
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal centers and aromatic moieties in Schiff base complexes: impact on G-quadruplex stabilization and oncogene downregulation†
Aurane
Froux
 ab,
Luisa
D'Anna
a,
Aurianne
Rainot
ac,
Camille
Neybecker
b,
Angelo
Spinello
a,
Riccardo
Bonsignore
a,
Raphaël
Rouget
b,
Guillaume
Harlé
b,
Alessio
Terenzi
a,
Antonio
Monari
c,
Stéphanie
Grandemange
*b and
Giampaolo
Barone
*a
ab,
Luisa
D'Anna
a,
Aurianne
Rainot
ac,
Camille
Neybecker
b,
Angelo
Spinello
a,
Riccardo
Bonsignore
a,
Raphaël
Rouget
b,
Guillaume
Harlé
b,
Alessio
Terenzi
a,
Antonio
Monari
c,
Stéphanie
Grandemange
*b and
Giampaolo
Barone
*a
aUniversità di Palermo, Department of Biological, Chemical and Pharmaceutical Sciences and Technologies Viale delle Scienze, Edificio 17, 90128 Palermo, Italy. E-mail: giampaolo.barone@unipa.it
bUniversité de Lorraine, CNRS, CRAN, F-54000 Nancy, France. E-mail: stephanie.grandemange@univ-lorraine.fr
cUniversité Paris Cité and CNRS, ITODYS, F-75006 Paris, France
First published on 18th July 2024
Abstract
We present the synthesis and characterization of novel square planar transition metal complexes of Schiff base ligands, which act as guanine quadruplex binders and stabilizers. The complexes stabilize quadruplexes related to telomere stability or present in oncogene regulatory sequences, as determined by optical spectroscopy, pointing out the emergence of selectivity towards specific structures or sequences. These results are supported and rationalized by molecular modeling simulations. Furthermore, we show that the treatment of cancer cell lines with our complexes is associated with the increase in the number of nuclear guanine quadruplexes and the downregulation in the expression of the considered oncogenes. Remarkably, only very moderate cytotoxicity can be observed for all complexes. These results pave the way for the development of selective anticancer treatment by metal compounds targeting the expression of specific oncogenes.
Introduction
Guanine quadruplexes (G4) are nowadays recognized as fundamental nucleic acid motifs with important regulatory and protective functions in cells.1 As such, their deregulation has also been linked to the emergence of debilitating and life-threatening diseases, including cancer and neurodegenerative disorders.2–4 Their presence in the genome of microorganisms such as viruses and bacteria has also been recently pointed out.5–8 G4s could represent ideal pharmacological targets to finely tune and regulate cellular responses.4,9 Yet, the widespread use of G4-targeting drugs is still hampered by a general lack of selectivity, which may correlate with increased, and in some cases heavy, secondary effects. Clearly the rational design of drugs aimed at controlling the G4 cellular level will greatly benefit from a deeper understanding of the structural interplay between the nucleic acids and the chosen drugs. Indeed, from a structural point of view, G4s share a common rigid core, constituted by a superposition of tetramers composed of four guanines stabilized by Hoogsten hydrogen bond patterns.1,10 The G4 superstructure is also highly dependent on π-stacking interactions and shows the concomitant presence of a rigid core and flexible backbone and loop units. The flexibility of the loops and the possible orientation of the glycosidic bond also lead to the well-known G4 polymorphisms reflected in the presence of parallel, antiparallel, and hybrid structures.9,10G4 structures can form in both RNA and DNA sequences and may arise from single or multiple nucleic acid strands.1 However, single-stranded DNA-based G4s are by far the more commonly encountered, as also shown by the larger number of solved structures. They are found in guanine-rich regions of the genome, and different bioinformatic tools exist to predict their presence from the analysis of a given sequence.11–13 In eukaryotic cells the presence of G4s is particularly important in telomeres, where they play a protective role on the chromosome ends, and in non-coding gene expression regions, where they may be seen as regulatory check-points.14 Indeed, telomere G4 stabilization, impeding their unfolding, is effective at inhibiting telomerase activity, thus preventing the emergence of immortality phenotypes in cancer cells.4 G4s have also been found in several oncogene promoters, such as kRAS, BCL2, or cMYC, which are all importantly involved in different steps of carcinogenesis.15–17 However, the G4 regulatory role in gene-expression is highly complex. While G4 stabilization is usually related to a downregulation of gene expression,18–21 in some cases G4 may recruit transcription factors that lead to upregulation of their expression.9,22 Furthermore, in some crucial oncogene promoting regions, such as c-KIT, multiple G4s exist in close spatial proximity, giving rise to a strong coupling, which may even influence their individual topology and biological role.23,24
Despite these difficulties, the development of G4 stabilizers is still considered a promising therapeutic strategy, and indeed some G4-targeting drugs have reached the clinical test stage.25,26 Effective stabilizers need to show a strong G4-binding selectivity compared to their propensity toward double-helical DNA (B-DNA), to avoid global genomic instability.27 Furthermore, a preferential selectivity for only a limited number of G4 sequences, presenting a peculiar structure and topology, would be desirable.
Given the global structural features of G4s, square planar metal complexes are among the motifs of choice in the design of G4 stabilizers.14,28,29 Indeed, the presence of extended π-conjugated systems allows for a favourable end-stacking with the G4 tetrads, while minimizing the competitive intercalation into DNA structures. Furthermore, peripheral positive charges are clearly beneficial in increasing water solubility and in favouring the stabilization of the G4/ligand aggregate by developing electrostatic stabilization with the nucleic acid backbone phosphate groups. Different complexes of transition metal ions have been proposed as suitable G4 stabilizers30 which have been largely tested on DNA31–36 and RNA oligomers, as well as in vitro.26 As for the ligand counterpart, salphen moieties have proven their efficacy in providing an ideal π-stacking scaffold and, most importantly, in increasing their selectivity versus G4 compared to B-DNA.14,32,34,37
However, the effects of the extension of the ligand π-conjugation on the G4 binding and selectivity might be explored in more depth, together with the fine role of the positively charged substituents in improving the electrostatic stabilization. Motivated by these considerations, we present a systematic study on square planar transition metal complexes of Schiff bases as G4 binders. In particular, we assess the binding selectivity towards different relevant G4 sequences present in human oncogenes and telomeres. Furthermore, we also examine the often-overlooked role of the central metal ion in promoting G4 binding and selectivity towards a specific sequence. Finally, we also show that G4 stabilization nicely correlates with the downregulation of the corresponding specific oncogene, even when cytotoxic effects are rather limited. Therefore, our results provide a general framework for the development of G4-oriented therapies specifically targeting the expression of selected oncogenes, thus reducing systemic toxicity and side effects of chemotherapeutic approaches.
Results and discussion
Synthesis and characterization
Salphen-like metal complexes have already proven their efficiency in promoting selective binding to G4 structures. Here, we have focused on improving their potentiality by acting on two structural/electronic factors, namely: (i) the introduction of a 4-methyl-pyridinium group to bring peripheral positive charges on the chelating ligand and maximize electrostatic interactions with the DNA backbone, and (ii) varying the extension of the π-conjugated system on the N–N bridge, tuning the π-stacking with the G4 quartets. The main proposed compounds together with their synthetic routes are presented in Scheme 1. Note that our proposed modifications of the aldehyde moiety have led to the development of an extensive and modulable library of potential ligands, all bearing two formally positive charges on the lateral substituents. To enlarge the chemical space of our potential G4 binders we have, indeed, considered complexation with different metal ions, namely Zn(II), Ni(II), Cu(II), Pd(II), and Pt(II) (Scheme 1). Furthermore, the obtained compounds were grouped based on the extension of the π-conjugated substituents belonging to the salen, salphen and salnaphen families, respectively. | ||
| Scheme 1 Reaction pathways for the synthesis of compound B and complexes 1–12. | ||
An efficient synthetic route was devised to allow the production of our extensive library of compounds. To this aim, 5-chloromethylsalicylaldehyde was functionalized with 4-methylpyridine, via a nucleophilic substitution reaction conducted in the tetrahydrofuran solvent, yielding the positively charged pyridinium B (Scheme 1), which represents the fundamental building block for obtaining the whole library of metal complexes.
The final compounds, belonging to the class of Schiff base metal complexes, have been obtained by template synthesis, during which B is condensed, in the presence of the given metal salt, with three different diamines: o-phenylenediamine to give the salphen family, ethylenediamine to provide the salen family, and 2,6-naphthalenediamine to afford the salnaphen family.
Metal complexes 1–12 were obtained in a one-step reaction, without previous isolation of the ligand, based on similar reported procedures.38–41 Indeed, aldehyde B was directly condensed with the chosen primary diamine to obtain the Schiff base ligand, which was deprotonated with a strong base, before coordination with the corresponding metal salt (Scheme 1). The explicit structure of each complex is listed in Fig. S1.†
For the synthesis and purification of the Pt(II) complex 5, we opted for a successive precipitation and counterion exchange strategy as described in the Experimental section. The chloride salt was firstly exchanged with sodium hexafluorophosphate. The final Pt(II) product was obtained after ion exchange with the Amberlite Cl-form resin, to give the Pt(II) complex 5 as a chloride salt. While this strategy induced a reduction of the global reaction yield, it significantly improved the purity of the final product. The salen Pt(II) and salnaphen Pd(II) and Pt(II) complexes were not obtained with the required purity and were discarded for this study.
Intermediate B and complexes 1–12 have been characterized by 1H-NMR spectroscopy (Fig. S2†), except for the paramagnetic copper(II) complexes, as well as mass spectrometry (Fig. S3†) and elemental analysis (Experimental section). Concerning the NMR signals of the symmetric metal complexes, each peak obviously corresponds to double the number of protons, as specified in the labelling schemes presented in each spectrum of Fig. S2.†
DNA-binding in solution
The binding strength of our metal complexes towards different G4 sequences, adopting different topologies, has been investigated using Förster Resonance Energy Transfer (FRET) melting assays. In addition, the selectivity towards G4 was also assessed by comparing the affinity of our complexes for either G4s or for a self-complementary double-stranded DNA (dsDNA) mimicking the double-helical B-DNA conformation. The chosen G4 sequences correspond to a variety of biologically relevant G4 motifs, including the human telomers (hTelo)42,43 and oncogene promoters BCL2,44kRAS,45cMYC,46 and RET.47 Furthermore, the three G4 units present in the whole cKIT24,48 sequence have been used. The sequences of all G4s used are listed in Table S1.†To assess G4 stabilization, we labelled each oligomer with a FRET active fluorophore and monitored the changes in the melting temperature (TM) after the addition of our complexes in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 [DNA]:[metal complex]. An increase in TM reveals stabilization induced by complex binding.
:5 [DNA]:[metal complex]. An increase in TM reveals stabilization induced by complex binding.
The data in Fig. 1A summarize the changes in the melting temperature (ΔTM) of each DNA sequence bound to the corresponding metal complex (see also Table S2† for the numerical values of ΔTM). Importantly, none of the 12 compounds presented significant stabilization of dsDNA, while a statistically significant increase in the melting temperature was observed for the G4s. As of note, and mainly in the presence of compounds 1, 3, 10 and 12 the stabilization achieved using a 1:5 ratio was so strong that the instrumental maximum of ΔTM = 30 °C was exceeded. This is particularly noticeable for compound 10 interacting with cKit2, BCL2, cMYC, and RET G4s.
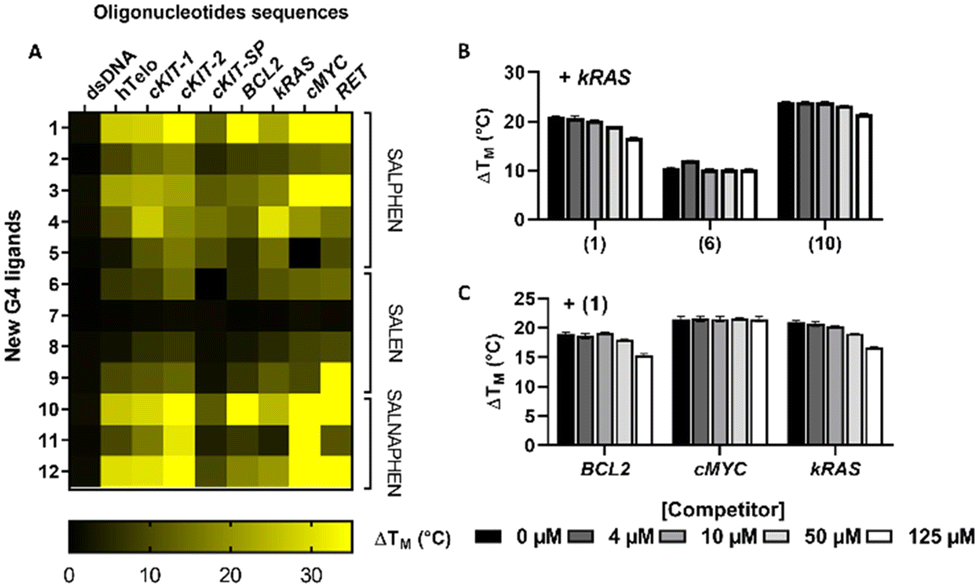 | ||
| Fig. 1 (A) Heatmap of the stabilizing behaviour of the 12 metal complexes on the selected G4-folded oligonucleotides obtained by the FRET melting curve assay. The darkest region corresponds to the absence of stabilization and the brightest yellow to the highest induced stabilization ΔTM (°C). (B) Stabilization of kRAS G4 by the three Ni(II) compounds 1 (salphen), 6 (salen) and 10 (salnaphen), in the presence of increasing concentrations of duplex DNA competitor (ctDNA). (C) Stabilizing ability of compound 1 towards BCL2, cMYC and kRAS G4, in the presence of increasing concentrations of the ctDNA competitor. DNA was dissolved in 0.2 μM solutions, in the presence of 1 μM of the metal complex, in 60 mM or 10 mM potassium cacodylate buffer at pH 7.4. | ||
Some additional global tendencies can also be extracted from our data. The salen family members (6–9) appear as the weakest G4 stabilizers both in term of melting temperature increase and of the number of targeted G4 which are stabilized. Conversely, the salnaphen group (10–12) clearly outstands the other families in promoting the strongest G4 stabilization. This result is not surprising and clearly highlights the beneficial role of the extended conjugation pattern in promoting favourable interactions with the guanine tetrads.
Ni(II) metal complexes (complexes 1, 6 and 10) appear to be the most efficient G4 stabilizers compared to other analogues. This fact is particularly evident when comparing compound 1 to the isostructural Pd(II) and Pt(II) compounds 4 and 5, indicating the important role of the metal centre. Interestingly, the affinity trend obtained by FRET measurements of salphen complexes 1, 4 and 5, decreases with the increase of the atomic number of group 10 elements, in the order Ni > Pd > Pt. The stronger affinity of Ni compound 1, compared to the analogous Pd and Pt compounds 4 and 5, could be attributed to the higher positive charge on the center of the metal complex, determined by the higher ionic character of the Ni–N and Ni–O bonds, compared to that of the analogous Pd/Pt–N and Pd/Pt–O bonds, being characterized by a higher covalent nature.49
Finally, while neither specificity nor selectivity toward a particular G4 sequence emerged unambiguously from our study, our results suggest a preference of the metal complexes for parallel topologies. Indeed, parallel G4 sequences (BCL2, kRAS, c-MYC, RET and c-KIT2) are stabilized by most of the metal complexes. Conversely, the anti-parallel c-KIT-SP (Fig. 1A) is the least stabilized sequence.
Thus, these results clearly show that our metal compounds, particularly 1, 3, 10 and 12, may be classified as potentially strong G4 stabilizers.
To better assess their selectivity, we have evaluated the specificity of some G4 stabilizers with kRAS sequence versus dsDNA. To this aim, FRET melting assays were performed in the presence of increasing concentrations of the unlabelled B-DNA competitor (calf thymus DNA, ctDNA) and Ni(II) complexes 1, 6 and 10. As indicated in Fig. 1B, even in the presence of both G4 and dsDNA, the three compounds induce a strong and specific stabilization of the kRAS G4. Indeed, at the maximum concentration of dsDNA, complex 1 showed a reduction of the stabilization capability towards G4 of around 20%. The other complexes were even less affected by the presence of the competitor DNA. Indeed, complex 10 presents a reduction of stabilization amounting to 10%, while remarkably, the stabilization potential of complex 6 was almost unaffected and has been reduced by only 2%. The specificity of 1 for different G4 structures, namely, BCL2, cMYC, and kRAS, has been deeply analysed. A competitive FRET assay showed a reduction of about 20% of the stabilization of BCL2 and kRAS G4s in the presence of 125 μM dsDNA (Fig. 1C). Interestingly, cMYC G4 was very strongly stabilized by complex 1 at an initial ratio of 1:5, with the consequence that just a minor increase in FAM fluorescence was observed when the temperature is increased (see Fig. S4†). This exceptional stabilization did not allow us to determine the G4 melting temperature upon interaction with complex 1 under these experimental conditions. For this reason, the compound concentration was reduced from 1.0 to 0.60 μM to reach a [G4 DNA]/[complex 1] of 1:3. Even under these conditions, no reduction of cMYC G4 stabilization was observed also at a higher concentration of double helical DNA (<0.2%).
We have also investigated the G4 binding ability of compounds 1, 6 and 10 by UV-vis and circular dichroism (CD) spectroscopy, using again ctDNA as a comparison.
For the binding studies, two different double-stranded DNA sequences were used: UV-vis and CD experiments were performed using calf-thymus DNA, while FRET experiments were conducted in the presence of dsDNA, a self-complementary 20-mer oligonucleotide bearing a central linker (see Table S1†). Despite this difference, both sequences adopt the same conformation and display the same spectral signals.50
The evolution of DNA topologies upon increasing the amounts of metal complexes was monitored by circular dichroism measurements. After checking the correct folding of the parallel quadruplexes kRAS, BCL2 and cMYC by the negative and positive ellipticities at 240 and 260 nm, respectively, we evaluated how increasing amounts of 1, 6 and 10 would perturb such patterns. As shown in Fig. 2, the addition of 1 and 10 led to a general hypochromic effect at 260 nm in all the G4 spectra. This is coherent with significant binding, and so confirms the FRET experiment. Interestingly, CD spectra of ctDNA were strongly modified by the addition of compounds 1 and 10 (Fig. 2A and E). A strong interaction is suggested by the consequent hyperchromic shift of the 270 nm band for both compounds 1 and 10. Notably, the increase of 1 corresponds to an induced positive band at 366 nm and a negative one at 306 nm, while 10 caused the appearance of a positive band around 360 nm and the accentuation of the ctDNA negative one at 250 nm. Such results suggest, on the one hand, that the topology of the G4 is globally maintained, since no major changes are observed in the CD spectra. On the other hand, the changes in the ctDNA CD spectrum are probably connected with the presence of a CD band induced by the metal complexes and/or with a stronger alteration of the DNA structure. Finally, compound 6 did not yield significant changes in the CD spectra of ctDNA or G4s (Fig. S5†). The lack of spectral changes of the Ni(II) salen complex 6 suggests that an external binding mode, different from intercalation, is occurring here. On the other hand, the perturbation of the CD spectra of ctDNA shown in Fig. 2A and E, in the presence of salphen and salnaphen Ni(II) complexes 1 and 10, points out the occurrence of consistent structural modification of the double helix DNA after intercalation. The strong DNA binding of compounds 1 and 10 is also witnessed by the appearance of induced CD bands at around 300 and 450 nm, respectively.
 | ||
| Fig. 2 Compounds 1 and 10 strongly interact with DNA. Circular dichroism spectra of ctDNA (a and e), kRAS (b and f), BCL2 (c and g) and cMYC (d and h) with increasing concentrations of complex 1 (a–d) or complex 10 (e–h), in 50 mM Tris HCl, 100 mM KCl, pH 7.4. | ||
The characterization of the G4 binding ability of Ni(II) compounds 1, 6 and 10 was extended by determining their intrinsic DNA-binding constants (Kb), by titrating each complex with increasing amounts of nucleic acid. We monitored the UV-vis spectra of the corresponding complex in the 300–400 nm region, where the complex exhibits a characteristic absorption band (1: 366 nm; 6: 316 nm; 10: 350 nm) and where none of the investigated oligomers absorb (Fig. S6–S8†). Intrinsic binding constants could be determined by applying the Thordarson equation and are listed in Table 1.51 The three tested compounds present higher Kb values with ctDNA than G4 structures. The high Kb values obtained from ctDNA titration are consistent with the important changes in the ctDNA CD spectra. Importantly, the absence of stable double helix stabilization observed in the FRET assay does not necessarily indicate a lack of interaction.
| Binding constant KB (M−1) | |||
|---|---|---|---|
| 1 | 6 | 10 | |
| ctDNA | (3.83 ± 0.06) × 104 | (1.68 ± 0.06) × 104 | (1.17 ± 0.06) × 105 |
| kRAS | (5.89 ± 0.12) × 103 | (5.74 ± 0.16) × 103 | (4.79 ± 0.05) × 103 |
| BCL2 | (2.40 ± 0.02) × 102 | (3.89 ± 0.02) × 103 | (7.67 ± 0.06) × 103 |
| cMYC | (4.25 ± 0.17) × 104 | (1.37 ± 0.02) × 104 | (7.78 ± 0.15) × 103 |
Moreover, the small binding constants obtained by the UV-vis titration of G4 sequences consistently correlate with the poor hypochromism observed for the CD spectra of the titrated G4. It is worth pointing out that the significant stabilization of G4 induced by our compounds, as shown by FRET experiments, does not align with the value of the binding constants obtained by UV-Vis and the impact on the CD signals of the used sequences. While a perfect agreement among the three methods, FRET, UV-vis, and CD, used to characterize intermolecular interactions would be ideal, such coherence may be unrealistic due to the intrinsic differences in the experimental approaches. Specifically, the significant increase in ΔTM observed in FRET experiments may arise from interactions that do not markedly alter the conformation of G4s, and therefore do not significantly impact their CD spectra. This phenomenon can be primarily attributed to system stabilization via external electrostatic attraction between the negative phosphate groups and the positive charges of the metal compounds, rather than specific non-covalent interactions like π–π stacking. In contrast, the pronounced changes in the CD spectra of ctDNA upon interaction with compounds 1 and 10 (Fig. 2A and E) suggest that these compounds intercalate ctDNA, leading to substantial conformational changes such as base stacking disruption. Such structural modification does not occur after the interaction of our metal complexes with G4s, which possess a rigid core as also evidenced by MD simulations (vide infra). This clearly points to the occurrence of a top-stacking binding mode with the drug candidate overhanging on the peripheral tetrad.
In silico characterization
To better understand the binding capacity of the different transition metal complexes towards G4s, we have performed all-atom molecular dynamics simulation to study the interaction of Ni-salphen (1) and Ni-salnaphen (10) complexes with kRAS and BCL2's G4s, respectively.As shown in Fig. 3 and 4 the formation of a stable complex maintained by favourable π-stacking and electrostatic interactions is observed in the early stage of the unbiased MD simulation and maintained during the entire μs time-span, especially for the interaction with kRAS. This can be most notably observed by the time evolution of the root mean square deviation (RMSD) and of the distance between the centre of mass of the transition metal and the G4. Interestingly, as it can be also observed from the representative snapshot reported in Fig. 3, the interaction with the transition metal complex involves the interaction with both the tetrads and the G4 loops. This behaviour seems particularly important for the interaction with BCL2's G4. Indeed, a rather large structural reorganization taking place in the 500 ns range is observed for this structure while interacting with the salphen complex (1). This is also coherent with the lower binding constant experimentally determined. In the case of the salnaphen system, while a persistent aggregate is observed, the latter is not forming an optimal π-stacking with the tetrad (Fig. 4) and is, instead, opportunistically interacting with the extruded loop bases, suggesting a less pronounced stabilization of the G4. Conversely, for both G4s the interaction with the ligand does not induce any important structural deformation of the G4 and is only partially affecting the dynamics of the flexible loop present in the kRAS oncogene, in coherence with the CD spectra. Interestingly, interactions with the loops are also favourable for maintaining the aggregate as shown in Fig. 3. Globally, MD simulations confirm that the stabilization of kRAS's G4 by our transition metal complexes is more favourable than for its BCL2 counterpart. This is coherent with the experimental observations and is most probably due to the fact that kRAS's G4 allows a more optimal stacking scaffold to accommodate large and rigid metal complexes.
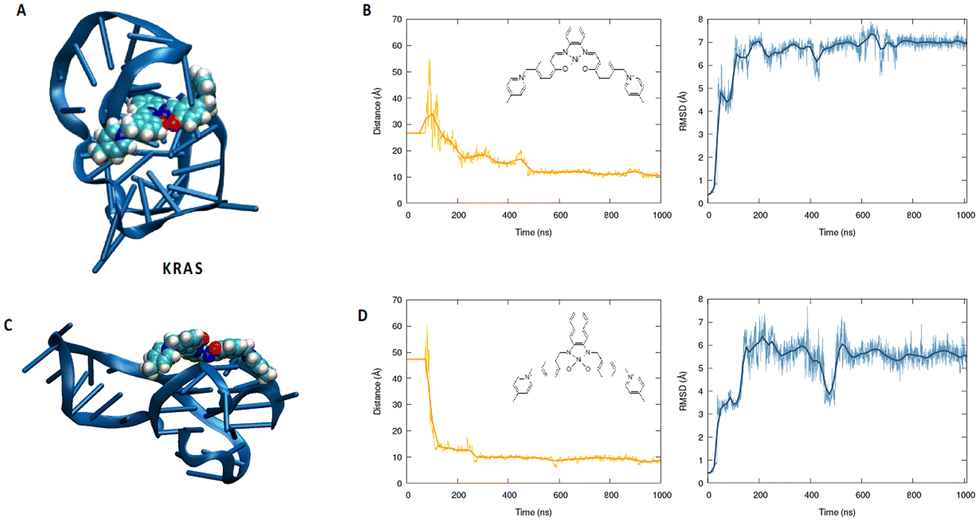 | ||
| Fig. 3 Representative snapshot of the interaction between Ni-salphen compound 1 (A) and Ni-salnaphen compound 10 (C) complex and the kRAS’ G4 as obtained by MD simulations. The corresponding time evolution of the RMSD and of the distance between the centres of mass of the G4 and the complex are reported in panel (B) and (D), respectively. | ||
 | ||
| Fig. 4 Representative snapshot of the interaction between Ni-salphen 1 (A) and Ni-salnaphen 10 (C) complex and the BCL2‘s G4 as obtained by MD simulations. The corresponding time evolution of the RMSD and of the distance between the centres of mass of the G4 and the complex are reported in panel (B) and (D), respectively. | ||
G4 stabilization in cells
Encouraged by the promising results obtained in solution and in silico, we have investigated the effect of G4 stabilization in cells. Notably, the ability of compounds 1, 6 and 10 to affect the G4 landscape in cancer cell lines was assessed by the immunocytofluorescence assay. We hypothesize that our compounds should induce an increase in the number of cellular G4s. Thus, pancreatic cancer T3M4 cells were treated with increasing concentrations of compounds 1, 6 or 10, as shown in Fig. 5. Nuclear G4 foci were counted and the median number of foci for each condition was compared with the control, which was treated with the DMSO vehicle. While compound 1 affected G4 numbers solely at 50 μM, compounds 6 and 10 had more important effects. The counted nuclear foci increased with the concentration of the metal complex, revealing a dose-dependent response of the G4 presence in cells treated with compounds 6 and 10. Particularly, compound 10 provoked a strong increase in the nuclear G4 numbers at 5 μM, as compared to the control (Fig. 5). Thus, our results suggest that these compounds are efficient at shaping the amount of G4, and thus the genome landscape in cancer cells. Furthermore, these results confirm that our complexes induce G4 stabilization in a cellular environment, coherent with the in vitro results. | ||
| Fig. 5 Immunodetection of nuclear G4 in cancerous cells and the impact of G4 stabilizers. T3M4 cells were treated for 24 hours with 5, 10 or 50 μM of compound 1, 6 or 10. Nuclear G4s were detected by immunocytofluorescence, using BG4 antibodies (red channel). Nuclear BG4 foci were enumerated considering 100 cells. Mean ± SEM. (n = 3); **p < 0.01; ***p < 0.001; ****p < 0.0001. One-way ANOVA. | ||
Oncogene expression regulation
Since we have achieved the stabilization of G4 sequences involved in the modulation of gene expression, we investigated if the transcription level of the studied kRAS, cMYC, BCL2, and RET oncogenes was altered in the presence of our compounds.While kRAS and cMYC expression levels were evaluated using T3M4 cells, BCL2 and RET were quantified on T47D cells, according to the basal gene expression level, specific to each cell line previously investigated (data not shown). The cells were treated for 6 hours with 50 μM of each complex, and the transcript level was assessed by RT-qPCR. The results are expressed relative to the control (Fig. 6). A downregulation of the kRAS transcript level is significantly observed after the treatment of the cells with 6 of the 12 complexes (Fig. 6A). However, the Zn(II) compounds, especially 2 and 11, showed a strong reduction in kRAS expression by around 3-fold. In T3M4 cells, cMYC was also downregulated, notably by compounds 1, 2, 5, 10 and 11 by about 1.4-fold and even more strongly by compound 12, which resulted in a 1.9-fold reduction (Fig. 6A).
 | ||
| Fig. 6 The expression level of the considered oncogene was evaluated by RT-qPCR after treating cells for 6 hours with 50 μM of the corresponding compound. (A) kRAS and cMYC transcript levels were evaluated on T3M4 cells, while BCL2 and RET (B) were assessed on T47D cells. Data are shown as expression level, relative to the control conditions (reported for 1). For representation, the control (CTRL) is the mean of different controls. Significance for each treated condition is expressed compared to its internal control. Mean ± SEM. (n = 4) t-test. | ||
Conversely, BCL2 transcripts were downregulated only by complex 5 (Fig. 6B) with a fold-change of approximately 1.6 and were globally unaltered by the other compounds. Finally, RET was more affected compared to the other transcripts since 8 of the 12 complexes led to its downregulation by around 2-fold (Fig. 6B).
If these results do not correlate perfectly with the G4 stabilization pattern observed in solution (Fig. 1A), they confirm the global trend. Namely they confirm that our complexes can stabilize the G4 folding, and thus, modulate the expression level of the investigated oncogenes, mostly leading to their significant downregulation.
We tested the expression of a large range of G4s contained in oncogene promoters and demonstrated the ability of our complexes to strongly stabilize them. The list of the oncogene expression evaluation is larger than those generally found in the literature and highlights their capacity to efficiently downregulate a wide panel of G4-driven oncogenes. No real specificity towards a particular oncogene has been observed (except for BCL2 being downregulated solely by compound 5), but a kind of parallel conformation preference emerges. If those results are at a previous stage in the characterization of the oncogene modulator capacity of those new compounds, major efforts could be made for a deeper and larger identification of the gene targets, through RNA sequencing analysis after treating the cells with some G4 stabilizers. This approach would allow a strong characterization and identification of the molecular targets under G4 landscape modulation.
Effect on cell viability and proliferation
The observed stabilization of G4 and the inhibition of oncogene expression by our metal complexes led us to determine their impact on cell viability and proliferation in aggressive cancer models. Indeed, kRAS, RET and cMYC proteins play key roles in regulating cell proliferation and the anti-apoptotic protein BCL2 is particularly involved in cell survival.15,27,52–54Cell viability after treatment with the G4 stabilizers 1–12 has been evaluated on three different human cancer cell lines, derived from human pancreatic ductal adenocarcinoma (T3M4), triple-negative (MDA-MB-231) and hormone-dependent (T47D) breast ductal carcinoma. The half maximal inhibitory concentration (IC50) of the 12 complexes was determined after 48 hours of incubation, by exposing the cells to a range of concentrations spanning 1 to 200 μM (Fig. 7A and S9–S11†). For some complexes (e.g.2 on MDA-MB-231 cells), the inhibition of 50% of the cellular population was not reached under such conditions (Fig. S11†). Therefore, the concentration of 50 μM for a duration of 48 hours was chosen to determine the viability of the three cell lines. The majority of our compounds significantly reduced the viability of the three cell lines tested (Fig. 7B–D). However, MDA-MB-231 cells appeared less sensitive to the treatment compared to T3M4 and T47D cells. As a matter of fact, 5 complexes (2, 3, 6, 9 and 11) showed IC50 values higher than 200 μM for MDA-MB-231 cells, while only one (9) for T3M4 and none for T47D cells. This difference could be associated with the lower cell line doubling time of the MDA-MB-231 cell line (44 ± 4.6 hours) compared with T3M4 (20 ± 0.5 hours) and T47D (30 ± 1.3 hours) models (Fig. S12†). Indeed, it is widely reported that G4 formation is dependent of cell cycle dynamics. G4 structures are described to preferentially form during the S phase, due to the replication fork.55,56
 | ||
| Fig. 7 Half maximal inhibitory concentration IC50 determination on T3M4, MDA-MB-231 and T47D cell lines. (A) IC50 of the 12 compounds on T3M4, T47D and MDA-MB-231 cell models at 48 hours of treatment, determined by the crystal violet assay. Mean ± SEM (n = 5). Relative viability of pancreatic cancer cells T3M4 (B), hormonal-dependent breast cancer cells T47D (C) and triple-negative breast cancer cells MDA-MB-231 (D) upon treatment with 50 μM of the corresponding compound for 48 hours, determined by crystal violet (n = 4). Mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. One-way ANOVA. | ||
The results obtained point out that most of the 12 compounds have strong stabilizing effects in solution, except for zinc(II) compound 7. In the cellular environment, the stabilizing activity was confirmed by the analysis of oncogene expression, pointing out that only compound 9 does not repress gene expression on any of the four tested oncogenes. To confirm these results, we have chosen three compounds to analyze by immunocytofluorescence their impact on the number of G4 foci in cells. As indicated in Fig. 5, while the increase of the G4 foci is more relevant with compound 10 than with compound 1, the three nickel(II) compounds 1, 6 and 10 show significant stabilizing activities for G4 structures in a cellular environment. Thus, although in solution data indicate strong stabilization activity of most of compounds, it appears that in a cellular context the binding/stabilizing activity of complexes can be different because of their entry into the cells and/or the nucleus. However, the use of a large variety of experiments in solution and in cells, allowed us to highlight the compounds with the most relevant activity.
Furthermore, the real time proliferation rate of the treated cancer cells with increasing concentrations (0, 1, 10 and 50 μM) of our metal complexes was followed for 72 hours. Ni(II) compounds 1, 6 and 10 showed a strong antiproliferative capacity at 50 μM (Fig. 8), generally after 48 hours of treatment. No dose-dependent response was observed between 10 and 50 μM. Interestingly, some complexes showed complete inhibition of cell proliferation over the time-span, such as 1 on T3M4 (Fig. 8A) and T47D (Fig. 8B), suggesting a total arrest of the cell cycle. The proliferation inhibition rate at 48 and 72 hours of the other complexes is reported in Table S3.† Globally, we may conclude that while our G4 stabilizers affect the proliferation and viability of all the three tested cancer cell line models, they were poorly cytotoxic as underlined by the need to use high doses (approx. 50 μM) to induce a significant proliferation inhibition.
 | ||
| Fig. 8 The proliferation of T3M4 (upper panel), T47D (middle panel) and MDA-MB-231 (lower panel) cell lines was followed by the real-time cell imager Cytonote (IPRASENSE®), over 72 hours while treated with different concentrations of nickel(II) compounds 1 (A–C), 6 (D–F) and 10 (G–I). Mean ± SEM (n = 3); *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Two-way ANOVA. | ||
Those effects at 50 μM over time are in perfect coherence with the ones obtained from the evaluation of oncogene expression (Fig. 6). Indeed, compounds 1, 6 and 10 significantly downregulate kRAS, cMYC and RET oncogenes, and are known as the key regulators of cell proliferation.27,52
On the other hand, our results on T3M4 cells treated with compound 1 suggest a total arrest in the cell cycle progression. Further experiments and analysis of the cell cycle should be performed to characterize these results.
Experimental section
Synthetic procedures
:1 (5 mL), and solid NaOH (40.0 mg; 1 mmol) was added before stirring at room-temperature for 20 minutes. Parallelly, an aqueous solution of 1,2-phenylenediamine (54.1 mg; 0.5 mmol) was prepared with M(ClO4)2·6H2O (0.55 mmol) (with M = Ni, Zn or Cu) and dropwise added to the first solution. The resulting mixture was left stirring at room-temperature for 4 hours. The obtained precipitate was filtered under reduced pressure and washed with cold water, ethanol and diethyl ether. Salen metal compounds (6, 7, and 8) were synthesized as the salphen compounds, but pure ethylenediamine (33.4 μL; 0.5 mmol) was added instead of 1,2-phenylenediamine. Salnaphen compounds (10, 11, and 12) were obtained as the salphen complexes but o-phenylenediamine was replaced with 2,6-naphthalenediamine (79.2 mg; 0.5 mmol). The obtained salnaphen solids were recrystallized in a solution of acetonitrile, acetone and water 1:2:3 to obtain the final compounds.
:1 (30 mL), and NaPF6 (67.2 mg; 0.4 mmol) was added to the solution. The mixture was stirred at room-temperature, under an inert atmosphere for 2 hours, before adding the same quantity of NaPF6 and allowed to react for 24 hours. The resulting solid was harvested by filtration and washed with abundant cold water and diethyl ether. The red powder was then exchanged with Amberlite® IRA-402 Cl form ion exchange resin (Thermo Fisher Scientific) in water, under an inert atmosphere for 24 hours. The day after, the resin was washed with abundant cold water and ethanol. The solvents were eliminated by lyophilization. A reddish powder was recovered (37.6 mg; 47.4% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.55 (s, 1H), 9.04 (d, J = 6.1 Hz, 2H), 8.56–8.34 (m, 1H), 8.02 (d, J = 6.7 Hz, 3H), 7.70 (dd, J = 9.0, 2.2 Hz, 1H), 7.48 (dd, J = 6.4, 3.2 Hz, 1H), 7.16 (d, J = 8.9 Hz, 1H), 5.79 (s, 2H), 2.62 (s, 3H). Elemental analysis of C34H30Cl2N4O10Pt·8H2O (5). Calculated: C 43.60%; H 4.95%; N 5.98%. Found: C 43.57%; H 4.58%; N 6.53%. ESI-MS (m/z). Calculated for (C34H30N4O2Pt)2(Cl)2 [M]2+ 757.17; found 757.17.
DNA-binding in solution
:drug ratio equal to 1:5. DMSO never exceeded 0.1% of the final volume. Finally, FAM emission was acquired from 25 °C to 95 °C, with a stepwise increase of 1 °C every 30 s. Data were normalized from 0 to 1, and measurements were made in duplicate. The melting temperature of the G4 oligonucleotides in the presence of the metal complex was compared to the melting temperature of the G4 oligonucleotide alone, to give ΔTM. Also, the evolution of the stabilizer behaviour of 1, 6 or 10 towards G4 oligonucleotides (0.2 μM) was also monitored in a FRET melting competitive assay, with increasing amounts of non-fluorescent ctDNA.
Biological activity
:1) for 10 min, at −20 °C. After PBS 1× washes, blocking was performed using 3% BSA in PBS 1× at RT for 1 h, before incubation with a mouse anti DNA/RNA G-quadruplex antibody BG4 (Absolute Antibody, Ab00174-1.1) diluted 1:300 in the blocking solution, for 2 hours at RT. Cells were incubated with an anti-mouse Alexa Fluor™ 594-conjugated antibody (Invitrogen, A11032), diluted 1:2500 in PBS 1×, for 1 hour at RT. Nuclei were stained with Hoechst 33342 (Invitrogen) 1:10000 in PBS 1× for 5 min at RT. Coverslips were mounted with the FluorSave™ reagent (Millipore). After each step, cells were washed 3 times with PBS 1×, for 5 min each. Images were recorded on a hybrid fluorescence microscope (Revolve, ECHO). Nuclear focus quantification was established through biologically independent triplicate tests, where BG4-nuclear foci were detected and counted by automated focus detection/quantification from 100 nuclei under each condition using FiJI software macro FindFoci.68
 . The obtained relative viability was plotted against the decimal logarithm of the molecule concentration log([molecule]). The IC50 was determined using a non-linear regression on GraphPad Prism 8 software.
. The obtained relative viability was plotted against the decimal logarithm of the molecule concentration log([molecule]). The IC50 was determined using a non-linear regression on GraphPad Prism 8 software.
Conclusions
The designed and synthesized transition metal complexes are strong G4 stabilizers. CD and optical spectroscopy measurements in solution highlight their stabilization of specific G4 present in relevant oncogene promoters. Stabilization is considerably more pronounced for G4 than for double-stranded DNA. Moreover, the metal centre plays a pivotal role in modulating the affinity and stabilization of the nucleic acid folding. In particular, the stabilizing effect of Ni(II) compounds 1, 6 and 10 is on the average larger than that of the other considered metal complexes. In parallel, the size increase of the π-conjugated moiety on the N–N bridge enhances the stabilization of the G4 tetrads. In fact, the G4 stabilization strength is always in the order salnaphen > salphen > salen complexes. These results, confirmed by molecular modelling, highlight the establishment of a persistent binding between G4 and the considered metal complexes, in particular 1 and 10. Both computational simulations and experimental measurements, in particular the CD spectra, show that the G4-binding of the metal complexes does not induce noticeable structural deformations of the nucleic acid conformation, while it involves an interplay between binding with tetrads and loops.Tested on different cell lines, all metal complexes show rather limited cytotoxicity, witnessed by the high IC50 values, although partial inhibition of cell proliferation has been evidenced. On the other hand, Ni(II) compounds 1, 6 and 10 induce an increase in the number of nuclear G4s in the treated cell lines. This effect is dose-dependent and may be associated with their capacity to modulate the G4 landscape at the cellular level. Going a step further, the modulation of such structures can be associated with the downregulation of the tested oncogenes. However, despite G4 stabilization by the considered metal compounds, which has been proven in solution and in cells, we cannot exclude other targets that exist in the complex intracellular system. Moreover, the synthesized metal compounds may also play roles unrelated to oncogene downregulation. Considering that G4 can influence several cellular processes, including DNA replication, the induction of G4 structure stabilization by our molecules could also lead to a decrease in cell proliferation, without direct influence on oncogene regulation. Although in the absence of an absolute one-to-one correspondence between differential G4 stabilization and downregulation of oncogenes, our results prove that the use of metal complexes is beneficial for modulating gene expression. Furthermore, some specific selectivity can be achieved by targeting some genes of interest, e.g. the Pt(II) salphen compound 5 with BCL2 and Pd(II) salen compound 9 with RET. Such properties, accompanied by the low observed cytotoxicity, are important in reducing secondary effects of the possible treatments. Overall, our results suggest that the modulation of the equilibrium between the interaction with the loops and/or with the tetrad could be a factor to be exploited for a rational drug design aimed at increasing G4 selectivity.
Author contributions
Aurane Froux: Investigation, methodology, formal analysis, visualization, writing – original draft, and review & editing. Luisa D'Anna: in silico investigation and formal analysis. Aurianne Rainot: in silico investigation and formal analysis. Camille Neybecker: in cellulo investigation and methodology. Angelo Spinello: methodology and review & editing. Riccardo Bonsignore: Methodology and review & editing. Raphaël Rouget: Investigation and methodology. Guillaume Harlé: Methodology and review & editing. Alessio Terenzi: Conceptualization, methodology, formal analysis, writing – original draft, and review & editing. Antonio Monari: Conceptualization, methodology, formal analysis, writing – original draft, and review & editing. Stéphanie Grandemange: Conceptualization; methodology, formal analysis, writing – original draft, review & editing, and supervision. Giampaolo Barone: Conceptualization; methodology, formal analysis, writing – original draft, review & editing, and supervision.Data availability
- Fitting of the UV-visible spectroscopic data has been performed using the freely available software provided at the following link: https://app.supramolecular.org/bindfit/.- Molecular dynamics simulations have been performed using the NAMD free software (https://www.ks.uiuc.edu/Research/namd/) and the data were analysed and visualized using VMD free software (https://www.ks.uiuc.edu/Research/vmd/).
- Immunocytofluorescence analysis has been carried out using the freely available Fiji software (https://imagej.net/software/fiji/).
- Chemical structures have been drawn using the ChemDraw software (https://revvitysignals.com/products/research/chemdraw).
- Plots of spectral measurements have been produced using the GraphPad Prism software (https://www.graphpad.com/features).
- NMR spectra have been analyzed using TopSpin Bruker NMR software (https://www.bruker.com/it/products-and-solutions/mr/nmr-software/topspin.html).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
All authors thank GENCI and Explor computing centers and the Platform P3MB for computational resources, as well as ANR and CGI for financial support of this work through Labex SEAM ANR 11 LABEX 086, ANR 11 IDEX 05 02. The authors thank Prof A. Palumbo Piccionello, University of Palermo, for his support with mass spectrometry, and Prof C. Kowol and Dr W. Kandioller, University of Vienna, and Dr Y. Bernhard, L2CM - UMR CNRS 7053, University of Lorraine, for their support with elemental analysis. FRET experiments were performed at the AteN Center of the University of Palermo. The support of the IdEx “Université Paris 2019” ANR-18-IDEX-0001 is also acknowledged. AR is thankful for the co-founding of her Ph.D. program by the Labex Seam of Université Paris Cité and by the University of Palermo. The financial support of the European Union – NextGenerationEU through the Italian Ministry of University and Research under PNRR – M4C2-I1.3 Project PE_00000019 “HEAL ITALIA” CUP (B73C22001250006) is gratefully acknowledged.References
- D. Varshney, J. Spiegel, K. Zyner, D. Tannahill and S. Balasubramanian, The regulation and functions of DNA and RNA G-quadruplexes, Nat. Rev. Mol. Cell Biol., 2020, 21(8), 459–474, DOI:10.1038/s41580-020-0236-x.
- Y. Wang, J. Yang and A. T. Wild, et al., G-quadruplex DNA drives genomic instability and represents a targetable molecular abnormality in ATRX-deficient malignant glioma, Nat. Commun., 2019, 10(1), 943, DOI:10.1038/s41467-019-08905-8.
- R. Hänsel-Hertsch, A. Simeone and A. Shea, et al., Landscape of G-quadruplex DNA structural regions in breast cancer, Nat. Genet., 2020, 52(9), 878–883, DOI:10.1038/s41588-020-0672-8.
- N. Kosiol, S. Juranek, P. Brossart, A. Heine and K. Paeschke, G-quadruplexes: a promising target for cancer therapy, Mol. Cancer, 2021, 20(1), 40, DOI:10.1186/s12943-021-01328-4.
- G. Liu, W. Du and X. Sang, et al., RNA G-quadruplex in TMPRSS2 reduces SARS-CoV-2 infection, Nat. Commun., 2022, 13(1), 1444, DOI:10.1038/s41467-022-29135-5.
- E. Ruggiero and S. N. Richter, Targeting G-quadruplexes to achieve antiviral activity, Bioorg. Med. Chem. Lett., 2023, 79, 129085, DOI:10.1016/j.bmcl.2022.129085.
- L. D'Anna, T. Miclot and E. Bignon, et al., Resolving a guanine-quadruplex structure in the SARS-CoV-2 genome through circular dichroism and multiscale molecular modeling, Chem. Sci., 2023, 14(41), 11332–11339, 10.1039/D3SC04004F.
- T. Miclot, C. Hognon and E. Bignon, et al., Structure and Dynamics of RNA Guanine Quadruplexes in SARS-CoV-2 Genome. Original Strategies against Emerging Viruses, J. Phys. Chem. Lett., 2021, 12(42), 10277–10283, DOI:10.1021/acs.jpclett.1c03071.
- T. Tian, Y. Q. Chen, S. R. Wang and X. Zhou, G-Quadruplex,: A Regulator of Gene Expression and Its Chemical Targeting, Chem, 2018, 4(6), 1314–1344, DOI:10.1016/j.chempr.2018.02.014.
- J. Spiegel, S. Adhikari and S. Balasubramanian, The Structure and Function of DNA G-Quadruplexes, Trends Chem., 2020, 2(2), 123–136, DOI:10.1016/j.trechm.2019.07.002.
- E. Puig Lombardi and A. Londoño-Vallejo, A guide to computational methods for G-quadruplex prediction, Nucleic Acids Res., 2020, 48(3), 1603, DOI:10.1093/nar/gkaa033.
- V. Brázda, J. Kolomazník and J. Lýsek, et al., G4Hunter web application: a web server for G-quadruplex prediction, Bioinformatics, 2019, 35(18), 3493–3495, DOI:10.1093/bioinformatics/btz087.
- M. Barshai, B. Engel, I. Haim, Y. Orenstein and F. M. Fadlelmola, G4mismatch: Deep neural networks to predict G-quadruplex propensity based on G4-seq data, PLoS Comput. Biol., 2023, 19(3), e1010948, DOI:10.1371/journal.pcbi.1010948.
- N. H. Campbell, N. H. A. Karim and G. N. Parkinson, et al., Molecular Basis of Structure–Activity Relationships between Salphen Metal Complexes and Human Telomeric DNA Quadruplexes, J. Med. Chem., 2012, 55(1), 209–222, DOI:10.1021/jm201140v.
- N. Alegret, J. A. Valdez and A. Rodríguez-Fortea, et al., HMGB1 binds to KRAS promoter G-quadruplex: a new player in oncogene transcriptional regulation?, Chem. Commun., 2016, 52(1), 64–67, 10.1039/x0xx00000x.
- J. G. Keller, K. M. Hymøller and M. E. Thorsager, et al., Topoisomerase 1 inhibits MYC promoter activity by inducing G-quadruplex formation, Nucleic Acids Res., 2022, 50(11), 6332–6342, DOI:10.1093/nar/gkac482.
- H. Sun, J. Xiang and Y. Shi, et al., A newly identified G-quadruplex as a potential target regulating Bcl-2 expression, Biochim. Biophys. Acta, Gen. Subj., 2014, 1840(10), 3052–3057, DOI:10.1016/j.bbagen.2014.07.014.
- M. Falabella, J. E. Kolesar and C. Wallace, et al., G-quadruplex dynamics contribute to regulation of mitochondrial gene expression, Sci. Rep., 2019, 9(1), 5605, DOI:10.1038/s41598-019-41464-y.
- J. Jana, S. Mondal and P. Bhattacharjee, et al., Chelerythrine down regulates expression of VEGFA, BCL2 and KRAS by arresting G-Quadruplex structures at their promoter regions, Sci. Rep., 2017, 7(1), 40706, DOI:10.1038/srep40706.
- A. M. Psaras, R. K. Carty, J. T. Miller, L. N. Tumey and T. A. Brooks, Indoloquinoline-Mediated Targeted Downregulation of KRAS through Selective Stabilization of the Mid-Promoter G-Quadruplex Structure, Genes, 2022, 13(8), 1440, DOI:10.3390/genes13081440.
- M. H. Hu and J. H. Lin, New Dibenzoquinoxalines Inhibit Triple-Negative Breast Cancer Growth by Dual Targeting of Topoisomerase 1 and the c-MYC G-Quadruplex, J. Med. Chem., 2021, 64(10), 6720–6729, DOI:10.1021/acs.jmedchem.0c02202.
- J. Shen, D. Varshney and A. Simeone, et al., Promoter G-quadruplex folding precedes transcription and is controlled by chromatin, Genome Biol., 2021, 22(1), 143, DOI:10.1186/s13059-021-02346-7.
- E. Bignon, A. Spinello and T. Miclot, et al., Predicting the Three-Dimensional Structure of the c- KIT Proto-Oncogene Promoter and the Dynamics of Its Strongly Coupled Guanine Quadruplexes, J. Phys. Chem. Lett., 2023, 14(20), 4704–4710, DOI:10.1021/acs.jpclett.3c00765.
- C. Ducani, G. Bernardinelli, B. Högberg, B. K. Keppler and A. Terenzi, Interplay of Three G-Quadruplex Units in the KIT Promoter, J. Am. Chem. Soc., 2019, 141(26), 10205–10213, DOI:10.1021/jacs.8b12753.
- M. Ohanian, M. L. Arellano and M. Y. Levy, et al., A Phase 1a/b Dose Escalation Study of the MYC Repressor Apto-253 in Patients with Relapsed or Refractory AML or High-Risk MDS, Blood, 2021, 138(Supplement 1), 3411, DOI:10.1182/blood-2021-150049.
- J. Hilton, K. Gelmon and P. L. Bedard, et al., Results of the phase I CCTG IND.231 trial of CX-5461 in patients with advanced solid tumors enriched for DNA-repair deficiencies, Nat. Commun., 2022, 13(1), 3607, DOI:10.1038/s41467-022-31199-2.
- I. Alessandrini, M. Recagni, N. Zaffaroni and M. Folini, On the Road to Fight Cancer: The Potential of G-Quadruplex Ligands as Novel Therapeutic Agents, Int. J. Mol. Sci., 2021, 22(11), 5947, DOI:10.3390/ijms22115947.
- S. Neidle, Quadruplex Nucleic Acids as Novel Therapeutic Targets, J. Med. Chem., 2016, 13(59), 5987–6011, DOI:10.1021/acs.jmedchem.5b01835.
- A. Lauria, A. Terenzi and R. Bartolotta, et al., Does ligand symmetry play a role in the stabilization of DNA G-quadruplex host-guest complexes?, Curr. Med. Chem., 2014, 23(21), 2665–2690, DOI:10.2174/0929867321666140217155156266.
- J. Zegers, DNA G-quadruplex-stabilizing metal complexes as anticancer drugs, J. Biol. Inorg. Chem., 2023, 28, 117–138, DOI:10.1007/s00775-022-01973-0.
- G. Farine, C. Migliore and A. Terenzi, et al., On the G–Quadruplex Binding of a New Class of Nickel(II), Copper(II), and Zinc(II) Salphen–Like Complexes, Eur. J. Inorg. Chem., 2021, 2021(14), 1332–1336, DOI:10.1002/ejic.202100067.
- L. D’Anna, S. Rubino and C. Pipitone, et al., Salphen metal complexes as potential anticancer agents: interaction profile and selectivity studies toward the three G-quadruplex units in the KIT promoter, Dalton Trans., 2023, 52, 2966–2975 Search PubMed.
- R. Bonsignore, E. Trippodo and A. P. Carreca, et al., Novel half Salphen cobalt(III) complexes: synthesis, DNA binding and anticancer studies, Dalton Trans., 2024, 14(53), 6311–6322, 10.1039/D4DT00092G.
- R. Bonsignore, F. Russo and A. Terenzi, et al. The interaction of Schiff Base complexes of nickel(II) and zinc(II) with duplex and G-quadruplex DNA, J. Inorg. Biochem., 2018,(178), 106–114 Search PubMed.
- W. Streciwilk, A. Terenzi and R. Misgeld, et al. Metal NHC Complexes with Naphthalimide Ligands as DNA-Interacting Antiproliferative Agents, ChemMedChem, 2017,(12), 214–225, DOI:10.1002/cmdc.201600557.
- W. Streciwilk, A. Terenzi and F. Lo Nardo, et al., Synthesis and Biological Evaluation of Organometallic Complexes Bearing Bis-1,8-naphthalimide Ligands, Eur. J. Inorg. Chem., 2018, 3104–3112, DOI:10.1002/ejic.201800384.
- A. Arola and R. Vilar, Stabilisation of G-Quadruplex DNA by Small Molecules, Curr. Top. Med. Chem., 2008, 8(15), 1405–1415, DOI:10.2174/156802608786141106.
- A. Terenzi, D. Lötsch and S. van Schoonhoven, et al., Another step toward DNA selective targeting: Ni II and Cu II complexes of a Schiff base ligand able to bind gene promoter G-quadruplexes, Dalton Trans., 2016, 45(18), 7758–7767, 10.1039/C6DT00648E.
- A. Terenzi, R. Bonsignore and A. Spinello, et al., Selective G-quadruplex stabilizers: Schiff-base metal complexes with anticancer activity, RSC Adv., 2014,(63), 33245–33256, 10.1039/C4RA05355A.
- G. Barone, N. Gambino, A. Ruggirello, A. Silvestri, A. Terenzi and V. T. Liveri, Spectroscopic study of the interaction of NiII-5-triethyl ammonium methyl salicylidene ortho-phenylendiiminate with native DNA, J. Inorg. Biochem., 2009, 103(5), 731–737, DOI:10.1016/j.jinorgbio.2009.01.006.
- A. W. Kleij, D. M. Tooke, A. L. Spek and J. N. H. Reek, A Convenient Synthetic Route for the Preparation of Nonsymmetric Metallo-salphen Complexes, Eur. J. Inorg. Chem., 2005, 4626–4634, DOI:10.1002/ejic.200500628.
- A. Ambrus, D. Chen, J. Dai, T. Bialis, R. A. Jones and D. Yang, Human telomeric sequence forms a hybrid-type intramolecular G-quadruplex structure with mixed parallel/antiparallel strands in potassium solution, Nucleic Acids Res., 2006, 34(9), 2723–2735, DOI:10.1093/nar/gkl348.
- J. Dai, C. Punchihewa, A. Ambrus, D. Chen, R. A. Jones and D. Yang, Structure of the intramolecular human telomeric G-quadruplex in potassium solution: a novel adenine triple formation, Nucleic Acids Res., 2007, 35(7), 2440–2450, DOI:10.1093/nar/gkm009.
- J. Dai, D. Chen, R. A. Jones, L. H. Hurley and D. Yang, NMR solution structure of the major G-quadruplex structure formed in the human BCL2 promoter region, Nucleic Acids Res., 2006, 34(18), 5133–5144, DOI:10.1093/nar/gkl610.
- S. Cogoi and L. E. Xodo, G-quadruplex formation within the promoter of the KRAS proto-oncogene and its effect on transcription, Nucleic Acids Res., 2006, 34(9), 2536–2549, DOI:10.1093/nar/gkl286.
- A. Ambrus, D. Chen, J. Dai, R. A. Jones and D. Yang, Solution structure of the biologically relevant G-quadruplex element in the human c-MYC promoter. Implications for G-quadruplex stabilization, Biochemistry, 2005, 44(6), 2048–2058, DOI:10.1021/bi048242p.
- X. Tong, W. Lan, X. Zhang, H. Wu, M. Liu and C. Cao, Solution structure of all parallel G-quadruplex formed by the oncogene RET promoter sequence, Nucleic Acids Res., 2011, 39(15), 6753–6763, DOI:10.1093/nar/gkr233.
- A. Kotar, R. Rigo, C. Sissi and J. Plavec, Two-quartet kit* G-quadruplex is formed via double-stranded pre-folded structure, Nucleic Acids Res., 2018, 47, 2641–2653, DOI:10.1093/nar/gky1269.
- V. Butera, L. D'Anna and S. Rubino, et al., How the Metal Ion Affects the 1 H NMR Chemical Shift Values of Schiff Base Metal Complexes: Rationalization by DFT Calculations, J. Phys. Chem. A, 2023, 127(44), 9283–9290, DOI:10.1021/acs.jpca.3c05653.
- A. De Rache and J. L. Mergny, Assessment of selectivity of G-quadruplex ligands via an optimised FRET melting assay, Biochimie, 2015, 115, 194–202, DOI:10.1016/j.biochi.2015.06.002.
- D. Brynn Hibbert and P. Thordarson, The death of the Job plot, transparency, open science and online tools, uncertainty estimation methods and other developments in supramolecular chemistry data analysis, Chem. Commun., 2016, 52(87), 12792–12805, 10.1039/C6CC03888C.
- T. A. Brooks, S. Kendrick and L. Hurley, Making sense of G-quadruplex and i-motif functions in oncogene promoters: G-quadruplex and i-motif in oncogene promoters, FEBS J., 2010, 277(17), 3459–3469, DOI:10.1111/j.1742-4658.2010.07759.x.
- S. Cogoi, V. Rapozzi, S. Cauci and L. E. Xodo, Critical role of hnRNP A1 in activating KRAS transcription in pancreatic cancer cells: A molecular mechanism involving G4 DNA, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861(5), 1389–1398, DOI:10.1016/j.bbagen.2016.11.031.
- W. Wang, S. Hu and Y. Gu, et al., Human MYC G-quadruplex: From discovery to a cancer therapeutic target, Biochim. Biophys. Acta, Rev. Cancer, 2020, 1874(2), 188410, DOI:10.1016/j.bbcan.2020.188410.
- G. Biffi, D. Tannahill, J. McCafferty and S. Balasubramanian, Quantitative visualization of DNA G-quadruplex structures in human cells, Nat. Chem., 2013, 5(3), 182–186, DOI:10.1038/nchem.1548.
- A. De Magis, M. Kastl, P. Brossart, A. Heine and K. Paeschke, BG-flow, a new flow cytometry tool for G-quadruplex quantification in fixed cells, BMC Biol., 2021, 19(1), 45, DOI:10.1186/s12915-021-00986-6.
- S. J. Angyal, P. J. Morris, J. R. Tetaz and J. G. Wilson, The Sommelet reaction. Part III. The choice of solvent and the effect of substituents, J. Chem. Soc., 1950, 2141–2145 Search PubMed.
- W. Kandioller, J. Theiner, B. K. Keppler and C. R. Kowol, Elemental analysis: an important purity control but prone to manipulations, Inorg. Chem. Front., 2022, 9(3), 412–416, 10.1039/D1QI01379C.
- J. Marquevielle, M. V. Vasantha Kumar, J. L. Mergny and G. F. Salgado, 1H, 13C, and 15N chemical shift assignments of a G-quadruplex forming sequence within the KRAS proto-oncogene promoter region, Biomol. NMR Assignments, 2018, 12, 123–127, DOI:10.1007/s12104-017-9793-0.
- S. Bielskutė, J. Plavec and P. Podbevšek, Oxidative lesions modulate G-quadruplex stability and structure in the human BCL2 promoter, Nucleic Acids Res., 2021, 49(4), 2346–2356, DOI:10.1093/nar/gkab057.
- R. Galindo-Murillo, J. C. Robertson and M. Zgarbová, et al., Assessing the Current State of Amber Force Field Modifications for DNA, J. Chem. Theory Comput., 2016, 12(8), 4114–4127, DOI:10.1021/acs.jctc.6b00186.
- P. Mark and L. Nilsson, Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K, J. Phys. Chem. A, 2001, 105(43), 9954–9960, DOI:10.1021/jp003020w.
- P. Li and K. M. Merz, MCPB.py: A Python Based Metal Center Parameter Builder, J. Chem. Inf. Model., 2016, 56(4), 599–604, DOI:10.1021/acs.jcim.5b00674.
- C. W. Hopkins, S. Le Grand, R. C. Walker and A. E. Roitberg, Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning, J. Chem. Theory Comput., 2015, 11(4), 1864–1874, DOI:10.1021/ct5010406.
- J. C. Phillips, R. Braun and W. Wang, et al., Scalable molecular dynamics with NAMD, J. Comput. Chem., 2005, 26(16), 1781–1802 Search PubMed.
- J. C. Phillips, D. J. Hardy and J. D. C. Maia, et al., Scalable molecular dynamics on CPU and GPU architectures with NAMD, J. Chem. Phys., 2020, 153(4), 044130, DOI:10.1063/5.0014475.
- W. Humphrey, A. Dalke and K. Schulten, VMD: visual molecular dynamics, J. Mol. Graphics, 1996, 14(1), 22–28, DOI:10.1016/0263-7855(96)00018-5.
- A. D. Herbert, A. M. Carr, E. Hoffmann and M. Lichten, FindFoci: A Focus Detection Algorithm with Automated Parameter Training That Closely Matches Human Assignments, Reduces Human Inconsistencies and Increases Speed of Analysis, PLoS One, 2014, 9(12), e114749, DOI:10.1371/journal.pone.0114749.
- M. W. Pfaffl, A new mathematical model for relative quantification in real-time RT-PCR, Nucleic Acids Res., 2001, 29(9), 45e–445, DOI:10.1093/nar/29.9.e45.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qi01394h |
| This journal is © the Partner Organisations 2024 |