
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pyridyl–triazole ligands enable in situ generation of a highly active dihydride iridium(III) complex for formic acid dehydrogenation†
Miriam
Abán‡
 ,
J. Marco
Cuenca
,
Irene
Embid
,
Alba
de Toro
,
Pilar
Gómez-Sal
,
Ernesto
de Jesús
,
Marta
Valencia
* and
Cristina G.
Yebra
*
,
J. Marco
Cuenca
,
Irene
Embid
,
Alba
de Toro
,
Pilar
Gómez-Sal
,
Ernesto
de Jesús
,
Marta
Valencia
* and
Cristina G.
Yebra
*
Departamento de Química Orgánica y Química Inorgánica, Instituto de Investigación Química “Andrés M. del Río”, Universidad de Alcalá, Campus Universitario, 28805 Alcalá de Henares, Madrid, Spain. E-mail: cgarcia.yebra@uah.es; marta.valencia@uah.es
First published on 2nd October 2024
Abstract
The reaction of [Cp*IrCl2]2 with 1-pyridyl-1,2,3-triazoles La–Ld in the presence of sodium triflate yielded the complexes [Cp*Ir(k2-NN)(Cl)][OTf] (1a–1d) in which the k2-NN ligand coordinates to iridium through the pyridyl nitrogen and the N2 nitrogen of the triazole ring, leaving the more basic N3 atom free. These complexes serve as efficient catalytic precursors for the dehydrogenation of formic acid in the presence or absence of an external solvent, achieving turnover frequencies (TOFmax) of up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 703 h−1 and producing a 1:1 mixture of hydrogen and carbon dioxide with no detectable carbon monoxide. The catalysts can be reused after a single reaction cycle, as they retain their activity. This retention of activity has allowed a cumulative turnover number (TON) of 26876 to be reached after six reloading cycles of neat formic when using sodium formate as a base. Kinetic and 1H NMR studies reveal that the active catalyst, [Cp*Ir(CO)H2] (2), forms in situ from precursors 1a–1d under the reaction conditions. Thus, complexes 1a–1d function as stable precatalysts, simplifying the use of the otherwise unstable complex 2. The reaction mechanism is likely to involve mono- and dihydride species. The decarboxylation of the coordinated formate is the rate-determining step at high formic acid concentrations, according to the kinetic profiles and kinetic isotope effect (KIE) values obtained.
703 h−1 and producing a 1:1 mixture of hydrogen and carbon dioxide with no detectable carbon monoxide. The catalysts can be reused after a single reaction cycle, as they retain their activity. This retention of activity has allowed a cumulative turnover number (TON) of 26876 to be reached after six reloading cycles of neat formic when using sodium formate as a base. Kinetic and 1H NMR studies reveal that the active catalyst, [Cp*Ir(CO)H2] (2), forms in situ from precursors 1a–1d under the reaction conditions. Thus, complexes 1a–1d function as stable precatalysts, simplifying the use of the otherwise unstable complex 2. The reaction mechanism is likely to involve mono- and dihydride species. The decarboxylation of the coordinated formate is the rate-determining step at high formic acid concentrations, according to the kinetic profiles and kinetic isotope effect (KIE) values obtained.
Introduction
The use of hydrogen as an energy carrier presents challenges in terms of storage due to low gravimetric and volumetric density and transportation.1,2 Addressing these challenges necessitates the development of safe and cost-effective technologies that ensure a stable hydrogen supply. Additionally, the variability of carbon monoxide content accompanying hydrogen in fuel cells can be detrimental to electrode materials, emphasizing the need for high-purity hydrogen with minimal carbon monoxide content, typically below 10 ppm.3Physical storage of H2 gas requires materials with special properties, resistant to hydrogen corrosion and capable of withstanding high pressures to ensure safety during transport.4 Liquid hydrogen storage is not a competitive alternative due to its high energy demand, involving extremely low temperatures and very high pressures, with daily losses of 3–6% due to vaporization.5 Chemical hydrogen storage offers an alternative approach using materials with high hydrogen content that are easy to transport and store. These materials can release their hydrogen content through a chemical process, often catalyzed.6 Among liquid organic hydrogen carriers (LOHCs), formic acid (FA) has been considered a hydrogen storage material for decades as it is a weak, biodegradable acid, liquid at room temperature, and relatively low in toxicity. It can reversibly convert into hydrogen (H2) and carbon dioxide (CO2) using catalysts (HCOOH(l) → H2(g) + CO2(g), ΔG° = −32.9 kJ mol−1).7,8
Dehydrogenation of formic acid using heterogeneous catalysts typically requires high temperatures and produces carbon monoxide as a result of competitive FA dehydration (HCOOH(l) → H2O(l) + CO(g), ΔG° = −12.4 kJ mol−1).7,9–11 Therefore, the development of catalysts that allow the production of high-purity hydrogen from formic acid, under milder conditions, becomes an important issue.
Homogeneous catalysts are particularly interesting as highly versatile coordination complexes that enable formic acid dehydrogenation under milder conditions and facilitate the proceeding of the reaction and understanding of the reaction mechanisms,12,13 allowing for the isolation and study of their reaction intermediates.14 On the other hand, the dehydrogenation of formic acid in the absence of solvent is an environmentally sustainable process, producing only H2 and CO2 as the final reaction products. However, the number of organometallic complexes capable of efficiently catalysing the dehydrogenation of formic acid under these conditions is limited and the use of additives is generally needed (Chart 1). Investigations employing ruthenium complex A, as detailed by Milstein,15 lead to remarkable turnover numbers (TON) of up to 1.7 million over 24 runs. The [Ir(tBu2PCH2(2-py))(cod)]+ precatalyst (B), as described by Williams, has been examined for both high efficiency (turnover frequency, TOF, up to 13320 h−1) and robustness.16 Analysis of the gaseous mixture in these cases reveals the presence of variable amounts of carbon monoxide. Experiments conducted utilizing the rhodium catalysts stabilized by pincer CNC ligands C, perform with maximum TOF over 10000 h−1 in the presence of sodium formate and water.17 Research conducted with the iridium system D, indicates that the maximum TOF value is reached at 5 hours, albeit necessitating the addition of 30 mol% sodium formate and 100 mol% water.18 Other iridium systems only provide initial TOF values (lasting few minutes or seconds), lacking information on the process rate over extended durations (E).19 Cp*IrIII-derivatives developed by Fischmeister (F) dehydrogenate neat formic acid with initial TOF of up to 13292 h−1 at 100 °C.20 Xiao also reports impressive TOF values of up to 147000 h−1, based on a few seconds of reaction, for the dehydrogenation of neat 5:2 azeotropic HCOOH/NEt3 mixtures using their Cp*Ir-systems (G).21
 | ||
| Chart 1 Efficient organometallic catalysts for solventless formic acid dehydrogenation; H2O, NEt3, or HCOONa are used as additives when specified. | ||
Catalysts containing the organometallic fragment Cp*Ir (Cp* = pentamethyl-η5-cyclopentadienyl) and k2-N,N ligands, show high activity and stability in aqueous media.12,22–31 Recently, He and co-workers have prepared complexes [Cp*Ir(k2-NN)(OH2)]n+ (k2-NN being pyridyl-1,2,3-triazole and pyridyl-1,2,4-triazole ligands, and n = 1, 2),32,33 in which the 1,2,3-triazole heterocycles coordinate to iridium through their more electron-rich N3 atom.34–38 These complexes efficiently catalyse formic acid dehydrogenation in aqueous solution.
Herein, we present the preparation and characterization of a family of [Cp*Ir(k2-NN)(Cl)]+ complexes, featuring 1-pyridyl-1,2,3-triazole ligands coordinated to the iridium centre through the N atom of the pyridine and the N2 atom of the triazole. These complexes serve as precatalysts for the dehydrogenation of formic acid in the presence or absence of solvent. They exhibit competitive activities under neat conditions, including TOF values of up to 10703 h−1 and accumulated TON exceeding 26800. 1H NMR analysis of the catalytic residues confirms the in situ formation of the catalytically active species, identified as [Cp*Ir(CO)H2], which forms upon the release of the pyridyl–triazole ligand under the reaction conditions.
Results and discussion
Synthesis of complexes 1a–1d
Iridium(III) complexes 1a–1d have been prepared in 70–85% yield by reaction of [Cp*IrCl2]2 with two equiv. of the corresponding 1-pyridyl-1,2,3-triazole ligand La–Ld in the presence of sodium triflate (Scheme 1).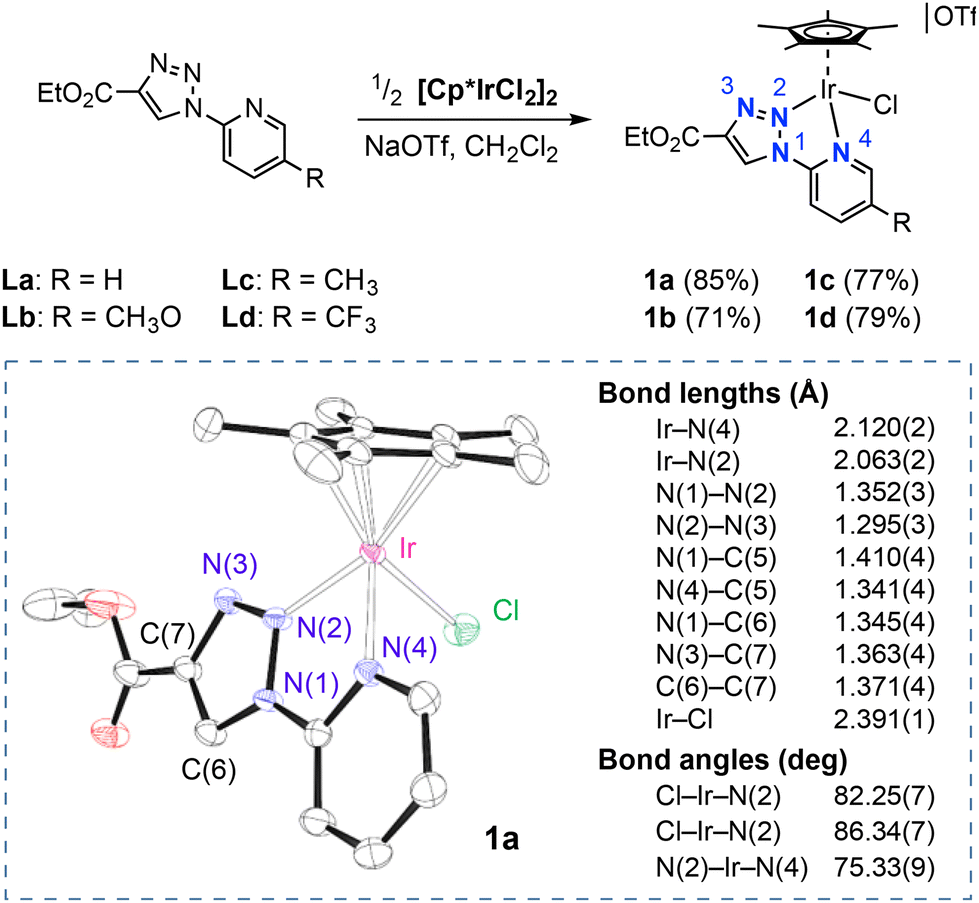 | ||
| Scheme 1 Synthesis of complexes [Cp*Ir(k2-NN)(Cl)][OTf] (1a–1d) and ORTEP diagram of the cationic unit of 1a (50% probability ellipsoids; H atoms omitted). | ||
These ligands adopt the less common inverse triazole complex-type k2-(N,N)-coordination mode,34,39 in which the N2 nitrogen of the triazole ring and the pyridyl nitrogen coordinate to iridium, leaving free the more basic N3 atom of the triazole ring (see structure of 1a obtained by X-ray diffraction analysis in Scheme 1). Otherwise, complex 1a displays a typical piano stool geometry with three ledges formed by the chlorido and the bidentate pyridyl–triazole ligand. The Ir–triazole distance (Ir–N(2) = 2.063(2) Å) is shorter than the Ir–pyridine (Ir–N(4) = 2.120(2) Å) distance and also slightly shorter than that previously reported for related inverse triazole iridium complexes (2.07–2.08 Å).36 Complete characterization details for complexes 1a–1d are given in the ESI.†1H NMR spectra of the complexes reveal the highly acidic character of the triazolium proton, that appears as a singlet at around 9.5 ppm for 1a–1c and is shifted to a higher frequency for 1d (9.77 ppm), which agrees with the presence of a p-CF3 substituted pyridine in the latter. The observation of 13C resonances at around 128 ppm for the CH of the triazole moiety confirms the coordination of the triazole moiety through the N2 atom.
FA dehydrogenation activity of 1a–1d in toluene
The catalytic performance of these new iridium(III) complexes in the dehydrogenation of formic acid was initially evaluated in organic solvents with addition of triethylamine as a base. At a temperature of 70 °C and using 1 mol% metal loading, complex 1a showed no activity in polar solvents such as THF or DMF. However, it gave a quantitative conversion of formic acid to an equimolar mixture of CO2 and H2 in toluene, under the same conditions (entries 1–5, Table S1†). Temperatures below 70 °C resulted in poor reaction rates (entries 6 and 7, Table S1†), while the improved performance obtained by raising the temperature to 90 °C allowed to reduce the catalyst loading to 0.5 mol%, resulting in a TOF at 50% of conversion (TOF50) of 733 h−1 (entry 11, Table S1†). Analogous experiments showed that complexes 1b–1d were also able to achieve nearly quantitative formic acid conversions, reaching turnover numbers (TONs) close to 90 at 70 °C with 1 mol% of Ir loadings (entries 1–4, Table S2†). However, the TOF50 values observed at 70 °C (in the range of 72 to 158 h−1) reflected the existence of notable differences in the catalytic behaviour of the four complexes. These discrepancies vanished when the reactions were conducted at higher temperatures (TOF50 values between 635 and 760 h−1 at 90 °C; entries 5–8, Table S2†). The reaction profiles show the presence of an induction period. The duration of this period depends on the reaction temperature and on the nature of the catalytic precursor, ranging from only a few minutes for 1d to over 30 minutes for 1b at 70 °C (Fig. 1). The differences described above for the TOF50 values obtained with complexes 1a–1d at this temperature are a consequence of the remarkable differences observed in the induction periods (1b > 1c > 1a > 1d), rather than of significant differences in catalytic activity after catalyst activation. At 90 °C, the induction period is reduced to a few minutes for all the complexes, and the differences between them are no longer distinguishable. An interpretation of these results will be presented in a subsequent section of this text. | ||
| Fig. 1 Reaction profiles for the dehydrogenation of formic acid in toluene, in the presence of triethylamine, using complexes 1a–1d as precatalysts, at 70 °C (1 mol% of Ir), and 90 °C (0.5 mol% of Ir). | ||
Notably, there is no induction period in subsequent reaction cycles, when fresh reactants (FA and NEt3) are added to the residue from a previous catalytic batch. Fig. 2 depicts one such experiment, in which complex 1a was subjected to four reloading experiments with the objective of evaluating the durability of the catalytic system. While the complete dehydrogenation of FA took 1 hour in the initial cycle, this time was reduced to 25 minutes in the first recharge, reaching 50% conversion in only 5 minutes (TOF50 = 664 h−1) due to the absence of an induction period. The catalytic activity decreases slightly in the third cycle and sharply in the fourth cycle (TOF50 of 416 and 36 h−1, respectively; Table S3†).
 | ||
| Fig. 2 Turnover numbers (TON) vs. time for the dehydrogenation of formic acid (FA) using complex 1a as catalytic precursor. Following the initial cycle (cycle 1), three subsequent recharges were conducted by the addition of fresh FA and NEt3 to the residue obtained from the previous catalytic batch. Cycle 1: HCOOH (10 μL, 0.264 mmol), NEt3 (11 mol%), complex 1a (1 mol%) in toluene (1 mL), 70 °C. Cycles 2–4: subsequent additions of HCOOH (10 μL, 0.264 mmol) and NEt3 (11 mol%) at 70 °C. | ||
FA dehydrogenation activity of 1a–1d in neat formic acid
In light of the promising results obtained with complexes 1a–1d in toluene, we decided to evaluate their performance in the solventless dehydrogenation of neat formic acid. In the temperature range tested (between 70 and 100 °C), gas evolution occurred only after the addition of a base such as triethylamine. Thus, consistent TONs between 680 and 790, and maximum TOF values (TOFmax) of up to 3300 h−1 were obtained at 100 °C, with a 0.1 mol% loading of 1a–1d after addition of a 11 mol% of triethylamine (entries 1–4, Table S4†). An improved TOFmax value of 9332 h−1 was attained by reducing the loading of 1a to 0.01 mol%, (entry 5, Table S4; Fig. S3†).When using complex 1d, the durability of the catalytic system was further investigated by adding successive fresh charges of HCOOH/NEt3 mixtures. After three hours and five cycles of reaction at 100 °C, a cumulative turnover number of 4350 was reached when a 0.1 mol% of 1d was used in the initial cycle (Fig. 3a). However, there was a noticeable decline in the catalytic activity with each reloading cycle. This was evidenced by the reduction in the measured TOF50 values from 2465 h−1 in the first cycle to 1237 h−1 in the fifth cycle (Table S5†).
 | ||
| Fig. 3 Temporal evolution of the turnover number during the dehydrogenation of neat formic acid at 90 or 100 °C in the presence of triethylamine (a) or sodium formate (b and c). In each case, cycle 1 (red plots) was initiated by the addition of the specified amount of the iridium complex 1d and (a) HCOOH (2.64 mmol)/NEt3 (0.29 mmol, 11 mol%); (b) and (c) HCOOH (2.35 mmol)/HCOONa (0.24 mmol, 10 mol%). Subsequently, further cycles of recharging with fresh formic acid (2.64 mmol) were conducted (blue plots). In experiment (a), triethylamine was added in each cycle (5 × 0.29 mmol, 11 mol%). In experiments (b) and (c), sodium formate was added only in the initial cycle (C1). | ||
Furthermore, the performance of complex 1d was examined in the dehydrogenation of formic acid/sodium formate mixtures under the same conditions (Fig. 3b). This approach has the advantage of significantly reducing the total reaction volume, which in turn increases the energy density. Additionally, the elimination of volatile organic solvents prevents any potential damage to the materials in fuel cell electrodes. The reactor was charged with a 10:1 formic acid/sodium formate mixture and a 0.1 mol% of 1d in the first cycle, and exclusively with formic acid in subsequent cycles. A rapid evolution of gases was observed when reagents and precatalyst were initially mixed at a temperature of 100 °C (C1; Fig. 3b). This resulted in a turnover number (TON) of 634 and a maximum turnover frequency (TOFmax) of 1749 h−1 within just 16 minutes (entry C1, Table S6†). Following the addition of fresh, neat formic acid, a TOFmax of 4253 h−1 was achieved at the same temperature (entry C2, Table S6†). Subsequently, the temperature was reduced to 90 °C and neat formic acid was added for eight additional runs, with both TOFmax and TOF50 values consistently exceeding 2000 h−1. An accumulated TON of 7702 was achieved after overall ten cycles of formic acid dehydrogenation. By lowering the iridium loading to 0.02 mol% (Fig. 3c), the accumulated TON increased to 26876 over six runs (entries 1-C1 to 1-C6 in Table S7†). A further reduction in the metal-loading to 0.01 mol% over two runs resulted in an improved TOF value of up to 10703 h−1 (entries 2-C1 and 2-C2, Table S7; Fig. S4†).
Identification of catalytic active species
To gain insight into the identity of the organometallic species involved in these catalytic transformations, the evolution of complex 1a was monitored by 1H NMR spectroscopy under conditions closely resembling those used in the dehydrogenation of formic acid in toluene. The iridium loading was increased to 5 mol%, and the temperature was fixed at 65 °C in deuterated toluene. A more detailed analysis of the course of the reaction and the intermediate species identified is given in Section 5.1 of the ESI.† Only the most relevant results will be highlighted here.The starting complex 1a is almost completely converted in about 1 h into two hydride complexes, neither of which contains the pyridyl–triazole moiety La (complexes D and E in Fig. S5a†). Complex D, probably a monohydride, is characterized by 1H resonances at 1.71 ppm (Cp*) and at −15.43 ppm (Ir–H). Complex E corresponds to the dihydride carbonyl compound [Cp*Ir(CO)H2] (2) previously reported by Heinekey and co-workers (Scheme 2).40 The latter complex was unambiguously identified by the HMBC correlation found between the carbonyl 13C resonance at about 176 ppm and the 1H resonances of both the Cp* ring at 1.94 ppm and the hydride ligand at −15.91 ppm (Fig. S7†). A new 1H NMR spectrum of the reaction mixture recorded after 18 h at room temperature, shows hydride 2 and free ligand to be the only species in solution (Fig. S6†). Similar observations were made in a related study with precatalyst 1d. In this case, the pyridyl–triazole ligand Ld was completely decoordinated after heating the reaction mixture at 70 °C for less than 20 min, and the complex 1d was fully converted to the hydride 2 after another 18 h of reaction at room temperature (Fig. S8†). The formation of the same complex [Cp*Ir(CO)H2] (2) from 1a and 1d is confirmed by comparison of the corresponding 1H NMR spectra after 18 h of reaction (Fig. S9†).
 | ||
| Scheme 2 Formation of [Cp*Ir(CO)H2] (2) by treatment of complexes 1a and 1d with HCOOH/NEt3. | ||
An open question at this stage was whether complex 2 was also formed under the solvent-free conditions used in the experiments of Fig. 3a. This concern was addressed by analysing the residue obtained from the dehydrogenation of neat HCOOH/NEt3 using 0.1 mol% of 1d at 100 °C, where complex 2 again emerged as the main organometallic species (Fig. S10†).
The formation of the complex [Cp*Ir(CO)H2] (2) involves an initial stage where the pyridyl–triazole ligand is released from the corresponding complex 1. This release is likely driven by the significant nucleophilicity of the N3 atom in the triazole ring (Fig. 4),41 making it more reactive towards electrophiles,36,42–44 particularly the proton of formic acid. Subsequent steps in the formation of 2 probably involve the disproportionation of formate intermediates to carbonyl complexes, as previously documented for transition metal complexes,45–48 including iridium.45,49
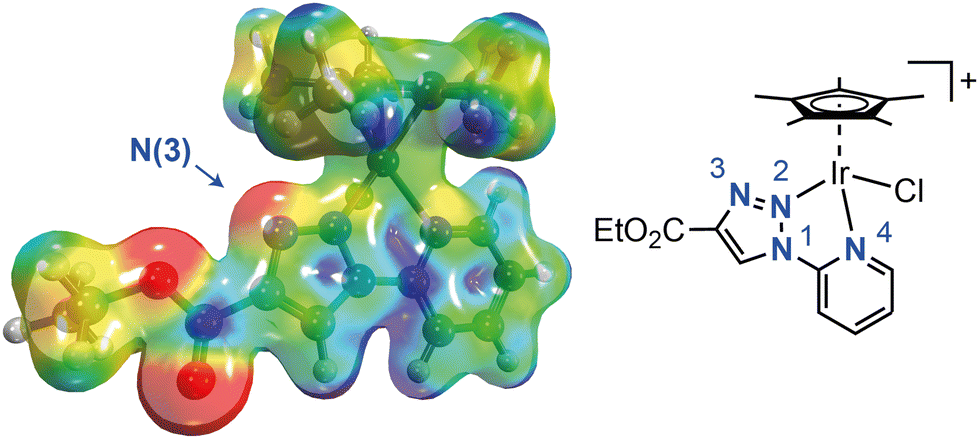 | ||
| Fig. 4 Molecular electrostatic potential surface of complex 1a calculated by DFT (M06/Def2TZVP). The electron density isovalue used to define the outer boundary of the molecule is 0.020 au. The surface potential ranges from 0.10 au (red) to 0.25 au (blue). The N3 atom is the most nucleophilic part of the molecule, along with the oxygen atoms of the CO2Et substituent. | ||
Complex 2 as FA dehydrogenation catalyst
To further confirm the role of 2 as an active FA dehydrogenation catalyst, the complex was prepared by treating [Cp*Ir(CO)Cl2] with NaBH4 and methanol in toluene. It is noteworthy that complex 2 tends to lose hydrogen, making it difficult to isolate in the solid state. Consequently, we chose to store it in solution under a hydrogen atmosphere, which effectively maintained the complex's stability for extended periods. The dehydrogenation of 2 gives rise to the formation of the dimer [Cp*Ir(CO)]2 along with other compounds. While complex 2 can be regenerated by reacting this dimer with hydrogen, the reaction takes several months to complete at room temperature under hydrogen pressures of 1–2 bar (Fig. S31†).Complex 2 was tested in the dehydrogenation of HCOOH/NEt3 (11 mol%) in toluene. The catalytic profile shows no discernible induction period, as anticipated from the results discussed above. Under conditions analogous to those used in Fig. 2 for precatalyst 1a (70 °C and 1 mol%), complex 2 reduced the time to reach 50% conversion from 25 minutes to approximately 2 min. This improvement is primarily due to the elimination of the induction period when comparing the performance of complex 2 with that of 1a in a subsequent catalytic cycle. In that second cycle, 1a has already been converted to 2 (Fig. S2†). However, complex 2 is thermally unstable, releasing dihydrogen and eventually forming the dimer [Cp*Ir(CO)]2. Consequently, and despite the introduction of an induction period, complexes 1a–1d may be useful precatalysts for FA dehydrogenation due to their ability to generate in situ the highly active, albeit thermally unstable, catalyst 2.
Fig. 5a delineates two hypotheses regarding the mechanism of action of 2 in the dehydrogenation of formic acid.7,50 The depicted IrIII–IrI mechanism is initiated by the reductive elimination of dihydrogen from 2, which is then followed by the oxidative addition of formic acid to the iridium(I) intermediate A. The subsequent decarboxylation of the resulting formate B regenerates the initial dihydride complex 2. However, it is anticipated that intermediate A will readily dimerize in this cycle, thereby generating the more stable complex [Cp*Ir(CO)]2 (C).40 This off-cycle species might re-enter the catalytic cycle by hydrogenation, but as discussed above, this process is exceedingly slow (Fig. S31†). Therefore, the catalyst should accumulate rapidly in the form of the off-cycle species C, leading to a decrease in catalytic activity, which is inconsistent with the observed data. In addition, dimer C was found to be inactive as a catalyst for the FA dehydrogenation at 90 °C under the otherwise standardized conditions that were used in this work. The mechanism shown on the right-hand side of Fig. 5a, which exclusively involves IrIII species, seems more plausible. The first step in such cycle is the release of hydrogen by the protonation of 2 with formic acid to form the monohydride B.
 | ||
| Fig. 5 Formic acid dehydrogenation catalysed by complex [Cp*Ir(CO)H2] (2). (a) Simplified mechanistic proposals for an IrIII/IrI cycle and a cycle based on IrIII species. (b) The experimental reaction profile obtained at 60 °C using a 0.6 mol% loading of 2 (red plot) is compared with that calculated using the COPASI software and a chemical model based on the IrIII species (blue plot).51 | ||
We have conducted a preliminary kinetics study to gain further insight into the reaction mechanism. As shown in Fig. 5b, the reaction profile is nearly linear up to approximately 80% conversion. Consequently, the reaction rate remains largely unaffected by the formic acid concentration up to that point. Assuming the IrIII cycle as the operating mechanism, the decarboxylation of B should be the rate-determining step. As illustrated in Fig. 5b, a satisfactory fit was obtained between the experimental and calculated reaction profile using a chemical model based on this IrIII cycle and a least squares regression analysis of the complete kinetic profile using the COPASI software (see ESI† for details).51 The analysis yielded precise values for the rate constant of the decarboxylation step across a temperature range of 40 to 90 °C (Table S12†). These rate constants, in conjunction with the Eyring equation, enabled the determination of the activation energy parameters for the decarboxylation step: ΔH‡ = 13.3 ± 1.0 kcal mol−1 and ΔS‡ = −21.8 ± 3.1 cal mol−1 K−1.
Additionally, we performed H/D Kinetic Isotope Effect (KIE) measurements to support a mechanistic proposal involving a rate-determining decarboxylation step. These experiments used complex 1d as the precatalyst for the dehydrogenation of neat formic acid, employing sodium formate (10 mol%) at 100 °C with no solvent. The observed maximum turnover frequency (TOFmax) decreased from 1749 h−1 to 641 h−1 when the HCOOH/HCOONa mixture was replaced with DCOOH/DCOONa (Table S8†). This change corresponds to a KIE value of 2.7, which is consistent with the proposed rate-determining step, as decarboxylation involves the cleavage of a C–H bond in the formate ligand. Furthermore, as observed experimentally, a negative variation of the activation entropy can be expected from the changes in the geometry of the complex that must occur to activate the C–H bond.
Finally, we analysed the gas mixtures present in the reactor headspace in four representative experiments, two using species 1a and 2 for the dehydrogenation of HCOOH/NEt3 solutions in toluene, and two using precatalyst 1d for the dehydrogenation of neat HCOOH/HCOONa mixtures (Table S9†). The CO content was below the detection limit of the GC analyser in all four cases. This result is consistent with observations made by other authors that the presence of a CO ligand in the structure of Cp*Ru active species prevents the competitive dehydration of formic acid, which produces carbon monoxide.52
Conclusions
In summary, a family of cationic Ir(III) complexes [Cp*Ir(Cl)(k2-(N,N))L][OTf] featuring pyridyl-1,2,3-triazole ligands 1a–d has been successfully synthesized. Compared to previous systems (Chart 1), the iridium-based precursors described in this study have proven to be effective promoters for the dehydrogenation of neat formic acid, producing a 1:1 mixture of H2 and CO2 with no detectable CO. The stability of these systems leads to competitive TOF values across successive catalytic cycles, showing no signs of deactivation and achieving TON values of up to 26876 over six runs and TOF values over 10700 h−1.
Complexes 1a–1d are not directly active in the dehydrogenation of formic acid. The catalytic profile of the reactions shows an induction period during which the pyridyl-1,2,3-triazole ligand is released and the dihydride complex [Cp*Ir(CO)H2] (2) is formed. This complex is responsible for the observed catalytic activity regardless of the catalytic precursor used. The handling of complex 2 is challenging due to its inherent instability towards dehydrogenation. This highlights the practicality of using complexes 1a–1d as stable precatalysts for the dehydrogenation of formic acid.
Author contributions
Miriam Abán, J. Marco Cuenca, Irene Embid and Alba de Toro contributed equally to methodology, validation, investigation and formal analysis of results. Pilar Gómez-Sal conducted the X-ray diffraction studies (formal analysis). Ernesto de Jesús, Marta Valencia and Cristina G. Yebra contributed to formal analysis, resources, writing – review & editing and funding acquisition. Marta Valencia and Cristina G. Yebra were responsible for conceptualization, writing – original draft, visualization, supervision and project administration.Data availability
The data supporting this article have been included as part of the ESI.†Crystallographic data for 1a has been deposited at the CCDC under 2362559 and can be obtained from https://www.ccdc.cam.ac.uk.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Spanish Ministerio de Ciencia e Innovación and by the European Union “NextGeneration EU/PRTR” with projects PID2020-114637GB-I00 and TED2021-129634B-I00. Marta Valencia acknowledges the award from the GOT TALENT 2019/004 Program of Research and Innovation Horizon 2020 from the European Union. We thank Dr M. González Lucas and F. González Pernas, from the Centro de Química Aplicada y Biotecnología at the Universidad de Alcalá, for their assistance with the analysis of gas mixtures by gas chromatography.References
- L. Schlapbach and A. Züttel, Hydrogen-storage materials for mobile applications, Nature, 2001, 414, 353–358, DOI:10.1038/35104634.
- N. E. Carpenter, Chemistry of Sustainable Energy, CRC Press, New York, 1st edn, 2014, DOI:10.1201/b16687.
- J. J. Baschuk and X. Li, Carbon monoxide poisoning of proton exchange membrane fuel cells, Int. J. Energy Res., 2001, 25, 695–713, DOI:10.1002/er.713.
- F. Marques, M. Balcerzak, F. Winkelmann, G. Zepon and M. Felderhoff, Review and outlook on high-entropy alloys for hydrogen storage, Energy Environ. Sci., 2021, 14, 5191–5227, 10.1039/D1EE01543E.
- U. Eberle, M. Felderhoff and F. Schüth, Chemical and Physical Solutions for Hydrogen Storage, Angew. Chem., Int. Ed., 2009, 48, 6608–6630, DOI:10.1002/anie.200806293.
- M. R. Usman, Hydrogen storage methods: Review and current status, Renewable Sustainable Energy Rev., 2022, 167, 112743, DOI:10.1016/j.rser.2022.112743.
- K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Homogeneous Catalysis for Sustainable Hydrogen Storage in Formic Acid and Alcohols, Chem. Rev., 2018, 118, 372–433, DOI:10.1021/acs.chemrev.7b00182.
- J. Guo, C. K. Yin, D. L. Zhong, Y. L. Wang, T. Qi, G. H. Liu, L. T. Shen, Q. S. Zhou, Z. H. Peng, H. Yao and X. B. Li, Formic Acid as a Potential On-Board Hydrogen Storage Method: Development of Homogeneous Noble Metal Catalysts for Dehydrogenation Reactions, ChemSusChem, 2021, 14, 2655–2681, DOI:10.1002/cssc.202100602.
- Y. Wang, Y. Qi, D. Zhang and C. Liu, New Insight into the Decomposition Mechanism of Formic Acid on Pd(111): Competing Formation of CO2 and CO, J. Phys. Chem. C, 2014, 118, 2067–2076, DOI:10.1021/jp410742p.
- C. Wan, L. Zhou, L. Sun, L. Xu, D.-G. Cheng, F. Chen, X. Zhan and Y. Yang, Boosting visible-light-driven hydrogen evolution from formic acid over AgPd/2D g-C3N4 nanosheets Mott-Schottky photocatalyst, Chem. Eng. J., 2020, 396, 125229, DOI:10.1016/j.cej.2020.125229.
- C. Wan, L. Zhou, S. Xu, B. Jin, X. Ge, X. Qian, L. Xu, F. Chen, X. Zhan, Y. Yang and D.-G. Cheng, Defect engineered mesoporous graphitic carbon nitride modified with AgPd nanoparticles for enhanced photocatalytic hydrogen evolution from formic acid, Chem. Eng. J., 2022, 429, 132388, DOI:10.1016/j.cej.2021.132388.
- N. Onishi, R. Kanega, H. Kawanami and Y. Himeda, Recent Progress in Homogeneous Catalytic Dehydrogenation of Formic Acid, Molecules, 2022, 27, 455, DOI:10.3390/molecules27020455.
- A. Kumar, P. Daw and D. Milstein, Homogeneous Catalysis for Sustainable Energy: Hydrogen and Methanol Economies, Fuels from Biomass, and Related Topics, Chem. Rev., 2022, 122, 385–441, DOI:10.1021/acs.chemrev.1c00412.
- M. A. Esteruelas, C. García-Yebra, J. Martín and E. Oñate, Dehydrogenation of Formic Acid Promoted by a Trihydride-Hydroxo-Osmium(IV) Complex: Kinetics and Mechanism, ACS Catal., 2018, 8, 11314–11323, DOI:10.1021/acscatal.8b02370.
- S. Kar, M. Rauch, G. Leitus, Y. Ben-David and D. Milstein, Highly efficient additive-free dehydrogenation of neat formic acid, Nat. Catal., 2021, 4, 193–201, DOI:10.1038/s41929-021-00575-4.
- J. J. A. Celaje, Z. Lu, E. A. Kedzie, N. J. Terrile, J. N. Lo and T. J. Williams, A prolific catalyst for dehydrogenation of neat formic acid, Nat. Commun., 2016, 7, 11308, DOI:10.1038/ncomms11308.
- P. Hermosilla, A. Urriolabeitia, M. Iglesias, V. Polo and M. A. Casado, Efficient solventless dehydrogenation of formic acid by a CNC-based rhodium catalyst, Inorg. Chem. Front., 2022, 9, 4538–4547, 10.1039/D2QI01056A.
- A. Iturmendi, M. Iglesias, J. Munarriz, V. Polo, V. Passarelli, J. J. Pérez-Torrente and L. A. Oro, A highly efficient Ir-catalyst for the solventless dehydrogenation of formic acid: the key role of an N-heterocyclic olefin, Green Chem., 2018, 20, 4875–4879, 10.1039/C8GC02794C.
- S. Cohen, V. Borin, I. Schapiro, S. Musa, S. De-Botton, N. V. Belkova and D. Gelman, Ir(III)-PC(sp3)P Bifunctional Catalysts for Production of H2 by Dehydrogenation of Formic Acid: Experimental and Theoretical Study, ACS Catal., 2017, 7, 8139–8146, DOI:10.1021/acscatal.7b02482.
- S. Wang, H. Huang, T. Roisnel, C. Bruneau and C. Fischmeister, Base-Free Dehydrogenation of Aqueous and Neat Formic Acid with Iridium(III) Cp*(dipyridylamine) Catalysts, ChemSusChem, 2019, 12, 179–184, DOI:10.1002/cssc.201802275.
- J. H. Barnard, C. Wang, N. G. Berry and J. Xiao, Long-range metal–ligand bifunctional catalysis: cyclometallated iridium catalysts for the mild and rapid dehydrogenation of formic acid, Chem. Sci., 2013, 4, 1234–1244, 10.1039/C2SC21923A.
- T. Shimbayashi and K.-I. Fujita, Iridium-Catalyzed Dehydrogenative Reactions, Top. Organomet. Chem., 2021, 69, 1–65, DOI:10.1007/3418_2020_56.
- H. Kawanami, M. Iguchi and Y. Himeda, Ligand Design for Catalytic Dehydrogenation of Formic Acid to Produce High-pressure Hydrogen Gas under Base-free Conditions, Inorg. Chem., 2020, 59, 4191–4199, DOI:10.1021/acs.inorgchem.9b01624.
- Y. Himeda, Highly efficient hydrogen evolution by decomposition of formic acid using an iridium catalyst with 4,4′-dihydroxy-2,2′-bipyridine, Green Chem., 2009, 11, 2018–2022, 10.1039/B914442K.
- N. Onishi, R. Kanega, E. Fujita and Y. Himeda, Carbon Dioxide Hydrogenation and Formic Acid Dehydrogenation Catalyzed by Iridium Complexes Bearing Pyridyl-pyrazole Ligands: Effect of an Electron-donating Substituent on the Pyrazole Ring on the Catalytic Activity and Durability, Adv. Synth. Catal., 2019, 361, 289–296, DOI:10.1002/adsc.201801323.
- R. Kanega, N. Onishi, L. Wang, K. Murata, J. T. Muckerman, E. Fujita and Y. Himeda, Picolinamide-Based Iridium Catalysts for Dehydrogenation of Formic Acid in Water: Effect of Amide N Substituent on Activity and Stability, Chem. – Eur. J., 2018, 24, 18389–18392, DOI:10.1002/chem.201800428.
- A. Matsunami, S. Kuwata and Y. Kayaki, A Bifunctional Iridium Catalyst Modified for Persistent Hydrogen Generation from Formic Acid: Understanding Deactivation via Cyclometalation of a 1,2-Diphenylethylenediamine Motif, ACS Catal., 2017, 7, 4479–4484, DOI:10.1021/acscatal.7b01068.
- C. Trotta, V. Langellotti, I. Manco, G. M. Rodriguez, L. Rocchigiani, C. Zuccaccia, F. Ruffo and A. Macchioni, Boosting Effect of Sterically Protected Glucosyl Substituents in Formic Acid Dehydrogenation by Iridium(III) 2-Pyridineamidate Catalysts, ChemSusChem, 2024, 17, e202400612, DOI:10.1002/cssc.202400612.
- L. Tensi, A. V. Yakimov, C. Trotta, C. Domestici, J. De Jesus Silva, S. R. Docherty, C. Zuccaccia, C. Copéret and A. Macchioni, Single-Site Iridium Picolinamide Catalyst Immobilized onto Silica for the Hydrogenation of CO2 and the Dehydrogenation of Formic Acid, Inorg. Chem., 2022, 61, 10575–10586, DOI:10.1021/acs.inorgchem.2c01640.
- G. Menendez Rodriguez, F. Zaccaria, L. Tensi, C. Zuccaccia, P. Belanzoni and A. Macchioni, Understanding the Deactivation Pathways of Iridium(III) Pyridine-Carboxiamide Catalysts for Formic Acid Dehydrogenation, Chem. – Eur. J., 2021, 27, 2050–2064, DOI:10.1002/chem.202003911.
- G. Menendez Rodriguez, C. Domestici, A. Bucci, M. Valentini, C. Zuccaccia and A. Macchioni, Hydrogen Liberation from Formic Acid Mediated by Efficient Iridium(III) Catalysts Bearing Pyridine-Carboxamide Ligands, Eur. J. Inorg. Chem., 2018, 2018, 2247–2250, DOI:10.1002/ejic.201701458.
- S. Ge, L. Gong, P. Yi, X. Mo, C. Liu, X.-Y. Yi and P. He, N-Site Regulation of Pyridyltriazole in Cp*Ir(N^N)(H2O) Complexes Achieving Catalytic FA Dehydrogenation, Inorg. Chem., 2023, 62, 18375–18383, DOI:10.1021/acs.inorgchem.3c01649.
- S. Ge, J.-H. Liu, L. Gong, X.-F. Mo, C. Liu, X.-Y. Yi and P. He, Dehydrogenation of Aqueous Formic Acid by Iridium and Ruthenium Complexes with Nitrogen-rich Diamino-bis(1H-1,2,4-triazole), Eur. J. Inorg. Chem., 2024, 27, e202300509, DOI:10.1002/ejic.202300509.
- M. C. Joseph, A. J. Swarts and S. F. Mapolie, Transition metal complexes of click-derived 1,2,3-triazoles as catalysts in various transformations: An overview and recent developments, Coord. Chem. Rev., 2023, 493, 215317, DOI:10.1016/j.ccr.2023.215317.
- W. K. Chu, C. K. Rono and B. C. E. Makhubela, New Triazolyl N^N Bidentate Rh(III), Ir(III), Ru(II) and Os(II) Complexes: Synthesis and Characterization, Probing Possible Relations between Cytotoxicity with Transfer Hydrogenation Efficacy and Interaction with Model Biomolecules, Molecules, 2022, 27, 2058, DOI:10.3390/molecules27072058.
- M. Valencia, H. Müller-Bunz, R. A. Gossage and M. Albrecht, Enhanced product selectivity promoted by remote metal coordination in acceptor-free alcohol dehydrogenation catalysis, Chem. Commun., 2016, 52, 3344–3347, 10.1039/C6CC00267F.
- G. A. Burley, Y. Boutadla, D. L. Davies and K. Singh, Triazoles from N-Alkynylheterocycles and Their Coordination to Iridium, Organometallics, 2012, 31, 1112–1117, DOI:10.1021/om201157g.
- C. Hua, K. Q. Vuong, M. Bhadbhade and B. A. Messerle, New Rhodium(I) and Iridium(I) Complexes Containing Mixed Pyrazolyl–1,2,3-Triazolyl Ligands As Catalysts for Hydroamination, Organometallics, 2012, 31, 1790–1800, DOI:10.1021/om201171b.
- M. Moreno-Latorre, M. C. de la Torre, H. Gornitzka, C. Hemmert and M. A. Sierra, Mono- and Dinuclear 1-(2-Pyridyl)-4-phenyl-1,2,3-triazole-Based Ir(III) and Rh(III) Complexes, Organometallics, 2024, 43, 1128–1136, DOI:10.1021/acs.organomet.4c00079.
- D. M. Heinekey, D. A. Fine, T. G. P. Harper and S. T. Michel, Dinuclear dihydride complexes of iridium: a study of structure and dynamics, Can. J. Chem., 1995, 73, 1116–1125, DOI:10.1139/v95-138.
- M. Frutos, M. Gómez-Gallego, E. A. Giner, M. A. Sierra and C. Ramírez de Arellano, Triazole vs. triazolium carbene ligands in the site-selective cyclometallation of o-carboranes by M(III) (M = Ir, Rh) complexes, Dalton Trans., 2018, 47, 9975–9979, 10.1039/C8DT02296H.
- R. Maity and B. Sarkar, Chemistry of Compounds Based on 1,2,3-Triazolylidene-Type Mesoionic Carbenes, JACS Au, 2022, 2, 22–57, DOI:10.1021/jacsau.1c00338.
- Á. Vivancos, C. Segarra and M. Albrecht, Mesoionic and Related Less Heteroatom-Stabilized N-Heterocyclic Carbene Complexes: Synthesis, Catalysis, and Other Applications, Chem. Rev., 2018, 118, 9493–9586, DOI:10.1021/acs.chemrev.8b00148.
- M. Valencia, A. Pereira, H. Müller-Bunz, T. R. Belderraín, P. J. Pérez and M. Albrecht, Triazolylidene-Iridium Complexes with a Pendant Pyridyl Group for Cooperative Metal–Ligand Induced Catalytic Dehydrogenation of Amines, Chem. – Eur. J., 2017, 23, 8901–8911, DOI:10.1002/chem.201700676.
- A. Anaby, M. Feller, Y. Ben-David, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon and D. Milstein, Bottom-Up Construction of a CO2-Based Cycle for the Photocarbonylation of Benzene, Promoted by a Rhodium(I) Pincer Complex, J. Am. Chem. Soc., 2016, 138, 9941–9950, DOI:10.1021/jacs.6b05128.
- M. L. Scheuermann, S. P. Semproni, I. Pappas and P. J. Chirik, Carbon Dioxide Hydrosilylation Promoted by Cobalt Pincer Complexes, Inorg. Chem., 2014, 53, 9463–9465, DOI:10.1021/ic501901n.
- T. Schaub and R. A. Paciello, A Process for the Synthesis of Formic Acid by CO2 Hydrogenation: Thermodynamic Aspects and the Role of CO, Angew. Chem., Int. Ed., 2011, 50, 7278–7282, DOI:10.1002/anie.201101292.
- M. A. McLoughlin, N. L. Keder, W. T. A. Harrison, R. J. Flesher, H. A. Mayer and W. C. Kaska, Interaction of Carbon Dioxide with [IrH2{(t,Bu2)PCH2CH2CHCH2CH2P(tBu2)}], Inorg. Chem., 1999, 38, 3223–3227, DOI:10.1021/ic980722f.
- S. Park, D. Bézier and M. Brookhart, An Efficient Iridium Catalyst for Reduction of Carbon Dioxide to Methane with Trialkylsilanes, J. Am. Chem. Soc., 2012, 134, 11404–11407, DOI:10.1021/ja305318c.
- Y. Pan, C.-L. Pan, Y. Zhang, H. Li, S. Min, X. Guo, B. Zheng, H. Chen, A. Anders, Z. Lai, J. Zheng and K.-W. Huang, Selective Hydrogen Generation from Formic Acid with Well-Defined Complexes of Ruthenium and Phosphorus–Nitrogen PN3-Pincer Ligand, Chem. – Asian J., 2016, 11, 1357–1360, DOI:10.1002/asia.201600169.
- COPASI: Biochemical System Simulator, 2024, Version 4.43, https://copasi.org.
- H. Fujita, S. Takemoto and H. Matsuzaka, Tin–Ruthenium Cooperative Catalyst for Disproportionation of Formic Acid to Methanol, ACS Catal., 2021, 11, 7460–7466, DOI:10.1021/acscatal.1c01344.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis of ligands and complexes; description of catalytic experiments, including tables, reaction profiles, and kinetic studies; NMR and mass spectra; crystallographic data and computational details. CCDC 2362559. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qi01818d |
| ‡ Present address: IMDEA Energy, Avda. Ramón de la Sagra 3, 28935 Móstoles (Madrid), Spain. |
| This journal is © the Partner Organisations 2024 |