
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering uranyl sites into MOFs for efficient and highly selective photocatalytic CO2 reduction†
Zhi-Heng
Zhou‡
ab,
Xiao-Bo
Li‡
a,
Zhi-Wei
Huang
a,
Qun-Yan
Wu
 a,
Jun-Xi
Wang
ab,
Zhi-Hui
Zhang
c,
Ji-Pan
Yu
a,
Lei
Mei
a,
Fu-Qiu
Ma
*b,
Kong-Qiu
Hu
*a and
Wei-Qun
Shi
*a
a,
Jun-Xi
Wang
ab,
Zhi-Hui
Zhang
c,
Ji-Pan
Yu
a,
Lei
Mei
a,
Fu-Qiu
Ma
*b,
Kong-Qiu
Hu
*a and
Wei-Qun
Shi
*a
aLaboratory of Nuclear Energy Chemistry, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing, 100049, China. E-mail: hukq@ihep.ac.cn; shiwq@ihep.ac.cn
bYantai Research Institute, Harbin Engineering University, Yantai, 264006 Shandong, China. E-mail: mafuqiu@hrbeu.edu.cn
cJiangsu Key Laboratory of Advanced Catalytic Materials and Technology, Advanced Catalysis and Green Manufacturing Collaborative Innovation Center, Changzhou University, Changzhou, 213164, China
First published on 14th August 2024
Abstract
Utilizing sunlight for the photocatalytic reduction of CO2 to CO and other high value-added products represents a pivotal strategy for environment protection and mitigation of the energy crisis. Herein, we have designed and prepared a uranium-based organic-framework (MOF), IHEP-101, featuring a uranyl photocatalytic active center, engineered for the efficient photocatalytic reduction of CO2. Demonstrating exceptional activity, IHEP-101 achieves a CO production rate of up to 458 μmol g−1 h−1. The mechanism underlying IHEP-101's photocatalytic CO2 reduction is thoroughly detailed through in situ Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFTS) and theoretical calculations. This study underscores the effectiveness of UO22+ cations as active sites for the photocatalytic reduction of CO2, introducing an innovative method for designing and synthesizing highly efficient photocatalysts aimed at CO2 reduction.
Introduction
In recent decades, with the massive consumption of fossil fuels, the concentration of CO2 in the atmosphere has gradually increased, leading to significant environmental problems such as global warming and the energy crisis.1–5 At present, numerous methods for CO2 conversion exist, including photocatalytic reduction,6 electrochemical reduction,7,8 thermochemical conversion,9,10 adsorption,11 and biological conversion.12,13 Among these, the photocatalytic reduction of CO2 into high value-added products using sunlight is considered one of the most promising solutions due to its cleanness and environmental friendliness. However, CO2 is extremely stable, presenting a challenge of overcoming the high dissociation energy of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond to achieve efficient conversion.14–18 Synthesizing efficient and highly selective photocatalysts to reduce the thermodynamic energy barrier is an urgent problem that needs to be addressed.15,19,20
O bond to achieve efficient conversion.14–18 Synthesizing efficient and highly selective photocatalysts to reduce the thermodynamic energy barrier is an urgent problem that needs to be addressed.15,19,20
Metal–organic frameworks (MOFs) have broad applications in the field of photocatalysis, offering well-defined coordination environments, unique functional ligands, high specific surface areas, and unsaturated metal sites for excellent catalytic performances.21–39 Furthermore, as heterogeneous catalysts, MOFs can be easily separated from the reaction system for recycling, thus extending the catalyst's service life and preventing pollution. The introduction of different metal ions into MOFs directly affects their structures, physical and chemical properties, and, consequently, their photocatalytic CO2 reduction performance. Currently, most MOF materials used for the photocatalytic CO2RR are based on transition metal elements, with fewer examples of actinide-based MOFs and even rarer use of actinides as catalytic active centers. In recent years, the field of carbon dioxide fixation has increasingly adopted actinide functional materials, notably actinide-based MOFs. For example, Huang et al. prepared a supported polymetallic oxygen cluster mesoporous/microporous porphyrin metal–organic framework POMs@IHEP-20, which achieved a CO yield of 970 μmol g−1 h−1.19 In addition, Huang et al. utilized thorium ions, known for their large ionic radius and high coordination numbers, as metal nodes to precisely regulate the rapid electron transport pathway that forms between the photoactive motifs of porphyrin MOFs, thereby influencing the photocatalytic CO2RR performance.40 It is important to note, however, that in these MOFs, actinides serve as the structural components of the metal nodes rather than directly acting as catalytic centers.
Uranium is widely recognized for its pivotal role in nuclear power generation. However, the focus also extends to the recycling of depleted uranium, the principal component of spent fuel, which has attracted much attention.41–44 The uranyl ion exhibits exceptional photochemical properties, notably a peak absorption wavelength near 470 nm, allowing it to be excited by blue light ranging from 450 to 495 nm.45–50 In addition, the excited states of uranyl showcase a fluorescence lifetime in the microsecond range, and complexes containing uranyl enable efficient electron transfer from the photoexcited *UO22+ to organic molecules.51–55 These distinctive photochemical properties, combined with varied structural configurations, establish uranyl complexes as exemplary candidates for the development of new photocatalysts. Acting as a photocatalyst, the uranyl ions can be activated through the ligand-to-metal charge transfer (LMCT) process. Within this process, electrons within the OUO unit are transferred from the O 2p orbital to the U 5f orbital, effectively leading to electron promotion from the highest occupied molecular orbital (HOMO) to the non-bonded orbital. This induces the formation of a U(V) center and an oxygen radical, thereby facilitating direct catalysis of the conversion and synthesis of a diverse array of compounds.54,56,57 By integrating the uranyl cation within a MOF as a catalytic active site, its catalytic functionality is further refined, offering a more controllable approach to catalysis.
The development of salen ligands and their metal complexes has been rapid due to their simple synthesis methods and their structures, which are both diverse and easily modifiable.58 Salen ligands, featuring an [N2O2] coordination cavity, can coordinate with various metal ions to form M(Salen) (M = Cu, Co, Zn, etc.) complexes, exhibiting excellent charge transfer capabilities and abundant catalytic active sites. Consequently, they have been widely used in photocatalysis.59–63 For example, Su and coworkers synthesized two coordinatively unsaturated Co–Salen complexes that demonstrated high activity and CO selectivity for visible-light-driven CO2 reduction in a water-containing system.64 Azam and collaborators found that a uranyl salen coordination complex could capture visible light and selectively photocatalytically reduce CO2 to MeOH.65
In this work, two novel uranium-based MOFs, [(CH3)2NH2][UO2][UO2(L1)(DMF)][UO2(L1)(H2O)]0.5·(DMF)1.5 (IHEP-101, H4L1 = 5,5′-((1E,1′E)-((2,2-dimethylpropane-1,3-diyl)bis(azaneylylidene))bis(methaneylylidene)) bis (4-hydroxy-3-methoxybenzoic acid)) and [UO2(DMF)][UO2(L2)(DMF)] (IHEP-102, H4L2 = 5,5′-((1E,1′E)-(ethane-1,2-diylbis(azaneylylidene))bis (methaneylylidene)) bis (4-hydroxy-3-methoxybenzoic acid)), were synthesized by the solvothermal reaction of the uranyl cation and the two previously mentioned salen ligands. IHEP-101 features a three-dimensional (3D) framework composed of adjacent two-dimensional (2D) cellular networks that are stacked through π–π interactions. IHEP-102 possesses a compact three-dimensional (3D) structure formed from adjacent one-dimensional (1D) chains stacked by π–π interactions. The photocatalytic CO2RR results revealed that IHEP-101 could achieve a CO yield of 458 μmol g−1 h−1 without requiring the addition of a sacrificial agent. Moreover, the mechanism underlying the photocatalytic CO2RR facilitated by IHEP-101 was comprehensively elucidated through in situ Diffuse Reflectance Infrared Fourier Transform (DRIFTS) spectroscopy and theoretical analyses.
Experimental
Caution! Uranyl nitrate hexahydrate UO2(NO3)2·6H2O is a radioactive and chemically toxic reactant, and precautions with suitable care and protection for handling such substances should be followed although it was used in the experiment. UO2(NO3)2·6H2O was dissolved in ultra-pure water (50 mL) to obtain a uranyl nitrate stock solution (0.50 M).3-Formyl-4-hydroxy-5-methoxybenzoic acid was synthesized according to a published procedure.66 H4L1 and H4L2 were synthesized according to a modified version of a published procedure.67 Other required chemicals are derived from commercial products and can be used directly without further purification.
Synthesis
Results and discussion
Structural description of IHEP-101
The single crystal X-ray diffraction study of IHEP-101 reveals that it crystallizes in the monoclinic space group C2/c (Table S1†). The asymmetric unit of the framework structure contains three UO22+ cations, 3/2 L14− ligands, one DMF molecule, and one H2O molecule as shown in Fig. 1a. The lengths of the U–O bonds on the equatorial plane are in the range of 2.229(12)–2.509(10) Å, and the lengths of the axial U–O bonds are in the range of 1.737(13)–1.774(8) Å, which are similar to the literature reports (Table S2†).68–70 U1 and U3 adopt a similar pentagonal bipyramidal geometry, bound to two phenoxy atoms and two imine nitrogen atoms of L14− ligands and one oxygen atom of DMF or H2O, respectively, on the equatorial plane. Each U2 atom coordinates to six oxygen atoms from three carboxylic groups on the equatorial plane. Two adjacent U2 atoms are bridged together by one assembled [UO2(L1)]2− ligand to generate a twisted two-dimensional (2D) honeycomb-like network (Fig. 1c). Then adjacent layers are stacked into a three-dimensional (3D) framework through π–π interactions (Fig. 1d). In addition, the single layer of IHEP-101 has a 35.17 Å diameter pore which is partially obscured by interlamellar stacking, but IHEP-101 has formed a pore with a diameter of approximately 27.35 Å along the c-axis (Fig. S5†). The PLATON calculation result indicates that the pore volume ratio of IHEP-101 reached 46.4%.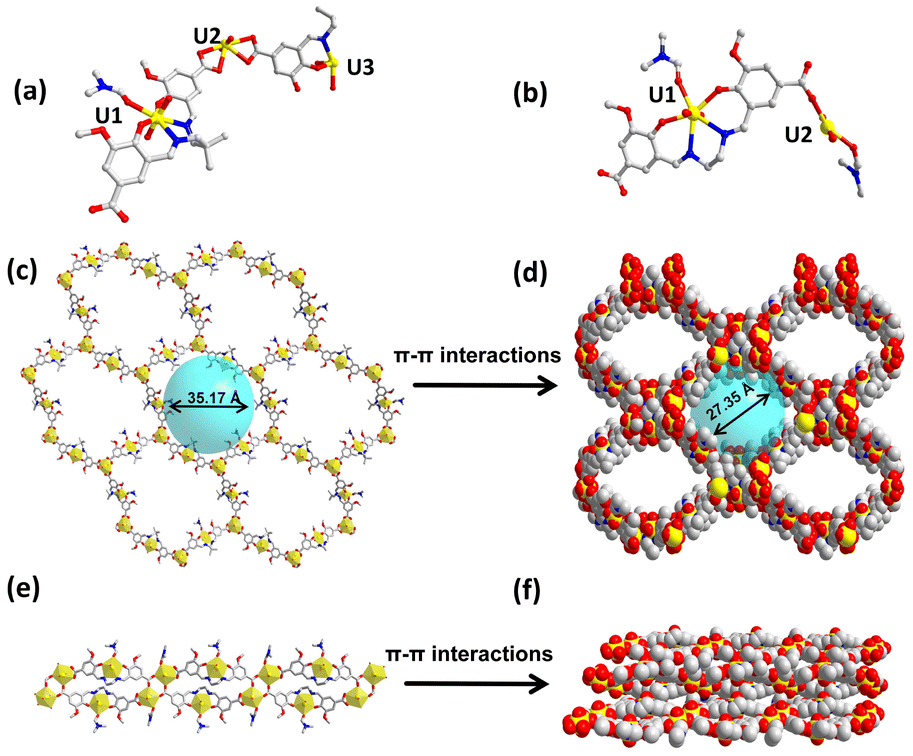 | ||
| Fig. 1 (a) Asymmetric unit of IHEP-101. (b) Asymmetric unit of IHEP-102. (c) Single layer of IHEP-101. (d) Complete structure of IHEP-101. (e) Single strand of IHEP-102. (f) Complete structure of IHEP-102. Color scheme: U, yellow; C, gray; O, red; N, blue. Hydrogen atoms were omitted for clarity. | ||
Structural description of IHEP-102
Single crystal X-ray diffraction reveals that IHEP-102 crystallizes in the monoclinic space group P21/c. The asymmetric unit contains two UO22+ cations, one L24− anion, and two DMF molecules, as shown in Fig. 1b. U1 locates in the L24− [N2O2] cavity and coordinates to two phenoxy atoms and two imine nitrogen atoms of an L24− ligand, as well as one oxygen atom of DMF, thus forming a metallized ligand [UO2(L2)]2−. Each U2 atom coordinates to four oxygen atoms from three carboxyl groups and one oxygen atom of DMF on the equatorial plane. Therefore, each [UO2(L2)]2− ligand bridges three U2 atoms and two adjacent U2 atoms are bridged together by two [UO2(L2)]2− ligands to form a regular one-dimensional (1D) chain structure (Fig. 1e). Then, adjacent chains are stacked into a dense three-dimensional (3D) structure through π–π interactions (Fig. 1f).Characterization of coordination compounds
The phase purity and structural stability of IHEP-101 and IHEP-102 were assessed through powder X-ray diffraction (PXRD) analysis. The PXRD patterns of IHEP-101 and IHEP-102 were found to be consistent with the patterns simulated from their CIF files, indicating that both materials exhibit high phase purity (Fig. S7†). In addition, PXRD analyses confirmed that IHEP-101 and IHEP-102 display stability in conventional solvents (Fig. 2a and b). | ||
| Fig. 2 (a) PXRD patterns of IHEP-101 after soaking in multiple solvents. (b) PXRD patterns of IHEP-102 after soaking in multiple solvents. (c) N2 adsorption/desorption isotherms of IHEP-101. (d) CO2 adsorption/desorption isotherms of IHEP-101. | ||
The thermal stability of IHEP-101 and IHEP-102 was evaluated through thermogravimetric analysis (TGA), as shown in Fig. S8.† IHEP-101 exhibits a loss of dimethylamine molecules and H2O molecules, as well as some solvent molecules, within the temperature range of 30–250 °C. Loss of DMF molecules occurred between 250 and 373 °C. Subsequently, the framework began to decompose, with the weight decreasing continuously until complete decomposition noted at 630 °C, leaving behind 52.8% U3O8 in the product. IHEP-102 shows a 5% weight loss before 230 °C, attributed to the release of solvent molecules. The loss of the coordinated DMF molecule was noted from 230 to 405 °C. The framework then commenced disassembly after 405 °C, with the weight further decreasing until stabilization, with the remaining U3O8 fragment stable upon further heating until 800 °C. As shown in Fig. 2c, the N2 adsorption–desorption isotherm reveals that the Brunauer–Emmett–Teller (BET) specific surface area of IHEP-101 reaches 681.79 cm2 g−1, and the corresponding pore size distribution also indicates the presence of several uniform mesoporous/microporous channel structures (Fig. S10†). Furthermore, the CO2 adsorption–desorption isotherm of IHEP-101 reveals that it possesses a high CO2 adsorption capacity of 40.28 cm3 g−1 (Fig. 2d).
The UV-vis absorption spectra of IHEP-101 and IHEP-102 were examined to evaluate their capacity for absorbing visible light. As shown in Fig. S11a and S12a,† IHEP-101 and IHEP-102 exhibit similar UV-vis absorption spectra, with strong absorption in both the ultraviolet and visible regions. Based on the Tauc plots, the band gap of IHEP-101 was determined to be 2.10 eV, and for IHEP-102, it is 2.09 eV (Fig. S11b and S12b,† respectively). The Mott–Schottky plots were utilized to ascertain the conduction band (CB) potentials of IHEP-101 and IHEP-102 (Fig. S11c and S12c†). The results of Mott–Schottky analyses conducted at frequencies of 1200, 1400 and 1600 Hz reveal that both materials are typical n-type semiconductors.30,71 Consequently, the LUMO levels were determined to be −0.60 V for IHEP-101 and −0.553 V for IHEP-102 (vs. NHE). From the band gaps and conduction band (CB) potential data, the valence band (VB) potentials of IHEP-101 and IHEP-102 were calculated as 1.50 V and 1.537 V (vs. NHE), respectively. These findings underscore the potential of both IHEP-101 and IHEP-102 as promising photocatalysts (Fig. S11d and S12d†). As depicted in Fig. S13,† under full-spectrum irradiation, IHEP-101 rapidly achieved a stable photocurrent density of 0.38 μA cm−2 with a reversible response during on–off cycling, whereas IHEP-102 exhibited a photocurrent density of 0.36 μA cm−2. The similar photocurrent responses suggest comparable charge separation efficiencies in both materials.
Performance of photocatalytic CO2 reduction
The CO2RR experiments were carried out under visible-light irradiation (λ > 420 nm) in a CH3CN/H2O mixture (4/1, volume ratio), with IHEP-101 or IHEP-102 prepared as the catalyst, and [Ru(bpy)3]Cl2·6H2O (bpy = 2,2′-bipyridine) as the photosensitizer without an additional sacrificial agent. As shown in Fig. 3a, the CO2RR result of IHEP-101 shows a nearly linear increase in CO production over 3 h corresponding to a rate of 458 μmol g−1 h−1. The photocatalytic selectivity for CO production was 98.6%, with CH4 the only other gaseous product identified (Fig. 3b). In addition, the 1H NMR results indicate that no liquid products (HCOOH, CH3OH, etc.) produced from the photocatalytic reduction of CO2 were detected (Fig. S15†). When substituting CO2 with Ar or conducting the reaction under dark conditions, only trace amounts of gaseous products can be detected, suggesting that CO is indeed produced from the photocatalytic reduction of CO2, not from the degradation of IHEP-101. Remarkably, even without the introduction of a photosensitizer, IHEP-101 is capable of achieving CO2 photocatalytic reduction, with a CO production rate of 115.79 μmol g−1 h−1 and a selectivity of 95.2%. After three photocatalytic cycles, IHEP-101 retains similar catalytic activity and high crystallinity, demonstrating that the material has excellent stability during the photocatalytic process (Fig. 3d and Fig. S16a†). Fourier transform infrared spectroscopy (FT-IR) analysis further verifies the stability of IHEP-101 post-photocatalysis (Fig. S16b†). Although IHEP-101 and IHEP-102 exhibit similar photoelectrochemical properties, the photocatalytic CO2RR performance of the latter significantly lags behind that of the former. This discrepancy can likely be attributed to the low porosity of IHEP-102, which hinders the diffusion of CO2 to the metal active sites. | ||
| Fig. 3 (a) Time-dependent evolution of CO2 reduction products over IHEP-101. (b) Catalytic efficiency of IHEP-101 under different conditions. (c) The effect of additive amount of photosensitizer on photocatalytic performance. (d) The recycling performance of IHEP-101 for the photocatalytic reduction of CO2. | ||
Mechanisms of photocatalytic CO2 reduction
The reaction intermediates of the photocatalytic CO2 reduction process can be determined by in situ DRIFTS measurement, as shown in Fig. 4. Under dark conditions, the spectral signals remain inactive. However, upon exposure to light irradiation, several new peaks can be observed, which can be assigned to a variety of intermediate states potentially involved in the CO2 reduction process. The observed peaks at 1635 cm−1 (CO2*) and 1600 cm−1 (*COOH) are eventually converted to the reduction product CO. Furthermore, the *CO intermediates are promptly eliminated from the reaction interface, culminating in the formation of CO. Therefore, only a very faint peak corresponding to *CO can be observed at 2077 cm−1.40,72 The characteristic peak at 1040 cm−1 is attributed to *CH3O, which is an intermediate product of CH4.40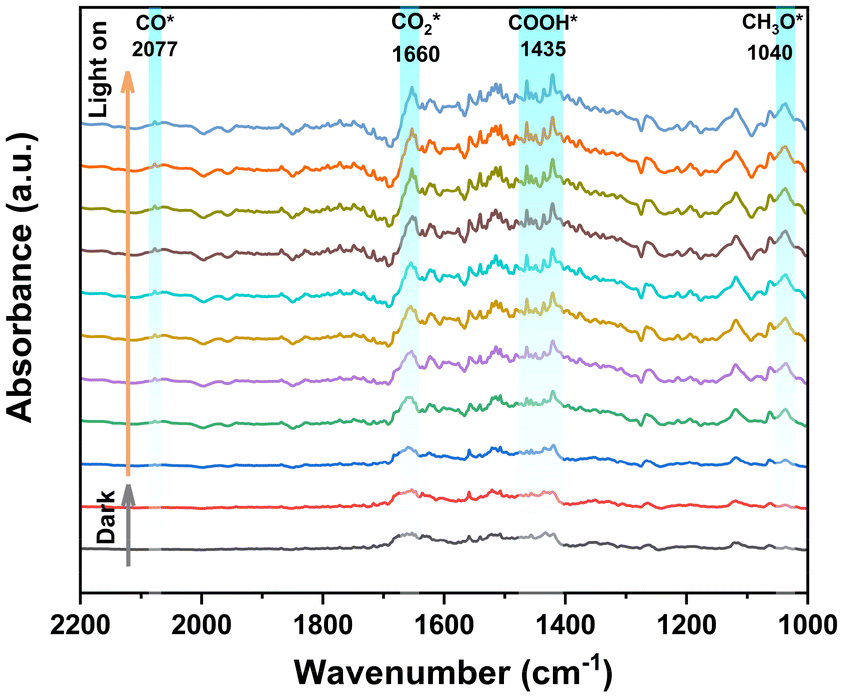 | ||
| Fig. 4 In situ DRIFTS spectra of IHEP-101 at 1000–2200 cm−1. A representative spectrum was selected at 5 min intervals, with the 10th minute being the start time of light irradiation. | ||
DFT calculations were performed to further uncover the CO2RR mechanism of IHEP-101, as shown in Fig. 5. First of all, under excitation by visible light, U(VI) enriched photogenerated electrons to form U(V). The IHEP-101 fragment combines with [UVO2]+ ion through two equal U–N bonds and two equal U–O bonds, where the U–N and U–O bonds are 2.616 and 2.378 Å, respectively. In addition, the average U–Oyl bond distance is 1.839 Å in the U(V) complex. The formation of the intermediate complex (IC) by the addition of a CO2 molecule in the U(V) complex is an endothermic process with a value of 0.40 eV. In the process under investigation, a new U–O3 bond in the intermediate complex (IC) with a bond length of 2.966 Å is formed, leading to two unequal U–N bonds (2.617 vs. 2.627 Å) and two slightly differing U–O bond lengths (2.372 vs. 2.385 Å). Additionally, within the IC, the CO2 molecule manifests two different C–O bond lengths at 1.175 Å and 1.165 Å, and an angle of 175° between them, which deviates from its typical structure in an isolated CO2 molecule. The transition from IC to the intermediate (INT) is an endothermic process with an energy requirement of 0.93 eV and involves overcoming an energy barrier of 0.96 eV. During the reaction sequence from IC to INT, the U–O3 bond length decreases progressively, measured at 2.435 Å in the transition state (TS) and further to 2.374 Å in INT. Concurrently, the average length of the U–Oyl bonds also follows this tendency. This change is counterbalanced by the elongation of the U–N1 bond, increasing from 2.617 Å (IC) to 2.656 Å (TS) and finally to 2.674 Å (INT). Similarly, the U–O1 bond shortens progressively from 2.372 Å in IC to 2.323 Å in TS to 2.293 Å in INT. The bonding dynamics among U–N2 and U–O2 mirror these trends, respectively showing progressive elongation and shortening. The elongation of U–N1 and U–N2 bond lengths, alongside the diminished lengths of U–O1 and U–O2 bonds, are influenced by the decreasing length of the U–O3 bond. Correspondingly, the CO2 molecule undergoes structural changes; the C–O bonds are elongated, especially the C–O3 bond, more so than the C–O4 bond, and the angle ∠O3CO4 gradually decreases from 175° in IC to 142° in TS and further to 132° in INT. These modifications signal the activation of the CO2 molecule in the progression from IC to INT, noted by the elongation of the two U–N bonds and the shortening of the U–Oyl, U–O1, U–O2, and U–O3 bonds. Crucially, the shortening of the U–Oyl bond reveals that the oxidation state of U is changed from +V to +VI, which can be demonstrated by the spin density analysis discussed below. Subsequently, COOH is formed by introducing H+ into INT, which is an exothermic process with an energy release of −1.05 eV. The U–O3 bond lengthens from 2.374 (INT) to 2.678 Å (COOH), while the average U–Oyl bond is slightly shortened from 1.791 to 1.787 Å. From INT to COOH, the observed elongation of U–N1 and U–N2 bonds and the shortening of U–O1 and U–O2 bonds are likely due to elongation of the U–O3 bond. Concurrently, the C–O3 bond shortens, and the C–O4 bond elongates, resulting from the formation of the O4–H bond. The angle ∠O3CO4 slightly decreases by 2° from INT to COOH. The subsequent formation of CO from COOH by the addition of H+ and e− is accompanied by the release of a water molecule, leading to a significant exothermic process with an energy change of −3.34 eV. During this process, the U–O3 bond further elongates, while the U–Oyl, U–N1, U–N2, U–O1, and U–O2 bonds all shorten. The elongated U–O3 bond (3.070 Å) in the CO complex indicates a weakened ability of the CO molecule to coordinate to the U atom. In contrast, the stronger coordination ability of DMF compared to the CO molecules leads to the formation of the U(VI) complex, with the release of the CO molecule and an exothermic reaction of −0.47 eV. The longer U–N1, U–N2, U–O1, and U–O2 bonds in the U(VI) complex compared to those in the CO complex may be attributed to the shorter U–ODMF bond of the former. The change in the Gibbs free energy (ΔG) from the U(V) complex to the U(VI) complex is −3.53 eV, indicating that this reaction is thermodynamically favorable. Spin density analysis can elucidate the change in the oxidation state of U, as reported by Gorantla and Mallik.73 As shown in Fig. S17,† the spin density on the U atom for the U(V) and IC structures is approximately 1.08 a.u., confirming the oxidation state of U as +V. The spin density of the U atom obviously decreases from 1.084 (IC) to 0.441 (TS) and then to 0.103 a.u. (INT), while the total spin density on the CO2 fragment considerably increases from 0 to 0.598 to 0.893 a.u., revealing that the single electron of the U atom on the IC complex gradually transfers to the CO2 fragment and the oxidation state of U gradually changes from +V (IC) to +VI (INT). The spin density on the U atom further reduces from 0.103 a.u. in IC to 0.019 a.u. in COOH, while the total spin density on the CO2 fragment increases from 0.893 a.u. to 0.969 a.u., indicating that the oxidation state of the U atom in COOH is conclusively +VI. These results demonstrate that the activation of CO2 by IHEP-101 is associated with the transformation of the oxidation state of the U atom from +V to +VI, coinciding with the progressive shortening of the average U–Oyl bond.
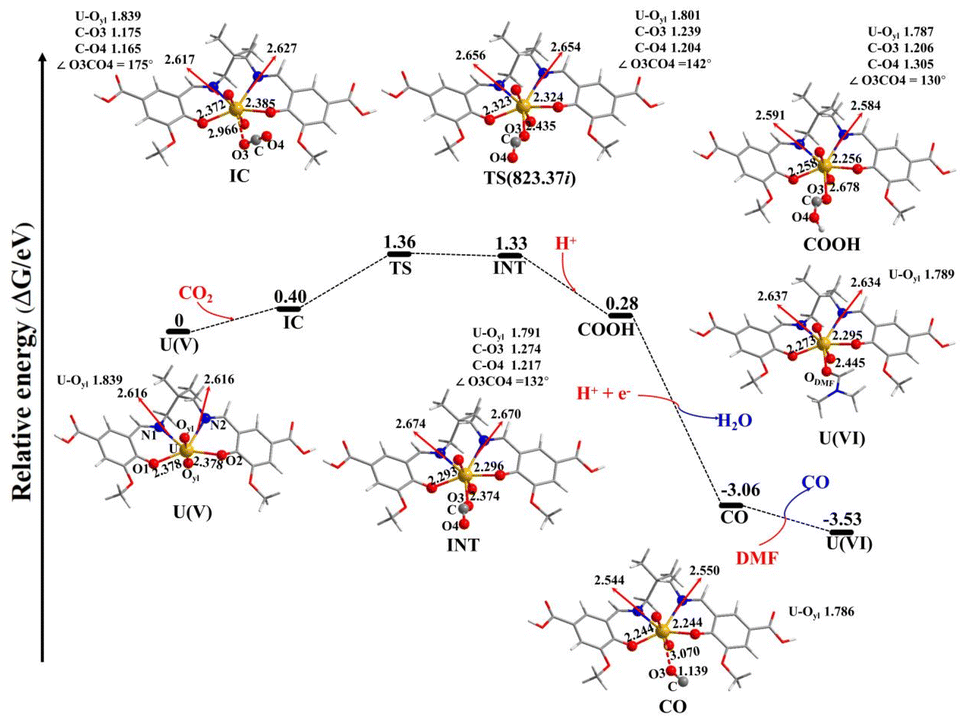 | ||
| Fig. 5 Potential energy profile for the possible mechanism of CO2 activation by IHEP-101. Values in parentheses are TS imaginary frequencies (cm−1). | ||
Based on existing experimental data and literature reports, a potential reaction pathway for the photocatalytic reduction of CO2 by IHEP-101 is proposed, as shown in Fig. 6. Initially, the photosensitizer [Ru(bpy)3]Cl2·6H2O (bpy = 2,2′-bipyridine) and the L1 ligand generate photogenerated electrons and holes upon exposure to visible light. The holes are utilized for the oxidation of H2O, and the photogenerated electrons are transferred from both the photosensitizer and the L1 ligand to the uranyl active sites in the unsaturated coordination in the MOF. This transfer reduces the hexavalent uranyl to the pentavalent uranium. Subsequently, through a two-electron transfer process, electrons are transferred to CO2, achieving the photocatalytic reduction of CO2 to CO, while the uranium is re-oxidized from pentavalent back to hexavalent.
 | ||
| Fig. 6 Schematic mechanism of the photocatalytic CO2 reduction by IHEP-101. | ||
Conclusions
In summary, two novel uranium-based MOFs, IHEP-101 and IHEP-102, have been synthesized using the coordination of salen ligands with UO22+ cations. IHEP-101 exhibits a high specific surface area and demonstrates a remarkable efficiency in the photocatalytic reduction of CO2. The mechanism of photocatalytic CO2 reduction by IHEP-101 is clearly elucidated using in situ DRIFTS and theoretical calculations. DFT calculations have confirmed that the activation of CO2 by IHEP-101 is both thermodynamically and kinetically feasible, accompanied by a cyclic transition in the oxidation state of the U atom. This work highlights the potential of UO22+ cations as active sites in the photocatalytic reduction of CO2, which provides valuable insights for further studies on the photocatalytic performance of the uranyl group and the design of new MOFs for photocatalysis.Author contributions
Z.H. Zhou conceived all experiments and wrote the manuscript. X.B. Li performed the theoretical calculations. Z.W. Huang, Z.H. Zhang, Q.Y. Wu, J.P. Yu and J.X. Wang assisted in performing the tests. K.Q. Hu, L. Mei and W.Q. Shi provided search ideas, and reviewed and edited the manuscript. F.Q. Ma, and W.Q. Shi provided revision guidance, suggestions and part of the financial support.Data availability
Crystallographic data for IHEP-101 and IHEP-102 have been deposited at CCDC under 2358409 and 2358410† and can be obtained from https://doi.org/10.1039/d4qi01578a.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support from the National Science Fund for Distinguished Young Scholars (Grant 21925603) is acknowledged. We also acknowledge support by the National Natural Science Foundation of China (22076187 and 22122609) and the Natural Science Foundation of Beijing Municipality (2242026). This work was also funded by the postdoctoral fellowship program of CPSF (GZB20230277), the China Postdoctoral Science Foundation (2023M731457), and the Natural Science Foundation of Gansu Province (23JRRA1124).References
- N. Zhang, L. Wang, H. M. Wang, R. H. Cao, J. F. Wang, F. Bai and H. Y. Fan, Self-assembled one-dimensional porphyrin nanostructures with enhanced photocatalytic hydrogen generation, Nano Lett., 2018, 18, 560–566 CrossRef CAS PubMed.
- T. M. McDonald, J. A. Mason, X. Q. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocella, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. W. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Cooperative insertion of CO2 in diamine-appended metal-organic frameworks, Nature, 2015, 519, 303–308 CrossRef CAS PubMed.
- X. K. Wang, J. Liu, L. Zhang, L. Z. Dong, S. L. Li, Y. H. Kan, D. S. Li and Y. Q. Lan, Monometallic catalytic models hosted in stable metal–organic frameworks for tunable CO2 photoreduction, ACS Catal., 2019, 9, 1726–1732 CrossRef CAS.
- A. Prašnikar, D. L. Jurković and B. Likozar, Reaction path analysis of CO2 reduction to methanol through multisite microkinetic modelling over Cu/ZnO/Al2O3 catalysts, Appl. Catal., B, 2021, 292, 120190 CrossRef.
- Y. Yang, S. Zhao, F. Bi, J. Chen, Y. Wang, L. Cui, J. Xu and X. Zhang, Highly efficient photothermal catalysis of toluene over Co3O4/TiO2 p–n heterojunction: The crucial roles of interface defects and band structure, Appl. Catal., B, 2022, 315, 121550 CrossRef CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Photoelectrocatalytic reduction of carbon-dioxide in aqueous suspensions of semiconductor powders, Nature, 1979, 277, 637–638 CrossRef CAS.
- M. Halmann, Photoelectrochemical reduction of aqueous carbon dioxide on p-type gallium phosphide in liquid junction solar cells, Nature, 1978, 275, 115–116 CrossRef CAS.
- J. F. Xie, J. J. Chen, Y. X. Huang, X. Zhang, W. K. Wang, G. X. Huang and H. Q. Yu, Selective electrochemical CO2 reduction on Cu–Pd heterostructure, Appl. Catal., B, 2020, 270, 118864 CrossRef CAS.
- M. Aresta and A. Dibenedetto, Utilisation of CO2 as a chemical feedstock: Opportunities and challenges, Dalton Trans., 2007, 28, 2975–2992 RSC.
- P. Hongmanorom, J. Ashok, P. Chirawatkul and S. Kawi, Interfacial synergistic catalysis over Ni nanoparticles encapsulated in mesoporous ceria for CO2 methanation, Appl. Catal., B, 2021, 297, 120454 CrossRef CAS.
- L. Qin, Y. Li, F. L. Liang, L. J. Li, Y. W. Lan, Z. Y. Li, X. T. Lu, M. Q. Yang and D. Y. Ma, A microporous 2D cobalt-based MOF with pyridyl sites and open metal sites for selective adsorption of CO2, Microporous Mesoporous Mater., 2022, 341, 112098 CrossRef CAS.
- A. J. Bard and M. A. Fox, Artificial photosynthesis: Solar splitting of water to hydrogen and oxygen, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS.
- Y. Xiong, Z. M. Hou, H. P. Xie, J. Z. Zhao, X. C. Tan and J. S. Luo, Microbial-mediated CO2 methanation and renewable natural gas storage in depleted petroleum reservoirs: A review of biogeochemical mechanism and perspective, Gondwana Res., 2023, 122, 184–198 CrossRef CAS.
- B. Han, X. W. Ou, Z. Q. Deng, Y. Song, C. Tian, H. Deng, Y. J. Xu and Z. Lin, Nickel metal–organic framework monolayers for photoreduction of diluted CO2: Metal-node-dependent activity and selectivity, Angew. Chem., Int. Ed., 2018, 57, 16811–16815 CrossRef CAS PubMed.
- X. Y. Feng, Y. H. Pi, Y. Song, C. Brzezinski, Z. W. Xu, Z. Li and W. B. Lin, Metal–organic frameworks significantly enhance photocatalytic hydrogen evolution and CO2 reduction with earth-abundant copper photosensitizers, J. Am. Chem. Soc., 2020, 142, 690–695 CrossRef CAS PubMed.
- T. Zhang, Y. Jin, Y. Shi, M. Li, J. Li and C. Duan, Modulating photoelectronic performance of metal–organic frameworks for premium photocatalysis, Coord. Chem. Rev., 2019, 380, 201–229 CrossRef CAS.
- S. Ullah, E. C. Lovell, T. H. Tan, B. Xie, P. V. Kumar, R. Amal and J. Scott, Photoenhanced CO2 methanation over La2O3 promoted Co/TiO2 catalysts, Appl. Catal., B, 2021, 294, 120248 CrossRef CAS.
- S. H. Jia, L. M. Wu, L. Xu, X. F. Sun and B. X. Han, Multicomponent catalyst design for CO2/N2/NOx electroreduction, Ind. Chem. Mater., 2023, 1, 93–105 RSC.
- Z.-W. Huang, K.-Q. Hu, L. Mei, D.-G. Wang, J.-Y. Wang, W.-S. Wu, Z.-F. Chai and W.-Q. Shi, Encapsulation of polymetallic oxygen clusters in a mesoporous/microporous thorium-based porphyrin metal–organic framework for enhanced photocatalytic CO2 reduction, Inorg. Chem., 2022, 61, 3368–3373 CrossRef CAS PubMed.
- Y. Wang, Q. Zhou, Y. Zhu and D. Xu, High efficiency reduction of CO2 to CO and CH4 via photothermal synergistic catalysis of lead-free perovskite Cs3Sb2I9, Appl. Catal., B, 2021, 294, 120236 CrossRef CAS.
- K. Q. Hu, Z. W. Huang, Z. H. Zhang, L. Mei, B. B. Qian, J. P. Yu, Z. F. Chai and W. Q. Shi, Actinide-based porphyrinic MOF as a dehydrogenation catalyst, Chem. – Eur. J., 2018, 24, 16766–16769 CrossRef CAS PubMed.
- J. L. C. Rowsell and O. M. Yaghi, Metal-organic frameworks: A new class of porous materials, Microporous Mesoporous Mater., 2004, 73, 3–14 CrossRef CAS.
- N. W. Ockwig, O. Delgado-Friedrichs, M. O'Keeffe and O. M. Yaghi, Reticular chemistry: Occurrence and taxonomy of nets and grammar for the design of frameworks, Acc. Chem. Res., 2005, 38, 176–182 CrossRef CAS PubMed.
- L. Y. Wu, Y. F. Mu, X. X. Guo, W. Zhang, Z. M. Zhang, M. Zhang and T. B. Lu, Encapsulating perovskite quantum dots in iron-based metal–organic frameworks (MOFs) for efficient photocatalytic CO2 reduction, Angew. Chem., Int. Ed., 2019, 58, 9491–9495 CrossRef CAS PubMed.
- S. S. A. Shah, T. Najam, M. Wen, S. Q. Zang, A. Waseem and H. L. Jiang, Metal–organic framework-based electrocatalysts for CO2 reduction, Small Struct., 2022, 3, 2100090 CrossRef CAS.
- Z. B. Fang, T. T. Liu, J. X. Liu, S. Y. Jin, X. P. Wu, X. Q. Gong, K. C. Wang, Q. Yin, T. F. Liu, R. Cao and H. C. Zhou, Boosting interfacial charge-transfer kinetics for efficient overall CO2 photoreduction via rational design of coordination spheres on metal–organic frameworks, J. Am. Chem. Soc., 2020, 142, 12515–12523 CrossRef CAS PubMed.
- H. J. Lu, J. Xie, X. Y. Wang, Y. X. Wang, Z. J. Li, K. Diefenbach, Q. J. Pan, Y. Qian, J. Q. Wang, S. Wang and J. Lin, Visible colorimetric dosimetry of UV and ionizing radiations by a dual-module photochromic nanocluster, Nat. Commun., 2021, 12, 2798 CrossRef CAS PubMed.
- Y. L. Wang, W. Liu, Z. L. Bai, T. Zheng, M. A. Silver, Y. X. Li, Y. X. Wang, X. Wang, J. Diwu, Z. F. Chai and S. Wang, Employing an unsaturated Th4+ site in a porous thorium-organic framework for Kr/Xe uptake and separation, Angew. Chem., Int. Ed., 2018, 57, 5783–5787 CrossRef CAS PubMed.
- Y. X. Li, Z. X. Yang, Y. L. Wang, Z. L. Bai, T. Zheng, X. Dai, S. T. Liu, D. X. Gui, W. Liu, M. Chen, L. H. Chen, J. Diwu, L. Y. Zhu, R. H. Zhou, Z. F. Chai, T. E. Albrecht-Schmitt and S. Wang, A mesoporous cationic thorium–organic framework that rapidly traps anionic persistent organic pollutants, Nat. Commun., 2017, 8, 1354 CrossRef PubMed.
- Z. W. Huang, K. Q. Hu, L. Mei, C. Z. Wang, Y. M. Chen, W. S. Wu, Z. F. Chai and W. Q. Shi, Potassium ions induced framework interpenetration for enhancing the stability of uranium-based porphyrin MOF with visible-light-driven photocatalytic activity, Inorg. Chem., 2021, 60, 652–660 Search PubMed.
- Z. Jiang, X. H. Xu, Y. H. Ma, H. S. Cho, D. Ding, C. Wang, J. Wu, P. Oleynikov, M. Jia, J. Cheng, Y. Zhou, O. Terasaki, T. Y. Peng, L. Zan and H. X. Deng, Filling metal–organic framework mesopores with TiO2 for CO2 photoreduction, Nature, 2020, 586, 549–554 CrossRef CAS PubMed.
- Y. S. Kang, Y. Lu, K. Chen, Y. Zhao, P. Wang and W. Y. Sun, Metal–organic frameworks with catalytic centers: From synthesis to catalytic application, Coord. Chem. Rev., 2019, 378, 262–280 CrossRef CAS.
- Z. W. Huang, S. W. An, K. Q. Hu, X. B. Li, Z. N. Bin, Z. H. Zhou, L. Mei, Z. J. Guo, W. S. Wu, Z. F. Chai and W. Q. Shi, Modulating the coordination microenvironment of uranyl compounds to enhance photocatalytic CO2 reduction, Inorg. Chem. Front., 2023, 10, 4754–4762 RSC.
- Z. H. Zhou, X. J. Li, Z. W. Huang, L. Mei, F. Q. Ma, J. P. Yu, Q. Zhang, Z. F. Chai, K. Q. Hu and W. Q. Shi, Th6-based multicomponent heterometallic metal–organic frameworks featuring 6,12-connected topology for iodine adsorption, Inorg. Chem., 2023, 62, 15346–15351 CrossRef CAS PubMed.
- C.-L. Song, Z. Li, Y.-N. Zhang, G. Zhang and Y.-W. Yang, Hydrazide–pillar[5]arene-mediated silver nanoparticles for highly efficient reductive degradation of organic dyes, Supramol. Mater., 2023, 2, 100035 Search PubMed.
- X. Yu, Y. Hu, C. Guo, Z. Chen, H. Wang and X. Li, Discrete terpyridine-lanthanide molecular and supramolecular complexes, Supramol. Mater., 2022, 1, 100017 Search PubMed.
- X. H. Kong, Q. Y. Wu, L. Mei, L. W. Zeng, Z. W. Huang, J. P. Yu, C. M. Nie, J. K. Gibson, Z. F. Chai, K. Q. Hu and W. Q. Shi, Silver ion-induced formation of unprecedented thorium nonamer clusters via lacuna-construction strategy, CCS Chem., 2023, 5, 1144–1153 CrossRef CAS.
- Y. Y. Liang, L. Mei, Q. Y. Jin, J. S. Geng, J. Y. Wang, K. Liu, K. Q. Hu, J. P. Yu and W. Q. Shi, Hierarchical assembly of uranyl metallacycles involving macrocyclic hosts, Chin. Chem. Lett., 2022, 33, 3539–3542 CrossRef CAS.
- X. X. Li, Y. X. Wang, R. H. Wang, C. Y. Cui, C. B. Tian and G. Y. Yang, Designed assembly of heterometallic cluster organic frameworks based on Anderson-type polyoxometalate clusters, Angew. Chem., Int. Ed., 2016, 55, 6462–6466 CrossRef CAS PubMed.
- Z. W. Huang, K. Q. Hu, X. B. Li, Z. N. Bin, Q. Y. Wu, Z. H. Zhang, Z. J. Guo, W. S. Wu, Z. F. Chai, L. Mei and W. Q. Shi, Thermally induced orderly alignment of porphyrin photoactive motifs in metal–organic frameworks for boosting photocatalytic CO2 reduction, J. Am. Chem. Soc., 2023, 145, 18148–18159 CrossRef CAS PubMed.
- S. T. Liddle, The Renaissance of Non-Aqueous Uranium Chemistry, Angew. Chem., Int. Ed., 2015, 54, 8604–8641 CrossRef CAS PubMed.
- T. W. Hayton, J. M. Boncella, B. L. Scott, P. D. Palmer, E. R. Batista and P. J. Hay, Synthesis of imido analogs of the uranyl ion, Science, 2005, 310, 1941–1943 CrossRef CAS PubMed.
- X. Zhang, L. Zhang, T. Bo, S. N. Huang, Z. W. Huang and W. Q. Shi, Two tetravalent uranium silicate and germanate crystals with three membered single-ring by molten salt method: K2USi3O9 and Cs2UGe3O9, Chin. Chem. Lett., 2022, 33, 3527–3530 CrossRef CAS.
- T. Y. Xiu, S. M. Zhang, P. Ren, S. Y. Liu, S. Wang, H. Shehzad, J. Y. Wang, M. Zhang, G. A. Ye, C. S. Jiao, Q. Y. Wu, L. Y. Yuan, Z. F. Chai and W. Q. Shi, Efficacious selective separation of U(VI) over Mo(VI) using novel 2,9-diamide-1,10-phenanthroline ligands: Liquid–liquid extraction and coordination chemistry, Chin. Chem. Lett., 2023, 34, 108440 CrossRef CAS.
- B. T. McGrail, L. S. Pianowski and P. C. Burns, Photochemical water oxidation and origin of nonaqueous uranyl peroxide complexes, J. Am. Chem. Soc., 2014, 136, 4797–4800 CrossRef CAS PubMed.
- G. Nocton, P. Horeglad, V. Vetere, J. Pecaut, L. Dubois, P. Maldivi, N. M. Edelstein and M. Mazzanti, Synthesis, structure, and bonding of stable complexes of pentavalent uranyl, J. Am. Chem. Soc., 2010, 132, 495–508 CrossRef CAS PubMed.
- K. Takao and S. Tsushima, The oxidation of borohydrides by photoexcited [UO2(CO3)3]4−, Dalton Trans., 2018, 47, 5149–5152 RSC.
- L. Natrajan, F. Burdet, J. Pecaut and M. Mazzanti, Synthesis and structure of a stable pentavalent-uranyl coordination polymer, J. Am. Chem. Soc., 2006, 128, 7152–7153 CrossRef CAS PubMed.
- M. J. Sarsfield and M. Helliwell, Extending the chemistry of the uranyl ion: Lewis acid coordination to a U=O oxygen, J. Am. Chem. Soc., 2004, 126, 1036–1037 CrossRef CAS PubMed.
- Z. Wang, J. M. Zachara, P. L. Gassman, C. Liu, O. Qafoku, W. Yantasee and J. G. Catalano, Fluorescence spectroscopy of U(VI)-silicates and U(VI)-contaminated Hanford sediment, Geochim. Cosmochim. Acta, 2005, 69, 1391–1403 CrossRef CAS.
- Y. Li, J. Su, E. Mitchell, G. Zhang and J. Li, Photocatalysis with visible-light-active uranyl complexes, Sci. China: Chem., 2013, 56, 1671–1681 CrossRef CAS.
- S. Fortier and T. W. Hayton, Oxo ligand functionalization in the uranyl ion UO22+, Coord. Chem. Rev., 2010, 254, 197–214 CrossRef CAS.
- K. X. Wang and J. S. Chen, Extended structures and physicochemical properties of uranyl–organic compounds, Acc. Chem. Res., 2011, 44, 531–540 CrossRef CAS PubMed.
- D. Q. Hu and X. F. Jiang, Perspectives for uranyl photoredox catalysis, Synlett, 2021, 1330–1342 CAS.
- J. G. Knapp, M. Y. Livshits, J. C. Gilhula, S. L. Hanna, I. D. Piedmonte, N. T. Rice, X. J. Wang, B. W. Stein, S. A. Kozimor and O. K. Farha, Influence of linker identity on the photochemistry of uranyl–organic frameworks, ACS Appl. Mater. Interfaces, 2023, 15, 43667–43677 CrossRef CAS PubMed.
- M. Sundararajan, A. J. Campbell and I. H. Hillier, Catalytic cycles for the reduction of UO22+ by cytochrome c7 proteins proposed from DFT calculations, J. Phys. Chem. A, 2008, 112, 4451–4457 CrossRef CAS PubMed.
- S. Kannan, A. E. Vaughn, E. M. Weis, C. L. Barnes and P. B. Duval, Anhydrous photochemical uranyl(VI) reduction: Unprecedented retention of equatorial coordination accompanying reversible axial oxo/alkoxide exchange, J. Am. Chem. Soc., 2006, 128, 14024–14025 CrossRef CAS PubMed.
- A. Neshat, M. Kakavand, F. Osanlou, P. Mastrorilli, E. Schingaro, E. Mesto and S. Todisco, Alcohol oxidations by Schiff base manganese(III) complexes, Eur. J. Inorg. Chem., 2020, 480–490 CrossRef CAS.
- M. North, S. C. Z. Quek, N. E. Pridmore, A. C. Whitwood and X. Wu, Aluminum(salen) complexes as catalysts for the kinetic resolution of terminal epoxides via CO2 coupling, ACS Catal., 2015, 5, 3398–3402 CrossRef CAS.
- P. W. Roesky, A. Bhunia, Y. H. Lan, A. K. Powell and S. Kureti, Salen-based metal–organic frameworks of nickel and the lanthanides, Chem. Commun., 2011, 47, 2035–2037 RSC.
- A. Bhunia, Y. Lan, V. Mereacre, M. T. Gamer, A. K. Powell and P. W. Roesky, Salen-based coordination polymers of iron and the rare earth elements, Inorg. Chem., 2011, 50, 12697–12704 CrossRef CAS PubMed.
- A. M. Shultz, A. A. Sarjeant, O. K. Farha, J. T. Hupp and S. T. Nguyen, Post-synthesis modification of a metal–organic framework to form metallosalen-containing MOF materials, J. Am. Chem. Soc., 2011, 133, 13252–13255 CrossRef CAS PubMed.
- H. Y. Suo, G. A. Solan, Y. P. Ma and W. H. Sun, Developments in compartmentalized bimetallic transition metal ethylene polymerization catalysts, Coord. Chem. Rev., 2018, 372, 101–116 CrossRef CAS.
- C. Su, Z. L. Chen, Q. Feng, F. S. Wei, M. L. Zhang, A. N. Mo, H. H. Huang, H. C. Hu and D. C. Liu, Highly efficient visible-light-driven CO2-to-CO conversion by coordinatively unsaturated co-salen complexes in a water-containing system, Inorg. Chem., 2022, 61, 19748–19755 CrossRef CAS PubMed.
- M. Azam, U. Kumar, J. O. Olowoyo, S. I. Al-Resayes, A. Trzesowska-Kruszynska, R. Kruszynski, M. S. Islam, M. R. Khan, S. F. Adil, M. R. Siddiqui, F. A. Al-Harthi, A. K. Alinzi, S. M. Wabaidur, M. R. Siddiqui, M. R. Shaik, S. L. Jain, M. A. Farkhondehfal and S. Hernandez, Dinuclear uranium(VI) salen coordination compound: an efficient visible-light-active catalyst for selective reduction of CO2 to methanol, Dalton Trans., 2020, 49, 17243–17251 RSC.
- V. Béreau, H. Bolvin, C. Duhayon and J. P. Sutter, Bi-compartmental Schiff-base with peripheral ester functionalization: Synthesis and magnetic behavior of bimetallic Zn–Ln complexes (Ln = Dy, Tb, Gd), Eur. J. Inorg. Chem., 2016, 4988–4995, DOI:10.1002/ejic.201600924.
- V. Béreau, S. Dhers, J.-P. Costes, C. Duhayon and J.-P. Sutter, Syntheses, structures, and magnetic properties of symmetric and dissymmetric ester-functionalized 3d–4f Schiff base complexes, Eur. J. Inorg. Chem., 2018, 66–73 CrossRef.
- P. Thuery and J. Harrowfield, Structural variations in the uranyl/4,4′-biphenyldicarboxylate system. Rare examples of 2D → 3D polycatenated uranyl–organic networks, Inorg. Chem., 2015, 54, 8093–8102 CrossRef CAS PubMed.
- S. L. Hanna, X. Zhang, K.-I. Otake, R. J. Drout, P. Li, T. Islamoglu and O. K. Farha, Guest-dependent single-crystal-to-single-crystal phase transitions in a two-dimensional uranyl-based metal–organic framework, Cryst. Growth Des., 2019, 19, 506–512 CrossRef CAS.
- P. Thuéry and J. Harrowfield, Structural variations in the uranyl/4,4′-biphenyldicarboxylate system. Rare examples of 2D → 3D polycatenated uranyl–organic networks, Inorg. Chem., 2015, 54, 8093–8102 CrossRef PubMed.
- K. Q. Hu, Z. W. Huang, X. B. Li, Y. Cheng, X. H. Kong, L. Mei, L. W. Zeng, Z. H. Zhang, J. P. Yu, J. K. Gibson, Z. F. Chai, H. Z. Kou and W. Q. Shi, Tailored persistent radical-containing heterotrimetal–organic framework for boosting efficiency of visible/NIR light-driven photocatalytic CO2 reduction, Adv. Funct. Mater., 2022, 33, 2213039 CrossRef.
- J. W. Wei, S. M. Zhang, J. L. Sun, T. Liang, Z. J. Li, Z. H. Li, X. L. Yi, R. T. Xiong, J. Y. Deng, Z. B. Yu, S. F. Wang and Y. P. Hou, Z-scheme CoAl-layered double hydroxide/indium vanadate heterojunction for enhanced and highly selective photocatalytic reduction of carbon dioxide to carbon monoxide, J. Colloid Interface Sci., 2023, 629, 92–102 CrossRef CAS PubMed.
- K. R. Gorantla and B. S. Mallik, Mechanistic insights into cobalt-based water oxidation catalysis by DFT-based molecular dynamics simulations, J. Phys. Chem. A, 2022, 126, 3301–3310 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, characterization results and computational methods. CCDC 2358409 for (IHEP-101) and 2358410 for (IHEP-102). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qi01578a |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2024 |