
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ammonia activation using a heteroleptic stannylene and lithium stannylenoid formation facilitated by hemilabile iminophosphorane-based ligands†
David M. J.
Krengel
 a,
Nico
Graw
a,
Regine
Herbst-Irmer
a,
Dietmar
Stalke
a,
Oliver P. E.
Townrow
*b and
Malte
Fischer
*a
a,
Nico
Graw
a,
Regine
Herbst-Irmer
a,
Dietmar
Stalke
a,
Oliver P. E.
Townrow
*b and
Malte
Fischer
*a
aInstitut für Anorganische Chemie, Georg-August-Universität Göttingen, Tammannstraße 4, D-37077 Göttingen, Germany. E-mail: malte.fischer@uni-goettingen.de
bInorganic and Organometallic Chemistry, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstraße 1, D-91058 Erlangen, Germany. E-mail: oliver.townrow@fau.de
First published on 30th September 2024
Abstract
Heteroleptic stannylenes, featuring pendant hemilabile iminophosphorane functionalities and kinetically stabilizing terphenyl ligands, were synthesized straightforwardly through formal C–H activation. Subsequently, they were investigated for their ability to activate ammonia through N–H bond scission. By combining synthetic modifications of the ancillary ligand framework and computational analyses, detailed insights into the mechanism of NH3 activation by these systems were obtained, highlighting an activation pathway at tin without a change in oxidation state. Additionally, an observed by-product during these studies underscores the non-innocence of a lithium salt in the synthesis of the stannylene starting materials, providing access to a novel lithium stannylenoid.
Introduction
The activation of small molecules like ammonia (NH3) to construct complex nitrogen-containing value-added products is a significant and dynamic area of study, particularly considering both their widespread use as feedstocks due to large-scale synthesis in industrial processes and that a majority of homogeneous catalytic processes involves these molecules.1 In this context, the direct hydroamination of non-activated alkenes with NH3 has emerged as one of the ‘holy grails’ in catalysis.2 Whilst transition metal complexes have traditionally been the focus of small molecule activation and catalysis, they face challenges in activating the N–H bond of ammonia, which has a high gas-phase bond dissociation energy of over 99 kcal mol−1, and tend to form robust Werner type complexes.3 In this context, the selective formation of monomeric transition metal amido hydride complexes through the oxidative addition of NH3 has only been accomplished using two iridium-based pincer derivatives.4 Main group species are increasingly gaining traction in this field, following the groundbreaking discoveries of NH3 activation by cyclic (alkyl)(amino)carbenes (Scheme 1, I) and heavier alkyne analogues (Scheme 1, III).5–7 More recently, advancements in NH3 activation by main group compounds include thermoneutral and reversible ammonia splitting protocols using geometrically constrained phosphines,8 activation via single-electron transfer protocols employing either a carbene-stabilized dithiolene zwitterion or a bismuth(II) complex,9,10 and the pioneering report on catalytic NH3 activation and transfer by an aluminium–carbon-based ambiphile.11 While a considerable number of Group 14-based systems (Si, Ge, Sn) have been developed to activate the N–H bond of ammonia,5,6,12–14 further advancements are required to achieve the catalytic functionalization of unsaturated substrates by these systems. Thus, it is crucial to maintain a delicate balance in transferring the activated small molecule to a substrate while preserving the ability to activate the next small molecule (i.e. to form a catalytic cycle). | ||
| Scheme 1 Top: First reports on ammonia activation by Group 14 compounds I and III; bottom: Oxidative addition and reductive elimination sequence of ammonia by a bis(boryl)stannylene V (Dipp = 2,6-iPr-C6H3; DippTer = 2,6-(2,6-iPr-C6H3)2C6H3). | ||
Given this consideration, heavier low-valent Group 14 compounds such as stannylenes show great promise due to their inherently weak M–E bonds (M = heavier tetrel element; E = H, N) and an accessible SnII/SnIV redox couple. However, only one example of oxidative addition of NH3 by a stannylene (V) has been reported, capitalizing on strongly σ-donating boryl ligands (Scheme 1, V).14 Decomposition from the SnIV intermediate VI to N-borylated products and reduced tin clusters indicates that further development in ligand design must be explored to tackle the challenge of instability and aggregation of the corresponding complexes after substrate activation. This is especially true for the heavier tetrel elements, such as tin, for which the low-valent form and, consequently, the +II oxidation state are thermodynamically favoured.
In this study, we introduce a direct synthetic approach for a range of stannylenes, bolstered by ancillary hemilabile15 iminophosphorane ligands via an intermolecular C–H activation pathway. This strategy was designed to streamline activation processes at SnII, meanwhile furnishing a comprehensive mechanistic account of subsequent NH3 activation within these frameworks. Furthermore, we have devised a straightforward protocol for the preparation of a lithium stannylenoid. Initially detected as a by-product during the synthesis of heteroleptic stannylenes featuring pendant iminophosphorane functionalities, this method offers a reliable route to its controlled synthesis.
Results and discussion
Synthesis and characterisation of heteroleptic stannylenes with pendant iminophosphorane functionalities
Reactions of the heteroleptic terphenyl-/amido stannylene MesTerSn{N(SiMe3)2} (MesTer = 2,6-(2,4,6-CH3-C6H2)2C6H3) (1) with freshly prepared imino-phosphoranes Me3PNR (R = 2,6-iPrC6H3 (Dipp) (2a), R = 3,5-CH3C6H3 (Xyl) (2b), R = 1-adamantyl (Ad) (2c))16 in C6D6 at room temperature lead to the formation of bis(trimethylsilyl)amine (HN(SiMe3)2), as evident from its characteristic 1H NMR chemical shift (δ1H = 0.10 ppm), and overall clean formation of the base-stabilized stannylenes MesTerSnCH2PMe2NR (3a–c), each of which featuring a four-membered Sn,C,P,N heterocycle (Scheme 2A).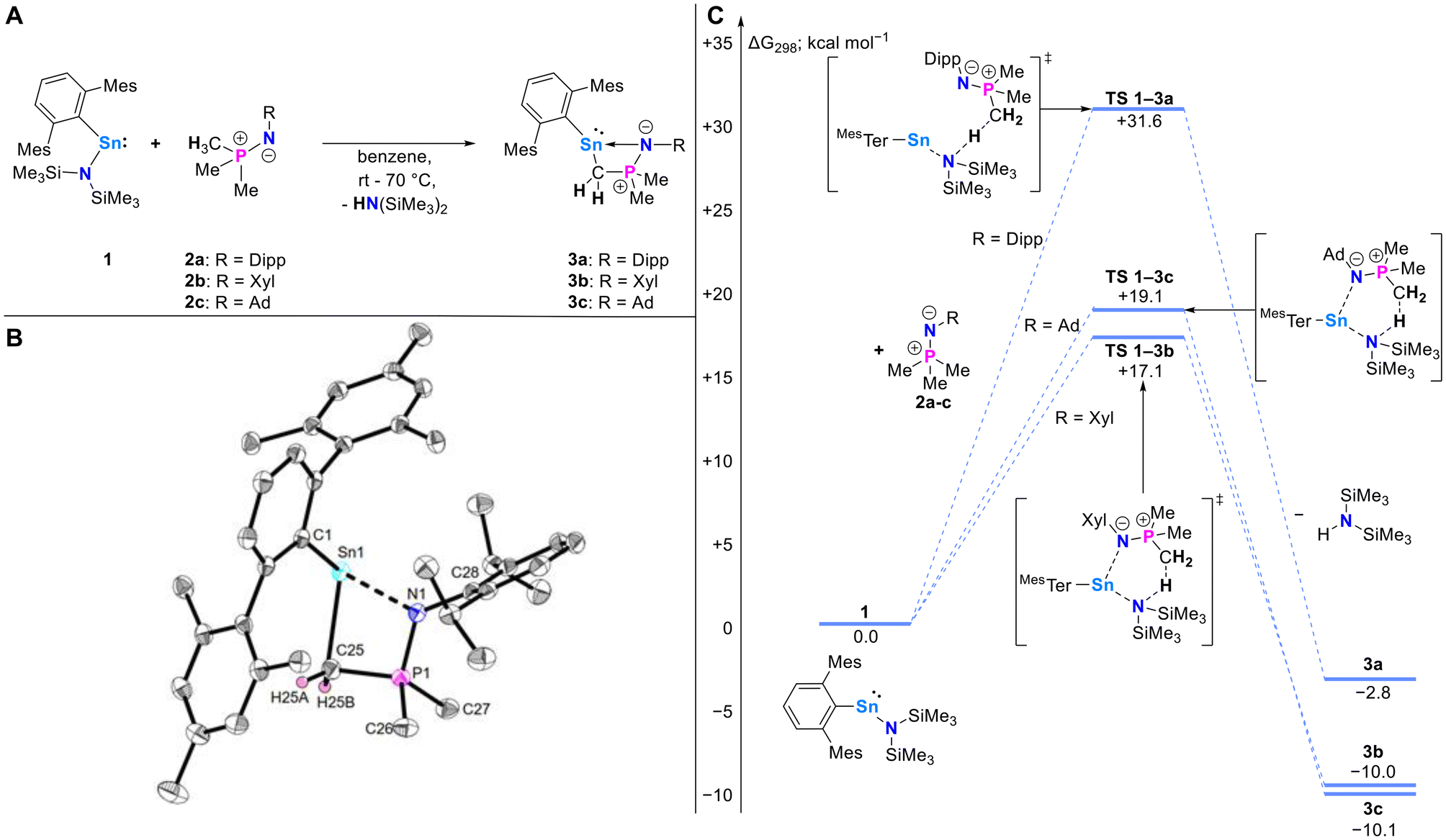 | ||
| Scheme 2 (A) Reactivity of stannylene 1 towards iminophosphoranes 2a–c to give stannylenes 3a–c. (B) Molecular structure of MesTerSnCH2PMe2NDipp (3a) determined by single crystal X-ray crystallography. Anisotropic displacement parameters are drawn at the 50% probability level (hydrogen atoms (except for H25A and H25B) and lattice solvent have been omitted for clarity). Selected bond lengths (Å) and angles (°): Sn1⋯N1 2.2877(14), Sn1–C1 2.2527(16), Sn1–C25 2.3220(17), P1–N1 1.6143(13), P1–C25 1.7503(17), P1–C26 1.8001(17), P1–C27 1.8050(17), N1–C28 1.426(2), C1–Sn1–C25 106.05(6), P1–C25–Sn1 91.90(7), C25–P1–N1 100.92(7), P1–N1–C28 127.42(11). (C) Computed mechanism for the formation of 3a–c from 1 and 2a–c (BP86-D3BJ/Def2-TZVP/Benzene(PCM)). | ||
The formation of 3a–c can be straightforwardly monitored by 31P NMR spectroscopy, which shows the formation of new singlet signals with tin satellites. These signals are shifted to lower-field when compared to the iminophosphoranes 2a–c (for an overview of characteristic NMR data of 2a–c and 3a–c, see Table 1).
| Compound | δ 31P, J119/117Sn,Pc | δ 119Sn, 2JSn,P | δ 1H(PCH2) | δ 1H(P(CH3)n)b |
|---|---|---|---|---|
| a δ values are given in ppm and J values in Hz. The data is given in benzene-d6 as solvent and at room temperature. b n = 3 for 2a–c and n = 2 for 3a–c. c Average value given. d The data is given in toluene-d8 as solvent and at 243 K. | ||||
| 2a | −8.2 | — | — | 0.93 |
| 2b | 1.4 | — | — | 0.95 |
| 2c | −14.5 | — | — | 1.00 |
| 3a | 40.4 | 422.0 | 0.24 | 0.72 |
| J 119/117Sn,P = 163.6 | 2 J Sn,P = 165.9 | |||
| 3a | — | — | 0.14 and 0.28 | 0.58 |
| 3b | 42.1 | 217.6 | 0.39 and 0.47 | 0.63 and 1.00 |
| J 119/117Sn,P = 142.3 | 2 J Sn,P = 145.7 | |||
| 3c | 33.8 | 213.3 | 0.39 and 0.62 | 0.62 and 0.74 |
| J 119/117Sn,P = 137.6 | 2 J Sn,P = 138.6 | |||
Notably, whilst conversion to 3b and 3c is quantitative within hours at room temperature, the formation of 3a is significantly slower. This can be mitigated by performing the reaction at 70 °C, leading to completion within 16 h. Overall, the multinuclear NMR spectroscopic data imply identical structures in solution, however, the Dipp-substituted derivative 3a shows signal broadening at room temperature and the 119Sn signal is low-field shifted, in comparison to 3b and 3c, by over 200 ppm.
The mechanisms for the formation of 3a–c were investigated using Density Functional Theory (DFT) at the BP86-D3BJ/Def2-TZVP/Benzene(PCM) level of theory (Scheme 2C), finding that in all cases the reactions are exergonic (ΔG298K: 3a, −2.8 kcal mol−1; 3b, −10.0 kcal mol−1; 3c −10.1 kcal mol−1; Scheme 2C). Comparison of the transition states (ΔG‡298K: 3a, +31.6 kcal mol−1; 3b, +17.1 kcal mol−1; 3c, +19.1 kcal mol−1) demonstrates the effect of higher steric bulk in 2a, where steric restriction prohibits the formation of a stabilizing Sn–N interaction, resulting in a higher activation energy when compared to 2b and 2c. This correlates with the experimental findings, where heating is required to obtain a similar rate of reaction for the formation of 3a, but not 3b and 3c (vide supra).
To elucidate any fluctional behaviour on the NMR timescale, a variable-temperature (VT) 1H NMR (600 MHz) experiment of compound 3a in toluene-d8 was performed (Fig. S20†), showing that the signals with the best resolution are observed at 243 K (Fig. S23†).17 At that temperature, the same signal pattern as for 3b and 3c is observed (e.g., one signal each for the –CH2PMe2– moiety, three signals for the terphenyl methyl groups etc.). In the temperature range 193–353 K, the respective 31P{1H} and 119Sn{1H} NMR signals shift by Δδ31P{1H} = 4.7 and Δδ119Sn{1H} = 49.2 ppm, respectively. This leads to the assumption that in solution more pronounced dative interactions between the nitrogen functionalities and the tin atoms in 3b,c when compared to 3a are responsible for the high-field shift in the 119Sn{1H} NMR spectra. Notably, compared to true two-coordinate stannylenes, the observed 119Sn{1H} NMR chemical shifts of 3a–c are significantly shifted upfield. For instance, the starting material 1 exhibits a 119Sn{1H} NMR chemical shift of δ119Sn{1H} = 1192.3 ppm (Fig. S83†).17
3a–c were purified by crystallization from aliphatic hydrocarbons and in the case of 3a and 3c, crystalline material suitable for single crystal X-ray diffraction (SCXRD) analysis was obtained, with the molecular structure of 3a being shown in Scheme 2B.17 The dative interaction between Sn1–N1 is suggested by relatively long distances (2.2877(14) Å (3a) and 2.269(2) Å (3c)), both significantly exceeding the respective single bond radii of the respective atoms (2.11 Å) by at least 0.15 Å.18 This dative interaction and the formation of respective four-membered Sn,C,P,N rings is further supported by almost perpendicular Sn1–C25–P1 angles of 91.90(7)° (3a) and 91.86(9)° (3c), respectively. By comparing the structures of the herein obtained free iminophosphoranes 2a,c with 3a,c, the respective nitrogen–phosphorus bond lengths are elongated by approximately 0.06 Å (1.6143(13) Å (3a) and 1.6116(17) Å (3c) vs. 1.5569(17) Å (2a) and 1.558(4) Å (2c)19), indicative of reduced charge transfer from the nitrogen atom to phosphorus but increased to the tin atom.20 The structural data is in good agreement to a previously reported five-membered Sn,C,C,P,N complex which was obtained through the reaction of the respective Sn–P Lewis pair with an organic azide (N–P 1.613(5)/1.608(4) Å; Sn–N 2.305(4)/2.292(4) Å).21
To obtain insights into the bonding within the core of 3a–c, DFT and Natural Bond Orbital (NBO) calculations were performed.
Inspection of the Kohn–Sham frontier molecular orbitals of 3a–c shows major contribution from the tin atomic orbitals, with a lone pair of electrons and orthogonal p-orbital in the HOMO and LUMO respectively (Fig. S84–S86†), which is typical for divalent Group 14 atoms.17 Further analysis of the Lewis structure by NBO 7.022 reveals a lone pair of electrons (s0.88p0.12 hybridised, 1.95 e−) in addition to two covalent, polar Sn–C single bonds around the tin atom (polarised 78% and 83% towards C), with no NBO describing a covalent Sn–N interaction, as expected from the elongated Sn⋯N interatomic distances found in the crystal structures (Fig. S89–S91†).17 Second Order Perturbation Theory analysis revealed a significant donor–acceptor interaction between the Sn and N atoms, with the nitrogen lone pair (LP) datively bonding into a valence lone vacant (LV) orbital at Sn with an energy of 40.7 kcal mol−1. This energy increases to 53.2 and 49.8 kcal mol−1 when the less sterically demanding xylyl or adamantyl substituents are present in 3b and 3c, respectively, aligning well with the observations from 119Sn NMR spectroscopy. In addition, the phosphorus bears four single bonds to each of its surrounding carbon and nitrogen neighbours, in line with the description of a zwitterionic –N–P+ moiety. The drain of N-charge density to the tin atom causes an elongation of the P–N bonds in comparison to the parent iminophosphoranes 2a–c.
Based on both the crystallographic and theoretical insights, it is most appropriate to describe this ligand moiety as zwitterionic, bonding covalently only via the carbon atom.20
Investigations on hemilability of the iminophosphorane-based ligand
To investigate the potential replacement of the nitrogen donor functionality with stronger Lewis bases and experimentally probe the hemilability of the selected ligand framework, we examined the reactivity of 3a–c towards the N-heterocyclic carbene (NHC) 1,3,4,5-tetramethyl-2-imidazol-2-ylidene (IMe4). Accompanied by subtle yet noticeable colour changes in the reaction mixtures, we observed the clean formation of the corresponding NHC-stabilized stannylenes MesTerSn(IMe4)CH2PMe2NR (4a–c) (Scheme 3A). Due to the coordination of the NHC to the tin atoms in 4a–c, the 31P and 119Sn resonances are observed at higher field (e.g., δ31P{1H} = 5.6 ppm; δ119Sn{1H} = −175.1 ppm for 4a; cf.Table 1). | ||
| Scheme 3 (A) Reactivity of 3a–c towards IMe4 to give the NHC-stabilized stannylenes 4a–c. (B) Molecular structure of MesTerSn(IMe4)CH2PMe2NDipp (4a) determined by single crystal X-ray crystallography. Anisotropic displacement parameters are drawn at the 50% probability level (hydrogen atoms, except for H25A and H25B, and second molecule have been omitted for clarity). Selected bond lengths (Å) and angles (°): Sn1⋯N1 3.571(3), Sn1–C1 2.269(3), Sn1–C25 2.263(3), Sn1–C40 2.287(3), N1–P1 1.573(3), P1–C25 1.766(3), C1–Sn1–C25 104.09(12), C1–Sn1–C40 92.65(11), C25–Sn1–C40 90.49(11). | ||
The molecular structure was verified crystallographically in 4a (Scheme 3B), revealing no tin–nitrogen donor–acceptor interaction (Sn1⋯N1 > 3.5 Å). The tin–carbene separation (Sn1–C40 2.287(3) Å) aligns with that observed in other stannylene carbene adducts.16 Consequently, the P1–N1 bond length of 1.573(3) Å in 4a is shorter than that observed in starting material 3a (1.6143(13) Å). The maintained electron density at the nitrogen atom now reinforces the Pδ+–Nδ− bond to almost the value in the free iminophosphorane 2a (1.5569(17) Å).
The electronic structure of the 3-coordinate tin compound 4a was investigated by NBO analysis, revealing a lone pair of electrons (s0.81p0.19 hybridized, 1.93 e−) situated at the SnII atom, in addition to three polar, covalent Sn–C single bonds (Fig. S92†). The IMe4 moiety adopts the imidazolium resonance form, characterized by one bonding (BD) NBO describing an external Sn–C σ-bond. NBO analysis further gives a 3-center-4-electron hyperbond between N97, C93, and N94 (across the NCN interface of the NHC), combining two NBOs (shown In Fig. S92†) in a ratio of 49.3 to 50.7 and a total electron count of 3.86.
In addition, as in the case of 3, the P–N functionality is zwitterionic, with the phosphorus atom forming four single bonds to each of its surrounding bonding partners, characterized by four bonding NBOs.
Ammonia activation with 3a–c
Comparison of the singlet–triplet energy difference and the magnitude of the HOMO–LUMO gap of divalent Group 14 atoms has been shown to be indicative for the tendency of such systems to undergo E–H oxidative addition (E = H, NR2, OR).23 Furthermore, considering the kinetics of E–H oxidative addition, it has been reported that the energy gap between the metallylene singlet ground state and the triplet excited state is inversely correlated with reactivity.23 Additionally, σ-donating substituents based on electropositive elements, in relation to the heavier tetrel elements, such as carbon, silicon, and boron have been demonstrated to reduce the HOMO–LUMO gap through destabilization of the HOMO.6,14,23,24 The boryl-substituted stannylene V and its NH3 adduct reported by Aldridge et al.,14 which enables the N–H oxidative addition of ammonia, has a DFT calculated singlet–triplet difference and HOMO–LUMO gap of −11.45 kcal mol−1 and −1.12 eV respectively (recalculated at the BP86-D3BJ/def2-TZVP level of theory to align with our methodology). 3a–c, on the other hand, bear carbon-based ligands, and possess comparatively large singlet–triplet energy differences and HOMO–LUMO gaps (3a: −35.0 kcal mol−1, +2.67 eV; 3b: −34.0 kcal mol−1, +2.98 eV; 3c: −41.7 kcal mol−1, +2.97 eV) (Fig. S84–S86†).17 This renders the oxidative addition of E–H-containing substrates such as ammonia a challenging task with these systems.We hypothesized however, that the pendant, hemilabile, iminophosphorane-based ligand might facilitate ammonia activation by combining a hydrogen bonding Nammonia–H⋯NLigand interaction and a donor–acceptor Nammonia → Sn interaction to form a six-membered transition state. This would thus allow for the oxidation-state-neutral NH3 activation with respect to the tin ambiphile.
Accordingly, 3a–c were reacted with 1.2 bar NH3 in C6D6 at room temperature (Scheme 4A). To our delight, clean reactions were observed by immediately monitoring the reaction progress using 1H and 31P NMR spectroscopy. Interestingly, these reactions varied depending on the substitution pattern at nitrogen. The Ad-substituted derivative 3c reacts cleanly within 30 minutes, and the formation of the free iminophosphorane 2c was observed by its characteristic 31P NMR chemical shift at δ31P = −14.5 ppm, with no detectable intermediates (Fig. S71†).17 In contrast, the reaction of the Xyl-substituted derivative 3b results in the consumption of the starting material and the observed signal broadening of the 31P NMR signal corresponding to 3b over the course of 24 hours at room temperature, leading to the formation of iminophosphorane 2b (Fig. S69†).17 The Dipp-substituted derivative 3a, under the same reaction conditions, exhibits a broad 31P resonance at δ31P = 16.3 ppm in addition to the observed 31P NMR signal resembling the free iminophosphorane 2a, which gradually decreased over approximately 16 hours to yield 2a as the sole P-containing species (Fig. S70†).17 In all cases, the tin containing product was identified to be the literature-known NH2 bridged dimer [MesTerSn(μ-NH2)]2 (5) as verified by SCXRD and multinuclear NMR spectroscopy (Scheme 4A and Fig. S73–S75†).17,25
 | ||
| Scheme 4 Mechanistic investigation of ammonia activation at complexes 3a and 3b. (A) Reactivity of 3a,b towards ammonia to give the bridged tin amide [MesTerSn(μ-NH2)]2 (5) and free iminophosphoranes 2a,b. (B) Calculated mechanisms (BP86-D3BJ/def2-TZVP) for the formation of 5 from 3a (red) or 3b (blue); An energy difference of 7.2 kcal mol−1 is an effect of the different Ar group used. | ||
To test our initial hypothesis and gain insight into the mechanism for the N–H activation of ammonia by aryl-substituted 3a and 3b, DFT investigations were conducted (Scheme 4B). Contrary to their formation, they were found to be almost identical. Two barrierless pathways were found for the approach of NH3 towards Sn, resulting in two different encounter complexes as possible intermediates (A). Whilst the intermediate A2, which features an intramolecular hydrogen bond, was found to be the energetically most favourable of the two (ΔG298: −7.5 kcal mol−1; +1.1 kcal mol−1), no pathway towards a lower energy activation product was found, and so it is most likely in equilibrium with the starting materials. This reflects the experimental data, where an intermediate with 1H and 31P NMR signals consistent with an ammonia complex is initially observed for 3a + NH3, whereas in the case of 3b + NH3, only some signal broadening can be seen.
Following the barrierless formation of encounter complex A1 (ΔG298: +0.6 kcal mol−1; +7.2 kcal mol−1) however, the ammonia N–H can then undergo metathesis across the Sn–C bond (ΔG‡298 +14.4 kcal mol−1; +19.4 kcal mol−1), in a similar concerted transition state to that in the formation of 3b (Scheme 2C) and its cooperative nature is also reminiscent of that found recently for the N–H metathesis of ammonia across a P–N bond.26
This produces the base-stabilized terphenyl SnII amido intermediate B (ΔG298: −3.0 kcal mol−1; +5.1 kcal mol−1) which undergoes a barrierless disassociation of 2, to form intermediate C (ΔG298: +1.8 kcal mol−1; +9.0 kcal mol−1), which finally dimerises to the crystallographically and NMR spectroscopically characterized 5, thus providing the thermodynamic driving force for the overall reaction (ΔG298 −13.3 kcal mol−1; −6.1 kcal mol−1). Due to the observed formation of an intermediate during the reaction of 3a with NH3 and to evidence our hypothesis of ammonia–adduct formation, a sample of 3a in toluene-d8 was reacted with 1.2 bar of NH3 whilst keeping the sample at low temperature (−80 °C) for its in situ characterization by multinuclear NMR spectroscopy.17 Indeed, the corresponding ammonia adduct MesTerSn(NH3)CH2PMe2NDipp (A2a) was successfully identified, and the observed signal pattern perfectly matched those of the stannylene–carbene adducts 4a–c reported in this study (Scheme 5).17 It is noteworthy that 4a–c did not react with ammonia even under harsher conditions (2 bar NH3, prolonged heating up to 100 °C), likely due to the strong coordination of the NHC to the tin atom, persistently blocking Lewis base coordination of NH3.
 | ||
| Scheme 5 A: Synthesis of intermediate A2a at −80 °C in toluene-d8; B: Stacked 1H NMR spectra of 3a and A2a at −80 °C in toluene-d8. | ||
Synthesis and characterization of the lithium stannylenoid 7
During the investigations of ammonia activation by 3a, a by-product (7) (up to 10%) was occasionally detected by 31P NMR spectroscopy at a chemical shift of δ31P = 30.5 ppm during its synthesis. We found that this occurred when 3a–c were synthesized from dried mother liquors of 1, rather than crystalline material, presumable due to small amounts of lithium bis(trimethylsilyl)amide (LiN(SiMe3)2) being carried through from its synthesis (Sn{N(SiMe3)2} + MesTerLi) reacting with 2a, generating a reactive intermediate.16To confirm this experimentally, we targeted independent synthetic routes to synthesize 7.
By reacting 2a with equimolar amounts of LiN(SiMe3)2 in benzene, complete consumption of 2a was observed, resulting in the formation of apparently two new species in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio according to 31P NMR spectroscopy: one broad singlet at δ31P = 11.3 ppm and one sharp singlet at δ31P = 25.9 ppm. Through analysis of the multinuclear NMR data and employing ECC-DOSY NMR spectroscopy (with adamantane as the internal standard), we identified the minor component of the product mixture as the lithium salt “LiCH2PMe2NDipp” (6).17,27,28 Despite its heterogeneity, this mixture could still serve as a readily accessible source of “LiCH2PMe2NDipp” (6) for the synthesis of compound 7.
:1 ratio according to 31P NMR spectroscopy: one broad singlet at δ31P = 11.3 ppm and one sharp singlet at δ31P = 25.9 ppm. Through analysis of the multinuclear NMR data and employing ECC-DOSY NMR spectroscopy (with adamantane as the internal standard), we identified the minor component of the product mixture as the lithium salt “LiCH2PMe2NDipp” (6).17,27,28 Despite its heterogeneity, this mixture could still serve as a readily accessible source of “LiCH2PMe2NDipp” (6) for the synthesis of compound 7.
By either reacting 1 with two equivalents of 6 or reacting 3a with one equivalent of the 6, clean conversions towards the species with the phosphorus resonance at δ31P = 30.5 ppm are observed (see Scheme 6A).
 | ||
| Scheme 6 (A) Synthesis of lithium stannylenoid 7. (B) Molecular structure of MesTerSn(CH2PMe2NDipp)2Li (7) in the crystal. Anisotropic displacement parameters are drawn at the 50% probability level (hydrogen atoms, except for H25A, H25B, H28A and H28B, have been omitted for clarity). Selected bond lengths (Å) and angles (°): Sn1–C1 2.242(3), Sn1–C25 2.293(3), Sn1–C28 2.288(3), N1–P1 1.598(2), N2–P2 1.596(2), Sn1⋯Li1 2.913(5), Li1–N1 1.921(5), Li1–N2 1.924(6), P1–C25 1.766(3), P2–C28 1.773(3), P1–C26 1.822(3), P1–C27 1.800(3), P2–C29 1.807(3), P2–C30 1.804(3), N1–C31 1.429(4), N2–C43 1.428(3), C1–Sn1–C25 106.51(10), C1–Sn1–C28 100.13(10), C25–Sn1–C28 91.72(11), N1–P1–C25 108.65(13), N2–P2–C28 109.55(13), N1–Li1–N2 147.1(3). | ||
Recrystallization from n-hexane at −30 °C clearly confirms the compound to be the lithium stannylenoid (lithium stannate(II)) [MesTerSn(CH2PMe2NDipp)]2Li (7), which was isolated as a colourless crystalline material (see Scheme 6B).
The molecular structure of 7 reveals the tin atom to be three-coordinate (Σ∢Sn = 298.4(3)°), with all tin–carbon bond lengths (Sn1–C1 2.242(3) Å, Sn1–C25 2.293(3) Å, Sn1–C28 2.288(3) Å) falling within the same range, typical of tin–carbon single bonds (single bond covalent radii: 2.15 Å (ref. 18)), and in good agreement with the molecular structures of 3a,c, and 4a reported herein. NBO analysis found a lone pair of electrons situated at the SnII atom (s0.81p0.19 hybridized, 1.91e−), in addition to three polar, covalent tin–carbon single bonds (Fig. S93†).17 The interatomic distances P1–N1 1.598(2) Å and P2–N2 1.596(2) Å fall between those of 3a and 4a. Accordingly, 7 bears two zwitterionic iminophosphine arms, which have similar electronic structures to those described for 3a (vide supra). The lithium–nitrogen bond lengths (mean value 1.923(6) Å) fall within the same range as observed for other lithium stannylenoids, e.g., in the lithium bis(imino)stannylenoid ClSn(NIPr)2Li (1.946(9) Å and 2.004(9) Å; NIPr = bis(2,6-(diisopropylphenyl)imidazolin-2-imino)).29 A lone pair of electrons on each nitrogen atom directs towards the lithium cation situated in the centre; however, as expected, no significant donor–acceptor interaction was found by Second Order Perturbation Theory analysis. The Sn1⋯Li1 distance of 2.913(5) Å clearly exceeds the sum of covalent radii (2.73 Å (ref. 18)) and accordingly, no bonding interaction between the lithium and the tin atom is detected.30
The 119Sn{1H} NMR spectrum displays a doublet resonance for the trigonal pyramidal coordinated tin atom due to coupling to phosphorus (2JSn,P = 167.0 Hz) at δ119Sn{1H} = 437.9 ppm, placing it in the same range as observed for 3a (Table 1). Additionally, a singlet is observed in the 7Li{1H} NMR spectrum at δ7Li{1H} = 1.3 ppm.
7 joins the ranks of scarce examples of structurally characterized and largely unexplored stannylenoids,31 thus representing a heavier congener of a carbenoid.32 Its synthesis has been proven to be straightforward, starting from the respective stannylenes 1 or 3a, respectively.
Conclusions
In conclusion, we present a comprehensive investigation into the activation of ammonia by heteroleptic stannylenes featuring pendant hemilabile N-donor iminophosphorane functionalities. These stannylenes are accessed via formal C–H activation at a C(sp3)–H moiety adjacent to the phosphorus atom in the employed free iminophosphoranes in reactions with the heteroleptic amido-/terphenyl-stannylene precursor. Our findings reveal that these systems react with ammonia, cleaving one N–H bond in a redox-neutral manner, thus tin maintains in its +II oxidation state throughout the process. Depending on the N-substituent, spectroscopic characterization of an ammonia–stannylene adduct is possible, and the mechanism of ammonia activation is rationalized computationally. Furthermore, we discovered the non-innocence of lithium hexamethyldisilazide, originating from the synthesis of the stannylene starting materials, leading to the development of a straightforward synthetic protocol for a novel lithium stannylenoid.Author contributions
D. M. J. K. and M. F. carried out the experimental work. N. G., R. H.-I., and D. S. were responsible for the SCXRD experiments. O. P. E. T. carried out the quantum chemical calculations and wrote the initial draft of the computational part of the manuscript. M. F. was responsible for the conceptualization, supervision of the experimental investigations, and wrote the initial draft of the manuscript. All authors contributed to the finalization of the manuscript and agreed to the submitted content.Data availability
The data that support the findings of this study are available in the ESI† of this manuscript or by contacting the authors. Included are synthetic and characterizing data, representative spectra, details of the X-ray crystal structures and quantum chemical calculations. X-ray data is further available through the CCDC. The authors have cited additional references within the ESI.†33–49Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Financial support (VCI Liebig fellowship for M.F., Alexander von Humboldt post-doctoral fellowship for O.P.E.T.) is gratefully acknowledged. M. F. further wishes to acknowledge the Georg-August-Universität Göttingen for financial support and Prof. Inke Siewert for her continuous support and guidance. The research group led by Prof. Sven Schneider, with special mention to Marc Neben, Felix Fuchs and Sven Rosendahl, is warmly acknowledged for granting access to their ammonia line and for their assistance in its utilization. We thank Franziska Rüttger (research group led by Prof. Dietmar Stalke) for assistance with the interpretation of the DOSY NMR data.References
- J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito, CA, 2010 Search PubMed.
- S. Streiff and F. Jérôme, Hydroamination of non-activated alkenes with ammonia: a holy grail in catalysis, Chem. Soc. Rev., 2021, 50, 1512–1521 RSC.
- M. J. Bezdek, S. Guo and P. J. Chirik, Coordination-induced weakening of ammonia, water, and hydrazine X–H bonds in a molybdenum complex, Science, 2016, 354, 730–733 CrossRef CAS.
- (a) J. Zhao, A. S. Goldman and J. F. Hartwig, Oxidative Addition of Ammonia to Form a Stable Monomeric Amido Hydride Complex, Science, 2005, 307, 1080–1082 CrossRef CAS PubMed; (b) E. Morgan, D. F. MacLean, R. McDonald and L. Turculet, Rhodium and Iridium Amido Complexes Supported by Silyl Pincer Ligation: Ammonia N–H Bond Activation by a [PSiP]Ir Complex, J. Am. Chem. Soc., 2009, 131, 14234–14236 CrossRef CAS.
- G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Facile splitting of hydrogen and ammonia by nucleophilic activation at a single carbon center, Science, 2007, 316, 439–441 CrossRef CAS PubMed.
- Y. Peng, B. D. Ellis, X. Wang and P. P. Power, Diarylstannylene Activation of Hydrogen or Ammonia with Arene Elimination, J. Am. Chem. Soc., 2008, 130, 12268–12269 CrossRef CAS PubMed.
- (a) P. P. Power, Main-group elements as transition metals, Nature, 2010, 463, 171–177 CrossRef CAS PubMed; (b) S. Yadav, S. Saha and S. S. Sen, Compounds with Low-Valent p-Block Elements for Small Molecule Activation and Catalysis, ChemCatChem, 2016, 8, 486–501 CrossRef CAS; (c) C. Weetman and S. Inoue, The Road Travelled: After Main-Group Elements as Transition Metals, ChemCatChem, 2018, 10, 4213–4228 CrossRef CAS; (d) L. Greb, F. Ebner, Y. Ginzburg and L. M. Sigmund, Element-Ligand Cooperativity with p-Block Elements, Eur. J. Inorg. Chem., 2020, 3030–3047 CrossRef CAS; (e) J. Abbenseth and J. M. Goicoechea, Recent developments in the chemistry of non-trigonal pnictogen pincer compounds: from bonding to catalysis, Chem. Sci., 2020, 11, 9728–9740 RSC; (f) F. Hanusch, L. Groll and S. Inoue, Recent advances of group 14 dimetallenes and dimetallynes in bond activation and catalysis, Chem. Sci., 2021, 12, 2001–2015 RSC; (g) J. M. Lipshultz, G. Li and A. T. Radosevich, Main Group Redox Catalysis of Organopnictogens: Vertical Periodic Trends and Emerging Opportunities in Group 15, J. Am. Chem. Soc., 2021, 143, 1699–1721 CrossRef CAS; (h) K. Oberdorf and C. Lichtenberg, Small molecule activation by well-defined compounds of heavy p-block elements, Chem. Commun., 2023, 59, 8043–8058 RSC; (i) T. J. Hannah and S. S. Chitnis, Ligand-enforced geometric constraints and associated reactivity in p-block compounds, Chem. Soc. Rev., 2024, 53, 764–792 RSC.
- (a) A. J. King and J. M. Goicoechea, Ligand Centered Reactivity of a Transition Metal Bound Geometrically Constrained Phosphine, Chem. – Eur. J., 2024, 30, e202400624 CrossRef CASFor NH3 activation with phosphorus-based compounds see also: (b) S. M. McCarthy, Y.-C. Lin, D. Devarajan, J. W. Chang, H. P. Yennawar, R. M. Rioux, D. H. Ess and A. T. Radosevich, Intermolecular N–H Oxidative Addition of Ammonia, Alkylamines, and Arylamines to a Planar σ3-Phosphorus Compound via an Entropy-Controlled Electrophilic Mechanism, J. Am. Chem. Soc., 2014, 136, 4640–4650 CrossRef CAS; (c) K. Cui, Y. Li, R. Ganguly, A. Inthirarajah, H. Hirao and R. Kinjo, Metal-Free σ-Bond Metathesis in Ammonia Activation by a Diazadiphosphapentalene, J. Am. Chem. Soc., 2014, 136, 16764–16767 CrossRef PubMed; (d) T. P. Robinson, D. M. De Rosa, S. Aldridge and J. M. Goicoechea, E–H Bond Activation of Ammonia and Water by a Geometrically Constrained Phosphorus(III) Compound, Angew. Chem., Int. Ed., 2015, 54, 13758–13763 ( Angew. Chem. , 2015 , 127 , 13962–13967 ) CrossRef CAS; (e) A. Hinz, A. Schulz and A. Villinger, Metal-Free Activation of Hydrogen, Carbon Dioxide, and Ammonia by the Open-Shell Singlet Biradicaloid [P(μ-NTer)]2, Angew. Chem., Int. Ed., 2016, 55, 12214–12218 ( Angew. Chem. , 2016 , 128 , 12402–12406 ) CrossRef CAS; (f) R. Streubel, A. Schmer, A. W. Kyri and G. Schnakenburg, 1,1′-Bifunctional Aminophosphane Complexes via N–H Bond Insertions of a Li/Cl Phosphinidenoid Complex and First Studies on N/P Mono Functionalizations, Organometallics, 2017, 36, 1488–1495 CrossRef CAS; (g) S. Volodarsky and R. Dobrovetsky, Ambiphilic geometrically constrained phosphenium cation, Chem. Commun., 2018, 54, 6931–6934 RSC; (h) F. Dankert, J.-E. Siewert, P. Gupta, F. Weigend and C. Hering-Junghans, Metal-Free N–H Bond Activation by Phospha-Wittig-Reagents, Angew. Chem., Int. Ed., 2022, 61, e202207064 ( Angew. Chem. , 2022 , 134 , e202207064 ) CrossRef CAS.
- Y. Wang, P. M. Tran, M. E. Lahm, Y. Xie, P. Wie, E. R. Adams, J. N. Glushka, Z. Ren, V. V. Popik, H. F. Schaefer III and G. H. Robinson, Activation of Ammonia by a Carbene-Stabilized Dithiolene Zwitterion, J. Am. Chem. Soc., 2022, 144, 16325–16331 CrossRef CAS PubMed.
- X. Yang, E. J. Reijerse, K. Bhattacharyya, M. Leutzsch, M. Kochius, N. Nöthling, J. Busch, A. Schnegg, A. A. Auer and J. Cornella, Radical Activation of N–H and O–H Bonds at Bismuth(II), J. Am. Chem. Soc., 2022, 144, 16535–16544 CrossRef CAS.
- F. Krämer, J. Paradies, I. Fernández and F. Breher, A crystalline aluminium–carbon-based ambiphile capable of activation and catalytic transfer of ammonia in non-aqueous media, Nat. Chem., 2024, 16, 63–69 CrossRef PubMed.
- Ammonia activation by selected Si compounds: (a) A. Jana, C. Schulzke and H. W. Roesky, Oxidative Addition of Ammonia at a Silicon(II) Center and an Unprecedented Hydrogenation Reaction of Compounds with Low-Valent Group 14 Elements Using Ammonia Borane, J. Am. Chem. Soc., 2009, 131, 4600–4601 CrossRef CAS PubMed; (b) A. Meltzer, S. Inoue, C. Präsang and M. Driess, Steering S–H and N–H Bond Activation by a Stable N-Heterocyclic Silylene: Different Addition of H2S, NH3, and Organoamines on a Silicon(II) Ligand versus Its Si(II)→Ni(CO)3 Complex, J. Am. Chem. Soc., 2010, 132, 3038–3046 CrossRef CAS; (c) D. Wendel, T. Szilvási, D. Henschel, P. J. Altmann, C. Jandl, S. Inoue and B. Rieger, Precise Activation of Ammonia and Carbon Dioixde by an Iminodisilene, Angew. Chem., Int. Ed., 2018, 57, 14575–14579 ( Angew. Chem. , 2018 , 130 , 14783–14787 ) CrossRef CAS PubMed; (d) D. Reiter, P. Frisch, D. Wendel, F. M. Hörmann and S. Inoue, Oxidation reactions of a versatile, two-coordinate, acyclic iminosiloxysilylene, Dalton Trans., 2020, 49, 7060–7068 RSC.
- Ammonia activation by selected Ge and Sn compounds: (a) C. Stanciu, S. S. Hino, M. Stender, A. F. Richards, M. M. Olmstead and P. P. Power, Synthesis and Characterization of Ge(II), Sn(II), and Pb(II) Monoamides with –NH2 Ligands, Inorg. Chem., 2005, 44, 2774–2780 CrossRef CAS; (b) A. Jana, I. Objartel, H. W. Roesky and D. Stalke, Cleavage of a N–H Bond of Ammonia at Room Temperature by a Germylene, Inorg. Chem., 2009, 48, 798–800 CrossRef CAS; (c) W. Wang, S. Inoue, S. Yao and M. Driess, Reactivity of N-Heterocyclic Germylene Toward Ammonia and Water, Reactivity of a N-Heterocyclic Germylene Toward Ammonia and Water, Organometallics, 2011, 30, 6490–6494 CrossRef CAS; (d) M. Usher, A. V. Protchenko, A. Rit, J. Campos, E. L. Kolychev, R. Tirfoin and S. Aldridge, A Systematic Study of Structure and E–H Bond Activation Chemistry by Sterically Encumbered Germylene Complexes, Chem. – Eur. J., 2016, 22, 11685–11698 CrossRef CAS; (e) D. C. H. Do, A. V. Protchenko, M. A. Fuentes, J. Hicks, P. Vasko and S. Aldridge, N–H cleavage vs. Werner complex formation: reactivity of cationic group 14 tetrelenes towards amines, Chem. Commun., 2020, 56, 4684–4687 RSC; (f) A. C. Phung, J. C. Fettinger and P. P. Power, Insertion Reactions of NH3 and H2O with the Ferriogermylenes ArGeFeCp(CO)2 (Ar = ArMe6 (–C6H3-(C6H2-2,4,6-Me3)2) or AriPr4 (–C6H3-(C6H3-2,6-iPr2)2); Cp = η5-C5H5): Structural Isomerism and Polymorphism in a Metallogermylene, Organometallics, 2021, 40, 3472–3479 CrossRef CAS.
- A. V. Protchenko, J. I. Bates, L. M. A. Saleh, M. P. Blake, A. D. Schwarz, E. L. Kolychev, A. L. Thompson, C. Jones, P. Mountford and S. Aldridge, Enabling and Proving Oxidative Addition and Reductive Elimination at a Group 14 Metal Center: Cleavage and Functionalization of E–H Bonds by a Bis(boryl)stannylene, J. Am. Chem. Soc., 2016, 138, 4555–4564 CrossRef CAS.
- Selected review articles on the concept of hemilability: (a) A. Bader and E. Lindner, Coordination chemistry and catalysis with hemilabile oxygen-phosphorus ligands, Coord. Chem. Rev., 1991, 108, 27–110 CrossRef CAS; (b) P. Braunstein and F. Naud, Hemilability of Hybrid Ligands and the Coordination Chemistry of Oxazoline-Based Systems, Angew. Chem., Int. Ed., 2001, 40, 680–699 CrossRef CAS.
- M. Fischer, M. M. D. Roy, L. L. Wales, M. A. Ellwanger, A. Heilmann and S. Aldridge, Structural Snapshots in Reversible Phosphinidene Transfer: Synthetic, Structural, and Reaction Chemistry of a Sn=P Double Bond, J. Am. Chem. Soc., 2022, 144, 8908–8913 CrossRef CAS.
- See the ESI.†.
- (a) P. Pyykkö and M. Atsumi, Molecular Single-Bond Covalent Radii for Elements 1–118, Chem. – Eur. J., 2009, 15, 186–197 CrossRef PubMed; (b) P. Pyykkö and M. Atsumi, Molecular Double-Bond Covalent Radii for Elements Li–E112, Chem. – Eur. J., 2009, 15, 12770–12779 CrossRef.
- The observed P–N bond lengths are in good agreement with other structurally characterized iminophosphoranes, and the following examples show P–N bond lengths from 1.54 Å to 1.60 Å: (a) E. Bohm, K. Dehnicke, J. Beck, W. Hiller, J. Strahle, A. Maurer and D. Fenske, Die Kristallstrukturen von Ph3PNPh, [Ph3PN(H)Ph][AuI2] und von 2,3-Bis(triphenylphosphoranimino)maleinsäure-N-methylimid, Z. Naturforsch., B: J. Chem. Sci., 1988, 43, 138–144 CrossRef; (b) A. Müller, M. Möhlen, B. Neumüller, N. Faza, W. Massa and K. Dehnicke, Die Kristallstrukturen der silylierten Phosphanimine Me3SiNP(c-C6H11)3 und (Me3SiNPPh2)2CH2, Z. Anorg. Allg. Chem., 1999, 625, 1748–1751 CrossRef; (c) B. Neumüller and K. Dehnicke, Phosphanimin- und Phosphaniminato-Komplexe von Beryllium. Kristallstrukturen von [BeCl2(HNPPh3)2], [BeCl(HNPPh3)2(Py)]Cl und [Be3Cl2(NPPh3)4], Z. Anorg. Allg. Chem., 2004, 630, 369–376 CrossRef; (d) K. E. Aldrich and A. L. Odom, A photochemical route to square planar, ruthenium(IV)-bis(imide), Chem. Commun., 2019, 55, 4403–4406 RSC; (e) J. Du, Y. Zhang, Z. Huang, S. Zhou, H. Fang and P. Cui, Heterobimetallic Pd(0) complexes with Pd→Ln (Ln = Sc, Y, Yb, Lu) dative bonds: rare-earth metal-dominated frustrated Lewis pair-like reactivity, Dalton Trans., 2020, 49, 12311–12318 RSC.
- The P–N bond shortening in iminophosphoranes is due to electrostatics and has been thoroughly investigated: (a) M. Bolboaca, T. Stey, A. Murso, D. Stalke and W. Kiefer, P–N Bond Length Alterations Monitored by Infrared Absorption and Fourier Transform Raman Spectroscopy in Combination with Density Functional Theory Calculations, Appl. Spectrosc., 2003, 57, 970–976 CrossRef CAS PubMed; (b) N. Kocher, D. Leusser, A. Murso and D. Stalke, Metal Coordination to the Formal P=N Bond of an Iminophosphorane and Charge-Density Evidence against Hypervalent Phosphorus(V), Chem. – Eur. J., 2004, 10, 3622–3631 CrossRef CAS.
- S. Freitag, K. M. Krebs, J. Henning, J. Hirdler, H. Schubert and L. Wesemann, Stannylene-Based Lewis Pairs, Organometallics, 2013, 32, 6785–6791 CrossRef CAS.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2018 Search PubMed.
- (a) Y. Wang and J. Ma, Silylenes and germylenes: The activation of H–H bond in hydrogen molecule, J. Organomet. Chem., 2009, 694, 2567–2575 CrossRef CAS; (b) Y. Peng, B. D. Ellis, X. Wang and P. P. Power, Diarylstannylene Activation of Hydrogen or Ammonia with Arene Elimination, J. Am. Chem. Soc., 2008, 130, 12268–12269 CrossRef CAS PubMed; (c) A. V. Protchenko, K. H. Birjkumar, D. Dange, A. D. Schwarz, D. Vidovic, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, A Stable Two-Coordinate Acyclic Silylene, J. Am. Chem. Soc., 2012, 134, 6500–6503 CrossRef CAS PubMed.
- D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Stable Carbenes, Chem. Rev., 2000, 100, 39–92 CrossRef CAS.
- Y. Peng, J.-D. Guo, B. D. Ellis, Z. Zhu, J. C. Fettinger, S. Nagase and P. P. Power, Reaction of Hydrogen or Ammonia with Unsaturated Germanium or Tin Molecules under Ambient Conditions: Oxidative Addition versus Arene Elimination, J. Am. Chem. Soc., 2009, 131, 16272–16282 CrossRef CAS.
- J. Abbenseth, O. P. E. Townrow and J. M. Goicoechea, Thermoneutral N–H Bond Activation of Ammonia by a Geometrically Constrained Phosphine, Angew. Chem., Int. Ed., 2021, 60, 23625–23629 ( Angew. Chem. , 2021 , 133 , 23817–23821 ) CrossRef CAS PubMed.
- (a) S. Bachmann, B. Gernert and D. Stalke, Solution structures of alkali metal cyclopentadienides in THF estimated by ECC-DOSY NMR-spectroscopy (incl. Software), Chem. Commun., 2016, 52, 12861–12864 RSC; (b) S. Bachmann, R. Neufeld, M. Dzemski and D. Stalke, New External Calibration Curves (ECCs) for the Estimation of Molecular Weights in Various Common NMR Solvents, Chem. – Eur. J., 2016, 22, 8462–8465 CrossRef CAS PubMed.
- A. Steiner and D. Stalke, Substituent-Controlled Reactions of Iminophosphoranes with Methyllithium, Angew. Chem., Int. Ed. Engl., 1995, 34, 1752–1755 ( Angew. Chem. , 1995 , 107 , 1908–1910 ) CrossRef CAS.
- T. Ochiai, D. Franz, X.-N. Wu, E. Irran and S. Inoue, A Tin Analogue of Carbenoid: Isolation and Reactivity of a Lithium Bis(imidazolin-2-imino)stannylenoid, Angew. Chem., Int. Ed., 2016, 55, 6983–6987 ( Angew. Chem. , 2016 , 128 , 7097–7101 ) CrossRef CAS PubMed.
- D. Reed, D. Stalke and D. S. Wright, Observation of a Direct Sn–Li Bond; The Crystal and Molecular Structure of Monomeric [Ph3SnLi·PMDETA] and the Detection of 119,117Sn–7Li NMR Coupling in Solution, Angew. Chem., Int. Ed. Engl., 1991, 30, 1459–1460 ( Angew. Chem. , 1991 , 103 , 1539–1540 ) CrossRef.
- (a) A. M. Arif, A. H. Cowley and T. M. Elkins, A bulky silyl derivative of tin(II), J. Organomet. Chem., 1987, 325, C11–C13 CrossRef CAS; (b) H. Arp, J. Baumgartner, C. Marschner and T. Müller, A Cyclic Disilylated Stannylene: Synthesis, Dimerization, and Adduct Formation, J. Am. Chem. Soc., 2011, 133, 5632–5635 CrossRef CAS; (c) C. Yan, Z. Li, X. Q. Xiao, N. Wei, Q. Lu and M. Kira, Reversible Stannylenoid Formation from the Corresponding Stannylene and Cesium Fluoride, Angew. Chem., Int. Ed., 2016, 55, 14784–14787 ( Angew. Chem. , 2016 , 128 , 15004–15007 ) CrossRef CAS PubMed; (d) H. Zhao, J. Li, X. Q. Xiao, M. Kira, Z. Li and T. Müller, Cation-Triggered Stannate(II)/Stannylenoid/Stannylene Conversion, Chem. – Eur. J., 2018, 24, 5967–5973 CrossRef CAS PubMed; (e) M. Auer, F. Diab, K. Eichele, H. Schubert and L. Wesemann, Reactivity of organogermanium and organotin trihydrides, Dalton Trans., 2022, 51, 5950–5961 RSC; (f) X.-X. Zhao, S. Fujimori, J. A. Kelly and S. Inoue, Isolation and Reactivity of Stannylenoids Stabilized by Amido/Imino Ligands, Chem. – Eur. J., 2023, 29, e202202712 CrossRef CAS.
- According to IUPAC, carbenoids are defined as “complexed carbene-like entities that display the reactivity characteristics of carbenes, either directly or by acting as sources of carbenes”.
- (a) N. Kuhn and T. Kratz, Synthesis of Imidazol-2-ylidenes by Reduction of Imidazole-2(3H)-thiones, Synthesis, 1993, 561–562 CrossRef CAS; (b) C. Barnett, M. L. Cole and J. B. Harper, Steric Properties of N-Heterocyclic Carbenes affect the Performance of Electronic Probes, Eur. J. Inorg. Chem., 2021, 47, 4954–4958 CrossRef.
- S. K. Chen, W.-Q. Ma, Z.-B. Yan, F.-M. Zhang, S.-H. Wang, Y.-Q. Tu, X.-M. Zhang and J.-M. Tian, Organo-Cation Catalyzed Asymmetric Homo/Heterodialkylation of Bisoxindoles: Construction of Vicinal All-Carbon Quaternary Stereocenters and Total Synthesis of (-)-Chimonanthidine, J. Am. Chem. Soc., 2018, 140, 10099–10103 CrossRef CAS.
- (a) Y. Liu, J. Du and L. Deng, Synthesis, Structure, and Reactivity of Low-Spin Cobalt(II) Imido Complexes [(Me3P)3Co (Nar)], Inorg. Chem., 2017, 56, 8278–8286 CrossRef CAS PubMed; (b) K. E. Aldrich and A. L. Odom, A photochemical route to a square planar, ruthenium(IV)-bis(imide), Chem. Commun., 2019, 55, 4403–4406 RSC.
- Y. M. Badiei, A. Krishnaswamy, M. M. Melzer and T. H. Warren, Transient Terminal Cu-Nitrene Intermediates from Discrete Dicopper Nitrenes, J. Am. Chem. Soc., 2006, 128, 15056–15057 CrossRef CAS PubMed.
- (a) E. Kogut, H. L. Wiencko, L. Zhang, D. E. Cordeau and T. H. Warren, A Terminal Ni(III)-Imide with Diverse Reactivity Pathways, J. Am. Chem. Soc., 2005, 127, 11248–11249 CrossRef CAS PubMed; (b) T. F. C. Cruz, L. F. Veiros and P. T. Gomes, Cobalt(I) Complexes of 5-Aryl-2-iminopyrrolyl Ligands: Synthesis, Spin Isomerism, and Application in Catalytic Hydroboration, Inorg. Chem., 2018, 57, 14671–14685 CrossRef CAS.
- Bruker AXS Inc., Bruker Apex CCD, SAINT v8.40B, Bruker AXS Inst. Inc., Madison, WI, USA, 2019 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS.
- M. Sevvana, M. Ruf, I. Usón, G. M. Sheldrick and R. Herbst-Irmer, Non-merohedral twinning: from minerals to proteins, Acta Crystallogr., Sect. D: Struct. Biol., 2019, 75, 1040–1050 CrossRef CAS.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, ShelXle: a Qt graphical user interface for SHELXL, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, Density-functional approximation for the correlation energy of the inhomogeneous electron gas, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, spectroscopic data, computational and crystallographic details. CCDC 2356897 (2a), 2356898 (2c), 2356899 (3a), 2356900 (3c), 2356901 (4a) and 2356902 (7). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qi02202e |
| This journal is © the Partner Organisations 2024 |