dictates the difference in rate constants (k) for the reaction of each enantiomer, allowing their discrimination and determination of the efficiency of the KR using the selectivity factor (s) values.4 The catalytic KR can be divided into enzymatic,5 which has long been a popular strategy, and non-enzymatic processes,6 which include both metal catalysis7 and organocatalysis.8
Within the plethora of reactions studied for the catalytic kinetic resolution of racemates, [3 + 2] cycloadditions have been scarcely studied. The first example was described by Fu's group in 2005, who carried out the kinetic resolution of azomethine imines with activated alkynes catalysed by a chiral copper complex, achieving high enantioselectivities of the recovered dipole and with selectivity factors from 15 to 96 (Scheme 1a).9 Subsequently, other authors have published metal-catalyzed [3 + 2] kinetic resolution processes of different racemic dipolarophiles, reaching high ee and s values.10 Regarding organocatalyzed [3 + 2] cycloadditions, to date, there are only two precedents. The first was reported by Xie's group in 2010, who carried out the [3 + 2] cycloaddition of azomethine ylides to nitroolefins catalyzed by Takemoto's organocatalyst. While this procedure provided the KR of racemic 2-nitro-2H-chromene derivatives, the enantioselectivities obtained were from low to moderate (top, Scheme 1b).11 The second example relates to the three-component kinetic resolution catalysed by chiral bisphosphoric acids between racemic 2,3-allenoates and in situ formed azomethine ylides. In this case, 3-methylenepyrrolidine derivatives were obtained with high enantioselectivities (up to 94% ee) with the (R)-2,3-allenoates recovered in excellent enantioselectivities (up to 99% ee) (bottom, Scheme 1b).12 It should be noted that in both precedents, the authors must make use of azomethine ylide precursors that bear two electron-withdrawing (two esters are present) groups in the methylene carbon of imine which, inevitably, limits the structure of the final products.
 | ||
| Scheme 1 Catalyzed kinetic resolution by dipolar cycloaddition reactions. | ||
On the other hand, furanones, a five-membered ring containing an oxygen atom, are a class of heterocyclic compounds of widespread interest in the organic, pharmacological and biological fields, containing diverse biological properties such as analgesic, anti-inflammatory, and anticancer properties among others.13
Some of the most used synthons that present the butenolide structure are the 5-alkoxyfuran-2(5H)-ones which allow achieving new approaches to acyclic and heterocyclic products.14,15 Feringa14c,16 and others15,17,18 have used butenolide derivatives with chiral auxiliaries such as menthol or sulfinyl groups to achieve diastereoselective asymmetric processes. Starting from these chiral precursors, many interesting structures with high diastereoselectivities have been synthesized. While demonstrative, green chemistry principles consider the use of chiral auxiliaries as not efficient or atom economical, emphasizing the need to develop effective catalytic processes.19 Therefore, the development of new catalytic procedures to carry out asymmetric reactions with 5-alkoxyfuran-2(5H)-ones would be highly desirable. Taking into account the scarce number of examples of organocatalyzed [3 + 2] kinetic resolution and the high synthetic value of furan-2(5H)-ones, we hypothesized that an efficient kinetic resolution could be achieved by following a match/mismatch20 process using a bifunctional organocatalyst and intramolecular hydrogen bond activation (Scheme 1c).21
Results and discussion
Initially, to determine the influence of the hydroxyl group at the imine, we carried out the cycloaddition of the 5-methoxy-2(5H)-furanone (±)-2a with imines 1a (R = H) and 1b (R = OH) in the presence of 20 mol% of Takemoto's catalyst 3a, obtaining better conversion and enantioselectivity with imine 1b (Scheme 2). In addition, the reaction with imine 1a led to a mixture of diastereoisomers (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) while with 1b only one diastereoisomer 4b was achieved. These results bring to light the important role of the hydroxyl group at the imine in the reactivity and stereoselectivity of this asymmetric process.21b,c,22 Furthermore, it is remarkable that the selectivity factor (s) and the conversion (c) are very high, highlighting the effectiveness of the kinetic resolution of (±)-2a (see Table 1, entry 1).
:1) while with 1b only one diastereoisomer 4b was achieved. These results bring to light the important role of the hydroxyl group at the imine in the reactivity and stereoselectivity of this asymmetric process.21b,c,22 Furthermore, it is remarkable that the selectivity factor (s) and the conversion (c) are very high, highlighting the effectiveness of the kinetic resolution of (±)-2a (see Table 1, entry 1).
 | ||
| Scheme 2 Proof of concept for the intramolecular H-bond activation: OH role. The reactions were run with 0.1 mmol of imine 1 and 0.1 mmol of (±)-2a in 0.3 mL of p-xylene ([0.33] M). | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat (mol%) | Solvent | Conv.b (%) | ee 4bc (%) | ee (−)-2ac (%) | s (c (%)) |
| a The reaction was run with 0.1 mmol of imine 1b and 0.1 mmol of (±)-2a in 0.3 mL of indicated solvent ([0.33] M). b Conversion determined by 1H NMR. c Determined by chiral SFC. d Calculated conversion (c) = eeSM/(eeSM + eePR), selectivity factor (s) = ln[(1 − c)(1 − eeSM)]/ln[(1 − c)(1 + eeSM)]. e [0.16] M instead of [0.33] M. f The reaction was scaled up to 1.0 mmol of imine 1b. g The reaction was carried out with 0.05 mmol of imine 1b. | ||||||
| 1 | 3a (20) | p-Xylene | 48 | 97 | 87 | 190 (47) |
| 2 | — | p-Xylene | <5 | — | — | — |
| 3 | 3b (20) | p-Xylene | 40 | 96 | 65 | 100 (40) |
| 4 | 3c (20) | p-Xylene | 10 | — | — | — |
| 5 | 3d (20) | p-Xylene | 20 | — | — | — |
| 6 | 3e (20) | p-Xylene | 18 | — | — | — |
| 7 | 3f (20) | p-Xylene | 19 | — | — | — |
| 8 | 3g (20) | p-Xylene | <5 | — | — | — |
| 9 | 3a (20) | CH2Cl2 | 34 | 86 | 50 | 22 (37) |
| 10 | 3a (20) | Et2O | 47 | 95 | 91 | 120 (49) |
| 11 | 3a (20) | MTBE | 45 | 91 | 82 | 50 (47) |
| 12 | 3a (20) | Toluene | 48 | 92 | 98 | 110 (51) |
| 13e | 3a (20) | p-Xylene | 45 | 95 | 81 | 100 (46) |
| 14 | 3a (15) | p-Xylene | 42 | 97 | 76 | 150 (44) |
| 15 | 3a (10) | p-Xylene | 38 | 97 | 55 | 110 (36) |
| 16f | 3a (20) | p-Xylene | 48 | 97 | 92 | >200 (49) |
| 17g | 3a (20) | p-Xylene | 40 | 95 | 68 | 80 (41) |

Having determined that using imine 1b affords high selectivity, we continued looking for the optimal reaction conditions. Thus, different bifunctional organocatalysts (20 mol%) were studied, with Takemoto's thiourea catalyst 3a giving the highest selectivity for this cycloaddition reaction (compare entry 1 with entries 3–8, Table 1). Notably, a racemic background reaction was not operative, as no conversion to the product was obtained in the absence of catalyst 3a (entry 2). The use of squaramide-based catalysts led to a dramatic loss in conversion (entries 4 and 8). Interestingly, the pseudo-enantiomer catalyst 3e gave very low conversion in comparison with the organocatalyst 3b. Having identified the most promising catalyst, different solvents were screened (entries 9–12). The use of dichloromethane and tert-butylmethylether (entries 9 and 11, respectively) afforded reduced product conversions and enantioselectivities, and hence lower selectivity factors (s) than with p-xylene (entry 1). However, the reaction in diethylether and toluene (entries 10 and 12) gave rise to good conversion and enantioselectivity, but reduced s. Considering the data obtained, p-xylene was selected as the best solvent to continue the reaction screening. Subsequently, reaction concentration was also tested, with the highest selectivity at [0.33] M (entry 1) instead of [0.16] M (entry 13) (see the ESI‡). Finally, the catalyst loading was studied, with reduced product conversions and selectivity factors (s) observed when using 15 and 10 mol% catalyst (entries 14 and 15). The reaction was also scaled up to 1.0 mmol, with no detrimental effect on the stereoselectivity and obtaining the best results in terms of both enantiomeric excess and selectivity factor (4b: 47% and 2a: 46% yield, entry 16). Additionally, to determine if the use of a lower amount of imine 1b could provide similar results to that shown in entry 1, we carried out the reaction starting from 0.05 mmol of 1b (entry 17). However, the results obtained were worse, achieving only a 68% enantiomeric excess for furanone (−)-2a and a lower selectivity factor for the kinetic resolution.
With the optimized conditions in hand (c = 47%, s = 190, Table 1, entry 1), the scope of the dipolar cycloaddition and the efficiency of the KR for (±)-2a were evaluated (Scheme 3). For this purpose, a large assortment of imines 1 bearing an ortho-hydroxyl group at the aromatic ring and with different substituents were tested. When electron-donating groups are present at the aromatic ring of the imine such as the methyl (1c and 1d) or methoxy (1e and 1f) group, cycloaddition products 4c–f were obtained with high conversions and excellent enantioselectivities (94–99% ee). Moreover, the (R)-5-methoxyfuran-2(5H)-one (−)-2a could be recovered with high yields and good enantioselectivities (86–90% ee), achieving excellent selectivity factor values (s > 200) for 1c–e, while for 1f a reduced but still significant value was obtained (s = 90). However, the presence of a diethylamino group (1g) maintained the enantioselectivity of the cycloaddition (98% ee, s = 170) but led to decreased conversion (c = 25%). Furthermore, halogens at para- (1h–i) and meta-positions (1j) were employed. Cycloaddition products 4h–j were obtained with good conversions and high enantioselectivities (up to 95% ee for 4i). Regrettably, enantioselectivities of the recovered starting material (−)-2a were from moderate to good (up to 87% ee), affording reasonable selectivity factors (s = 28–80) for the resolution of (±)-2a. When a nitro group is present at the meta position (1k), the cycloadduct 4k and the furanone (−)-2a were obtained with excellent enantiomeric excess (95 and 90% ee, respectively) and, more importantly, the conversion (c = 49%) and the selectivity factor (s = 120) were very high. An ester group at the meta position (1l) afforded the cycloaddition product 4l with good enantioselectivity (91% ee), but with a decreased conversion and selectivity factor (c = 38%, s = 37). Two iminoesters with a naphthyl group (1m and 1n) were reacted at room temperature with the dipolarophile (±)-2a under the standard reaction conditions without obtaining the corresponding cycloadducts. To our delight, this limitation could be overcome by increasing the temperature to 50 °C, achieving thus 4m and 4n with excellent enantioselectivity values (93 and 97% ee, respectively) and with a high selectivity factor for both reactions (s = 90). Notably, alpha-substituted imines 1o and 1p, with a methyl or a phenyl group, respectively, worked well and allowed the asymmetric synthesis of 4-substituted pyrrolidines 4o and 4p, bearing a quaternary stereocentre, with good enantioselectivities (87 and 71% ee, respectively), high or complete diastereoselectivity (for 4p: 5:1 dr) and increased conversion (up to 51%). Nevertheless, regarding the selectivity, the resolution was more effective with the α-methyl imine 1o (s = 45) than with the α-phenyl ring 1p (s = 11). Different electron-withdrawing groups at the imine moiety, such as N,N-dimethyl amide 1q, ethyl ester 1r, and Weinreb's amide 1s, also led to the corresponding bicyclic adducts 4q–s in good to high enantioselectivities (84–93% ee) and high conversions. The best results in terms of selectivity factor were achieved from Weinreb's amide 1s (s = 100), which allowed recovery of the (R)-5-methoxyfuran-2(5H)-one (−)-2a with excellent enantioselectivity (99% ee). The absolute configuration of the stereogenic centres of the hydroxylated cycloadducts 4 was assigned by X-ray crystallographic analysis of a monocrystal of 4j (3S,3aR,4S,6R,6aS)23 (middle, Scheme 3) and assuming the same stereochemical outcome for the rest of the products.
 | ||
| Scheme 3 Scope of the [3 + 2] cycloaddition kinetic resolution. Reaction conditions: 1 (0.1 mmol), (±)-2 (0.1 mmol), 3a (20 mol%), p-xylene (0.3 mL), rt for 24 h unless otherwise noted. Isolated yields are shown. Enantiomeric ratio was measured by SFC. Calculated conversion (c) = eeSM/(eeSM + eePR). Selectivity factor (s) = ln[(1 − c)(1 − eeSM)]/ln[(1 − c)(1 + eeSM)]. aReaction was carried out at 50 °C for 2 days. | ||
Finally, to demonstrate the versatility of our [3 + 2] cycloaddition kinetic resolution, we studied the influence of the incorporation of a variety of substituents at C(3) and C(5) within the furan-2(5H)-one as well as the use of 2-(5H)-pyrrolones (Scheme 4). Varying the substituent at C(5) from the methoxy to ethoxy group gave excellent conversion to the product, giving an s factor >200 for the kinetic resolution, and provided resolved (R)-5-ethoxyfuran-2(5H)-one (−)-2b in excellent enantiomeric excess.
 | ||
| Scheme 4 Scope of the [3 + 2] cycloaddition kinetic resolution regarding furanone and its derivatives. Reaction conditions: 1 (0.1 mmol), (±)-2 (0.1 mmol), 3a (20 mol%), p-xylene (0.3 mL), rt for 24 h unless otherwise noted. Isolated yields are shown. Enantiomeric ratio was measured by SFC. Calculated conversion (c) = eeSM/(eeSM + eePR). Selectivity factor (s) = ln[(1 − c)(1 − eeSM)]/ln[(1 − c)(1 + eeSM)]. aReaction was carried out at 0 °C in toluene. | ||
Introducing a thiolate group resulted in the successful formation of the cycloadduct 4u with a high degree of enantioselectivity (92% ee). However, the furanone 2c was obtained with no enantioselectivity. This observation can be attributed to the high acidity of the thioacetal proton (H-5) in furanone 2c, which can lead to complete epimerization in the presence of organocatalyst 3a, resulting in a racemic mixture, followed by auto-selfcondensation as it was previously described in the literature.24 The reaction with a more reactive pseudo ester such as the 3-bromo-5-methoxyfuran-2(5H)-one (±)-2d was studied under the optimized conditions, achieving the cycloadduct 4v with good enantioselectivity (87% ee) and moderate conversion (c = 35%) and selectivity. The reactions with the aromatic derivative (±)-2e worked well, giving rise to the cycloadduct 4w with good selectivity, while the heteroaromatic species (±)-2f gave the cycloadduct 4x in excellent enantioselectivity (s = 120), but with low conversion, accounting for the recovery of the pyrrole derivative (−)-2f in low enantiomeric excess. With the incorporation of an alkenyl group the cycloaddition reaction was facile, leading to the formation of the corresponding adduct 4y and the pseudoester (−)-2g with excellent enantioselectivities and yields. Notably, in these recent examples, it was possible to generate a quaternary carbon stereocenter in a precisely controlled manner. In contrast, we also studied the reactivity of 5-methoxypyrrol-2(5H)-ones containing an acetyl substituent at the nitrogen atom. Regrettably, under the specified conditions the acetyl derivative (±)-2h reacted to give the lactam 4z and 2-(5H)-pyrrolone 2h in high yield, but as a racemic mixture. Furthermore, attempts were made to conduct [3 + 2] cycloaddition reactions using 5-methoxyfuran-2(5H)-ones containing a methyl substituent at the alpha position. However, no conversion was observed in these reactions.
Considering the excellent results obtained in the kinetic resolution of 5-methoxyfuran-2(5H)-one (±)-2a and to showcase the utility of the developed process, we envisioned the use of the enantioenriched isomer (−)-2a as the starting material for new asymmetric reactions (Scheme 5). Thus, the crude reaction product obtained during the [3 + 2] cycloaddition was submitted to a one-pot procedure. Therefore, several Michael additions14g and a [3 + 2] cycloaddition18b were carried out to demonstrate the applicability of our methodology (Proc. A, Scheme 5). Thus, the addition of (R)-1-phenylethan-1-amine or thiophenol to the crude reaction mixture from the cycloaddition (equations a and b) afforded the corresponding aza-Michael or thio-Michael products (5 and 6, respectively) with excellent enantio- and diastereo-selectivity and in high yields (only 50% of the final product can be obtained). Furthermore, the addition of 11H-dibenzo[b,e]azepine 5-oxide led to the cycloadduct 7 with excellent results (equation c).
 | ||
| Scheme 5 Further derivatization of (−)-2a in one pot and direct procedures. One-pot (Proc. A) and direct (Proc. B) procedures: aaza-Michael addition of (S)-1-phenylethanamine to resolved (−)-2a. bthio-Michael addition of thiophenol to resolved (−)-2a. c[3 + 2] cycloaddition of nitrone to resolved (−)-2a. Reactions were carried out at the 0.1 mmol and 0.05 mmol scale for procedure A and B, respectively. | ||
Finally, to corroborate the configurational stability of furanone, the same reactions showed before, starting from the cycloaddition reaction crude products, were conducted from the (R)-5-methoxyfuran-2(5H)-one (−)-2a obtained by kinetic resolution and the subsequent purification by flash chromatography on silica gel (Proc. B, Scheme 5). The enantiomeric excesses achieved in these transformations brought to light that the 5-methoxyfuran-2(5H)-one (−)-2a obtained by kinetic resolution is configurationally stable and can be isolated in the enantioenriched form without any racemization.
Mechanistic proposal
Once the scope of this organocatalyzed [3 + 2] asymmetric cycloaddition and the synthetic applicability of this kinetic resolution were demonstrated, we wished to rationalise the stereochemical outcome of this process. Taking into account the absolute configuration of final adducts 4, an endo approach of ylide 1 in its anti-conformation to the less hindered face of the furanone 2 (anti-approach) takes place. To corroborate this proposal, quantum chemistry calculations were carried out to obtain further theoretical insights into the enantioselectivity of the reaction.25 We initially considered the possible relative orientations between catalyst 3a and reactants 1b and (±)-2a. Thus, we followed a similar strategy to that previously proposed,26 which consists of a systematic exploration of the potential energy surface (PES) using the GFN2-xTB method27 as implemented in the CREST code.28 From the most stable structures found with this method, the PES was refined using density functional theory (DFT) calculations, by combining the B3LYP functional and the 6-31+G(d,p) basis set, and including dispersion forces through the D3 version of Grimme's method with the Becke–Johnson damping.29 These calculations were carried out with the Gaussian 16 code.30The reaction was studied considering the endo approach for the two enantiomers of 2 (exo approaches were discarded since they are less favourable than the endo ones,15c,21b see the ESI‡).25 We considered a two-step process. The first step is the activation of the imine 1b by an H transfer from 1b to catalyst 3a. In the second step, new C–C bonds are formed, yielding the 5-membered ring (Fig. 1 and 2, respectively). For both enantiomers, the H transfer requires ∼18 kcal mol−1 from the initial pre-association complex, PAC (pre-H transfer: complex formed by the catalyst and the two reactants 1b and (±)-2a) (Fig. 1). However, the main difference was found in the second cycloaddition step (Fig. 2). While for the TS leading to the formed cycloadduct a low barrier of ∼3.5 kcal mol−1 is observed (black line), in the alternative case a transition state was located at a much higher energy, ∼9 kcal mol−1 (blue line), than the initial pre-association-complex (PAC, Fig. 2); i.e., even higher than the first TS for H transfer. Therefore, for the non-observed isomer, the much higher barrier for the cycloaddition prevents its formation, thus explaining the experimentally obtained adduct.
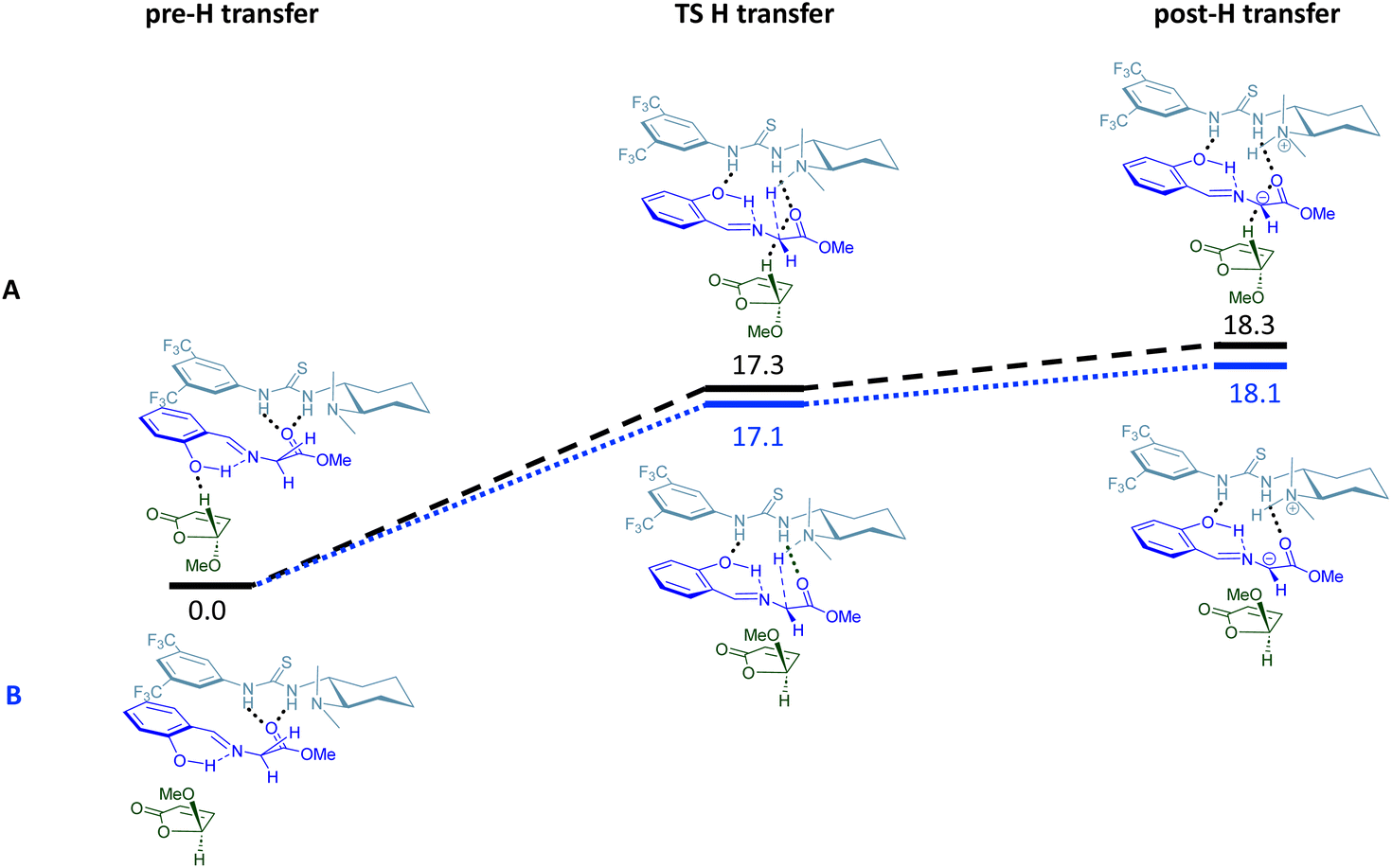 | ||
| Fig. 1 Potential energy surface for the H transfer reaction and the structures corresponding to the stationary states, both for the path leading to the observed product (A) and the path for the not observed enantiomer (B). Relative Gibbs free energies in kcal mol−1 are referred to the pre-H transfer complex. | ||
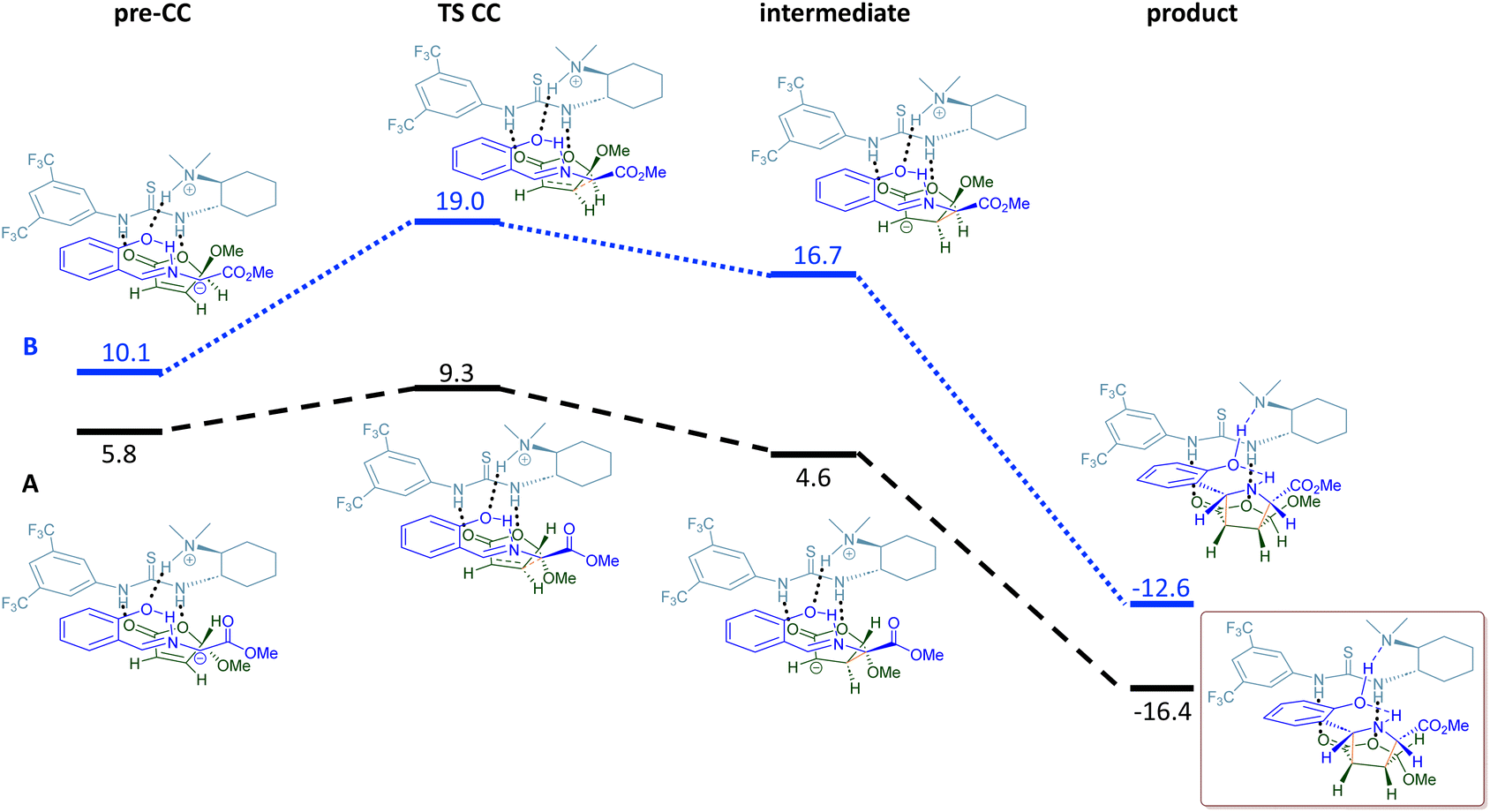 | ||
| Fig. 2 Potential energy surface for the CC bond formation and the structures corresponding to the stationary states, both for the path leading to the observed product (A, black line) and the path for the not observed enantiomer (B, blue line). Relative Gibbs free energies in kcal mol−1 are referred to the pre-H transfer complex. | ||
To obtain further molecular insights into the origin of the stereocontrol, we analysed the non-covalent interaction (NCI) at the second TS (TS CC, Fig. 3) by means of NCI plots, using the NCI code.31 NCI plots show the different interaction regions using a colour code to rank those interactions. Red is used for destabilizing interactions, blue for stabilizing interactions and green for delocalized weak interactions. The intensity of these colours is associated with the interaction strength.
 | ||
| Fig. 3 Noncovalent interaction (NCI) plot for the C–C bond formation transition states leading to the observed product (A) and the isomer (B). The green, blue, and red regions, respectively, represent attractive, strongly attractive, and repulsive interactions. | ||
For both TSs, NCI plots revealed weak delocalized interactions (green) between the three moieties interacting (Fig. 3). However, the orientations of 1b and both enantiomeric dipolarophiles 2a with respect to the catalyst are slightly different, leading to different non-covalent interactions in the C–C bond forming transition state. TS-A (observed reaction) presents a strong NH⋯O hydrogen bond (blue flat circular region) between the ammonium group, generated after the H transfer to the catalyst, and the hydroxy group of ylide 1b (top image in Fig. 3A). This leads to a stronger intramolecular hydrogen bond in 1b between the hydroxy group and the iminic nitrogen (actually it appears as H bonded to both atoms). While for TS-B, this hydrogen bond is formed with the iminic nitrogen of 1b (instead of the hydroxy group) and is weaker (only slightly blue) (top image in Fig. 3B). The significance of the hydroxyl group on the aromatic ring of the imine becomes apparent from these findings. It plays a crucial role in establishing a beneficial intermolecular interaction with the organocatalyst through hydrogen bonding. In contrast, the absence of this hydroxyl group in imine 1a prevents such an interaction, making it impossible to form a structured and organized complex between 1a and 3a. Consequently, this leads to low enantioselectivity.
Concerning the NCI between the catalyst and dipolarophile 2a, at TS-A there are two hydrogen bonds between the thiourea group in the catalyst and oxygen atoms 1 and 2 in (+)-2a, with 1 being the stronger one (bottom image in Fig. 3A). For TS-B (not-formed adduct), the different orientation of the methoxy group of (−)-2a allows the formation of a very weak hydrogen bond (green) between carbonyl oxygen atom 1 in the ylide 1b (bottom image in Fig. 3B) and the catalyst, displacing the furanone unit to the right.
This displacement leads to the formation of only one hydrogen bond between oxygen 2 in furanone (−)-2a (NH⋯O) and the catalyst, which is weaker than in TS-A (lighter blue color). In addition, for TS-B, there is a third CH⋯O bond (3), not present in TS-A, but it is very weak (green). The arrangement of the reactants at TS-B makes the region between the three moieties more crowded than in TS-A, hampering the approach of the two reactants to form the new C–C bond. This is reflected in a higher barrier for CC bond formation (TS-B energy is ∼10 kcal mol−1 higher than that of TS-A).
Conclusions
In conclusion, in this work we report an organocatalyzed [3 + 2] enantioselective cycloaddition of formal azomethine ylides with racemic furan-2(5H)-ones. This allows the generation of highly functionalized and versatile bicyclic adducts with up to 5 contiguous stereogenic centres in a stereocontrolled endo approach and with high enantioselectivities (up to 99% ee) due to the presence of the hydroxyl group at the aromatic ring of imines. Moreover, this cycloaddition reaction is paired with an efficient kinetic resolution of the furan-2(5H)-one, leading to the resolved substrate in an enantioenriched form (up to 99% ee). This kinetic resolution takes place with selectivity factors up to 200 and very high conversions. DFT calculations have demonstrated the great effectiveness of the organocatalyzed [3 + 2] asymmetric cycloaddition to reach a very efficient kinetic resolution and the crucial influence of the hydroxyl group at the imine aromatic ring on the deprotonation process to generate the reactive ylide and on the stereoselectivity.Data availability
Experimental details, general procedures, optimization of reaction conditions, characterization of products, copies of NMR and HPLC spectra of all products and computational details are provided in the ESI.‡Author contributions
A. F. and J. A. conceived, designed, and supervised this work. M. A. V.-A. and C. F. performed the experiments and the synthesis and characterization of new compounds. A. M.-S. and S. D.-T. performed the calculations and wrote the theoretical part of the article. A. F. and M. A. V.-A prepared the ESI.‡ A. F., J. A., M. A. V.-A and A. D. S. wrote the article. All authors contributed to the discussion of the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Spanish Ministry of Science and Innovation (projects PID2019-110091GB-I00, PID2021-122299NB-I00, PID2022-138470NB-I00, TED2021-130470B-I00, and TED2021-129999B-C32), “Comunidad de Madrid” for European Structural Funds (S2018/NMT-4367), Proyectos Sinérgicos I + D (Y2020/NMT-6469), and “María de Maeztu” (CEX2018-000805-M) Program for Centers of Excellence in R&D. A. M. S. thanks the Madrid Government (Comunidad de Madrid – Spain) under the Multiannual Agreement with Universidad Autónoma de Madrid in the line Support to Young Researchers, in the context of the V PRICIT (SI3-PJI-2021-00463). C. F. thanks Ministerio de Universidades for a Margarita Salas grant. In addition, the authors acknowledge the generous allocation of computer time at the Centro de Computación Científica at the Universidad Autónoma de Madrid (CCC-UAM).References
- (a) H. B. Kagan and J. C. Fiaud, in Topic in Stereochemistry, ed. E. L. Eliel and S. H. Wilen, John Wiley & Sons, Inc., 1988, ch. 4, vol. 18, pp. 249–330 Search PubMed; (b) Enantiomers, Racemates, and Resolutions, ed. J. Jacques, A. Collet and S. H. Wilen, John Wiley & Sons, Inc., 1981 Search PubMed; (c) Separation of Enantiomers: Synthetic Methods, ed. M. H. Todd, Wiley-VCH, Weinheim, Germany, 2014 Search PubMed.
- (a) X.-C. He and C.-Y. Qi, Practical Tactics in Resolution of Racemates via Diastereomeric Salt Formation, Chin. J. Chem., 2007, 25, 583–586 CrossRef CAS; (b) M. R. Maddani, J.-C. Fiaud and H. B. Kagan, Stoichiometric Kinetic Resolution Reactions, in Separation of Enantiomers: Synthetic Methods, ed. M. H. Todd, Wiley-VCH, Weinheim, Germany, 2014, ch. 2, pp 13–74 Search PubMed; (c) S.-Y. Hsieh, B. Wanner, P. Wheeler, A. M. Beauchemin, T. Rovis and J. W. Bode, Stereoelectronic Basis for the Kinetic Resolution of N-Heterocycles with Chiral Acylating Reagents, Chem. – Eur. J., 2014, 20, 7228–7231 CrossRef CAS.
- (a) H. Pellissier, Catalytic Kinetic Resolution, in Separation of Enantiomers: Synthetic Methods, ed. M. H. Todd, Wiley-VCH, Weinheim, Germany, 2014, ch. 3, pp. 75–122 Search PubMed; (b) E. R. Jarvo and S. J. Miller, Acylation Reaction, in Comprehensive Asymmetric Catalysis, Supplement 1, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer-Verlag Berlin, Heidelberg, 2004, ch. 43, pp. 189–206 Search PubMed; (c) J. M. Keith, J. F. Larrow and E. N. Jacobsen, Practical Considerations in Kinetic Resolution Reactions, Adv. Synth. Catal., 2001, 343, 5–26 CrossRef CAS.
- M. D. Greenhalgh, J. E. Taylor and A. D. Smith, Best Practice Considerations for Using the Selectivity Factor, s, as a Metric for the Efficiency of Kinetic Resolutions, Tetrahedron, 2018, 74, 5554–5560 CrossRef CAS.
- (a) A. Ghanem and H. Y. Aboul-Enein, Application of Lipases in Kinetic Resolution of Racemates, Chirality, 2005, 17, 1–15 CrossRef CAS PubMed; (b) M. T. Reetz, Biocatalysis in Organic Chemistry and Biotechnology: Past, Present, and Future, J. Am. Chem. Soc., 2013, 135, 12480–12496 CrossRef CAS; (c) C. E. Humphrey, M. Ahmed, A. Ghanem and N. J. Turnerx, Application of Enzymes in Kinetic Resolutions, Dynamic Kinetic Resolutions and Deracemization Reactions, in Separation of Enantiomers: Synthetic Methods, ed. M. H. Todd, Wiley-VCH, Weinheim, Germany, 2014, ch. 4, pp. 123–160 Search PubMed; (d) S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Biocatalysis: Enzymatic Synthesis for Industrial Applications, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS; (e) Q. H. Zhang, Y. Fang, W. F. Luo and L. N. Huang, Biocatalytic Kinetic Resolution of D,L-pantolactone by Using a Novel Recombinant D-Lactonase, RSC Adv., 2021, 11, 721–725 RSC; (f) Q. H. Zhang, L. Yang, Y. B. Tang, L. N. Huang and W. F. Luo, Industrial Kinetic Resolution of D,L-Pantolactone by an Immobilized Whole-Cell Biocatalyst, RSC Adv., 2021, 11, 30373–30376 RSC.
- (a) D. E. J. E. Robinson and S. D. Bull, Kinetic Resolution Strategies Using Non-Enzymatic Catalysts, Tetrahedron: Asymmetry, 2003, 14, 1407–1446 CrossRef CAS; (b) E. Vedejs and M. Jure, Efficiency in Nonenzymatic Kinetic Resolution, Angew. Chem., Int. Ed., 2005, 44, 3974–4001 CrossRef CAS; (c) H. Pellissier, Catalytic Non-Enzymatic Kinetic Resolution, Adv. Synth. Catal., 2011, 353, 1613–1666 CrossRef CAS.
- (a) A. H. Hoveyda and M. T. Didiuk, Metal-Catalyzed Kinetic Resolution Processes, Curr. Org. Chem., 1998, 2, 489–526 CrossRef CAS; (b) G. R. Cook, Transition Metal-Mediated Kinetic Resolution, Curr. Org. Chem., 2000, 4, 869–885 CrossRef CAS; (c) L. Deng, Y. Fu, S. Y. Lee, C. Wang, P. Liu and G. Dong, Kinetic Resolution via Rh-Catalyzed C–C Activation of Cyclobutanones at Room Temperature, J. Am. Chem. Soc., 2019, 141, 16260–16265 CrossRef CAS; (d) J. M. González, B. Cendón, J. L. Mascareñas and M. Gulías, Kinetic Resolution of Allyltriflamides through a Pd-Catalyzed C–H Functionalization with Allenes: Asymmetric Assembly of Tetrahydropyridines, J. Am. Chem. Soc., 2021, 143, 3747–3752 CrossRef.
- (a) R. Gurubrahamam, Y.-S. Cheng, W.-Y. Huang and K. Chen, Recent Advances in Organocatalytic Kinetic Resolution for the Synthesis of Functionalized Products, ChemCatChem, 2016, 8, 86–96 CrossRef CAS and references therein cited; (b) U. Farid, M. L. Aiello and S. J. Connon, Highly Enantioselective Catalytic Kinetic Resolution of α-Branched Aldehydes through Formal Cycloaddition with Homophthalic Anhydrides, Chem. – Eur. J., 2019, 25, 10074–10079 CrossRef CAS PubMed; (c) S. Qu, S. M. Smith, V. Laina-Martín, R. M. Neyyappadath, M. D. Greenhalgh and A. D. Smith, Isothiourea-Catalyzed Acylative Kinetic Resolution of Tertiary α-Hydroxy Esters, Angew. Chem., Int. Ed., 2020, 59, 16572–16578 CrossRef CAS; (d) J. Liu, L. Vasamsetty, M. Anwar, S. Yang, W. Xu, J. Liu, S. Nagaraju and X. Fang, Organocatalyzed Kinetic Resolution of α-Functionalized Ketones: The Malonate Unit Leads the Way, ACS Catal., 2020, 10, 2882–2893 CrossRef CAS; (e) S. Sun, Z. Wang, S. Li, C. Zhou, L. Song, H. Huang and J. Sun, An Organocatalytic Kinetic Resolution of Aziridines by Thiol Nucleophiles, Org. Lett., 2021, 23, 554–558 CrossRef CAS PubMed; (f) Y. Wu, M. Li, J. Sun, G. Zheng and Q. Zhang, Synthesis of Axially Chiral Aldehydes by N-Heterocyclic-Carbene-Catalyzed Desymmetrization Followed by Kinetic Resolution, Angew. Chem., Int. Ed., 2022, 61, e202117340 CrossRef CAS; (g) S. Barik, R. C. Das, K. Balanna and A. T. Biju, Kinetic Resolution Approach to the Synthesis of C–N Axially Chiral N-Aryl Aminomaleimides via NHC-Catalyzed [3+3] Annulation, Org. Lett., 2022, 24, 5456–5461 CrossRef CAS PubMed.
- A. Suárez, C. W. Downey and G. C. Fu, Kinetic Resolutions of Azomethine Imines via Copper-Catalyzed [3+2] Cycloadditions, J. Am. Chem. Soc., 2005, 127, 11244–11245 CrossRef.
- (a) H. Takayama, Z.-J. Jia, L. Kremer, J. O. Bauer, C. Strohmann, S. Ziegler, A. P. Antonchick and H. Waldmann, Discovery of Inhibitors of the Wnt and Hedgehog Signaling Pathways through the Catalytic Enantioselective Synthesis of an Iridoid-Inspired Compound Collection, Angew. Chem., Int. Ed., 2013, 52, 12404–12408 CrossRef CAS PubMed; (b) H. Xu, C. Golz, C. Strohmann, A. P. Antonchick and H. Waldmann, Enantiodivergent Combination of Natural Product Scaffolds Enabled by Catalytic Enantioselective Cycloaddition, Angew. Chem., Int. Ed., 2016, 55, 7761–7765 CrossRef CAS; (c) Y. Yuan, Z.-J. Zheng, L. Li, X.-F. Bai, Z. Xu, Y.-M. Cui, J. Cao, K.-F. Yang and L.-W. Xu, Silicon-based Bulky Group−Tuned Parallel Kinetic Resolution in Copper-Catalyzed 1,3-Dipolar Additions, Adv. Synth. Catal., 2018, 360, 3002–3008 CrossRef CAS; (d) C. Shen, Y. Yang, L. Wei, W.-W. Dong, L. W. Chung and C.-J. Wang, Kinetic Resolution of Alkylidene Norcamphors via a Ligand-Controlled Umpolung-Type 1,3-Dipolar Cycloaddition, iScience, 2019, 11, 146–159 CrossRef CAS PubMed; (e) H. Deng, T.-T. Liu, Z.-D. Ding, W.-L. Yang, X. Luo and W.-P. Deng, Kinetic resolution of 2H-azirines via Cu(I)-catalyzed asymmetric 1,3-dipolar cycloaddition of azomethine ylides, Org. Chem. Front., 2020, 7, 3247–3252 RSC.
- J.-W. Xie, L.-P. Fan, H. Su, X.-S. Li and D.-C. Xu, Efficient kinetic resolution of racemic 3-nitro-2H-chromene derivatives catalyzed by Takemoto's organocatalyst, Org. Biomol. Chem., 2010, 8, 2117–2122 RSC.
- J. Yu, W.-J. Chen and L.-Z. Gong, Kinetic Resolution of Racemic 2,3-Allenoates by Organocatalytic Asymmetric 1,3-Dipolar Cycloaddition, Org. Lett., 2010, 12, 4050–4053 CrossRef CAS PubMed.
- A. Husain, S. A. Khan, F. Iram, M. A. Iqbal and M. Asif, Insights into the chemistry and therapeutic potential of furanones: A versatile pharmacophore, Eur. J. Med. Chem., 2019, 171, 66–92 CrossRef CAS and references cited therein.
- (a) F. Fariña, M. V. Martín and F. Sánchez, Pseudoesters and Derivatives. XXIV. 1,3-Dipolar Cycloaddition of Diazomethane to 5-Methoxyfuran-2(5H)-ones, Heterocycles, 1986, 24, 2587–2592 CrossRef; (b) L. Fišera and P. Oravec, Stereoselectivity of Cycloadditions of Arylnitrile Oxides to 5-Alkoxy-2(5H)-furanone, Collect. Czech. Chem. Commun., 1987, 52, 1315–1324 CrossRef; (c) B. L. Feringa and B. de Lange, Asymmetric 1,4-Additions to 5-Alkoxy-2(5H)-furanones: An Efficient Synthesis of (R)- and (S,)-3,4-Epoxy-1-Butanol, Tetrahedron, 1988, 44, 7213–7222 CrossRef CAS; (d) B. L. Feringa and B. de Lange, 1,4-Additions of Amines to 5-Methoxyfuran-2(5H)-one; an Efficient Synthesis of Amino Diols, Heterocycles, 1988, 27, 1197–1205 CrossRef CAS; (e) E. Keller, B. de Lange, M. T. Rispens and B. L. Feringa, 1,3-Dipolar Cycloadditions to 5-Methoxy-2(5H)-furanone, Tetrahedron, 1993, 49, 8899–8910 CrossRef CAS; (f) F. Fariña, M. R. Martín, M. V. Martín and A. Martinez de Guereñu, Cycloaddition of Nitrile Oxides to 4-Oxobut-2-enoic Acid Derivatives, Heterocycles, 1994, 38, 1307–1316 CrossRef; (g) W. S. Faber, J. Kok, B. de Lange and B. L. Feringa, Catalytic Kinetic Resolution of 5-Alkoxy-2(5H)-furanones, Tetrahedron, 1994, 50, 4775–4794 CrossRef CAS.
- (a) J. L. García Ruano, A. Fraile, M. R. Martín and A. Núñez, First Asymmetric Cycloaddition of Carbonyl Ylides to Vinyl Sulfoxides and Furan-2(5H)-ones, J. Org. Chem., 2006, 71, 6536–6541 CrossRef; (b) A. Núñez Jr., M. R. Martín, A. Fraile and J. L. García Ruano, Abnormal Behaviour of Allenylsulfones under Lu's Reaction Conditions: Synthesis of Enantiopure Polyfunctionalised Cyclopentenes, Chem. – Eur. J., 2010, 16, 5443–5453 CrossRef; (c) J. L. García Ruano, A. Fraile, M. R. Martín, G. González, C. Fajardo and A. M. Martín-Castro, Asymmetric Synthesis of Pyrrolo[2,1-a]isoquinoline Derivatives by 1,3-Dipolar Cycloadditions of Stabilized Isoquinolinium N-Ylides with Sulfinyl Dipolarophiles, J. Org. Chem., 2011, 76, 3296–3305 CrossRef.
- (a) B. L. Feringa and J. C. de Jong, New Strategies in Asymmetric Synthesis Based on γ-Alkoxybutenolides, Bull. Soc. Chim. Belg., 1992, 101, 627–640 CrossRef CAS; (b) B. L. Feringa, B. de Lange, J. F. G. A. Jansen, J. C. de Jong, M. Lubben, W. Faber and E. P. Schudde, New Approaches in Asymmetric Synthesis Using γ-Alkoxybutenolides, Pure Appl. Chem., 1992, 64, 1865–1871 CrossRef CAS and references cited therein.
- (a) D. M. Cooper, R. Grigg, S. Hargreaves, P. Kennewell and J. Redpath, X=Y-ZH Compounds as Potential 1,3-Dipoles. Part 44.1 Asymmetric 1,3-Dipolar Cycloaddition Reactions of Imines and Chiral Cyclic Dipolarophiles, Tetrahedron, 1995, 51, 7791–7808 CrossRef CAS; (b) H. A. Dondas, R. Grigg and M. Thornton-Pett, Spiro(Pyrrolidinyl-2,3′-Benzodiazepines) Related to MK-329, Tetrahedron, 1996, 52, 13455–13466 CrossRef; (c) B. M. Trost and M. L. Crawley, A “Chiral Aldehyde” Equivalent as a Building Block Towards Biologically Active Targets, Chem. – Eur. J., 2004, 10, 2237–2252 CrossRef CAS PubMed; (d) R. Grigg, D. M. Cooper, S. Holloway, S. McDonald, E. Millingtona and M. A. B. Sarker, X=Y–ZH Systems as Potential 1,3-Dipoles. Part 61: Metal Exchanged Zeolites, Silver(I) Oxide, Ni(II) and Cu(I) Complexes as Catalysts for 1,3-Dipolar Cycloaddition Reactions of Imines Generating Proline Derivatives, Tetrahedron, 2005, 61, 8677–8685 CrossRef CAS.
- (a) J. L. García Ruano, A. Fraile and M. R. Martín, (Ss)-5-Ethoxy-3-p-Tolylsulfinylfuran-2(5H)-ones as Chiral Dipolarophiles: First Asymmetric Cycloaddition of Diazomethane to Vinyl Sulfoxides, Tetrahedron: Asymmetry, 1996, 7, 1943–1950 CrossRef; (b) J. L. García Ruano, J. I. Andrés Gil, A. Fraile, A. M. Martín Castro and M. R. Martín, Asymmetric 1,3-Dipolar Reactions of 3-Sulfinylfuran-2(5H)-ones with 11H-Dibenzo[b,e]azepine 5-Oxide. Synthesis of Pyrroloazepines via Isoxazoloazepines, Tetrahedron Lett., 2004, 45, 4653–4656 CrossRef; (c) J. L. García Ruano, A. Fraile, A. M. Martín Castro and M. R. Martín, The Role of the Sulfinyl Group on the Course of the Reactions of 3-p-Tolylsulfinylfuran-2(5H)-ones with Nitrones. Synthetic Uses of Cycloreversion Processes, J. Org. Chem., 2005, 70, 8825–8834 CrossRef PubMed; (d) J. L. García Ruano, A. Núñez Jr., M. R. Martín and A. Fraile, Totally Regio- and Stereoselective Behavior of Mono- and Diactivated Cyclic Alkenes in the Lu Reaction: Synthesis of Enantiopure Functionalized Cyclopentanes, J. Org. Chem., 2008, 73, 9366–9371 CrossRef PubMed.
- Green Chemistry: Theory and Practice, ed. P. T. Anastas and J. C. Warner, Oxford University Press, 1998 Search PubMed.
- D. G. Blackmond, Kinetic Resolution Using Enantioimpure Catalysts: Mechanistic Considerations of Complex Rate Laws, J. Am. Chem. Soc., 2001, 123, 545–553 CrossRef CAS PubMed.
- (a) A. Guerrero-Corrella, M. A. Valle-Amores, A. Fraile and J. Alemán, Enantioselective Organocatalyzed aza-Michael Addition Reaction of 2-Hydroxybenzophenone Imines to Nitroolefins under Batch and Flow Conditions, Adv. Synth. Catal., 2021, 363, 3845–3851 CrossRef; (b) F. Esteban, W. Cieslik, E. M. Arpa, A. Guerrero-Corella, S. Díaz-Tendero, J. Perles, J. A. Fernández-Salas, A. Fraile and J. Alemán, Intramolecular Hydrogen Bond Activation: Thiourea-Organocatalyzed Enantioselective 1,3-Dipolar Cycloaddition of Salicylaldehyde-Derived Azomethine Ylides with Nitroalkenes, ACS Catal., 2018, 8, 1884–1890 CrossRef CAS PubMed; (c) A. Guerrero-Corella, A. Fraile and J. Alemán, Intramolecular Hydrogen-Bond Activation: Strategies, Benefits, and Influence in Catalysis, ACS Org. Inorg. Au, 2022, 2, 197–204 CrossRef CAS PubMed.
- A. Guerrero-Corella, F. Esteban, M. Iniesta, A. Martín-Somer, M. Parra, S. Díaz-Tendero, A. Fraile and J. Alemán, 2-Hydroxybenzophenone as a Chemical Auxiliary for the Activation of Ketiminoesters for Highly Enantioselective Addition to Nitroalkenes under Bifunctional Catalysis, Angew. Chem., Int. Ed., 2018, 57, 5350–5354 CrossRef CAS.
- CCDC 2160961‡ (4i) contains the supplementary crystallographic data for this paper.
- F. Fariña and M. D. Parellada, Pseudoesters and Derivatives. 29. Regioselective Reactions of the 5-(Ethylthio)furan-2(5H)-one Anion with Electrophiles, J. Org. Chem., 1988, 53, 3330–3333 CrossRef.
- (a) All the optimized structures computed can be visualized and downloaded from https://dx.doi.org/10.19061/iochem-bd-8-10; (b) M. Álvarez-Moreno, C. de Graaf, N. Lopez, F. Maseras, J. M. Poblet and C. Bo, Managing the Computational Chemistry Big Data Problem: The ioChem-BD Platform, J. Chem. Inf. Model., 2015, 55, 95–103 CrossRef.
- I. Iribarren and C. Trujillo, Efficiency and Suitability when Exploring the Conformational Space of Phase-Transfer Catalysts, J. Chem. Inf. Model., 2022, 62, 5568–5580 CrossRef CAS.
- C. Bannwarth, S. Ehlert and S. Grimme, GFN2-xTB–An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- P. Pracht, F. Bohle and S. Grimme, Automated Exploration of the Low-Energy Chemical Space with Fast Quantum Chemical Methods, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the Damping Function in Dispersion Corrected Density Functional Theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2019 Search PubMed.
- (a) E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-García, A. J. Cohen and W. Yang, Revealing Noncovalent Interactions, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS; (b) J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, NCIPLOT: A Program for Plotting Non-Covalent Interaction Regions, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef.
Footnotes |
| † Dedicated to M. Rosario Martín Ramos for her great contributions to the 5-alkoxyfuran-2(5H)-one chemistry. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2160961. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qo01471a |
| This journal is © the Partner Organisations 2024 |