
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Vibrational analysis of auranofin complexes with cysteine and selenocysteine unveils distinct binding motifs and specific unimolecular reactivity†
Roberto
Paciotti‡
*a,
Davide
Corinti‡
 *b,
Cecilia
Coletti
a,
Nazzareno
Re
a,
Giel
Berden
c,
Jos
Oomens
c,
Simonetta
Fornarini
b and
Maria Elisa
Crestoni
b
*b,
Cecilia
Coletti
a,
Nazzareno
Re
a,
Giel
Berden
c,
Jos
Oomens
c,
Simonetta
Fornarini
b and
Maria Elisa
Crestoni
b
aDipartimento di Farmacia, Università G. D'Annunzio Chieti-Pescara, Via dei Vestini 31, Chieti I-66100, Italy. E-mail: r.paciotti@unich.it
bDipartimento di Chimica e Tecnologie del Farmaco, Università di Roma “La Sapienza”, I-00185 Roma, Italy. E-mail: davide.corinti@uniroma1.it
cRadboud University, Institute for Molecules and Materials, FELIX Laboratory, Toernooiveld 7, 6525ED Nijmegen, the Netherlands
First published on 23rd October 2024
Abstract
Auranofin (AF) is the most used gold(I)-containing drug, whose mechanism of action involves targeting sulfur and selenium-containing amino acids, including cysteine (Cys) and selenocysteine (Sec). These residues are present in the active sites of crucial proteins including thioredoxin reductase. Despite extensive exploration of AF-protein interactions over the years, experimental data at the molecular level are still limited. In this work, we studied the vibrational and structural features of solvent-free complexes obtained by the reaction of AF with Cys and Sec, [(Et3P)AuCys]+ and [(Et3P)AuSec]+ ions, respectively. Using tandem mass spectrometry and IR ion spectroscopy supported by density functional theory (DFT) calculations, we unveiled markedly different behaviors for the Cys and Sec bound complexes. In particular, our results indicate that, whereas the [(Et3P)AuSec]+ ions are mainly Se-bound, in agreement with the well-known affinity of Se for gold(I), the sampled [(Et3P)AuCys]+ ionic population is composed of both N- and S-bound isomers, with their ratio depending on the dielectric constant of the solvent used in the starting solution. Additionally, we found that the deamination process occurring in the gas-phase during collision-induced dissociation experiments significantly differ between the two complexes. This work's findings contribute to a deeper understanding of AF's reactivity with Cys and Sec-containing protein targets and highlight how the chemical environment may influence target selectivity of gold-containing drugs, which is crucial for their pharmacological activities.
1. Introduction
Gold-containing drugs have a long history spanning from traditional Chinese medicine and classical remedies based on gold amalgams to the entrance into clinical practice of gold(I)-based complexes used as antiarthritics.1 Auranofin (1-Thio-β-D-glucopyranosyltriethylphosphine gold-2,3,4,6-tetraacetate, AF, Scheme 1) represents the most widely used of these drugs, due to its appropriate oral bioavailability and decent risk-benefit ratio.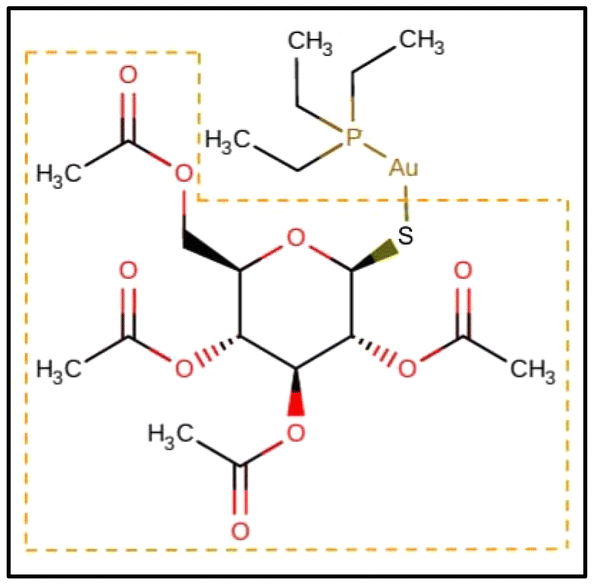 | ||
| Scheme 1 2D structure of auranofin (AF) retrieved from the Drugbank Online (https://go.drugbank.com). The thiosugar moiety, which is replaced by Cys or Sec leading to [(Et3P)AuCys]+ and [(Et3P)AuSec]+ complexes, respectively, is highlighted by the dashed orange box. | ||
Since its approval in 1985, the treatment of rheumatoid arthritis has advanced thanks to novel medicines, whose introduction has led to a decline in AF usage. On the other hand, AF and related gold(I)-containing drugs have lately attracted researchers’ attention for their activity towards other pathological targets, including different types of tumors, and parasitic, bacterial, and viral infections.2–11 Specifically, the possibilities offered by the repurposing of AF for gold-based chemotherapy have created new interest in this drug.12–19 Indeed, among the variety of transition elements that populate the periodic table and that have been tested for their potential anticancer activity, primarily platinum(II)-drugs, cisplatin and related complexes, have found their way into clinical therapy for cancer.20,21 Interestingly, platinum(II) and gold(I) complexes present differences in their mechanism of action: while cisplatin and congeners damage the DNA by binding with the nucleobases,21,22 gold complexes present a greater affinity for proteins.2,23 In particular, AF and related complexes favor binding to sulfur and selenium-containing amino acids, such as the cysteine (Cys) and selenocysteine (Sec) residues contained in proteins like albumin, the proteasome system, the NF-κB protein complex and thioredoxin reductase (TrxR).14,23–27 Among them, TrxR is believed to be the main target in AF's anticancer activity. Au(I) binds to the redox-active site of TrxR, finally causing oxidative stress that potentially leads to cellular death.13 For this reason, it is anticipated that auranofin and its derivatives may produce synergistic effects with current metal-based anticancer therapies.17,18,28,29
It is generally recognized that the mechanism of action of AF is based on the interaction of its cationic part, the [(Et3P)Au]+ ion, with the target nucleophile. Indeed, replacement of the thiosugar moiety with different ligands has not produced any significant alteration in the anticancer activity of the [(Et3P)Au]+ portion, suggesting the former to act as a carrier.19 Au(I) is highly thiophilic and readily reacts in solution with amino acids containing sulfur. Additionally, it shows a predilection for nucleophiles containing selenium. Indeed, the main protein target of AF, TrxR, features a Sec residue in its active site. Mass spectrometry experiments are particularly apt at identifying metalation sites in proteins,26,30–35 and have confirmed the formation of ionic species in which [(Et3P)Au]+ is bound to Sec and Cys.36 Mechanistic and structural studies on the interaction of AF and related gold(I)-complexes with proteins, peptides and amino acids have been based mainly on theory,25,37,38 but experimentally obtained data for their interaction at the molecular level are largely lacking. In fact, NMR data are only reported for models of biological thiol and selenol (benzenethiol and benzeneselenol, respectively).39
The present work presents a comprehensive characterization of the vibrational and structural features of complexes generated by reaction of AF with Cys and Sec, particularly the [(Et3P)AuCys]+ and [(Et3P)AuSec]+ ions, by using tandem mass spectrometry and IR ion spectroscopy backed by quantum-chemical calculations. This approach has been successfully applied to a variety of biomolecular metal–ligand complexes, including cisplatin,40,41 platinum(IV) complexes with anticancer activity42 and, more recently, dinuclear copper complexes specifically designed to interact with the phosphate groups in the backbone of DNA.43 IR ion spectroscopy, performed by monitoring the IR multiple photon dissociation (IRMPD) process at varying photon energies, allows the recording of the vibrational features of mass-selected ions in a mass spectrometer.44–46 Resulting vibrational signatures can be directly assigned to specific structural motifs, due to the absence of matrix effects on the experimental bands of mass-selected sampled ions.47 Results herein reported are compared with previous theoretical investigations on the reaction mechanism of substitution of the AF thiosugar moiety by Cys and Sec.25 Furthermore, the IRMPD spectra as well as the fragmentation mechanism occurring in the CID conditions are elucidated by density functional theory (DFT) calculations which reveal some interesting chemical reactivity differences between Se and S atoms in gas phase.
2. Experimental section
2.1. Materials
Research-grade auranofin and L-cysteine products were purchased from Merck s.r.l., Milan, Italy and used without further purification. L-Selenocysteine was directly obtained in solution (water/methanol 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) by reduction of L-selenocystine in the presence of dithiothreithol in a 1:4 proportion (solution A); both products were obtained from Merck s.r.l., Milan, Italy. A stock solution of auranofin was mixed with either a cysteine solution or solution A and diluted in water/methanol 1:1 to reach a final concentration of 10−5 M of each analyte.
:1) by reduction of L-selenocystine in the presence of dithiothreithol in a 1:4 proportion (solution A); both products were obtained from Merck s.r.l., Milan, Italy. A stock solution of auranofin was mixed with either a cysteine solution or solution A and diluted in water/methanol 1:1 to reach a final concentration of 10−5 M of each analyte.
2.2. MS and CID experiments
Electrospray ionization mass spectra and CID experiments were performed by a commercial linear ion trap (LTQ-XL, Thermo-Fisher). Samples were directly infused in the electrospray ionization source of the mass spectrometer with a 6 μL min−1 flow rate. ESI conditions were set as follows: sheath gas flow at 5 ml min−1, spray voltage at 5 Kv, capillary temp at 250 °C, capillary voltage at 22 V and tube lens voltage at 50 V. CID experiments were performed in the linear ion-trap through radiofrequency excitation (30 ms) with helium as buffer gas. MS3 experiments were performed on peaks arising from the first dissociation step to verify the fragmentation route.2.3. IRMPD spectroscopy
IR spectroscopy experiments of mass-selected ions were done at the Free Electron Laser for Infrared eXperiments (FELIX) Facility (Nijmegen, The Netherlands)48 employing a commercial 3D quadrupole ion trap mass spectrometer (Bruker Amazon speed ETD), which was modified to allow trapped ions to interact with the FEL light.49 Complexes of interest were mass-selected and irradiated by a single IR macropulse from the FEL to induce frequency-dependent infrared multiple-photon dissociation (IRMPD). Each ∼7 μs-long macropulse consists of a train of ∼5 ps-long micropulses spaced by 1 ns. For the aims of this work, FELIX was operated in two spectral ranges: the structurally diagnostic fingerprint (700 to 1800 cm−1) and the XH (X = C, N, O) stretching (1800 to 3600 cm−1) regions. In the fingerprint range, an energy of 10–75 mJ per macropulse was obtained, whereas in the XH stretching region the energy output was 10–40 mJ per macropulse (bandwidth 0.5% of IR frequency). In both cases, spectroscopic analysis was performed with steps of 5 cm−1 and with six replicate mass spectra averaged at each IR frequency. The spectra were recorded at several levels of laser-pulse energy attenuation to avoid excessive depletion of the parent ions and minimize the possibility of formation of fragment ions with m/z lower than the low-mass cut-off of the MS.50 To visualize the IRMPD spectra, the photofragmentation yield R (R = −ln[IP/(IP + ∑IF)], where IP and IF are the abundances of the precursor ion and of a fragment ion, respectively) was plotted as a function of the photon wavenumber.51 Additionally, a linear correction for the frequency-dependent variation in laser pulse energy was applied to R.522.4. Computational details
Quantum-chemical calculations were performed with two objectives: (1) to identify the most abundant species of [(Et3P)AuCys]+ and [(Et3P)AuSec]+ ions under the experimental conditions by calculating their theoretical vibrational frequencies for comparison with the IRMPD spectra; (2) to elucidate their fragmentation pathways observed in CID experiments.As shown in Fig. 1, in the [(Et3P)AuCys]+ and [(Et3P)AuSec]+ complexes, Cys and Sec can coordinate Au through three different groups, –SH/–SeH, NH2 and the carboxyl function (COOH), leading to the corresponding cSH, cN, cCOOH and uSeH, uN and uCOOH isomer for Cys and Sec, respectively (the first lowercase letters, “c” and “u”, represent the one-letter code of Cys and Sec, respectively).
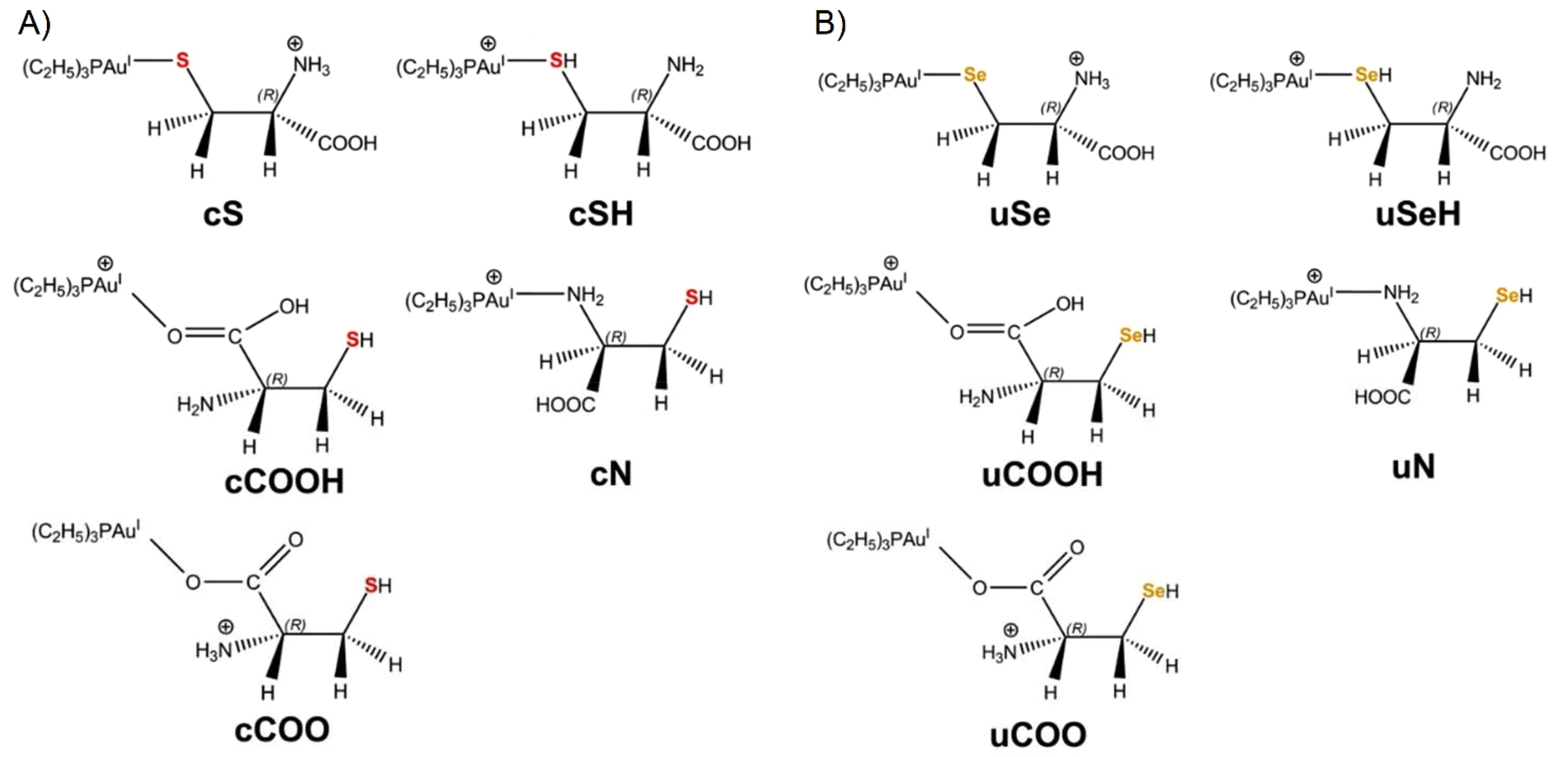 | ||
| Fig. 1 2D structures of (A) [(Et3P)AuCys]+ and (B) [(Et3P)AuSec]+ isomers investigated in this study, along with their corresponding labels (see the text for details). The S and Se atoms are colored in red and orange in [(Et3P)AuCys]+ and [(Et3P)AuSec]+ complexes, respectively. | ||
When the thiol/selenol and the COOH groups directly bind to the metal center, their acidity increases, leading to intramolecular proton transfer to the NH2 group. The corresponding isomers are therefore named as cS, cCOO and uSe, uCOO for Cys and Sec, respectively (Fig. 1).
In order to identify the most stable structures of [(Et3P)AuCys]+ and [(Et3P)AuSec]+ complexes, a conformational search study was performed for each isomer at the DFT tight-binding level of theory (GFN2-xTB method),53,54 using the iMTD-GC algorithm implemented in CREST software.55,56 A clustering analysis was then carried out using the CREGEN procedure available in CREST.
The most representative structures were preliminarily optimized at B3LYP/6-311+G(d,p) level of theory, using the LANL2DZ pseudopotential for Au, computing the vibrational frequencies and thermochemistry parameters (zero-point energy, thermal corrections, and entropies). After a visual inspection, the geometry of the lowest-energy conformers of each isomer were refined at the B3LYP level of theory adopting a basis set referred to as named BS1, consisting of 6-311+G(2df,pd) for H, C, N and O atoms, 6-311+G(3df) for P and S/Se, and the LANL2TZ-f pseudopotential for Au,57 to compute the thermochemistry parameters and the harmonic vibrational frequencies used for the interpretation of experimental IRMPD spectra.
To increase the accuracy of theoretical predictions, we computed the anharmonic spectra of the lowest energy structure of cN and cS isomers (cN-1 and cS-1), using the so-called hybrid approach,58 considering only the active modes falling in the range of 700–4000 cm−1, with the second-order perturbation theory, VPT2.58 Thus, the anharmonic corrections are evaluated at a less computationally demanding level of theory as B3LYP/6-311+G(d,p)/LANL2DZ and applied to harmonic frequencies computed using a larger basis set (B3LYP/BS1):
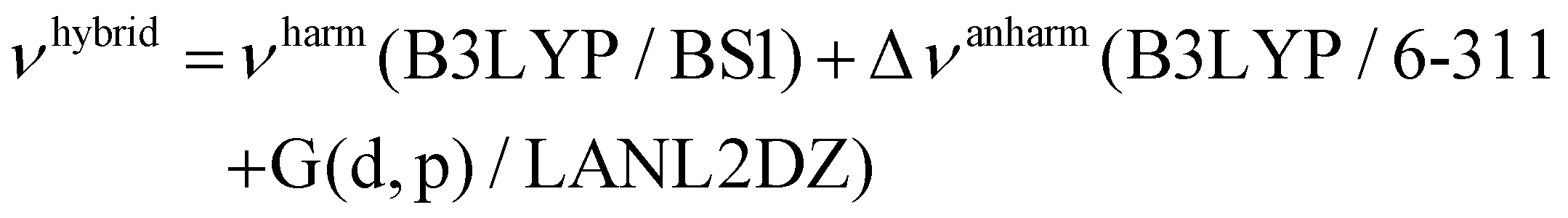 | (1) |
The relative stability of cN-1 and cS-1 in methanol, water and their mixture (50:50) were also performed at the B3LYP/BS1 level of theory using the CPCM59,60 and Onsager61 methods.
To shed light on the fragmentation processes occurring in CID experiments, we investigated the deamination products of uSe and cS, u*Se and c*S, respectively, by performing geometry optimization at the B3LYP/BS1 level of theory, including the calculation of thermochemistry properties and IR vibrational frequencies.
Harmonic frequencies computed at the B3LYP/BS1 level of theory and used for the interpretation of the IRMPD spectra were scaled by 0.985 and 0.955, in the 700–2000 and 2000–3800 cm−1 ranges, respectively. All the calculated spectra were convoluted assuming a Gaussian profile with a full width at half maximum of 15 cm−1 in the 700–2000 cm−1 range and 5 cm−1 in the 2000–3900 cm−1 frequency range.
In order to further characterize the possible dissociation pathway occurring during CID experiments, relaxed potential energy scans were performed at B3LYP/6-311+G(d,p)/LANL2DZ along the Au–Se/S and Cα–NH3+ bond reaction coordinates by using a step of 0.25 Å for a total of 45 steps.
All quantum chemical calculations were performed using the Gaussian 09 package.62
3. Results and discussion
3.1. Mass spectrometry and CID experiments
Samples containing auranofin and either cysteine or the mixed solutions prepared to obtain selenocysteine were mass analyzed in positive ion mode using a linear ion trap. Their mass spectra, reported in Fig. S1 and S2 in the ESI,† respectively, show the presence of complexes in which the thioglucose moiety of auranofin is substituted by the amino acids yielding the molecular formulas [(Et3P)AuCys]+ (m/z 436) and [(Et3P)AuSec]+ (isotopic cluster at m/z 480–486). Indeed, this evidence agrees with previously reported mass spectrometry experiments in which the gas-phase behavior of auranofin was studied as an isolated species in combination with cysteine and methylselenocysteine.36 Together with the complexes of interest, both mass spectra present several mass peaks that can be related to auranofin, in particular [auranofin + H]+ at m/z 679, [(Et3P)2Au]+ at m/z 433 and [(Et3P)Au + auranofin]+ at m/z 993. Additionally, the spectrum of the auranofin and cysteine solution presents a signal at m/z 750 corresponding to [((Et3P)Au)2(Cys–H)]+ and a similar ion appears also in the mass spectrum of the selenocysteine containing solution at m/z 794–800, in agreement with the formula [((Et3P)Au)2(Sec–H)]+.The fragmentation paths of mass-selected [(Et3P)AuCys]+ and [(Et3P)AuSec]+ have been examined by CID and verified by MS3 experiments in the linear ion trap as reported in Fig. S3 and S4,† respectively. Either reactant species yield primary product ions that result from ammonia loss, indicating that both S and Se atoms bind strongly to the gold-containing cation [(Et3P)Au]+. Sequential fragmentation paths result in the dissociation of the X–Au bond (X = S in Cys; Se in Sec) leading to the formation of [(Et3P)Au]+ (m/z 315). In the case of [(Et3P)Au(Sec–NH3)]+ (Fig. S4†), a distinct fragment is observed at m/z 393–395 by elimination of C3H4O2, which still retains the characteristic isotopic pattern of selenium and the [(Et3P)Au]+ ion, thus indicating a stronger Au–Se interaction relative to Au–S. In order to elucidate the effective binding site of both amino acids to [(Et3P)Au]+, the structural and vibrational features of the primary substitution products of auranofin by Cys and Sec, i.e., [(Et3P)AuCys]+ and [(Et3P)AuSec]+, will be investigated and described in the following section.
3.2. IRMPD spectroscopy and structure of [(Et3P)AuSec]+
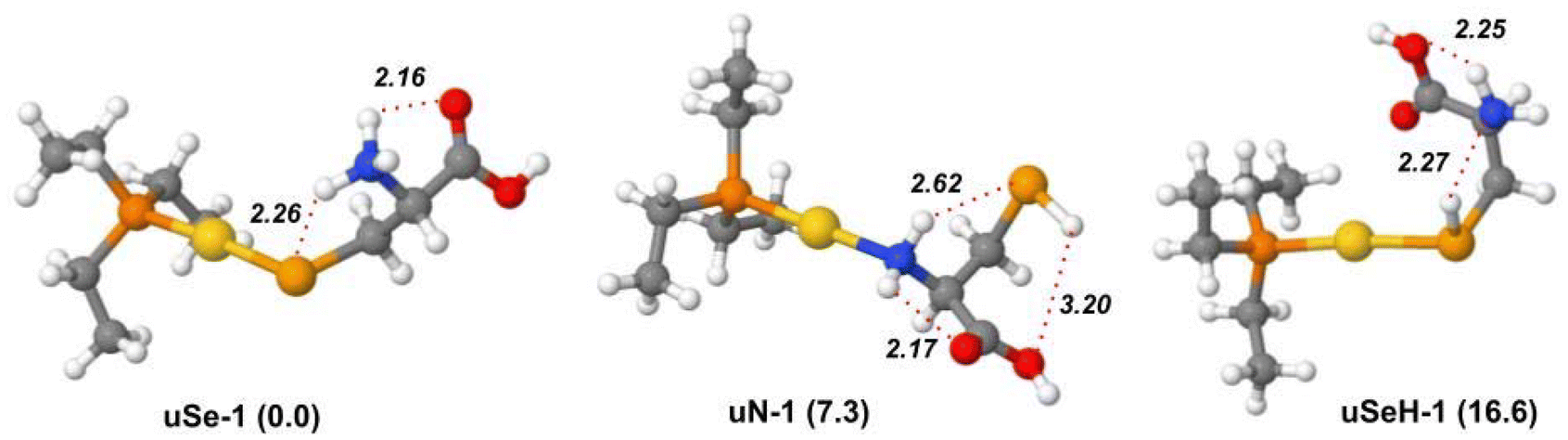
A DFT study at the B3LYP/BS1 level of theory was performed to locate the lowest energy structures of each possible isomer of [(Et3P)AuSec]+, named uSe, uSeH, uN, uCOO and uCOOH, based on the different functional groups of Sec coordinating to the metal center, and to calculate the corresponding IR signature to support the interpretation of the IRMPD spectrum.The lowest energy structure computed for [(Et3P)AuSec]+ is represented by the uSe-1 complex, where the amino group is protonated and the negatively charged Se atom coordinates Au(I), leading to zwitterionic Sec (Se−, NH3+), as shown in Fig. 2.
 | ||
| Fig. 2 Optimized geometries of the most stable isomers and conformers of the [(Et3P)AuSec]+ ion, computed in gas phase at the B3LYP/BS1 level of theory. Free energy values relative to uSe-1 are reported in parenthesis in kJ mol−1. Hydrogen bond distances (Å) are indicated by red dotted lines. | ||
The proton transfer from the selenol moiety of the Sec side chain (pKa = 5.4) to NH2 is facilitated by coordination with Au(I), which enhances the acidity of SeH function. The uSe-1 structure is characterized by a Se–Au bond distance of 2.47 Å and stabilized by two intramolecular H-bonds, established between the NH3+ group and Se and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the carboxylic group, located far from the metal center. This specific H-bond network was also found for Cys involved in the complex with cisplatin.40,41 When the COOH group is instead close to Au(I), maintaining the same H-bond pattern, the relative free energy is raised by 1.3 kJ mol−1 (uSe-2, Fig. S5†).
O of the carboxylic group, located far from the metal center. This specific H-bond network was also found for Cys involved in the complex with cisplatin.40,41 When the COOH group is instead close to Au(I), maintaining the same H-bond pattern, the relative free energy is raised by 1.3 kJ mol−1 (uSe-2, Fig. S5†).
Each ethyl moiety bonded to the P-atom is characterized by two single bonds for which a free rotation is possible leading to a great number of conformers close in energy, where Sec assumes the same binding conformations. For instance, the conformers uSe-3 and uSe-4 differ only by 0.1 kJ mol−1 due to the different conformation of the three ethyl tails (Fig. S5†). The free energy values relative to the global minimum, uSe-1, and the optimized geometries of other low-energy uSe conformers are reported in Table S1 and Fig. S5.†
The lowest-lying structure of the second most stable isomeric family uN, i.e.uN-1, lies at 7.3 kJ mol−1 of relative free energy and presents the Au(I) center coordinated to neutral cysteine at the NH2 functionality. The two hydrogen atoms of the NH2 group are in turn engaged in two different H-bonds with SeH and with CO, while the hydrogen of the selenol moiety is involved in a third H-bond with the oxygen of the carboxylic OH group. The breaking of the latter interaction leads to the conformer uN-2 (Fig. S6†), 4.7 kJ mol−1 higher in energy. Several less stable uN conformers were also identified, characterized by different H-bond patterns or distances, as shown in Fig. S6 and Table S1.†
In the lowest-energy conformer with a uSeH structure, uSeH-1 (16.6 kJ mol−1), the metal center coordinates the undissociated SeH function of neutral Sec, which benefits from a strong H-bond between the hydrogen of the SeH function and the lone pair of the amino group. Moreover, one of the NH2 hydrogen atoms establishes an H-bond with the carboxylic OH group, while the carbonyl oxygen points towards the Au(I) ion. Other higher in energy uSeH conformers, with different H-bonding networks or arrangements of the ethyl tails, are reported in Fig. S7 and Table S1.†
Isomers where the oxygen atom of the carboxylic group coordinates to the Au(I) center were also assessed (Fig. S8†). The most stable member of this family, i.e.uCOO-1 lying at 26.9 kJ mol−1, is characterized by [(Et3P)Au]+ binding with the zwitterionic Sec deprotonated at the carboxylic function. The corresponding isomer, where neutral Sec instead coordinates the Au(I) ion by the COOH group, namely uCOOH-1, is significantly higher in relative free energy (66.9 kJ mol−1). Conformers of both uCOO and uCOOH species lie much higher in energy than uSe-1 (Table S1†), allowing to rule out a significant contribution of these structures to the sampled ion population.
The theoretical IR frequencies were used to support the interpretation of the experimental IRMPD spectra and to identify the species sampled in the gas phase. The IRMPD spectrum of [(Et3P)AuSec]+ is shown in Fig. S9† and superimposed with calculated IR spectra of lowest energy isomers uSe-1, uN-1, and uSeH-1 in Fig. 3. To obtain the experimental spectrum, the ion at m/z 484 was mass-selected and submitted to FEL radiation. The photofragmentation products generated upon resonance agrees with the CID dissociation pathways, as usually observed for the IRMPD process, and include predominantly ammonia loss at m/z 467, but also elimination of C3H4O2 (m/z 395) and formation of [(Et3P)Au]+ (m/z 315). As shown in Fig. 3, the experimental spectrum of [(Et3P)AuSec]+ is well interpreted by the lowest-energy structure, uSe-1, both in the fingerprint (700–1900 cm−1) and X–H stretch (2000–3600 cm−1) ranges. In the first region, the most intense bands at 1775, 1390 (broad band), 1150 and 1090 cm−1 are well described by the vibrational modes computed at 1776, 1419/1406, 1165 and 1108 cm−1, respectively (Table S2†). The weaker bands located in the 765–1010 cm−1 region are also well described by theoretical frequencies computed for uSe-1 (see Table S2†). In the second region, 2000–3600 cm−1, the X–H stretch vibrations match fairly well if one takes into account that the DFT computed strong NH stretching mode at 2669 cm−1 is actually observed as a broad spectral band (2200–2850 cm−1) due to a dynamic H-bond of the NH group with the carbonyl function.63–65 The contribution of other low-energy uSe conformers likely explains the very structured bands in the fingerprint region of the IRMPD spectrum (Fig. S10 and S11†). Indeed, the position of the band found at 1040 cm−1 is reproduced with high accuracy by the theoretical spectrum computed for uSe-1 (1045 cm−1) while its high intensity can be rationalized by the contribution of additional uSe conformers also characterized by an absorption at ∼1045 cm−1.
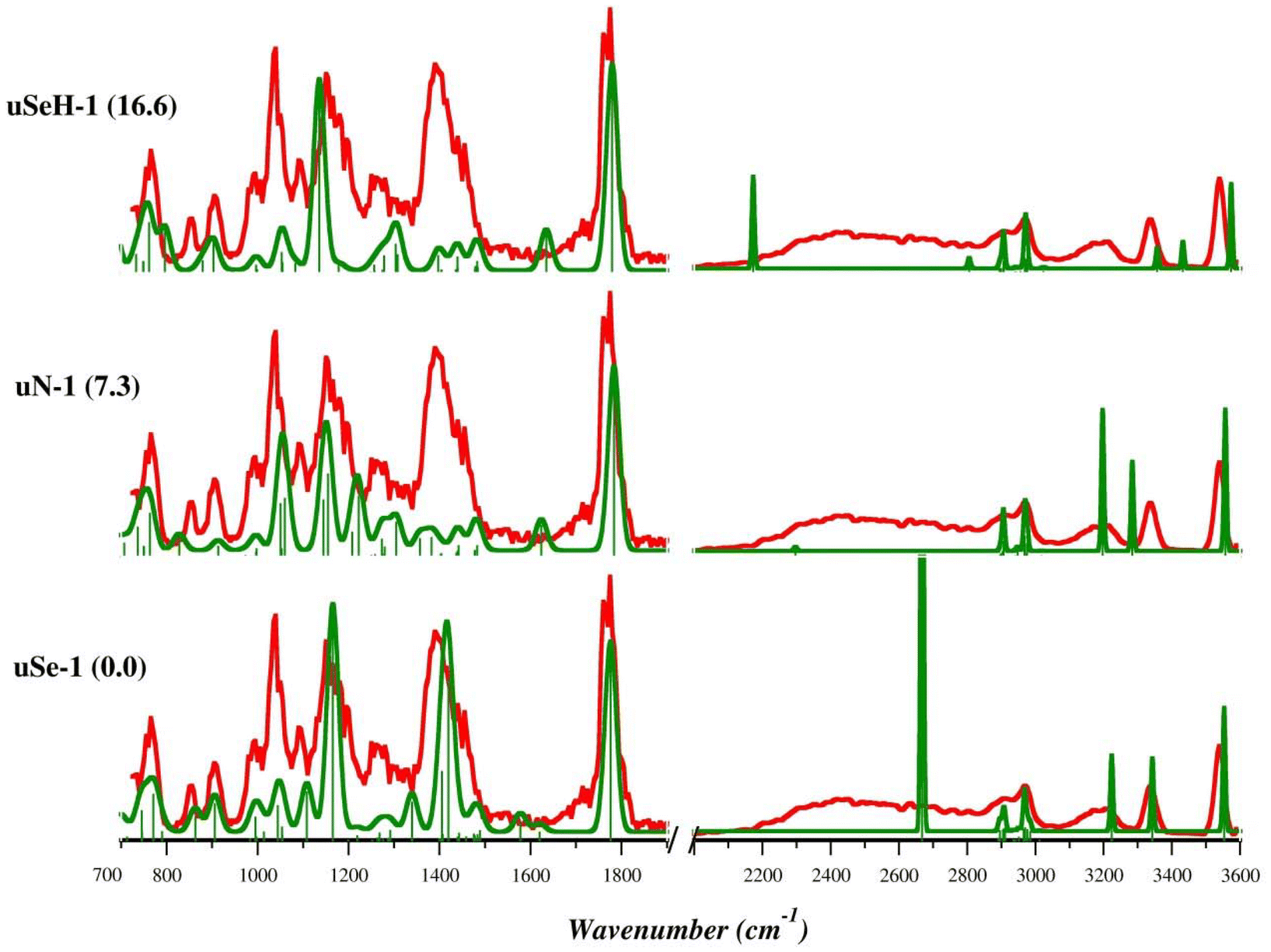 | ||
| Fig. 3 IRMPD spectrum of the [(Et3P)AuSec]+ ion (red profiles) and calculated harmonic IR spectra (green profiles) of uSe-1, uN-1, and uSeH-1, computed in the gas-phase at the B3LYP/BS1 level of theory. Free energy values relative to uSe-1 are reported in parenthesis in kJ mol−1. | ||
Notably, the intense band at 1390 cm−1 associated with the NH3-umbrella mode is critical for structural assignment. Although some important experimental bands find a correspondence in the theoretical spectrum of uN-1 (Table S3†), such as the CO stretching mode (1784 cm−1), NH2 bending modes and OH scissoring (1222, 1155/1145 cm−1) and the bending modes of ethyl tails (763 cm−1), the region between 1370 and 1500 cm−1 is only characterized by weak adsorptions, in contrast with the IRMPD spectrum, due to the absence of the NH3 moiety and therefore of the corresponding umbrella mode.
The X–H stretch region for uN-1 also presents only partial agreement with experimental signature. In particular, the stretching mode of the NH group H-bonded to the carbonyl is computed at 3284 cm−1, fairly red-shifted compared with the corresponding experimental band (3350 cm−1). The comparisons between the theoretical IR spectra of the remaining uN conformers and the IRMPD spectrum are reported in Fig. S12 and S13.† The marked differences between the experimental profile and the computed spectrum for uN-1 allow us to exclude a significant contribution of this isomer to the sampled ion population.
The experimental spectrum is also poorly described by the theoretical absorption bands computed for the uSeH-1 isomers (Fig. S14 and S15, Table S4†) and related conformers and there is no correspondence with the theoretical signatures of uCOO-1 and uCOOH-1 isomers, which are characterized by high relative free energies (Fig. S16†).
In summary, the comparison of theoretical and experimental spectra confirms that the most abundant species in the [(Et3P)AuSec]+ ion population is the zwitterionic uSe isomer (Se−, NH3+), characterized by the presence of a protonated amino group, whose associated umbrella mode gives rise to a highly diagnostic band in the structural assignment.
3.3. IRMPD spectroscopy and structure of [(Et3P)AuCys]+
Geometry optimization and harmonic frequency calculations were performed for the lowest-energy isomers and conformers of [(Et3P)AuCys]+ ion to support the elucidation of its IRMPD spectrum. The relative free energy values of the most stable isomers and conformers are reported in Table S5.†Unlike [(Et3P)AuSec]+, the lowest energy structure is represented by cN-1, characterized by the N-coordination of Au(I). As found in uN-1, the two H-atoms of the NH2 group are involved in two H-bonds with SH and the carbonyl oxygen. As shown in Fig. S17,†cN-2 has the same H bond pattern as uN-1 with the S⋯HN distance shorter than Se⋯HN (2.53 and 2.62 Å, respectively). As found for the corresponding [(Et3P)AuSec]+ complexes, the many conformations of the ethyl tails lead to many conformers close in energy, where Cys assumes the same conformation and the same non-covalent interactions (Fig. S17†).
Despite the high S affinity of the Au(I) ion, the cS isomer, where the zwitterionic Cys is characterized by –NH3+ and S− functions with the latter coordinating the metal center, is 8.3 kJ mol−1 higher in energy than cN-1 isomer. This result indicates that in the gas-phase N-coordination should be preferred to S−-coordination. Notably, cS-1 shows the same H-bond pattern found in uSe-1, where Cys adopts the same conformation as Sec, where the –NH3+ group is involved in two H-bonds with both the CO and S− functions (Fig. 4). This motif is present in the first ten lowest-energy conformers (Fig. S18†), which only differ by the ethyl tail conformation.
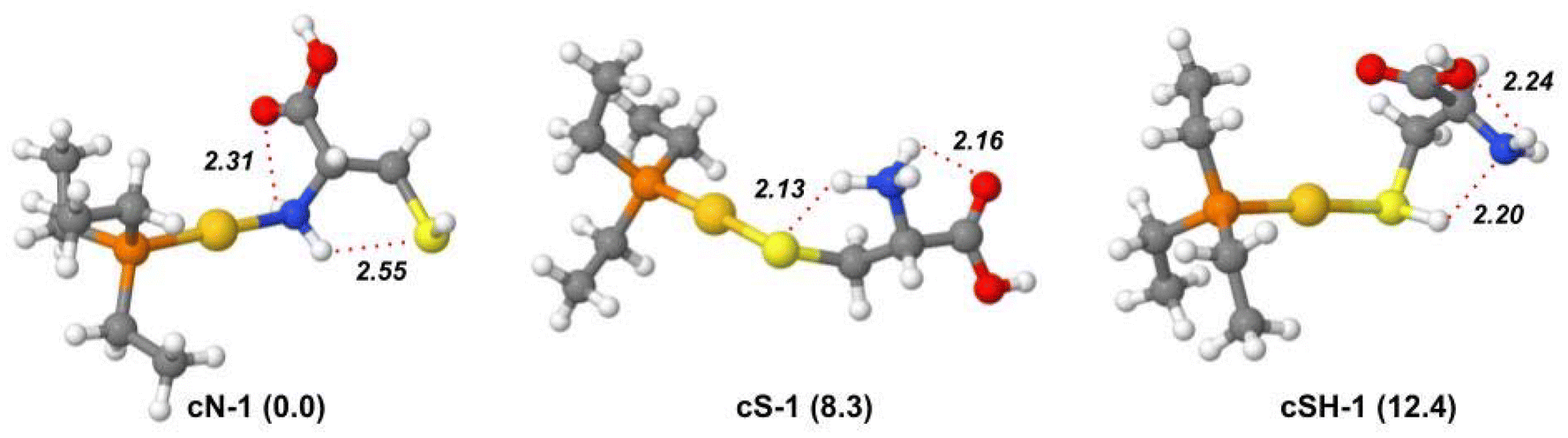 | ||
| Fig. 4 Optimized geometries of the most stable isomers and conformers of the [(Et3P)AuCys]+ ion, computed in gas phase at B3LYP/BS1 level of theory. Free energy values relative to cN-1 are reported in parenthesis in kJ mol−1. Hydrogen bond distances (Å) are indicated by red dotted lines. | ||
The most stable conformer of the cSH isomer is found at 12.4 kJ mol−1. The conformation of Cys as well as the intramolecular H-bonds are qualitatively like those observed in uSeH-1, with a strong H-bond between SH and N and the CO group pointing toward the metal center (Fig. 4). In cSH-2, the NH2 moiety is oriented towards Au(I) in place of CO, raising the relative energy to 14.7 kJ mol−1. Different H-bond interactions and conformations of the ethyl moieties are found in other cSH conformers that are higher in energy (Fig. S19†).
The isomer characterized by the coordination of the metal center by the deprotonated carboxylic function, cCOO-1, is found at 17.9 kJ mol−1, while the corresponding cCOOH-1 isomer, with Au coordinated by the neutral COOH group, is significantly less stable (42.8 kJ mol−1). The lowest energy conformers of cCOO and cCOOH isomers are shown in Fig. S20.†
Theoretical harmonic vibrational frequencies were computed for the most stable structures and used for the elucidation of the experimental IRMPD spectra. The comparison between the experimental IRMPD (orange profile) and theoretical spectra (blue profile) of the three low-lying isomers, cN-1, cS-1 and cSH-1 is shown in Fig. 5. The IRMPD spectrum covering the whole range from 700 to 3700 cm−1 is reported in Fig. S21† and shows no absorptions in the 1900–2700 cm−1 region (not reported in Fig. 5). As in the case of [(Et3P)AuSec]+, IRMPD of the ion at m/z 436, corresponding to [(Et3P)AuCys]+, produces the same fragmentation as observed with CID, i.e. mainly NH3 loss, but also the formation of [(Et3P)Au]+ at m/z 315.
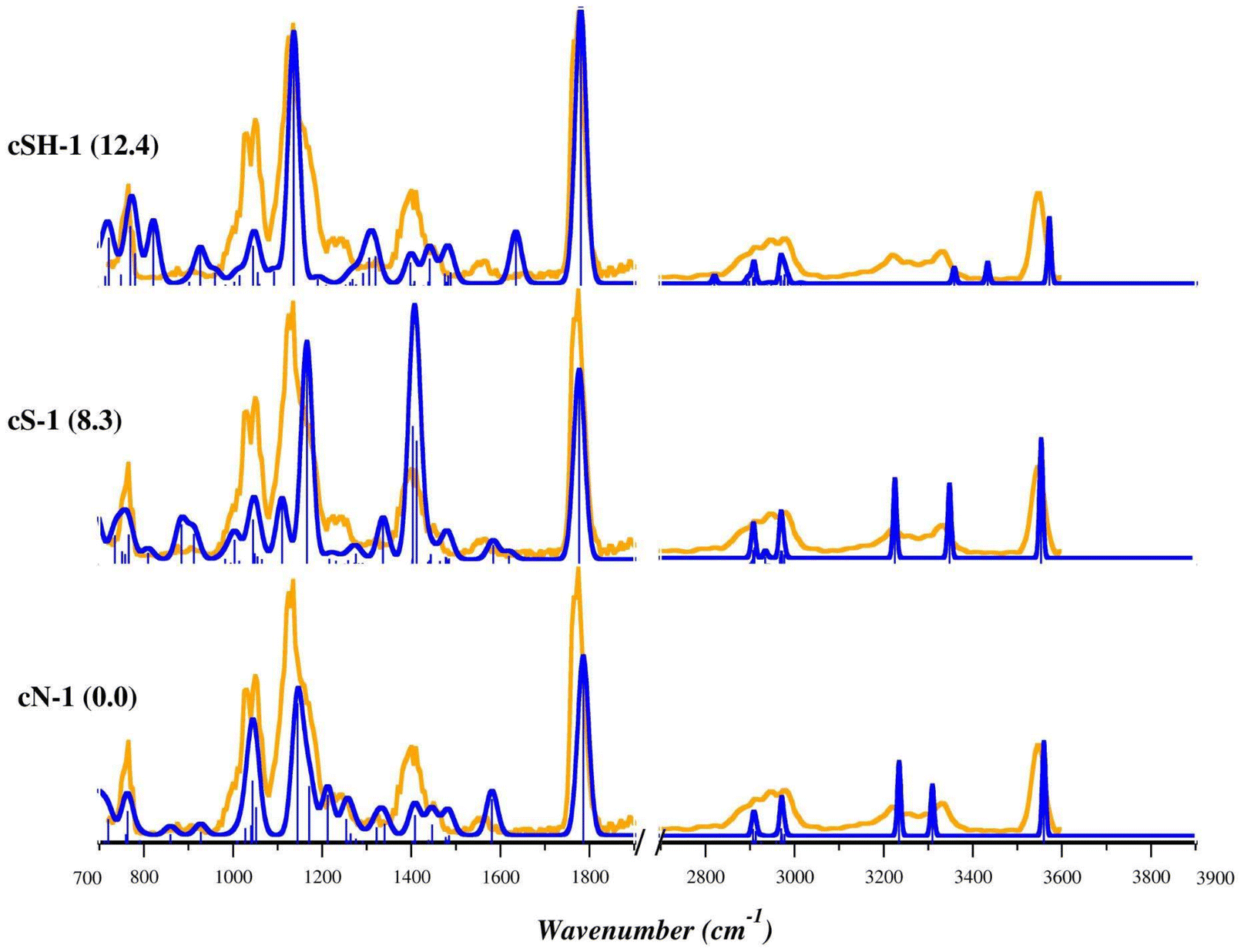 | ||
| Fig. 5 IRMPD spectrum of the [(Et3P)AuCys]+ ion (orange profiles) and calculated harmonic IR spectra of cN-1, cS-1, and cSH-1 (blue profiles) computed in gas phase at B3LYP/BS1 level of theory. Free energy values relative to cN-1 are reported in parenthesis in kJ mol−1. | ||
The 700–1300 cm−1 range as well as the sharp band at 1781 cm−1 of the IRMPD spectrum are well interpreted by the theoretical bands computed for cN-1, the lowest energy structure. In detail, the bands at 773, 1032/1053, 1142 and 1781 cm−1 are predicted at 763, 1044/1052, 1145/1171 and 1786 cm−1, respectively. They can be attributed to rocking of CH2/CH3 of the ethyl tails, stretching of Cα–N of Cys, scissoring of OH coupled with a bending mode of NH2 (wagging and twisting) and to stretching of the CO group, respectively (Table S6†). The X–H stretch region of the experimental spectrum is also well described by theoretical signatures computed for cN-1, with the sole exception of the band at 3340 cm−1 predicted at 3309 cm−1 and associated with antisymmetric stretching of NH2 group. However, the region of the experimental spectrum between 1320 and 1480 cm−1 is characterized by a structured band with a maximum at 1409 cm−1, only weakly present in the cN-1 spectrum for which only absorptions with intensity lower than 50 km mol−1 were computed. As discussed above, this region is typical of the umbrella mode of protonated alpha amino group. Indeed, the theoretical spectrum of cS-1, where Cys is in the zwitterionic state with S− and NH3+, is characterized by two intense bands in that region. They are computed at 1404 and 1412 cm−1 and are attributed to the umbrella mode of NH3+, coupled with the rocking of Cα–H and scissoring of OH function, respectively (Table S7†). As listed in Table S7,† other experimental bands are well described by theoretical frequencies computed for cS-1, especially in the X–H stretch region. However, in the fingerprint region the most intense bands show relative intensities quite different from the IRMPD spectrum, thus excluding an ion population of solely cS-1. The theoretical IR spectra of other stable cN and cS isomers are shown in Fig. S22–S25.†
Summarizing, the comparison of the experimental IRMPD spectra with the theoretical harmonic signatures computed for cN-1 and cS-1 indicates that the experimental envelope can be explained considering the contributions of both isomers to the sampled ion population.
The harmonic theoretical spectra computed for the lowest energy structure presenting canonical Cys bound to Au(I) through the S-atom, cSH-1, (Fig. 5 and Table S8†) and other cSH conformers (Fig. S26 and S27†) show a poor correspondence with the experimental spectrum in both regions of the spectrum. An even worse agreement was found with theoretical signatures computed for cCOO and cCOOH conformers as shown in Fig. S28.† This evidence, together with the computed relative free energy values, allows us to discard the presence of cSH, cCOO and cCOOH isomers in the ionic mixture sampled in the experiment.
To improve the accuracy of the theoretical prediction, we computed the anharmonic corrections at the B3LYP/6-311+G(d,p) level of theory for both cN-1 and cS-1 and then applied them to the harmonic vibrational frequencies computed at the B3LYP/BS1 level of theory. Bending modes of OH and NH2 groups can indeed present a high anharmonic nature.66 Moreover, in cS-1, these vibrations are coupled with the umbrella mode of NH3+, which affects the most intense bands falling in the fingerprint region. Therefore, the theoretical prediction is expected to greatly benefit from including anharmonic corrections. As shown in Fig. 6 and S29,† the anharmonic corrections significantly improve the correspondence with the experimental spectrum for both cN-1 and cS-1, allowing band attribution without applying any scaling factors. Notably, the region of the experimental spectrum between 700 and 1300 cm−1 is well interpreted and described by cN-1 absorptions, while the structured band with a peak at 1409 cm−1 can be clearly attributed to the participation of the cS-1 isomer. Furthermore, the harmonic bands computed for cS-1 at 1404 and 1412 cm−1 are red shifted by the anharmonic corrections to 1349 and 1394 cm−1, respectively. The larger splitting between these two bands leads to a less intense and more structured spectral envelope reproducing more accurately the experimental band at 1409 cm−1. The stretching mode of the CO group is instead well described by both theoretical spectra. The X–H stretch region also agrees with both anharmonic spectra, while the experimental band at 3340 cm−1 may be explained by only considering the presence of cS-1.
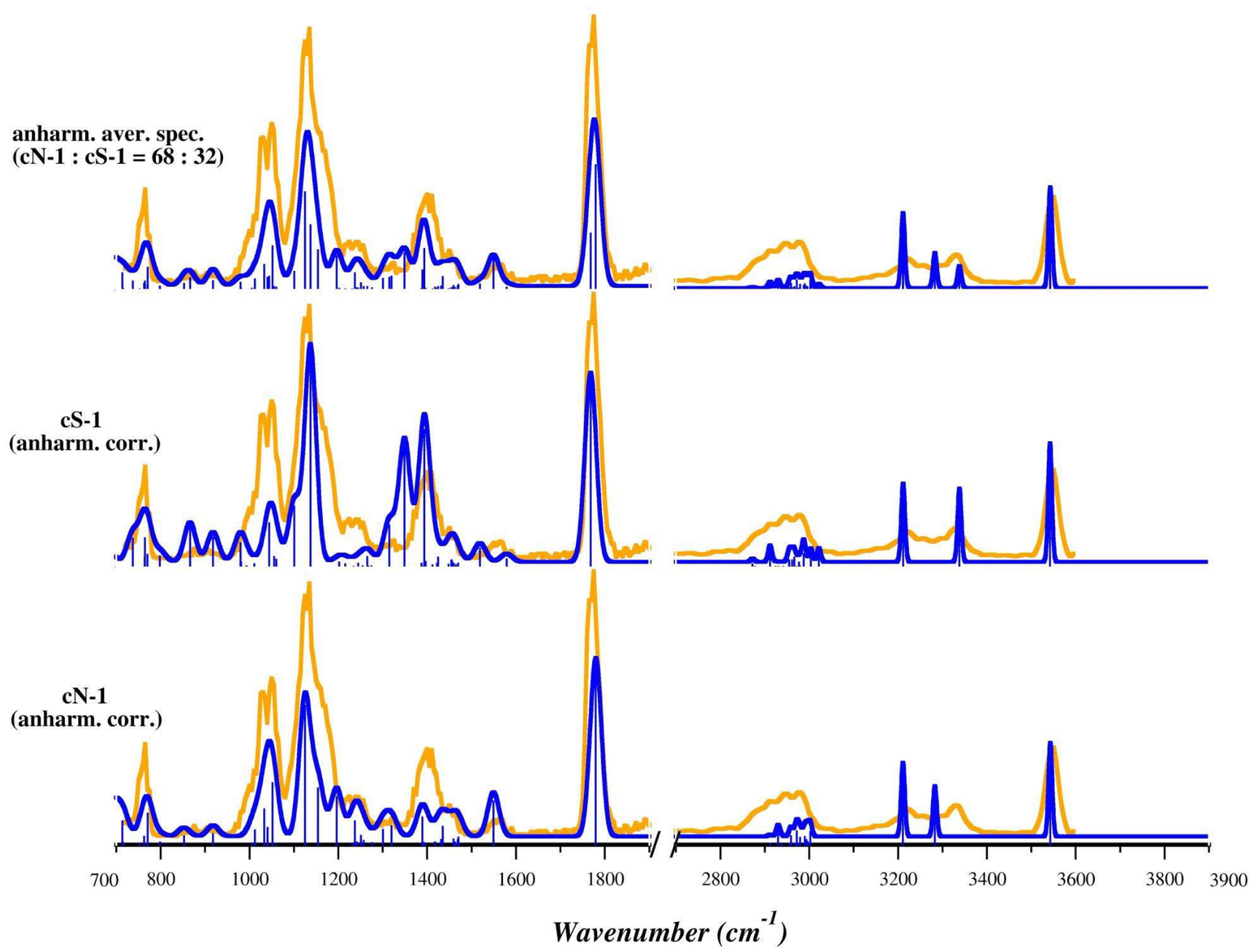 | ||
| Fig. 6 IRMPD spectrum (orange profiles) and calculated anharmonic IR spectra (blue profiles) of the lowest energy isomers of the [(Et3P)AuCys]+ ion, cN-1 and cS-1, computed in gas phase at B3LYP/BS1 level of theory. The anharmonic corrections were computed at B3LYP/6-311+G(d,p) using the LANL2DZ pseudopotential for AuI. The anharmonic averaged spectrum computed by considering contributions from 68% of cN-1 and 32% of cS-1 is shown in the top panel. No scaling factors have been applied. | ||
The comparison of the anharmonic corrected theoretical and the IRMPD spectra suggests that in the experimental conditions both isomers are present in the sampled ion population and therefore both species contribute to the experimental spectrum.
To shed some light on the relative abundance of the sampled cN and cS isomers, we studied the relative stability of cN-1 and cS-1 in the condensed phase, considering water and methanol as solvents (see the Experimental section). It is worth noting that in principle the ESI source, a soft ionization technique,67 transfers to the gas-phase the ionic species already present in solution.68 Thus, the composition of the ionic population in the gas-phase may reflect the relative stability of those species in solutions.
Considering that the starting solution was composed of water and methanol 50:50, we performed geometry optimization calculations at the B3LYP/BS1//CPCM level of theory of cS-1 and cN-1 using methanol and water as solvents to assess their relative stability in the condensed phase. As shown in Table 1, cN-1 is the lowest energy structure in methanol although cS-1 is very close in energy (1.4 kJ mol−1). In contrast, cS-1 is the most stable isomer in water, with cN-1 only 0.7 kJ mol−1 higher in energy. This result confirms that a more polar solvent (water, ε = 78.36) stabilizes the zwitterionic form of the cS-1 isomer more than cN-1. The latter is therefore more stable in less polar solvents such as methanol (ε = 32.61), where the calculated population is closer to that of the gas phase (ε = 1). The different reactivity of Cys with Au(I) determined by the solvent polarity, as suggested by DFT calculations, might have an impact also in the reactivity with biological targets, since the dielectric constant of a protein can vary depending on the considered region: low values (6–7) for the inner partition or hydrophobic regions, moderate values (20–30) for a protein's surface and high local ε for the charged residues.69
| [(Et3P)AuCys]+ isomer | ε = 1 (gas phase) | Relative free energy – Onsager method | ||
|---|---|---|---|---|
| ε = 32.61 (methanol) | ε = 78.36 (water) |
ε = 55.49b (water–methanol mixture 50:50) |
||
|
a Free energy value computed as [G(methanol) + G(water)/2] to estimate the relative free energies in a water–methanol mixture (50:50).
b Hypothetical dielectric constant of a water–methanol mixture (50:50) computed as [ε(methanol) + ε(water)]/2.
|
||||
| cN-1 | 0 | 0.0 | 0.0 | 0.0 |
| cS-1 | 8.3 | 3.5 | 3.2 | 3.3 |
To perform a first evaluation of the relative stability of cN-1 and cS-1 under our experimental conditions (water:methanol = 50:50), we computed their free energies as the average of their absolute free energies in methanol and water (Table 1). As a result, cN-1 appears to be the most stable structure, with cS-1 only 0.4 kJ mol−1 higher in energy. A similar analysis was also carried out by adopting the Onsager method, which allows using a specific value of dielectric constant, although it is a solvation method less accurate than CPCM. Thus, adopting ε = 55.49 (corresponding to average value of dielectric constant of water and methanol), we found that cN-1 is 3.6 kJ mol−1 more stable than cS-1 (Table 1). These results suggest that in a 50:50 methanol–water mixture, the cN and cS isomers might be present in almost the same amount or with a slight excess of cN. Therefore, a similar ratio may be expected in the gas-phase ion population assuming that kinetic trapping of the different isomers occurs during the desolvation process.
Considering a Boltzmann distribution, these relative free energy values correspond to a cN-1:cS-1 population, in a water–methanol mixture (50:50) and at room temperature, of 52%:48% and 68%:32% computed with CPCM and Onsager methods, respectively; a corresponding averaged anharmonic spectrum is shown in Fig. 6 and S29.† Of the two, the anharmonic spectrum for a 68:32 ratio accurately matches the experimental spectrum, not only in the positions of the bands (without frequency scaling), but also in their relative intensities, an additional indication that both isomers, cN and cS, are present in the sampled ion population, with an excess of cN.
Indeed, in real-case scenarios, amino acid residues in proteins will expose to auranofin only nucleophilic sites in their side chains, making the formation of cN-like structures unviable. However, the similar binding preference observed for N and S nucleophiles suggests possible competition for gold(I) binding between side-chain residues with amino functions, such as lysine and arginine, likely in low-polarity regions of proteins where their protonation is inhibited.
3.4. Theoretical interpretation of the CID behavior
As already described in section 3.1, CID studies of [(Et3P)AuCys]+ and [(Et3P)AuSec]+ were performed finding that both species primarily expel NH3. Their product ions then fragment to [(Et3P)Au]+ and to the neutral deaminated derivatives of Cys and Sec, indicating that the breaking of the Au–S/Se bond for both species is energetically more demanding than the loss of NH3. Indeed, NH3 can be released only by those isomers where the NH2 group is not involved in the chelation of the metal center, suggesting that the fragmentation process involves cSe and cS isomers for [(Et3P)AuSec]+ and [(Et3P)AuCys]+, respectively. Theoretical calculations in condensed phase and the comparison between IRMPD spectra of [(Et3P)AuCys]+ with theoretical predictions have however demonstrated that in the sampled ion mixture both cN and cS isomers are present, with an excess of the former. Therefore, an isomerization reaction from cN to cS should be invoked to interpret the CID behavior of the Cys-system.The formation and structural characterization of the deaminated complexes [(Et3P)Au(Sec–NH3)]+ and [(Et3P)Au(Cys–NH3)]+, namely u*Se and c*S, respectively, is discussed hereafter.
As shown in Fig. S30 and S31,† three isomers can be obtained after NH3 loss, named u*Se-n/c*S-n, with n = 1, 2 or 3. In detail, u*Se-1/c*S-1 is characterized by the presence of 3-sulfanylidenepropanoic acid or 3-selanylidenepropanoic acid obtained after NH3 loss and hydride attack from Cβ to Cα leading to a thio/selen-aldehyde (reaction path 1, Fig. S30A†). u*Se-2/c*S-2 can be obtained after a H+ capture by S−/Se− and the consequent formation of a double bond between Cα and Cβ leading to 3-selanylprop-2-enoic acid and 3-sulfanylprop-2-enoic acid for Sec and Cys, respectively (Fig. S30B†). Finally, the third isomer, u-Se-3/c*S-3, results from the nucleophilic attack of Se−/S− to Cα leading to three-member cyclic structures as 1-(selanylcyclopropan-2-yl)methanoic acid and 1-(thiacyclopropan-2-yl)methanoic acid (reaction path 3, Fig. S31†).
To identify the most stable structures of the deaminated isomers, we performed geometry optimizations as reported in Table S9.† Interestingly, we found that the most stable isomers are u*Se-3 for [(Et3P)AuSec*]+ and c*S-1 for [(Et3P)AuCys*]+, suggesting that Se and S exhibit different chemical reactivity when losing NH3. While Se acts as a nucleophilic group attacking directly the Cα carbocation, S facilitates the hydride transfer from Cβ to Cα with formation of a double bond between S and Cβ.
This different reactivity can be explained by considering that selenolate ions are more nucleophilic than thiolates due to the greater polarizability of selenium.70 In addition, one should consider the different energy stability of the π-bonds formed by S and Se with C atom. As is well known, the strength of π-bonds is larger when the involved atoms are first-row elements.71 Heavier elements, such as Se, are characterized by more diffuse p-orbitals leading to a weaker p-overlap, therefore favoring the formation of σ over π bonds.70 One exception is represented by the CS bond for which the energies of σ- and π-bonds are comparable,71 making the formation of both possible.
To shed some light on the reaction mechanism of the NH3 loss for Cys and Sec in the [(Et3P)AuSec]+ and [(Et3P)AuCys]+ ions, we performed relaxed potential energy scans simulating the NH3 detachment by increasing the Cα–NH3 distance by 0.25 Å for 45 steps for both uSe-1 and cS-1.
As shown in Fig. S32 and S33,† the NH3 loss by [(Et3P)AuSec]+ and [(Et3P)AuCys]+ leads to more stable products than the direct breaking of AuI–Se and AuI–S bonds suggesting that the deamination process is the favored reaction path from a thermodynamic point of view. On the contrary, the energy barrier estimated for the NH3 loss starting from uSe-1 is significantly lower than the value computed for cS-1 due to different reaction mechanisms.
It is worth noting that the formation of a 3-member ring structure (Fig. S34†) and the hydride transfer (Fig. S36†) spontaneously occur during the relaxed scan in correspondence of the highest energy step for the deamination process of uSe-1 (step 4, blue profile of Fig. S32†) and cS-1 (step 5, blue profile of Fig. S33†). These results confirm that, according to the reaction mechanism proposed in Fig. 7, u*Se-3 and c*S-1 species should be the most stable isomers after NH3 loss and then undergo AuI–Se/S dissociation in CID conditions.
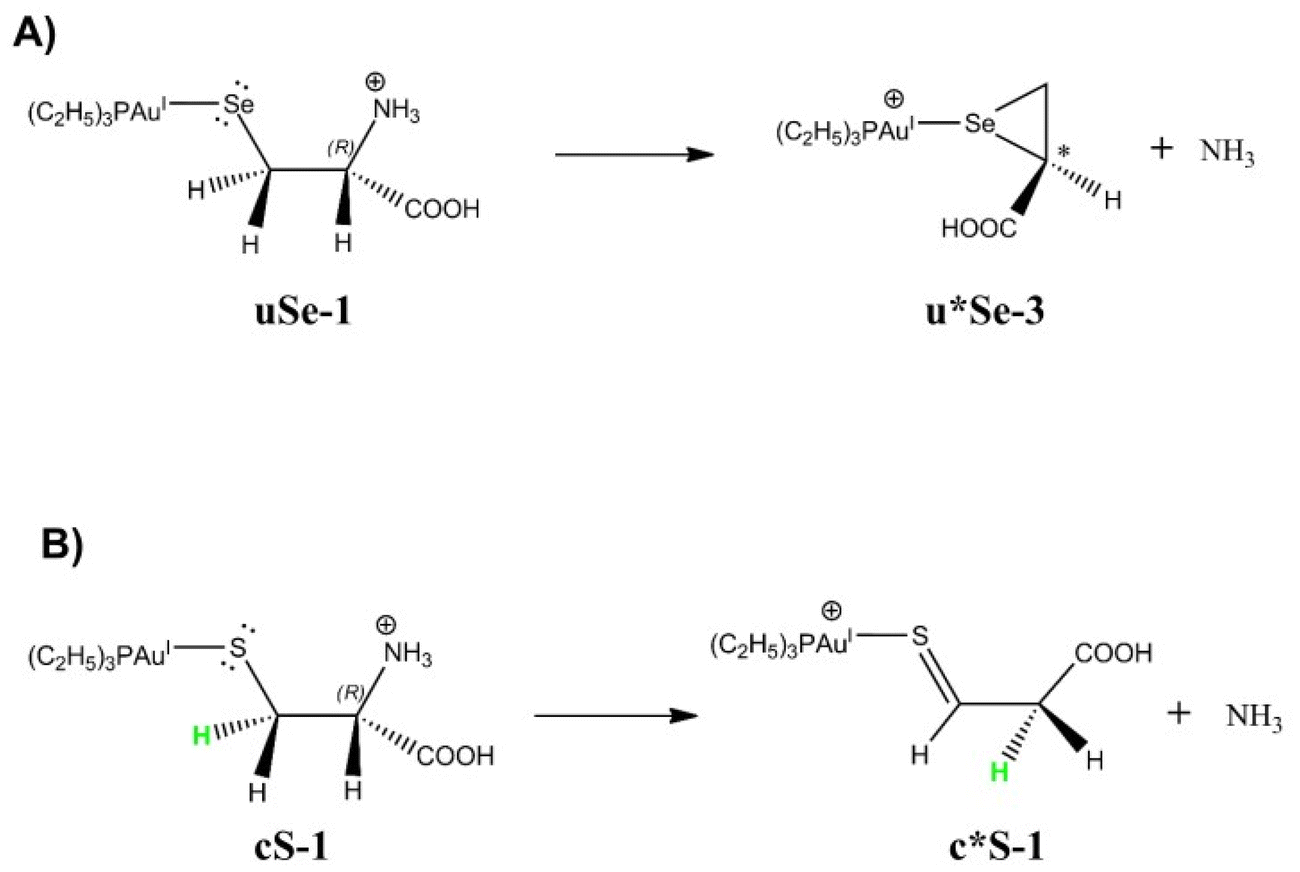 | ||
| Fig. 7 Most probable reaction paths leading to NH3 loss based on DFT calculations starting from (A) uSe-1 and (B) cS-1. The deaminated structures are indicated with an asterisk (*). | ||
In order to confirm this theoretical hypothesis, the IRMPD spectra of isolated deaminated [(Et3P)AuSec*]+ and [(Et3P)AuCys*]+ ions were recorded and compared to the harmonic IR signatures computed at the B3LYP/BS1 level of theory.
As shown in Fig. 8, the theoretical spectra computed for u*Se-3 (Table S10†) reproduce the IRMPD spectrum better than u*Se-1 (Table S11†), suggesting that intermediate species produced by the NH3 loss from uSe-1 might actually be u*Se-3, where Sec* is 1-(selanylcyclopropan-2-yl)methanoic acid. On the contrary, the IRMPD signature of [(Et3P)AuCys*]+ is well described by the theoretical spectrum computed for c*S-1 (Fig. 9, Table S12†), supporting the theoretical prediction that the most stable c*S isomer is represented by that in which Cys* is the 3-sulfanylidenepropanoic acid. The c*S-3 theoretical spectrum is however also in fair agreement with the experimental data (Fig. 9, Table S13†). As a consequence, it is not possible to conclusively determine the absence of c*S-3 in the assayed ionic population, despite its higher relative free energy (16.1 kJ mol−1).
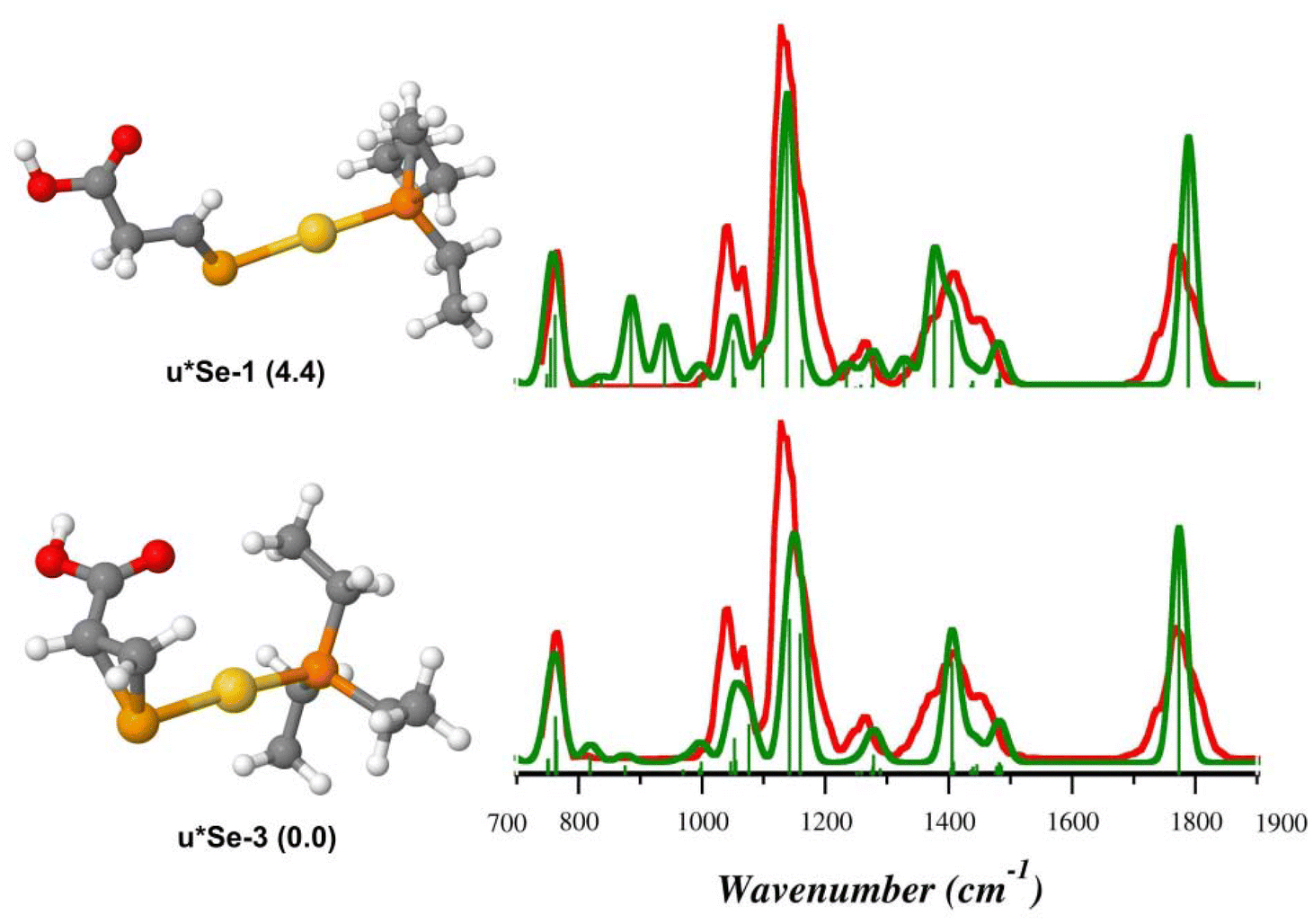 | ||
| Fig. 8 IRMPD spectrum of the [(Et3P)AuSec*]+ ion (red profiles) and calculated harmonic IR spectra (green profiles) along with optimized geometries of u*Se-3 and u*Se-1, computed in the gas-phase at the B3LYP/BS1 level of theory. Free energy values relative to u*Se-3 are reported in parenthesis in kJ mol−1. | ||
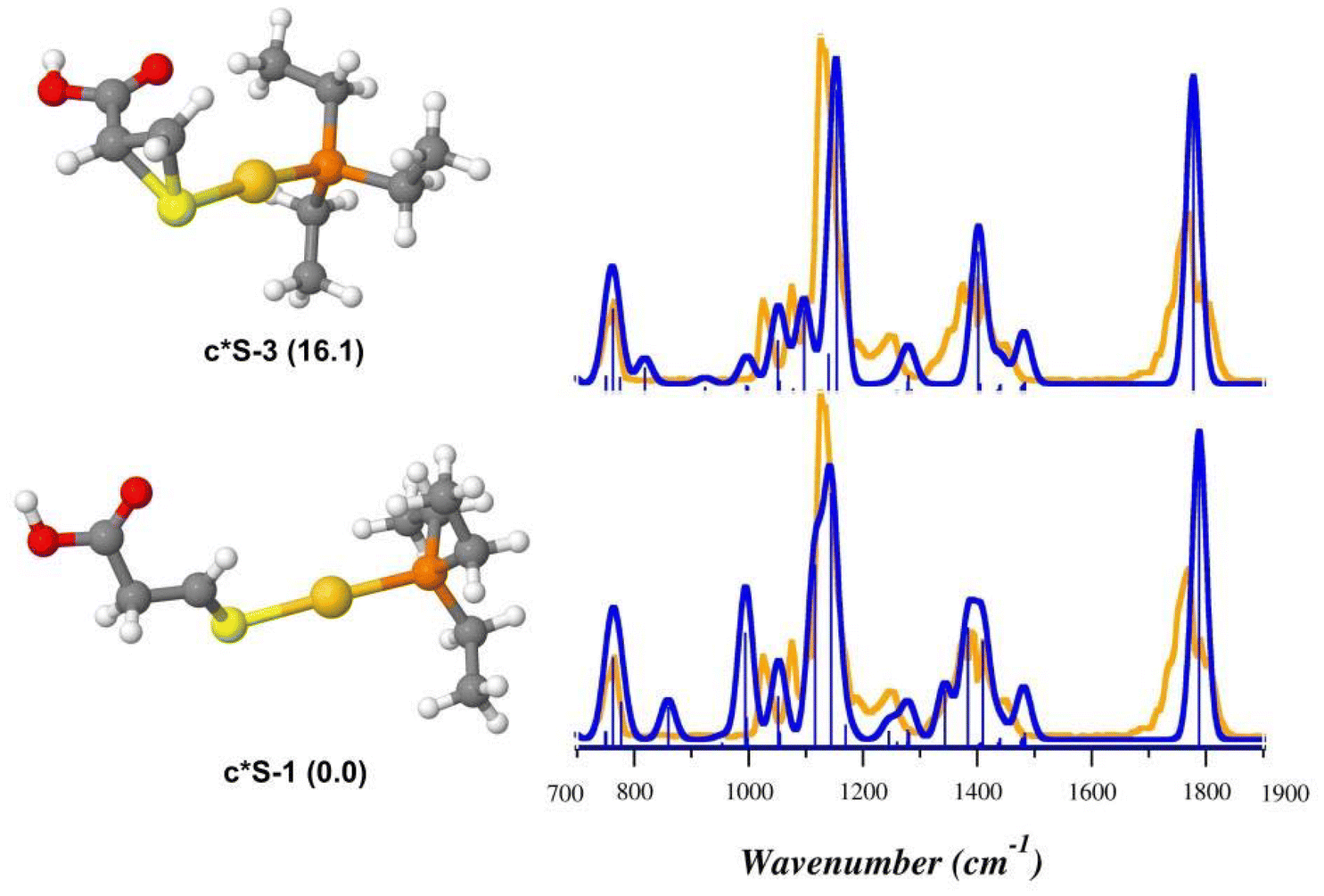 | ||
| Fig. 9 IRMPD spectrum of the [(Et3P)AuCys*]+ ion (orange profiles) and calculated harmonic IR spectra (blue profiles) along with optimized geometries of the c*S-1 and c*S-3, computed in the gas-phase at B3LYP/BS1 level of theory. Free energy values relative to c*S-1 are reported in parenthesis in kJ mol−1. | ||
Other isomers were also evaluated for both [(Et3P)AuSec*]+ and [(Et3P)AuCys*]+ ions, but their poor agreement with the IRMPD spectra allows to exclude their presence in the sampled ions (Fig. S38 and S39†), in agreement with relative free energy values (Table S9†).
DFT calculations and IRMPD experiments therefore allow to identify the most probable Au–Se/S dissociation mechanisms, confirming a different chemical reactivity for Se and S under our CID conditions.68–70
4. Conclusions
This work reports the first structural characterization of complexes generated by the interaction of auranofin with the proteinogenic amino acids cysteine and selenocysteine, [(Et3P)AuCys]+ and [(Et3P)AuSec]+, respectively, by means of mass spectrometry, IRMPD spectroscopy, and DFT calculations. Proteins, such as thioredoxin reductase, are recognized as relevant pharmacological targets of auranofin, and residues of cysteine and selenocysteine represent the most favorable binding sites for the drug.The complex generated by the substitution of the thiosugar moiety of auranofin by Sec, [(Et3P)AuSec]+, is well simulated by the theoretical spectrum of the lowest energy Se-bound isomer, in agreement with the strong tendency of gold(I) to bind soft nucleophiles, such as the selenolate function of Sec. Surprisingly, the assayed ion population of the corresponding complex generated in the presence of Cys, [(Et3P)AuCys]+, is found to be composed of both N- and S-bound isomers. The relative amount of the two isomers is likely related to their mutual stability in the condensed phase, which may be drastically modified by the solvent dielectric constant. Higher values stabilize the S-bound isomer in which the amino acid is in zwitterionic form. This information could be relevant when interpreting the binding of auranofin to proteins, if one considers the variety of local dielectric values present in different regions of these macromolecules. It is important to note that in proteins only the side-chain substituents are available for binding, and therefore, competing ligands to S-binding may be envisioned in the nucleophilic sites of amino acidic residues presenting amino groups, e.g. lysine and arginine.
The fragmentation behavior of [(Et3P)AuCys]+ and [(Et3P)AuSec]+ was studied and found to produce predominantly ammonia loss, a process that involves a preliminary isomerization reaction from the N-bound isomer of [(Et3P)AuCys]+. IRMPD spectra were also collected for these product ions, [(Et3P)Au(Cys–NH3)]+ and [(Et3P)Au(Sec–NH3)]+, respectively. Despite their similarities, comparison with calculations has shown that they present different structures. In particular, the deaminated cysteine bound to the cationic portion of auranofin presents a thioaldehyde-substituted structure, while deaminated selenocysteine cyclizes to 1-(selanylcyclopropan-2-yl)methanoic acid. Therefore, the two amino acids, despite differing only by the presence of sulfur or selenium in their structures shows a different unimolecular reactivity.
The evidence collected in this work may contribute to better model auranofin binding to its biological targets, aiding the understanding of its wide range of pharmacological activities and potentially adding new value from the repurposing of this drug. Moreover, these surprising results highlight the possibility of targeting different nucleophilic functions with gold-containing complexes depending on the dielectric constant of the medium and have shown that the unimolecular reactivity of cysteine and selenocysteine produces different results in the presence of a soft electrophile such as gold(I). Further studies are currently underway, focusing on the reaction products of auranofin with amino acids that feature nitrogen-containing side chains, as well as small peptides, to explore the potential for N-binding in proteins and to uncover binding preferences in increasingly complex model systems.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study has been funded by Sapienza Università di Roma-Progetti medi di Ateneo 2023, grant number RM123188F7509A0B and from LASERLAB-EUROPE (grant agreement No. 871124, European Union's Horizon 2020 research and innovation programme). We are grateful to the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO) for the support of the FELIX Laboratory. We acknowledge the CINECA award, under the ISCRA initiative, for the availability of high-performance computing resources and support (project IsCb5_G4MeFMO). N. Re acknowledges financial support funded by the European Union NextGenerationEU, under the National Recovery and Resilience Plan (NRRP), Mission 4 Component 2 M4C2, Investment 1.5 Call for tender No. 3277 of 30.12.2021, Italian Ministry of University, Award Number: ECS00000041, Project Title: “Innovation, digitalization and sustainability for the diffused economy in Central Italy”, Concession Degree No. 1057 of 23.06.2022 adopted by the Italian Ministry of University. CUP: D73C22000840006.References
- S. P. Pricker, Medical uses of gold compounds: Past, present and future, Gold Bull., 1996, 29, 53–60 CrossRef.
- J. M. Madeira, D. L. Gibson, W. F. Kean and A. Klegeris, The biological activity of auranofin: Implications for novel treatment of diseases, Inflammopharmacology, 2012, 20, 297–306 CrossRef CAS PubMed.
- A. Caroli, S. Simeoni, R. Lepore, A. Tramontano and A. Via, Investigation of a potential mechanism for the inhibition of SmTGR by Auranofin and its implications for Plasmodium falciparum inhibition, Biochem. Biophys. Res. Commun., 2012, 417, 576–581 CrossRef CAS PubMed.
- T. Marzo, D. Cirri, S. Pollini, M. Prato, S. Fallani, M. I. Cassetta, A. Novelli, G. M. Rossolini and L. Messori, Auranofin and its Analogues Show Potent Antimicrobial Activity against Multidrug-Resistant Pathogens: Structure–Activity Relationships, ChemMedChem, 2018, 13, 2448–2454 CrossRef CAS PubMed.
- N. P. Wiederhold, T. F. Patterson, A. Srinivasan, A. K. Chaturvedi, A. W. Fothergill, F. L. Wormley, A. K. Ramasubramanian and J. L. Lopez-Ribot, Repurposing auranofin as an antifungal: In vitro activity against a variety of medically important fungi, Virulence, 2017, 8, 138–142 CrossRef CAS PubMed.
- S. Yue, M. Luo, H. Liu and S. Wei, Recent Advances of Gold Compounds in Anticancer Immunity, Front. Chem., 2020, 8, 543 CrossRef CAS PubMed.
- A. AbdelKhalek, N. S. Abutaleb, H. Mohammad and M. N. Seleem, Antibacterial and antivirulence activities of auranofin against Clostridium difficile, Int. J. Antimicrob. Agents, 2019, 53, 54–62 CrossRef CAS PubMed.
- A. Loufouma Mbouaka, D. Leitsch, M. Koehsler and J. Walochnik, Antimicrobial effect of auranofin against Acanthamoeba spp., Int. J. Antimicrob. Agents, 2021, 58, 106425 CrossRef CAS PubMed.
- L. B. Rosa, R. L. Aires, L. S. Oliveira, J. V. Fontes, D. C. Miguel and C. Abbehausen, A “Golden Age” for the discovery of new antileishmanial agents: Current status of leishmanicidal gold complexes and prospective targets beyond the trypanothione system, ChemMedChem, 2021, 16, 1681–1695 CrossRef CAS PubMed.
- A. R. Sannella, A. Casini, C. Gabbiani, L. Messori, A. R. Bilia, F. F. Vincieri, G. Majori and C. Severini, New uses for old drugs. Auranofin, a clinically established antiarthritic metallodrug, exhibits potent antimalarial effects in vitro: Mechanistic and pharmacological implications, FEBS Lett., 2008, 582, 844–847 CrossRef CAS PubMed.
- G. Moreno-Alcántar, P. Picchetti and A. Casini, Gold Complexes in Anticancer Therapy: From New Design Principles to Particle-Based Delivery Systems, Angew. Chem., Int. Ed., 2023, 62, e202218000 CrossRef PubMed.
- N. Liu, Z. Guo, X. Xia, Y. Liao, F. Zhang, C. Huang, Y. Liu, X. Deng, L. Jiang, X. Wang, J. Liu and H. Huang, Auranofin lethality to prostate cancer includes inhibition of proteasomal deubiquitinases and disrupted androgen receptor signaling, Eur. J. Pharmacol., 2019, 846, 1–11 CrossRef CAS PubMed.
- I. Landini, L. Massai, D. Cirri, T. Gamberi, P. Paoli, L. Messori, E. Mini and S. Nobili, Structure-activity relationships in a series of auranofin analogues showing remarkable antiproliferative properties, J. Inorg. Biochem., 2020, 208, 111079 CrossRef CAS PubMed.
- F. Di Sarra, B. Fresch, R. Bini, G. Saielli and A. Bagno, Reactivity of auranofin with selenols and thiols - Implications for the anticancer activity of gold(I) compounds, Eur. J. Inorg. Chem., 2013, 2718–2727 CrossRef CAS.
- H. Li, J. Hu, S. Wu, L. Wang, X. Cao, X. Zhang, B. Dai, M. Cao, R. Shao, R. Zhang, M. Majidi, L. Ji, J. V. Heymach, M. Wang, S. Pan, J. Minna, R. J. Mehran, S. G. Swisher, J. A. Roth and B. Fang, Auranofin-mediated inhibition of PI3K/AKT/mTOR axis and anticancer activity in non-small cell lung cancer cells, Oncotarget, 2016, 7, 3548–3558 CrossRef PubMed.
- I. Momose, T. Onodera and M. Kawada, Potential anticancer activity of auranofin, Yakugaku Zasshi, 2021, 141, 315–321 CrossRef CAS PubMed.
- T. Gamberi, G. Chiappetta, T. Fiaschi, A. Modesti, F. Sorbi and F. Magherini, Upgrade of an old drug: Auranofin in innovative cancer therapies to overcome drug resistance and to increase drug effectiveness, Med. Res. Rev., 2022, 42, 1111–1146 CrossRef CAS PubMed.
- F. H. Abdalbari and C. M. Telleria, The gold complex auranofin: new perspectives for cancer therapy, Discover Oncol., 2021, 12, 42 CrossRef CAS PubMed.
- T. Marzo, D. Cirri, C. Gabbiani, T. Gamberi, F. Magherini, A. Pratesi, A. Guerri, T. Biver, F. Binacchi, M. Stefanini, A. Arcangeli and L. Messori, Auranofin, Et3PAuCl, and Et3PAuI Are Highly Cytotoxic on Colorectal Cancer Cells: A Chemical and Biological Study, ACS Med. Chem. Lett., 2017, 8, 997–1001 CrossRef CAS PubMed.
- D. Wang and S. J. Lippard, Cellular processing of platinum anticancer drugs, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- J. Reedijk, The mechanism of action of platinum antitumor drugs, Pure Appl. Chem., 1987, 59, 181–192 CrossRef CAS.
- S. M. Cohen and S. J. Lippard, Cisplatin: From DNA damage to cancer chemotherapy, Prog. Nucleic Acid Res. Mol. Biol., 2001, 67, 93–130 CAS.
- X. Zhang, K. Selvaraju, A. A. Saei, P. D'Arcy, R. A. Zubarev, E. S. Arnér and S. Linder, Repurposing of auranofin: Thioredoxin reductase remains a primary target of the drug, Biochimie, 2019, 162, 46–54 CrossRef CAS PubMed.
- A. Pratesi, D. Cirri, L. Ciofi and L. Messori, Reactions of Auranofin and Its Pseudohalide Derivatives with Serum Albumin Investigated through ESI-Q-TOF MS, Inorg. Chem., 2018, 57, 10507–10510 CrossRef CAS PubMed.
- J. A. S. Howell, DFT investigation of the interaction between gold(I) complexes and the active site of thioredoxin reductase, J. Organomet. Chem., 2009, 694, 868–873 CrossRef CAS.
- J. Lamarche, K. Bierla, L. Ouerdane, J. Szpunar, L. Ronga and R. Lobinski, Mass spectrometry insights into interactions of selenoprotein P with auranofin and cisplatin, J. Anal. At. Spectrom., 2022, 37, 1010–1022 RSC.
- I. Tolbatov and A. Marrone, Selenocysteine of thioredoxin reductase as the primary target for the antitumor metallodrugs: A computational point of view, J. Organomet. Chem., 2022, 965–966, 122330 CrossRef CAS.
- L. Messori, F. Scaletti, L. Massai, M. A. Cinellu, C. Gabbiani, A. Vergara and A. Merlino, The mode of action of anticancer gold-based drugs: A structural perspective, Chem. Commun., 2013, 49, 10100–10102 RSC.
- A. Casini and A. Pöthig, Metals in Cancer Research: Beyond Platinum Metallodrugs, ACS Cent. Sci., 2024, 10, 242–250 CrossRef CAS PubMed.
- M. Bernabeu De Maria, M. Matczuk, D. Tesauro, M. Saviano, J. Sikorski, G. Chiappetta, S. Godin, J. Szpunar, R. Lobinski and L. Ronga, Study of metalation of thioredoxin by gold(I) therapeutic compounds using combined liquid chromatography/capillary electrophoresis with inductively coupled plasma/electrospray MS/MS detection, Anal. Bioanal. Chem., 2024, 416, 2819–2833 CrossRef CAS PubMed.
- T. Gamberi, A. Pratesi, L. Messori and L. Massai, Proteomics as a tool to disclose the cellular and molecular mechanisms of selected anticancer gold compounds, Coord. Chem. Rev., 2021, 438, 213905 CrossRef CAS.
- C. Zoppi, L. Messori and A. Pratesi, ESI MS studies highlight the selective interaction of Auranofin with protein free thiols, Dalton Trans., 2020, 49, 5906–5913 RSC.
- H. Li, Y. Zhao, H. I. A. Phillips, Y. Qi, T. Y. Lin, P. J. Sadler and P. B. O'Connor, Mass spectrometry evidence for cisplatin as a protein cross-linking reagent, Anal. Chem., 2011, 83, 5369–5376 CrossRef CAS PubMed.
- N. Zhang, Y. Du, M. Cui, J. Xing, Z. Liu and S. Liu, Probing the Interaction of Cisplatin with Cytochrome c by Electrospray Ionization Fourier Transform Ion Cyclotron Resonance Mass Spectrometry, Anal. Chem., 2012, 84, 6206–6212 CrossRef CAS PubMed.
- M. Wenzel and A. Casini, Mass spectrometry as a powerful tool to study therapeutic metallodrugs speciation mechanisms: Current frontiers and perspectives, Coord. Chem. Rev., 2017, 352, 432–460 CrossRef CAS.
- T. Shoeib, D. W. Atkinson and B. L. Sharp, Structural analysis of the anti-arthritic drug Auranofin: Its complexes with cysteine, selenocysteine and their fragmentation products, Inorg. Chim. Acta, 2010, 363, 184–192 CrossRef CAS.
- H. F. Dos Santos, Reactivity of auranofin with S-, Se- and N-containing amino acids, Comput. Theor. Chem., 2014, 1048, 95–101 CrossRef CAS.
- I. Tolbatov, A. Marrone, C. Coletti and N. Re, Computational studies of au(I) and au(iii) anticancer metallodrugs: A survey, Molecules, 2021, 26, 1–27 CrossRef PubMed.
- F. Di Sarra, B. Fresch, R. Bini, G. Saielli and A. Bagno, Reactivity of auranofin with selenols and thiols - Implications for the anticancer activity of gold(I) compounds, Eur. J. Inorg. Chem., 2013, 2718–2727 CrossRef CAS.
- D. Corinti, R. Paciotti, C. Coletti, N. Re, B. Chiavarino, M. E. Crestoni and S. Fornarini, Elusive intermediates in cisplatin reaction with target amino acids: Platinum(II)-cysteine complexes assayed by IR ion spectroscopy and DFT calculations, J. Inorg. Biochem., 2022, 237, 112017 CrossRef CAS PubMed.
- D. Corinti, R. Paciotti, C. Coletti, N. Re, B. Chiavarino, G. Frison, M. E. Crestoni and S. Fornarini, IRMPD spectroscopy and quantum-chemical simulations of the reaction products of cisplatin with the dipeptide CysGly, J. Inorg. Biochem., 2023, 247, 112342 CrossRef CAS PubMed.
- D. Corinti, M. E. Crestoni, S. Fornarini, E. Dabbish, E. Sicilia, E. Gabano, E. Perin and D. Osella, A multi-methodological inquiry of the behavior of cisplatin-based Pt(IV) derivatives in the presence of bioreductants with a focus on the isolated encounter complexes, JBIC, J. Biol. Inorg. Chem., 2020, 25, 655–670 CrossRef CAS PubMed.
- M. Giampà, D. Corinti, A. Maccelli, S. Fornarini, G. Berden, J. Oomens, S. Schwarzbich, T. Glaser and M. E. Crestoni, Binding Modes of a Cytotoxic Dinuclear Copper(II) Complex with Phosphate Ligands Probed by Vibrational Photodissociation Ion Spectroscopy, Inorg. Chem., 2023, 62, 1341–1353 CrossRef PubMed.
- J. Lemaire, P. Boissel, M. Heninger, G. Mauclaire, G. Bellec, H. Mestdagh, A. Simon, S. Le Caer, J. M. Ortega, F. Glotin, P. Maitre, S. Le Caer, J. M. Ortega, F. Glotin and P. Maitre, Gas Phase Infrared Spectroscopy of Selectively Prepared Ions, Phys. Rev. Lett., 2002, 89, 273001–273002 CrossRef PubMed.
- C. N. Stedwell, J. F. Galindo, A. E. Roitberg and N. C. Polfer, Structures of Biomolecular Ions in the Gas Phase Probed by Infrared Light Sources, Annu. Rev. Anal. Chem., 2013, 6, 267–285 CrossRef CAS PubMed.
- J. Roithová, Characterization of reaction intermediates by ion spectroscopy, Chem. Soc. Rev., 2012, 41, 547–559 RSC.
- J. Martens, R. E. van Outersterp, R. J. Vreeken, F. Cuyckens, K. L. M. Coene, U. F. Engelke, L. A. J. Kluijtmans, R. A. Wevers, L. M. C. Buydens, B. Redlich, G. Berden and J. Oomens, Infrared ion spectroscopy: New opportunities for small-molecule identification in mass spectrometry - A tutorial perspective, Anal. Chim. Acta, 2020, 1093, 1–15 CrossRef CAS PubMed.
- D. Oepts, A. F. G. van der Meer and P. W. van Amersfoort, The Free-Electron-Laser user facility FELIX, Infrared Phys. Technol., 1995, 36, 297–308 CrossRef CAS.
- J. Martens, G. Berden, C. R. Gebhardt and J. Oomens, Infrared ion spectroscopy in a modified quadrupole ion trap mass spectrometer at the FELIX free electron laser laboratory, Rev. Sci. Instrum., 2016, 87, 103108 CrossRef PubMed.
- G. Berden, M. Derksen, K. J. Houthuijs, J. Martens and J. Oomens, An automatic variable laser attenuator for IRMPD spectroscopy and analysis of power-dependence in fragmentation spectra, Int. J. Mass Spectrom., 2019, 443, 1–8 CrossRef CAS.
- J. S. Prell, J. T. O'Brien and E. R. Williams, IRPD spectroscopy and ensemble measurements: Effects of different data acquisition and analysis methods, J. Am. Soc. Mass Spectrom., 2010, 21, 800–809 CrossRef CAS PubMed.
- F. A. M. G. van Geenen, R. F. Kranenburg, A. C. van Asten, J. Martens, J. Oomens and G. Berden, Isomer-Specific Two-Color Double-Resonance IR 2 MS 3 Ion Spectroscopy Using a Single Laser: Application in the Identification of Novel Psychoactive Substances, Anal. Chem., 2021, 93, 2687–2693 CrossRef CAS PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, GFN2-xTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, Extended tight-binding quantum chemistry methods, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11, e1493 CAS.
- P. Pracht, F. Bohle and S. Grimme, Automated exploration of the low-energy chemical space with fast quantum chemical methods, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- P. Pracht, S. Grimme, C. Bannwarth, F. Bohle, S. Ehlert, G. Feldmann, J. Gorges, M. Müller, T. Neudecker, C. Plett, S. Spicher, P. Steinbach, P. A. Wesołowski and F. Zeller, CREST—A program for the exploration of low-energy molecular chemical space, J. Chem. Phys., 2024, 160, 114110 CrossRef CAS PubMed.
- B. P. Pritchard, D. Altarawy, B. Didier, T. D. Gibson and T. L. Windus, New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community, J. Chem. Inf. Model., 2019, 59, 4814–4820 CrossRef CAS PubMed.
- V. Barone, M. Biczysko and J. Bloino, Fully anharmonic IR and Raman spectra of medium-size molecular systems: accuracy and interpretation, Phys. Chem. Chem. Phys., 2014, 16, 1759–1787 RSC.
- V. Barone and M. Cossi, Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, Energies, structures, and electronic properties of molecules in solution with the C–PCM solvation model, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- L. Onsager, Electric Moments of Molecules in Liquids, J. Am. Chem. Soc., 1936, 58, 1486–1493 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian Inc, Wallingford, CT, USA, 2009 Search PubMed.
- B. Martínez-Haya, J. R. Avilés-Moreno, F. Gámez, J. Martens, J. Oomens and G. Berden, Correlated proton dynamics in hydrogen bonding networks: the benchmark case of 3-hydroxyglutaric acid, Phys. Chem. Chem. Phys., 2024, 26, 198–208 RSC.
- F. Gámez, J. R. Avilés-Moreno, J. Martens, G. Berden, J. Oomens and B. Martínez-Haya, Vibrational signatures of dynamic excess proton storage between primary amine and carboxylic acid groups, J. Chem. Phys., 2024, 160, 094311 CrossRef PubMed.
- A. Sediki, L. C. Snoek and M. P. Gaigeot, N-H+ vibrational anharmonicities directly revealed from DFT-based molecular dynamics simulations on the Ala7H+ protonated peptide, Int. J. Mass Spectrom., 2011, 308, 281–288 CrossRef CAS.
- J. Oomens, J. D. Steill and B. Redlich, Gas-Phase IR Spectroscopy of Deprotonated Amino Acids, J. Am. Chem. Soc., 2009, 131, 4310–4319 CrossRef CAS PubMed.
- C. Bhardwaj and L. Hanley, Ion sources for mass spectrometric identification and imaging of molecular species, Nat. Prod. Rep., 2014, 31, 756–767 RSC.
- C. S. Ho, C. W. K. Lam, M. H. M. Chan, R. C. K. Cheung, L. K. Law, L. C. W. Lit, K. F. Ng, M. W. M. Suen and H. L. Tai, Electrospray ionisation mass spectrometry: principles and clinical applications., Clin. Biochem. Rev., 2003, 24, 3–12 CAS.
- L. Li, C. Li, Z. Zhang and E. Alexov, On the Dielectric “Constant” of Proteins: Smooth Dielectric Function for Macromolecular Modeling and Its Implementation in DelPhi, J. Chem. Theory Comput., 2013, 9, 2126–2136 CrossRef CAS PubMed.
- H. J. Reich and R. J. Hondal, Why Nature Chose Selenium, ACS Chem. Biol., 2016, 11, 821–841 CrossRef CAS PubMed.
- W. Kutzelnigg, Chemical Bonding in Higher Main Group Elements, Angew. Chem., Int. Ed. Engl., 1984, 23, 272–295 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qi02023e |
| ‡ The authors contributed equally. |
| This journal is © the Partner Organisations 2024 |