
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultielectron transfer and field-induced slow magnetic relaxation in opto-electroactive spin crossover cobalt(II) complexes: structure–function correlations†‡
Renato
Rabelo
ab,
Luminita
Toma
a,
Miguel
Julve
 a,
Francesc
Lloret
a,
Jorge
Pasán
c,
Danielle
Cangussu
b,
Rafael
Ruiz-García
a and
Joan
Cano
*a
a,
Francesc
Lloret
a,
Jorge
Pasán
c,
Danielle
Cangussu
b,
Rafael
Ruiz-García
a and
Joan
Cano
*a
aInstituto de Ciencia Molecular (ICMol), Universitat de València, 46980 Paterna, València, Spain. E-mail: joan.cano@uv.es
bInstituto de Química, Universidade Federal de Goiás, Av. Esperança Campus Samambaia, Goiânia, GO, Brazil
cLaboratorio de Materiales para Análisis Químico (MAT4LL), Departamento de Química, Facultad de Ciencias, Universidad de La Laguna, 38200 Tenerife, Spain
First published on 30th July 2024
Abstract
Designing and implementing multielectron transfer and single-molecule magnet properties in spin crossover compounds constitute a promising way to obtain a new class of multiresponsive and multifunctional materials. This contribution uses N-phenyl-substituted pyridine-2,6-diimine (PDI) ligands to explore a novel family of spin-crossover cobalt(II) complexes, with general formula [CoL2](ClO4)2·xH2O [L = 4-MePhPDI (1, x = 1), 4-MeOPhPDI (2, x = 0), 4-MeSPhPDI (3, x = 0), 4-Me2NPhPDI (4, x = 0.5), and 2,4-Me2PhPDI (5, x = 0)]. These mononuclear six-coordinate octahedral cobalt(II) bis(chelating) complexes incorporate tridentate PDI derivatives with various electron-donating substituents at para (X4 = Me, OMe, SMe, and Me2N) or ortho and para positions (X2, X4 = Me) of terminal phenyl rings. Our investigations reveal that these complexes exhibit both thermally-induced low-spin (LS)/high-spin (HS) transition and field-induced slow magnetic relaxation in the LS (S = 1/2) and HS (S = 3/2) states. Moreover, they display a dual multiredox behaviour featuring one-electron oxidation of the paramagnetic CoII ion to the diamagnetic CoIII ion and stepwise two-electron reduction of each PDI ligand to the corresponding imine-type PDI˙− π-radical anion and diimine-type PDI2− dianion, which positions them as candidates for prototypes of spin quantum transistors and capacitors, offering potential applications in quantum information processing.
Introduction
Multielectron transfer (MET) processes have significant biological and chemical functions1–5 and play a pivotal role in catalytic redox transformations6–9 and artificial energy storage10–12 and photosynthetic systems.13–15 Metal centres and radical cofactors embedded within specific proteins collaborate to orchestrate catalytic multielectron redox transformations of small substrates.4,5 Thus, the coordination chemistry of redox-active (“non-innocent”) ligands16–27 continues to attract attention because of their relevant role as biomimetic models of metalloenzymes with radical cofactors and their use as redox catalysts,28–36 redox flow batteries and capacitors,37,38 as well as photocatalysts and solar cells.39–42 Furthermore, it is critical in designing novel electroactive materials for molecular electronics and spintronics uses.43–45 In particular, spin crossover (SCO) transition metal complexes featuring potential metal- or ligand-based electroactivity are promising contenders for molecular spin quantum transistors.46–59Recently, mononuclear iron(II) and cobalt(II) complexes exhibiting both thermal-assisted HS/LS transitions and field-induced single-molecule magnet (SMM) behaviour, termed spin-crossover molecular nanomagnets, have caught our attention.60–72 In this context, our research on electroactive spin-crossover molecular nanomagnets is focused on the ability of pyridine-2,6-diimine (PDI) ligands to provide an additional reducible capacity to the oxidisable metal centre in their mononuclear cobalt(II) complexes.73–75 So, a recent study of ours demonstrated a proof-of-concept of a spin quantum electro-switch based on this unique class of nanomagnets,68 reporting the reversible electro-switching of the SMM behaviour through metal oxidation in an octahedral SCO cobalt(II)-PDI complex. In this paper, we propose to advance our investigation by focusing on ligand reductions instead of metal oxidation. Through this approach, we hope to develop a new type of molecular spin quantum capacitors based on this family of electroactive SCO/SMMs.
This report focuses on the details of the synthesis, structural and spectroscopic characterisation, static (dc) and dynamic (ac) magnetic studies, and redox properties of a series of cobalt(II) compounds of formula [Co(4-MePhPDI)2](ClO4)2·H2O (1), [Co(4-MeOPhPDI)2](ClO4)2 (2), [Co(4-MePhSPDI)2](ClO4)2 (3), [Co(4-Me2NPhPDI)2](ClO4)2·1/2H2O (4), and [Co(2,4-Me2PhPDI)2](ClO4)2 (5), as shown in Scheme 1. These mononuclear octahedral species feature tridentate PDI ligands with electron-donating group-substituted phenyl rings at para (X4 = Me, OMe, SMe, and Me2N) or both ortho and para positions (X2, X4 = Me). They exhibit thermally-induced spin-crossover and field-induced slow magnetic relaxation, similar to the reported related series with electron-withdrawing halogen-substituted phenyl rings at para [X4 = I (6), Br (7) and Cl (8)] and meta positions [X3 = Br (9)].70 Finally, we have analysed the influence of steric and electronic ligand features on this series’ MET, SCO, and SMM behaviours.
 | ||
| Scheme 1 General chemical formula of the cationic mononuclear octahedral cobalt(II) complexes with PDI ligands accounting with electron-donating substituents at the terminal phenyl rings. | ||
This combined spectroscopic, magnetic, and electrochemical investigation, along with the information obtained from the preceding paper,70 allowed us to establish appropriate structure–function correlations, further supported by density functional theory (DFT) and time-dependent DFT (TDDFT) calculations, which would aid the design and synthesis of multiresponsive and multifunctional SCO/SMM materials as potential candidates for multistable charge storage spintronic devices like molecular spin quantum transistors and capacitors.
Results and discussion
Ligand design and synthetic strategy
Except for 4-MeSPhPDI, all ligands used in this work and their coordination chemistry with transition metal ions are well-documented.73–84 These ligands were synthesised from the straightforward condensation of pyridine-2,6-diformaldehyde and the corresponding aniline derivative with electron-donating substituents in the para or para and ortho positions (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio) in the presence of acid as catalyst.70 All ligands were isolated as crystalline powders in good yields, and they were characterised by elemental analyses (C, H, N), Fourier-transform infrared (FT-IR) and proton nuclear magnetic resonance (1H NMR) spectroscopies (see Experimental section in the ESI‡).
:2 molar ratio) in the presence of acid as catalyst.70 All ligands were isolated as crystalline powders in good yields, and they were characterised by elemental analyses (C, H, N), Fourier-transform infrared (FT-IR) and proton nuclear magnetic resonance (1H NMR) spectroscopies (see Experimental section in the ESI‡).
Compounds 1–5 were synthesised from the reaction of cobalt(II) perchlorate hexahydrate and the previously isolated ligands (1:2 metal-to-ligand molar ratio), as reported previously for 6–9.70 All of them were isolated as X-ray quality single crystals in moderate yields by slow evaporation of ethanol (1) or methanol (4 and 5) solutions or by slow diffusion methods in a methanol/chloroform (2) or acetonitrile/ethyl acetate/chloroform (3) solvent mixtures. They were characterised by elemental analyses (C, H, N), Fourier-transform infrared (FT-IR), electronic absorption (UV-Vis), and electron paramagnetic resonance (EPR) spectroscopies (see Experimental section in the ESI‡). Single-crystal and powder X-ray diffraction determined the chemical identity of 1–5 (Fig. S1‡).
Description of the structures
Compounds 1–5 are not isostructural, reflecting thus variations in the electron-donating substituents’ nature, their ortho or para substitution, and occasionally, the presence of crystallisation water molecules. 1 and 3 crystallise in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, while 4 and 5 do in monoclinic P21/n and P21/c space groups, respectively, as occurs in the parent unsubstituted compound.772 crystallises in the tetragonal P43 space group. Tables S1 and S2‡ summarise crystallographic and selected structural data for 1–5.
space group, while 4 and 5 do in monoclinic P21/n and P21/c space groups, respectively, as occurs in the parent unsubstituted compound.772 crystallises in the tetragonal P43 space group. Tables S1 and S2‡ summarise crystallographic and selected structural data for 1–5.
These structures consist of either one (1, 3–5) or four (2) crystallographically independent mononuclear cobalt(II) complex cations, [CoIIL2]2+, perchlorate anions, and water molecules of crystallisation (1 and 4), as depicted in Fig. 1 and S2–S6.‡
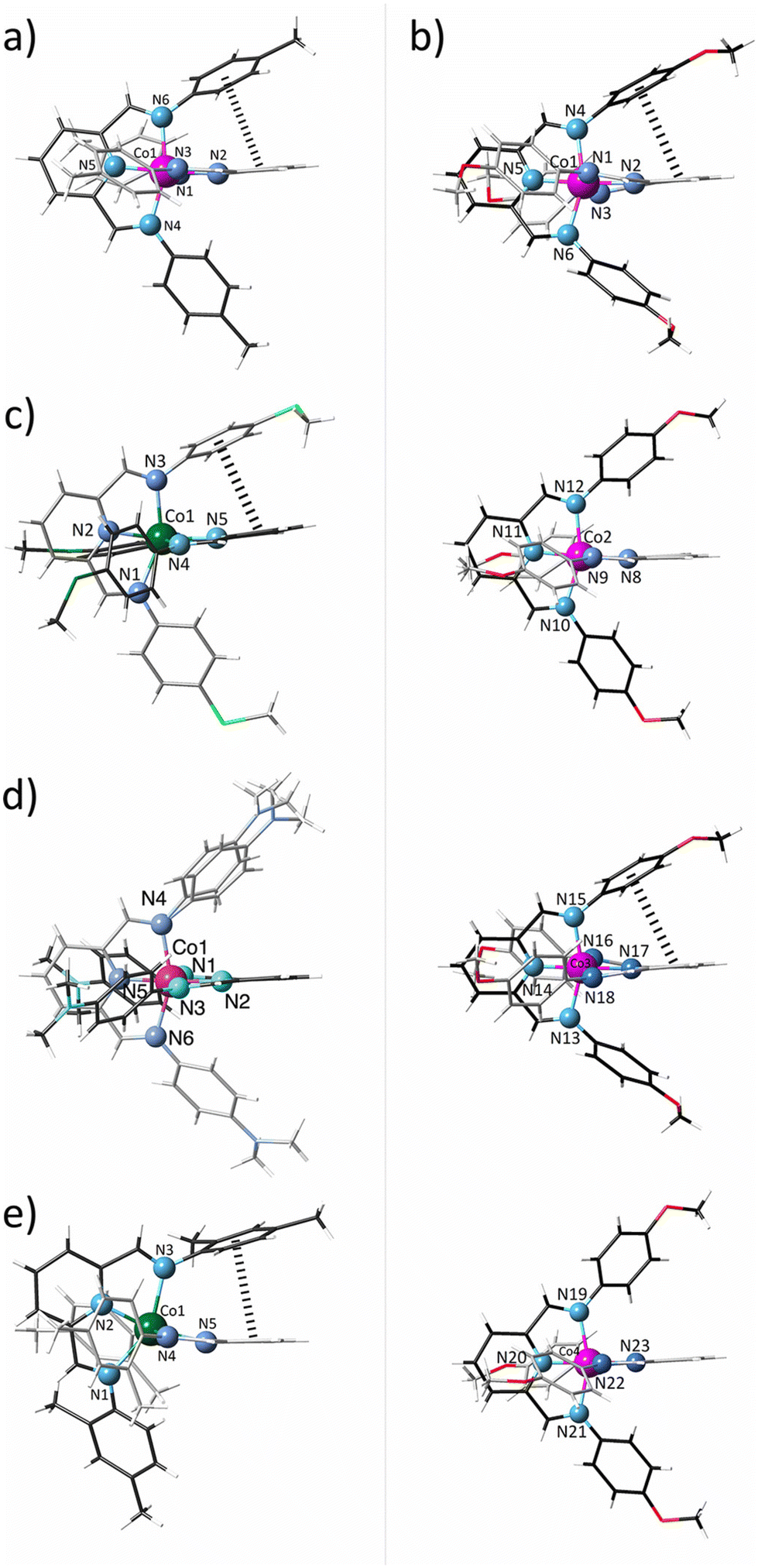 | ||
| Fig. 1 Perspective views of the crystallographically independent mononuclear complex cations of 1–5 (a–e) with the atom numbering scheme of the coordination sphere at the cobalt atom. The ligand backbones are drawn in grey and black colours for clarity whereas the pink and green cobalt atoms correspond to the LS and HS configuration. The π–π stacking interactions are drawn as dashed lines. | ||
The complex cations in 1–5 exhibit a distorted C2v molecular symmetry (Fig. 1). Nonetheless, in all cases, the PDI ligands are almost, but not perfectly, perpendicularly oriented (Φ = 72.2–90.0; Table S2‡), likely due to moderate to weak single (1, 2, and 5) or double (3) intramolecular pseudo-face-to-face π–π interactions between pyridine and phenyl rings of different ligands (Fig. 1), being the distance between the centroids of those rings and the dihedral angle between their mean planes [h = 3.61–3.94 Å with θ = 17.8–29.4°; Table S2‡] below or close to the limiting values for this kind of interaction (h < 4.4 Å and θ < 30°). However, two and one crystallographically independent complex cations in 2 and 4 do not show intramolecular contacts [h = 4.2–4.5 Å and θ = 47.8–59.1°; Table S2‡].
The two pairs of imine nitrogen atoms (Nim and N′im) from each PDI ligand define the equatorial plane of the cobalt octahedral coordination sphere, while the two pyridine nitrogen atoms (Npy) occupy the axial positions. Overall, the Co–N bonds are grouped into three different classes with two long Co–Nim [R1 = 2.085(4)–2.217(2) Å], two intermediate Co–N′im [R2 = 2.029(2)–2.186(2) Å], and two short Co–Npy distances [R3 = 1.872(5)–2.047(2) Å; Table S2‡]. This strong asymmetry makes difficult the spin state assignment, but the mean values in 1, 2, and 4 [R = 2.013–2.066 Å] are typical of LS CoII complexes whereas they correspond to HS in 3 and 5 [R = 2.130–2.150 Å; Table S2‡], according to a greater occupancy of the eg orbitals, which takes part in antibonding molecular orbitals with the ligands, in the HS (t2g5eg2) than LS (t2g6eg1) configuration. This situation is reflected in the axial and equatorial Co–N bond lengths for 1–9, which are almost linearly dependent on the electron-withdrawing ability of the X substituent in the ligand, expressed by the Hammett constant (σH),85 as shown in Fig. S7.‡ So, by removing or injecting electron density in the coordination regions of the PDI ligand, the antibonding nature of the molecular orbital is weakened or intensified and thus, the metal–ligand bonds are shortened or lengthened.
So, 1–5 exhibit a moderately rhombic distortion in their axially compressed coordination spheres, as evidenced by non-zero ΔR and δR parameters, being more pronounced in the LS configuration (Table S2‡), as anticipated due to the increased significance of the Jahn–Teller effect for 2Eg(t2g6eg1) and 4T1g(t2g5eg2) states. The differences in the rhombic distortion are also reflected in the metal-to-ligand bond angles, with the axial Npy–Co–Npy axis [176.1(1)–178.5(2) vs. 162.38(9)–164.3(8)°; Table S2‡] closer to the linearity in the LS state. However, all cases display systematic deviations from the ideal 90° angle in the equatorial plane [Nim–Co–N′im = 83.21(10)–92.1(2)° and N′im–Co–Nim = 92.4(2)–101.08(10)°; Table S2‡]. Besides, the two five-membered fused rings resulting from the tridentate coordination mode of the ligand impose a severe non-planar ruffling distortion, particularly on the HS ion, as reflected by its larger mean out-of-plane Nim displacements (δ = 0.555–0.560 vs. 0.395–0.430 Å; Table S2‡).
The PDI ligands in 1–5 adopt a helical conformation owing to the free rotation around the single phenyl carbon–imine nitrogen bond. Hence, the octahedral cobalt units are chiral, with both enantiomers occurring in the crystal lattice. The average torsional angle for the phenylimine ligand fragments is similar (φ = 37.7–46.1°; Table S2‡) except for 5, where it is significantly greater (φ = 63.3°; Table S2‡), more likely due to the steric hindrance between the ortho-methyl substituent on the phenyl rings and the methylene hydrogen atom from the imine groups.
In 1–5, the central pyridinediimine fragments exhibit average Cpy–Cim [d2 = 1.453(8)–1.474(4) Å; Table S2‡] and Cim–Nim bond lengths [d3 = 1.276(3)–1.291(7) Å; Table S2‡] typical for single carbon–carbon and double carbon–nitrogen bonds, while the average Cpy–Npy length [d1 = 1.337(3)–1.349(4) Å; Table S2‡] is intermediate between a single and a double bond, consistent with findings for the related complexes 6–9.70 Yet, the alternation bond parameter, defined as Δd = d2 − (d1 + d3)/2,74 reflects a slightly greater π–electron delocalisation within the pyridinediimine fragment for the LS ions (Δd = 0.135–0.150 vs. 0.156–0.167 Å; Table S2‡), as observed in Fig. S8.‡ Such a feature is likely attributed to the greater electron population of t2g shell for the LS (t2g6eg1) ions, which would eventually lead to enhanced π-backbonding interactions with the empty ligand orbitals.75
In the crystal lattices of 1–5, a segregated arrangement comprising complex cations and perchlorate layers are interconnected by electrostatic forces and van der Waals contacts (Fig. S2–S6‡). In 1 and 4, additional water molecules of crystallisation occupy the interlayer space, establishing moderate to weak intermolecular hydrogen-bonding interactions with the perchlorate anions [O⋯H–O = 2.922(5)–3.273(4) Å] and van der Waals contacts with the mononuclear cobalt(II) complex cations.
Mononuclear complexes in 1–5 interact with each other through moderate to weak intermolecular face-to-face (1–5) or edge-to-face (3) π–π stacking interactions between their phenyl rings (Fig. 2), leading to different supramolecular π-bonded motifs of variable dimensionality (nD, n = 0–2) and topology, including either discrete (5) or chain arrays (4) of dimers (Fig. 2e and d), as well as square (2 and 3) or rhombus (1) grids (Fig. 2a–c). Within each cationic layer of 4 and 5, the intradimer intermetallic distance through the face-to-face π-stacking [Co⋯Co = 10.402 (4) and 10.762 Å (5)] are comparable to those of the shortest interdimer ones [Co⋯Co = 10.401 (4) and 11.586 (5)]. Otherwise, the intralayer intermetallic distances through face-to-face [Co⋯Co = 8.664–13.110 (1), 9.359–9.759 (2), and 10.211 Å (3)] and edge-to-face π-stacking [Co⋯Co = 9.635 Å (3)] are similar, so that the mononuclear units are rather well-isolated from each other.
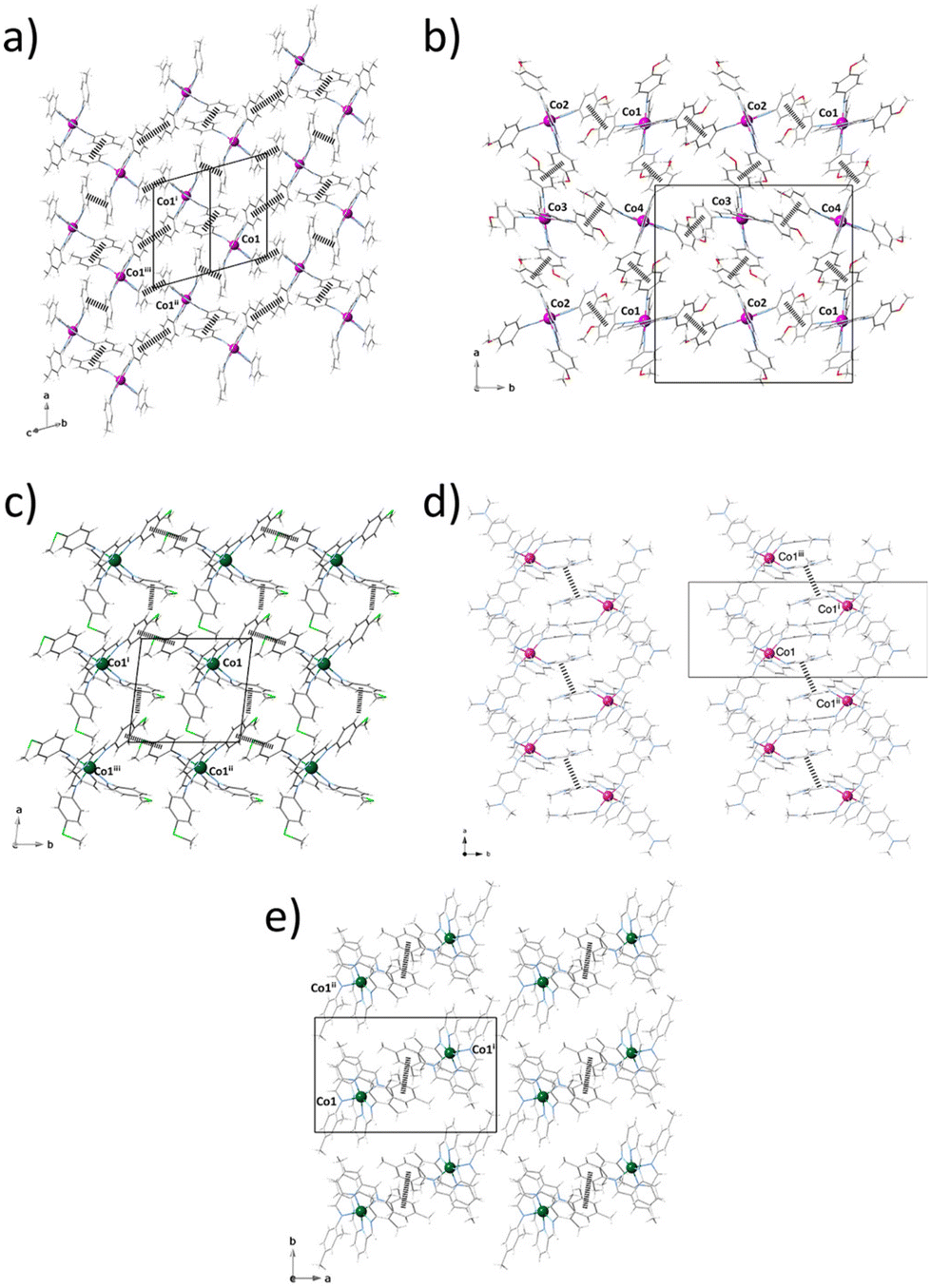 | ||
| Fig. 2 Projection views of the crystal packing of 1–5 (a–e) showing a layer of cationic mononuclear cobalt units in the array of π-bonded rhomb grids in 1 and 3 (a and c), square grids in 2 (b), dimers in 4 and 5 (d and e) among the mononuclear units [symmetry operation: (i) =1 − x, 1 − y, 1 − z; (ii) =1 + x, y, z; (iii) =1 − x, y, z (1 and 3); (i) =−x, −y, −z; (ii) =1/2 − x, 1/2 + y, 1/2 − z; (iii) =1/2 + x, 1/2 − y, 1/2 + z (2); (i) =−x, −y, −z; (ii) =1/2 − x, 1/2 + y, 1/2 − z; (iii) =1/2 + x, 1/2 − y, 1/2 + z (4 and 5)]. For clarity, grey and black colours are used for ligand backbones, pink and green colours for pink and green for LS and HS cobalt atoms, and dashed lines for π–π stacking interactions. | ||
Spectroscopic properties and theoretical calculations
The aim of this study was to investigate the spectroscopic properties of cobalt(II) complexes 1–9 with PDI ligands to analyse the effects of electronic nature of the substituents and their substitution pattern, since phenyl rings in ligands are electron-donating group-substituted in 1–5 but electron-withdrawing in 6–9.70PDI ligands show similar patterns in their 1H NMR spectra. The central pyridinediimine exhibits the characteristic doublet and triplet signals of the meta- and para-pyridine hydrogen atoms [δ(p-Hpy) = 8.21–8.32 ppm and δ(m-Hpy) = 7.86–7.95 ppm], together with a singlet signal corresponding to the imine hydrogen atom [δ(Him) = 8.57–8.74 ppm]. The para-substituted phenyl groups from 4-MePhPDI, 4-MeOPhPDI, 4-MeSPhPDI, 4-Me2NPhPDI, 4-IPhPDI, 4-BrPhPDI, and 4-ClPhPDI ligands show two doublet signals attributed to the ortho- and meta-hydrogen atoms [δ(o-HPh) = 7.06–7.40 ppm and δ(m-HPh) = 6.77–7.75 ppm], while a multiplet signal corresponding to the ortho-, meta-, and para-phenyl hydrogen atoms appears for the 2,4-Me2PDI [δ(o,m-HPh) = 7.04 ppm] and 3-BrPhPDI ligands [δ(o,m,p-HPh) = 7.30 ppm]. In addition, a singlet signal corresponding to the hydrogen atoms from the para-dimethylamino, para-methoxy, and para- or ortho-methyl substituents is observed for 4-MePhPDI [δ(p-HMe) = 2.39 ppm], 4-MeOPhPDI [δ(p-HOMe) = 3.85 ppm], 4-MeSPhPDI [δ(p-HSMe) = 3.85 ppm], 4-Me2NPhPDI [δ(p-HNMe2) = 3.01 ppm] and 2,4-Me2PhPDI [δ(p-HMe) = 2.35 ppm and δ(o-HMe) = 2.40 ppm].
These chemical shifts display a linear correlation with the substituent σH (Fig. S9‡). Hence, within the pyridinediimine fragment, δ(Him) and δ(m-Hpy) slightly decrease and increase with the electron-withdrawing character of the ligand substituents, whereas δ(p-Hpy) remains almost constant. In contrast, within the terminal phenyl rigs, δ(o-HPh) and δ(p-HPh) largely decrease, whereas δ(m-HPh) increases instead. Overall, these upfield or downfield shifts reflect the less efficient shielding effect of the substituent with the increasing distance from the hydrogen atom.
Calculations of 1H NMR chemical shifts on the ligands confirm these conclusions (Table S3‡). Thus, 4-MePhPDI and 4-Me2NPhPDI follow the observed correlations with the substituent's Hammett constant, but 2,4-Me2PhPDI deviates, showing a δ(imine) less than expected one for 4-MePhPDI. When a hydrogen atom replaces the ortho-methyl group in 2,4-Me2PhPDI, which is equivalent to building 4-MePhPDI keeping the most stable geometry of 2,4-Me2PhPDI, no effect is observed on δ(imine) and less on the pyridine hydrogen atoms. This last result is apparent evidence that the distortions originated from steric hindrances diminish the effect of electronic factors in 2,4-Me2PhPDI.
The FT-IR spectra of the PDI ligands and cobalt(II) complexes display a moderate band in the ranges of 1618–1626 and 1572–1608 cm−1, respectively, assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration. This vibrational frequency linearly decreases with the electron-withdrawing nature of the ligand substituent for the complexes (Fig. S10‡); but it remains almost constant through the ligand series. This fact may be likely explained by the increase of the aforementioned π-backbonding metal–ligand interactions with increasing the ligand's electron-withdrawing nature that would weaken the CN bond.
N stretching vibration. This vibrational frequency linearly decreases with the electron-withdrawing nature of the ligand substituent for the complexes (Fig. S10‡); but it remains almost constant through the ligand series. This fact may be likely explained by the increase of the aforementioned π-backbonding metal–ligand interactions with increasing the ligand's electron-withdrawing nature that would weaken the CN bond.
The electronic absorption spectra in acetonitrile solution of the 4-Me2NPhPDI and 4-ClPhPDI ligands and those of their cobalt(II) complexes 4 and 8, as representative examples of these two related series, are compared in Fig. 3.
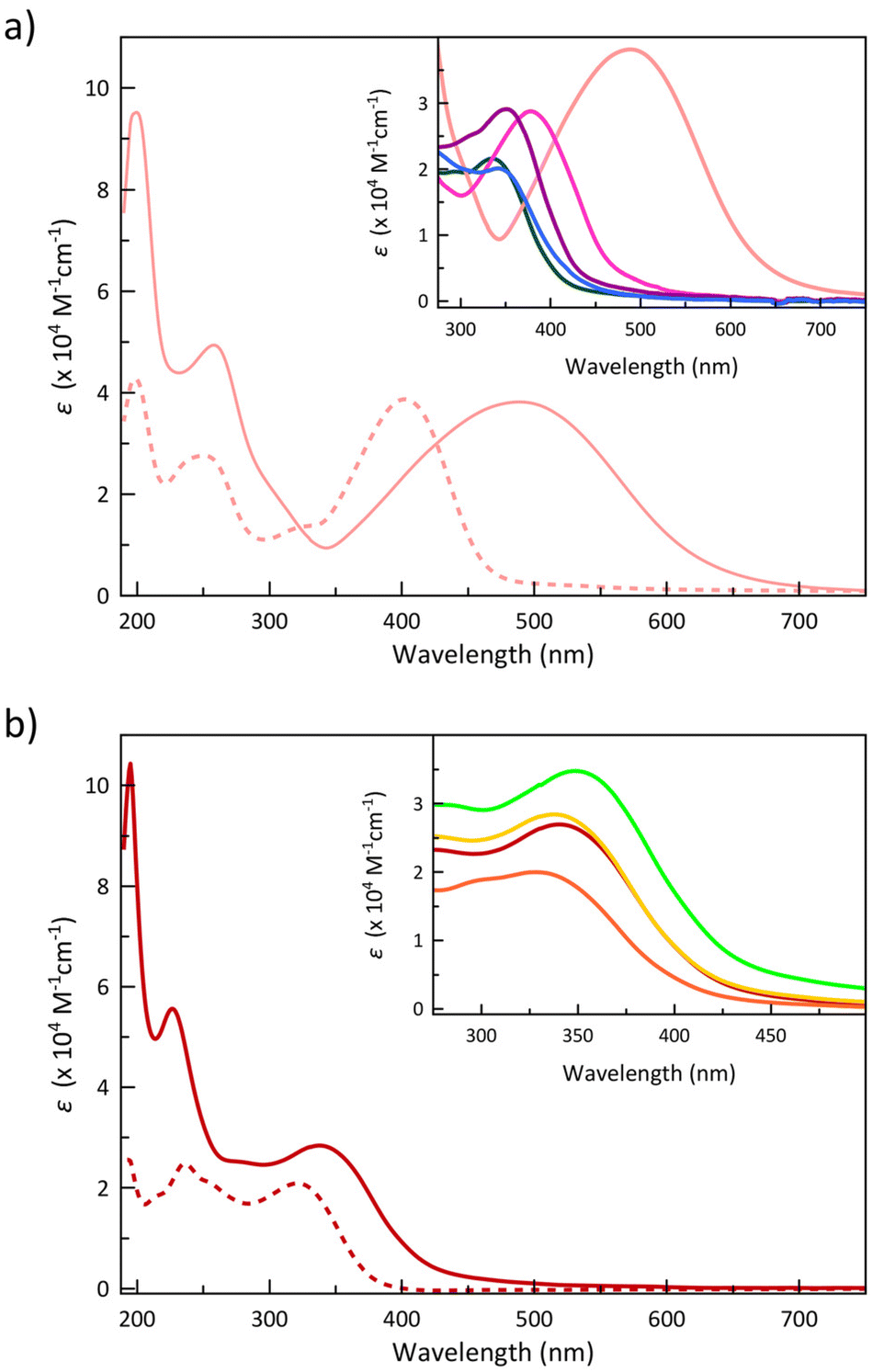 | ||
| Fig. 3 Electronic absorption spectra of (a) 4-Me2NPhPDI and (b) 4-ClPhPDI ligands (dashed lines) compared to the corresponding cobalt(II) complexes 4 and 8 (solid lines) in acetonitrile solution at room temperature. The insets show the lower energy UV-Vis band for 1 (purple), 2 (pink), 3 (green), 4 (pale pink), 5 (blue), 6 (light green), 7 (yellow), 8 (red), and 9 (orange). | ||
1–9 exhibit two very intense sharp absorption bands in the high-energy UV region (λ1 = 195–199 nm and λ2 = 218–258 nm), together with a broad, less intense low-energy UV band that extends into the visible region (λ3 = 335–489 nm) (insets of Fig. 3a and b).
The higher (λ1 = 195–200 nm and λ2 = 217–260 nm) and the lowest energy (λ3 = 335–489 nm) UV bands found for the ligands are formally attributed to intraligand (IL) π–π* and n–π* transitions, as it was done for the similar para-substituted phenyl- and azobenzene derivatives.85–89 Although the transition energies change along the series of ligands (Fig. 4), the most significant shift occurs for the lowest energy band (λ3), which moves to lower energies by increasing the electron-withdrawing character of the substituent, being confirmed by TDDFT calculations on the optimised geometries of MePhPDI and 4-NMe2PhPDI (Table S4‡).
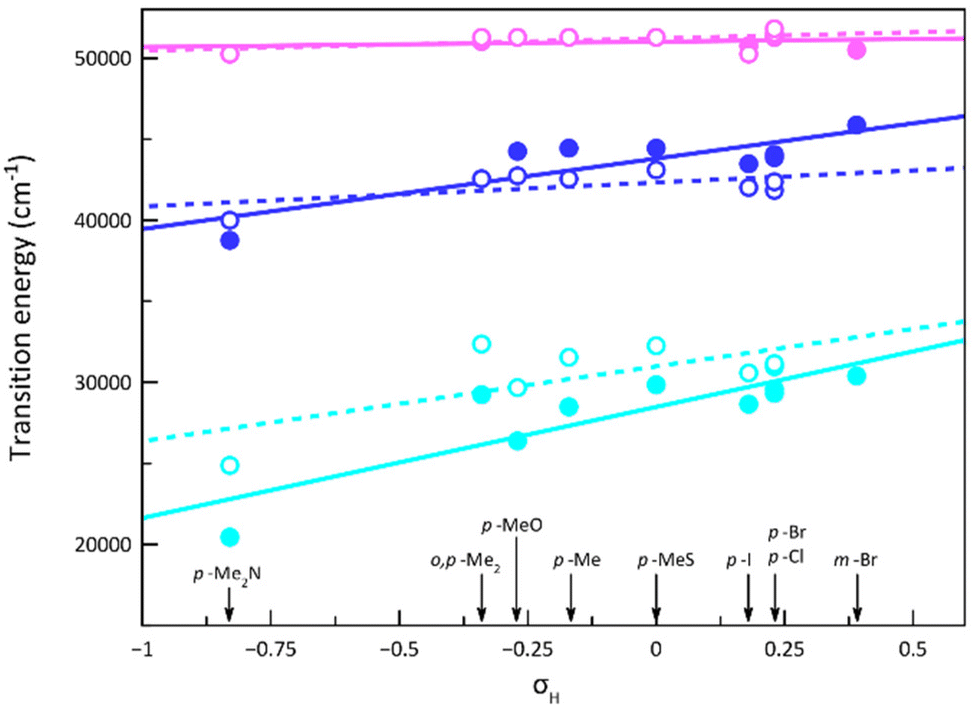 | ||
| Fig. 4 Variation of the transition energy values of the electronic transitions [λn with n = 1 (pink), 2 (blue), and 3 (cyan)] for the PDI ligands (○) and the corresponding cobalt(II) complexes 1–9 (●) with the Hammett constants (σH) of the ligand substituents. The identity of the ligand substituents is included for clarity. The dotted and solid lines are the linear fit curves for the ligands and complexes, respectively (see text). | ||
Upon the formation of the complexes, once again, this λ3 transition, particularly in 4, undergoes a redshift (Fig. 4). The resemblance of the spectra of 1–9 to those of their corresponding PDI ligands suggests that the recorded transitions have mainly IL character (Fig. 4), with shifts attributed to the involvement of the metal ion, possibly through ligand-to-metal (LMCT) or metal-to-ligand charge transfers (MLCT). However, in these electronic transitions involving partial IL charge transfer between the phenyl and pyridinediimine fragments (Fig. 5), the conformation adopted by the ligand into the complex may play a crucial role. Thus, a TDDFT calculation restricted to the conformation of the 4-Me2NPhPDI in the optimised geometry of its cobalt(II) complex (4) shows the experimentally observed energy displacement (Table S4‡), so that the contribution from the metal ion is not exclusively electronic. Still, it allows the approach of two PDI ligands, intensifying the steric effects already observed in the isolated ligand and thereby modifying the electronic transitions.
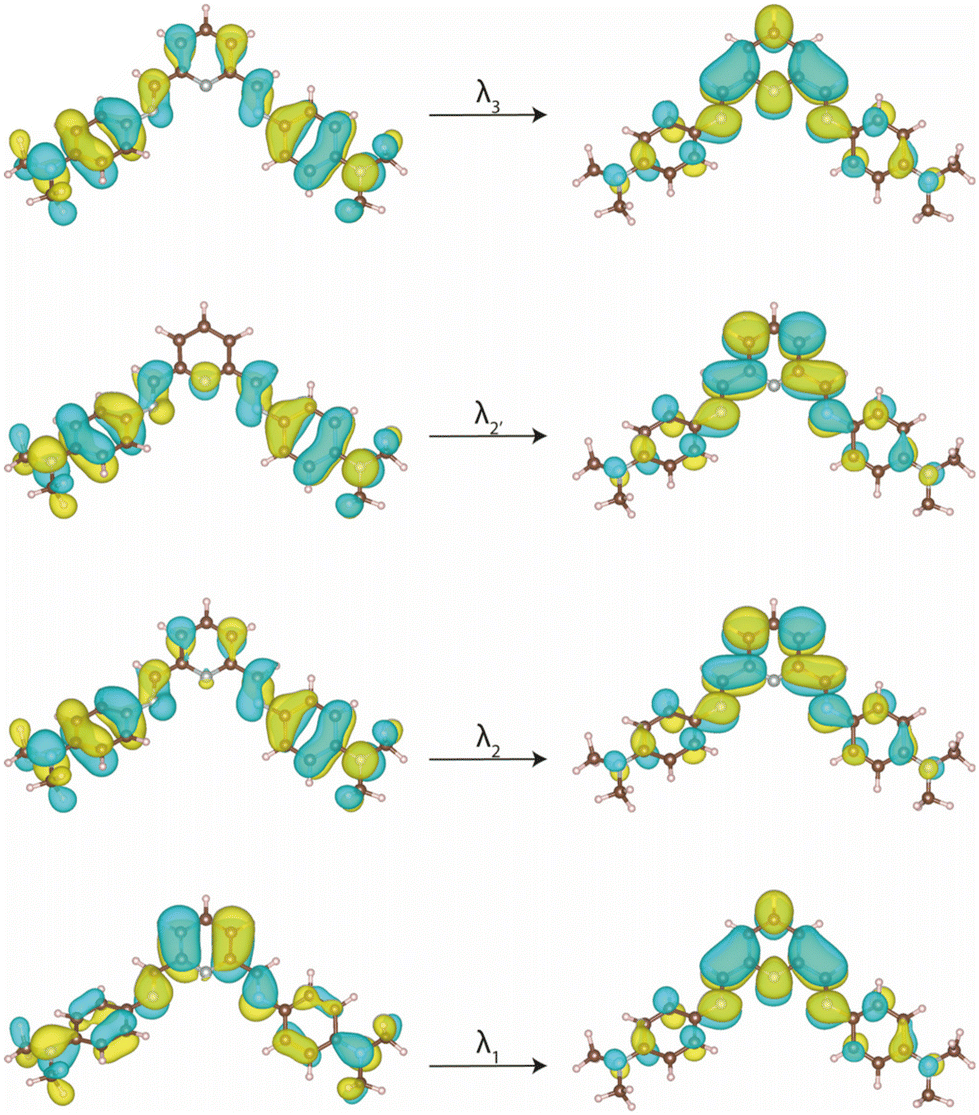 | ||
| Fig. 5 Natural transition orbitals (NTOs) involved in the more significant electronic transitions for 4-Me2NPhPDI in the optimised geometry of its LS complex 4. Isosurfaces are shown setting cut-off at 0.02 e per bohr3. | ||
Notably, Canton et al. recently documented a Jahn–Teller (JT) photoswitching behaviour in the related mononuclear cobalt(II)-2,2′:6′,2″-terpyridine (TERPY) complex.72 This intriguing phenomenon involving an ultrafast switching between the axially-elongated LS and axially-compressed HS forms occurs in aqueous solution at room temperature upon MLCT excitation under deep violet-blue light, evoking memory of the light-induced excited spin-state trapping (LIESST and reverse LIESST) initially observed by Decurtins et al. in octahedral mononuclear SCO iron(II) complexes,90,91 and later in related cobalt(II)-dioxolene complexes exhibiting reversible photo-induced valence tautomerism (VT).92 These findings open new exciting perspectives for implementing logic schemes in QIP through the optical control of quantum coherence (QC) properties in CoII SCO/SMMs as prototypes of optically-addressable qubits.
The Q-band EPR spectra of 1–5 at 4.0 K, depicted in Fig. 6 (see Experimental section in the ESI‡), reveal that, except for 3, all spectra comprise a strong signal at the high-field region around g = 2.0 (ca. 12000 G), typical for LS cobalt(II) complexes, which is split as a consequence of the high axiality and rhombicity present in these compounds. Additionally, 3 and 5 exhibit signals in the low-field region around g = 8.0 (ca. 3000 G), a signature of the presence of HS cobalt(II) complexes in an octahedral environment, which aligns with the incomplete spin transition or its absence observed by magnetometry (see discussion below).
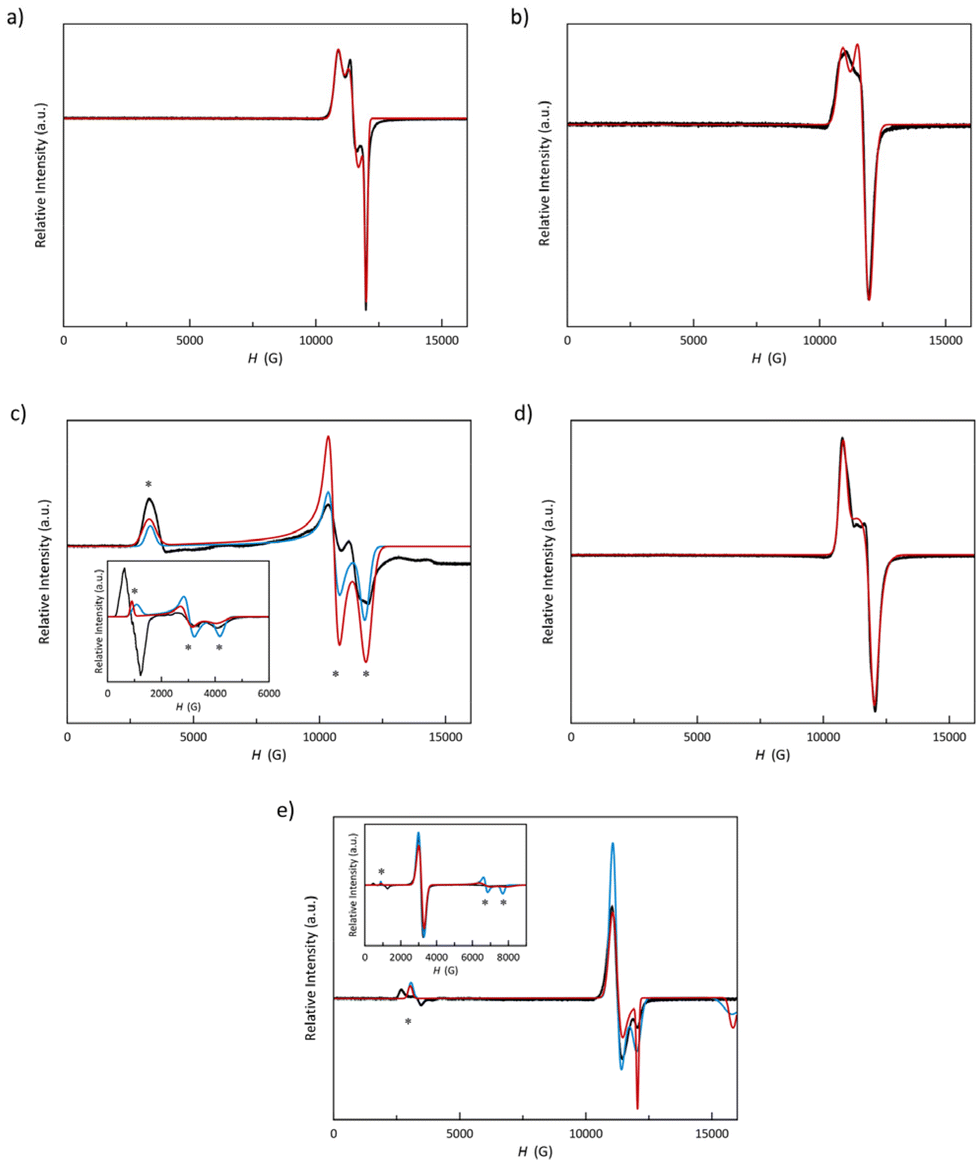 | ||
| Fig. 6 Experimental Q-band EPR spectra of 1–5 (a–e) in the solid state at 4.0 K (black solid lines). X-band spectra of 3 and 5 appear as insets. Red and blue solid lines are the simulated curves for Seff = 1/2 and anisotropic S = 3/2 approaches (see text). | ||
EPR spectra were simulated jointly for the LS and HS fractions. LS contributions were satisfactorily simulated considering a highly axial (gx = gy ≠ gz) for 2 and 5 and a rhombic octahedron (gx ≠ gy ≠ gz) for the rest. Somehow, there is an inversion of the g values for 5, in which gz value is the closest one to the free electron g-factor (2.0). This fact suggests that the magnetic orbital is defined the dz2 orbital instead of the dx2−y2 one, as in the other members of the series. Table S5‡ summarises the g values used in these simulations.
Among these five compounds, 3 and 5 exhibit an HS fraction at 4.0 K evidenced by the EPR signature with signals at very low fields in both Q- and X-band spectra. In 5, this signal is split most likely due to an effect of the field-modulation amplitude; and thus, the real g value is provided by its midpoint. The signals at gz = 7.5 (3) and 8.0 (5) are evidence of a uniaxial zfs (D < 0). In this scenario, the two remaining geff components are expected to adopt values so low that cannot be observed by Q-band EPR spectroscopy, but they do in X-band. In fact, the X-band spectra of 3 and 5 show two signals at high fields with gx = 1.65 (3)/0.85 (5) and gy = 2.30 (3)/1.01 (5). Moreover, when considering a zfs on a quartet spin state, we could only achieve good simulations for the X-band spectra with very large or moderate rhombicities. The parameters used in these simulations [gx = 2.23 (3)/2.15 (5), gy = 2.67 (3)/2.18 (5), gz = 2.90 (3)/2.65 (5), D < 0, and E/D = 0.33 (3)/0.15 (5)] accord with those found from ab initio calculations for 5 (see Experimental section in the ESI‡); but the g values slightly differ for 3 {geff = [1.35, 1.81, 8.09] (3)/[0.91, 1.06, 8.55] (5) or g3/2 = [2.01, 2.27, 2.89] (3)/[2.01, 2.17, 2.96] (5), D = −69.9 (3)/−79.8 (5) cm−1, and E/D = 0.252 (3)/0.152 (5)}.
Magnetic properties
To investigate the influence of phenyl substituents on the spin transition and spin dynamics of mononuclear cobalt(II)-PDI complexes, the dc and ac magnetic properties of compounds 1–5 were measured and compared to those of reference complexes 6–9.70 This comparison focused on the effects of the number of substituents, their electron-donating or electron-withdrawing nature, and their ortho, meta, or para position on the phenyl ring.The dc magnetic properties of 1–5 are depicted in Fig. 7 in the form of the χMT vs. T and M vs. H plots (χM and M being the dc molar magnetic susceptibility and the magnetisation and T and H, the absolute temperature and the applied magnetic field).
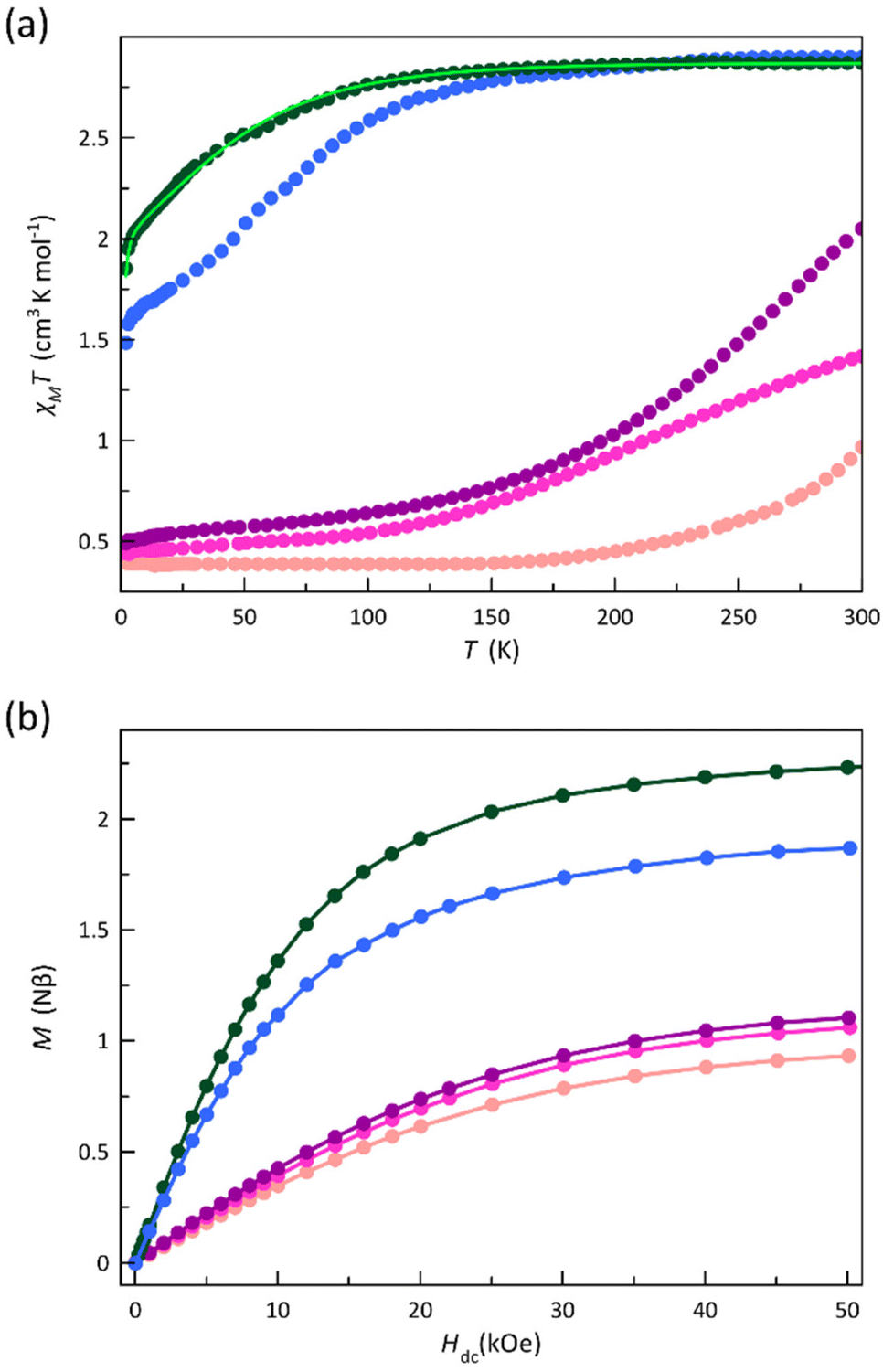 | ||
| Fig. 7 (a) Temperature dependence of χMT and (b) field dependence of M at 2.0 K for 1 (purple), 2 (pink), 3 (green), 4 (pale pink), and 5 (blue). The light green line is the best-fit curve for 3 (see text). | ||
The χMT vs. T plots (Fig. 7a) reveal the occurrence of distinct SCO behaviours for 1, 2, 4, and 5 as well as its absence for 3, similarly to what was found in 6–9.70
At room temperature, χMT for 3 and 5 (2.87 and 2.90 cm3 mol−1 K) is within the expected range for a HS CoII ion with an unquenched orbital momentum contribution, whereas it is rather lower for 1, 2, and 4. Upon cooling, χMT for 3 decreases smoothly down to 1.85 cm3 mol−1 K at 2.0 K, revealing the occurrence of a significant zfs arising from the spin–orbit coupling (SOC) of the HS CoII ion. In contrast, a partial spin transition occurs for 5, as revealed by the decrease of χMT around 100 K (T1/2 ≈ 75 K) until a value of 1.48 cm3 mol−1 K at 2.0 K that agrees with an approximate 2:1 HS/LS molar ratio. Instead, compounds 1, 2, and 4 show a complete and gradual spin transition, as evidenced by the smooth decrease of χMT upon cooling to reach a plateau around 100 (1 and 2) and 150 K (4) with values of 0.49 (1), 0.45 (2), and 0.40 cm3 mol−1 K (4) at 2.0 K, consistent with a LS CoII ion.
This scenario is further supported by the M vs. H plots at 2.0 K for 1–5 (Fig. 7b). Hence, M values at H = 50 kOe are close (1, 2, and 4) to the value of the saturation magnetisation for a LS CoII ion (1.10Nβ for g = 2.2). The reduced magnetisation curves for 2 and 4 in the temperature range of 2.0–10.0 K perfectly superimpose (Fig. S11b and S11d‡), as expected for an S = 1/2 state. On the other hand, the M value of 2.06Nβ at 50 kOe for 3 is close to the saturation magnetisation for an effective doublet spin state arising from a ground Kramers doublet that results from a zfs (2.10Nβ for Seff = 1/2 and g = 4.2). The maximum M value for 5 (1.87Nβ) is close to the calculated saturation magnetisation for a 2:1 HS/LS molar ratio (1.77Nβ). Moreover, the lack of superimposition below 10.0 K in the reduced magnetisation curves for 3 and 5 (Fig. S11c and S11e‡) supports the occurrence of a significant zfs on the quartet ground state.
Understanding the lack of SCO behaviour in 3 remains a challenge. While para-thiomethyl substituents are the least electron-donating in this series (based on Hammett parameters), favouring a less intense ligand field, this would not necessarily predict SCO behaviour. SCO depends heavily on both the molecule itself and how it packs in the crystal lattice, particularly the strength of intermolecular interactions that determine cooperativity. Studies on related cobalt(II) complexes with electron-withdrawing substituents or the family discussed here with electron-donating ones have not shown a clear correlation between ligand character and SCO.70 The only recurring observed feature is that weak X⋯X contacts exist exclusively between LS CoII complexes. Beyond halogen–halogen contacts, other electronic factors from intermolecular interactions undoubtedly play a crucial role.
The experimental magnetic data for 3 were effectively simulated by the simultaneous analysis of the magnetisation and magnetic susceptibility data by full-matrix diagonalisation, as implemented in the PHI program.93Eqn (1) defines the spin Hamiltonian used, where D and E are the axial and rhombic magnetic anisotropy parameters. Since the experimental measurements were carried out on finely crushed crystal samples, gx and gy components of the g-factor are indistinguishable. Therefore, only perpendicular and parallel components were preserved (gx = gy = g⊥ and gz = g∥). This approach usually leads to reasonable estimations of the g factor, the sign of D and even the E/D quotient, with the latter parameter carrying the most significant uncertainty in its determination. This fitting (Fig. 7a and S12‡) unveiled a large uniaxial zfs (D < 0) and a high magnetic rhombicity acting on the quartet ground state of the complex, consistent with the EPR study (as discussed earlier). Best-fit values of these parameters are g⊥ = 2.32, g∥ = 2.71, D = −59.4 cm−1 and E/D = 0.262.
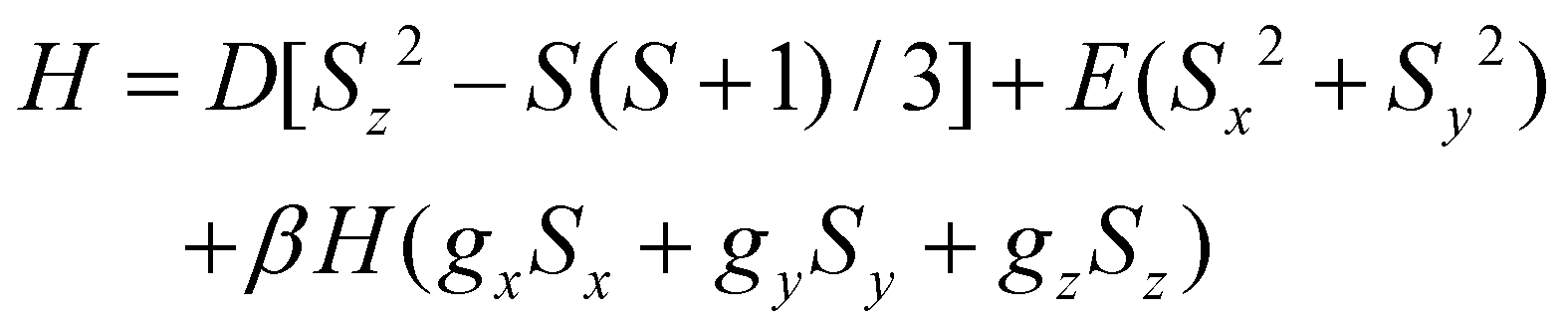 | (1) |
The ac magnetic properties of 1–5 in the presence of an applied dc magnetic field (Hdc) of 1.0 and 2.5 kOe are shown in Fig. S13–S17‡ in the form of the χ′M and χ″Mvs. T plots, where χ′M and χ″M represent the in-phase and out-of-phase ac molar magnetic susceptibility. In the absence of Hdc, no frequency dependence of χ′M and χ″M signals was observed (data not shown) more likely due to either fast quantum tunnelling of magnetisation (QTM)64 or intra-Kramer (IK) relaxation on the ground Kramers doublet of a LS or an easy-plane HS (D > 0) cobalt(II) complex.
Under a small dc magnetic field (Hdc = 1.0 or 2.5 kOe), both frequency- and field-dependent χ′M and χ″M signals were observed in all compounds, the last ones being only incipient at 1.0 kOe for 1, 2 and 4, but shifting towards higher temperatures at 2.5 kOe, whereas those curves for 3 and 5 remained almost unaltered. These features resemble the conclusions from a preceding contribution,70 in which the LS and HS Co(II) complexes accounted for a different field dependence of the slow magnetic relaxation behaviour. Besides the low-temperature χ″M incipient signals, a distinct shoulder developed for 1 at higher temperatures under both dc magnetic fields would suggest a double magnetic relaxation process.
The joint analysis of the χ′M and χ″Mvs. ν plots (Fig. S18–S22‡) through the generalised Debye model provided magnetic relaxation times (τ).94 The occurrence of two relaxation processes in 1 required a second contribution for its analysis. As the temperature increases, the significance of the second low-temperature (LT) process dimes until it becomes irrelevant at 4.0 K. So, the LT contribution was cut off from the analysis above this temperature.
The Arrhenius plots were built from these relaxation times, as displayed in Fig. S23.‡ In addition to the notable difference in τ between the LS and HS forms, while Hdc strongly influences it in the LS configuration (1, 2 and 4), its effect is weak in the HS one (3 and 5), as seen earlier for 6–9.70
In the LS configuration observed in 1, 2, and 4, characterised by a mS = ±1/2 ground Kramers doublet, the absence of an energy barrier governing spin reversal implies that neither Orbach nor quantum-tunnelling mechanisms will likely govern the magnetic relaxation. A comparable scenario may arise in a HS configuration for a CoII ion featuring an easy-plane zfs, as well as in cases with a uniaxial zfs combined with pronounced rhombicity, as exemplified by 3 and 5, where the E/D ratio approaches the limit of 1/3. Furthermore, the lnτ vs. lnT plots (Fig. 8) show linear dependencies in some temperature regions. These observations allow the conclusion that Raman, direct, or temperature-independent IK mechanisms are primarily involved in the magnetic relaxation of the studied cobalt(II)-PDI compounds. Consequently, parameters obtained from the model employed to analyse these plots are summarised in Table S6‡ and compared with those found for 6–9 in Fig. 9 and S24.‡70
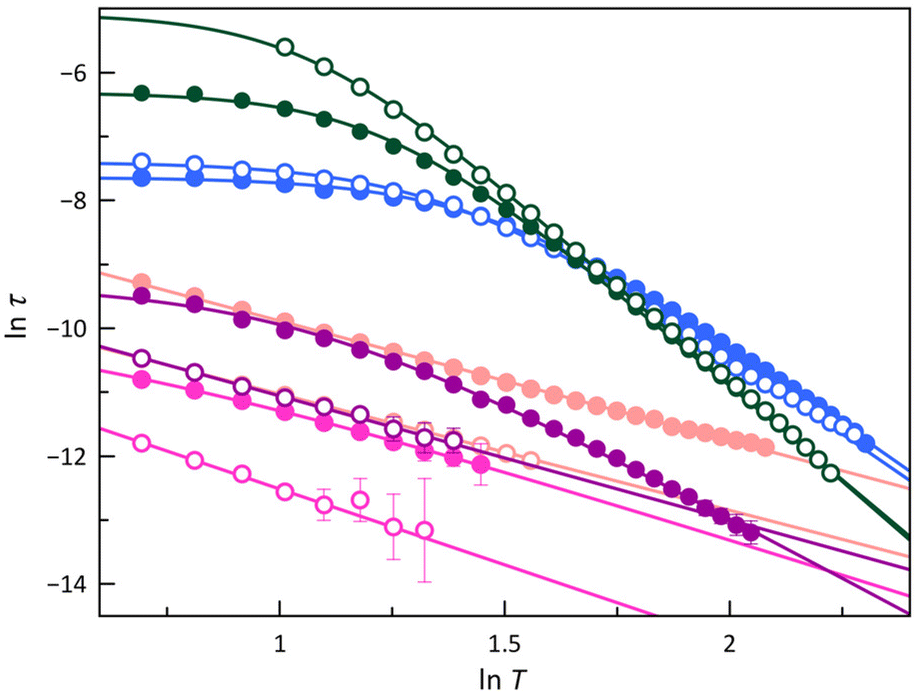 | ||
| Fig. 8 The lnτ vs. lnT plots for the calculated magnetic relaxation times (τ) of 1 (purple), 2 (pink), 3 (green), 4 (pale pink), and 5 (blue) under applied dc magnetic fields of 1.0 (○) and 2.5 kOe (●). Solid lines are the best-fit curves (see text). Vertical error bars denote the standard deviations. | ||
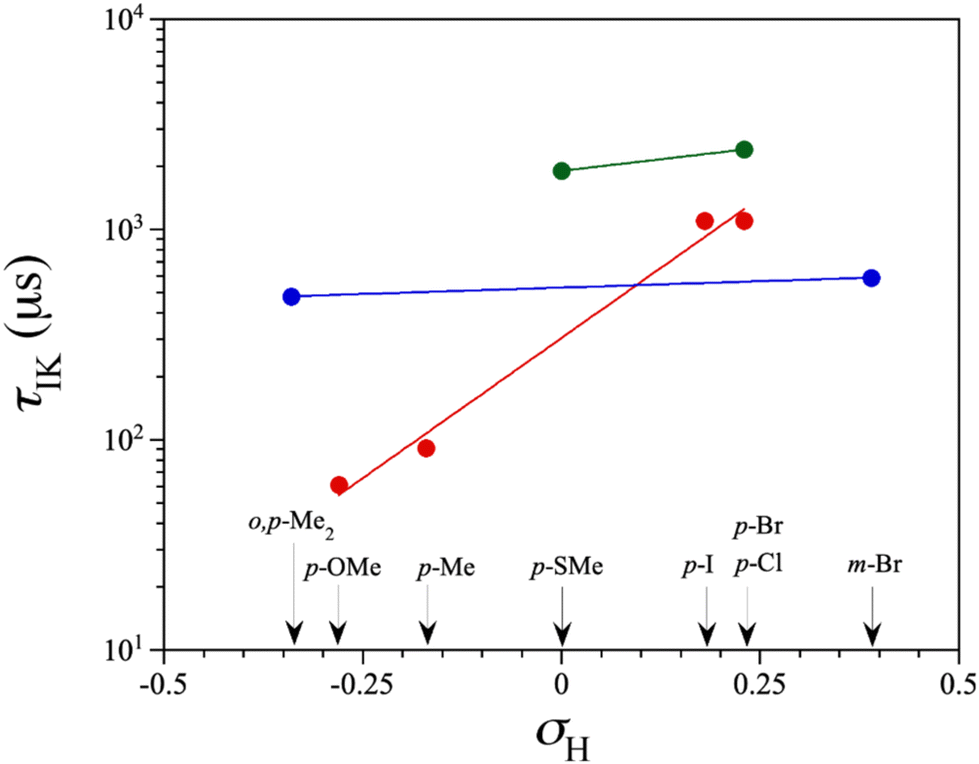 | ||
| Fig. 9 The magnetic relaxation time variation for an IK mechanism (τIK) under an applied dc magnetic field of 2.5 kOe, plotted against the Hammett constant (σH) of the ligand substituents across cobalt(II)-PDI complexes 1–3 and 5–9. For clarity, the identity of the ligand substituents is provided for each data point, which are drawn in red, green, and blue to distinguish LS, HS, and LS/HS complexes, respectively. The solid lines are the best-fit exponential curves (see text). | ||
At the lowest Hdc, the IK process prevails for the HS and mixed LS/HS complexes 3 and 5, respectively, with variable significance and relaxation rates at low temperatures, while in the LS complexes 1, 2, and 4, a Raman mechanism assisted by optical phonons with n ca. 2 govern the relaxation of the magnetisation at any applied Hdc. Conversely, acoustic phonons with n ca. 6 are responsible for the behaviour observed in the HS complex 3. An intermediate situation was observed for the mixed LS/HS complex 5, where n is close to 5, with optical and acoustic phonons competing to decide the magnetic relaxation without the possibility of discerning them. The second LT relaxation in 1 was simulated by considering a Raman mechanism involving optical phonons (n ≈ 2), although the narrow temperature range in which this process is relevant compromises this result (data not shown).
An analysis of the magnetic relaxation parameters for cobalt(II)-PDI complexes 1–9 reveals some correlations with the electron-withdrawing character of the ligand substituents expressed by the Hammett constant (Fig. 9), which depend on the spin state. That is, the relaxation mechanisms that operate may be different depending on the spin state, making it difficult to find correlations. For example, the exponential factor n in Raman relaxation differs significantly between 3 and 5 compared to 1, 2, and 4, suggesting the involvement of phonons of different natures. Therefore, correlations between parameters governing spin relaxations and Hammett constant should be analysed while considering the spin state. For LS, HS, or mixed LS/HS complexes, τIK at the highest Hdc, for which more data are available, increases exponentially with the σ-type inductive electron withdrawing of the para-substituent. So, the magnetic relaxation rate through the IK mechanism is slowed down when electron and spin densities are removed from the metal towards the ligand, independently of the spin configuration. This extensive delocalisation makes spin reversal more challenging, requiring a synchronised inversion of all local spin densities distributed throughout the complex.
However, this dependence is palpably more pronounced for the LS configuration. This situation likely arises from variances in spin delocalisation and spin polarisation effects on the ligand in LS and HS complexes. Consequently, the magnetic relaxation rate within the ground Kramers doublet (ms = ±1/2) is significantly slower than anticipated. So, our DFT study suggested that this phenomenon is attributable to a greater degree of electron and spin delocalisation in the HS than in the LS configuration, as reported earlier.70
Similar correlations are sighted when looking at the parameters of a Raman relaxation mechanism (Fig. S24‡). However, these results must be used with caution since, although the exponent n should take values close to 2 or in the range 7–9 depending on the type of phonons (optical or acoustic) involved in the process, sometimes intermediate values are found because of the impossibility of discerning the two competing mechanisms. This problem, therefore, translates to the values of C closely correlated with those of n.
These findings show the potential to manipulate SMM and, subsequently, QC properties of LS CoII ions using an external magnetic field, thus opening the possibility for their application as molecular spintronic devices in future spin-based quantum computing machines. Crucially, for a system with a doublet spin ground state, the magnetic relaxation time coincides with the spin–lattice relaxation time (T1 = τ), which acts as a limiting factor for the quantum phase memory time (Tm ≤ T1) varying nearly tenfold at 2.0 K in our LS cobalt(II)-PDI complexes (from around 10–100 μs to 0.1–1 ms for Hdc = 1.0 and 2.5 kOe, respectively). This significant change in relaxation dynamics offers a promising avenue for developing a new class of magnetic field-effect spin quantum transistors (MFE-SQT), which, in contrast to conventional electronic transistors in classic computers, relying on electric voltage for control, would function based on an external magnetic field.
Electrochemical properties and theoretical calculations
The electrochemical properties of 1–9 have been investigated by cyclic voltammetry to analyse the influence of the electron-donating or withdrawing nature of the phenyl substituents, as well as their ortho, meta, or para substitution pattern, on the metal- and ligand-centred, multiredox behaviour. A summary of the electrochemical data is listed in Table S7.‡The cyclic voltammograms in acetonitrile at room temperature show a pattern of qualitatively similar redox behaviour (Fig. 10 and S25‡). They exhibit one reversible oxidation wave (E1 from 0.14 to 0.59 V) and two reversible reduction waves (E2 from −0.52 to −0.82 V and E3 from −1.38 to −1.67 V) together with one (1 and 3–9) or two (2) completely irreversible reduction waves at very high negative formal potentials (E4 from −1.85 to −2.10 V and E5 = −2.19 V). In addition, one completely irreversible oxidation wave appears at very high positive formal potentials for 4 (data not shown).
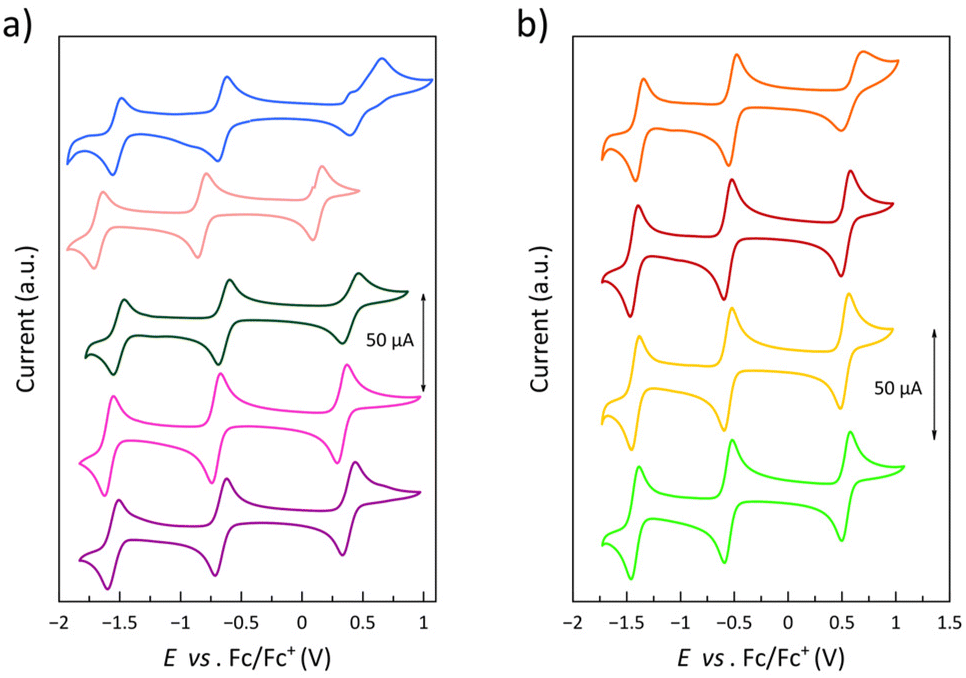 | ||
| Fig. 10 Cyclic voltammograms of (a) 1–5 (from purple to blue) and (b) 6–9 (from light green to orange) in acetonitrile (0.1 M nBu4NPF6) at 25 °C and 200 mV s−1. | ||
For all compounds, the anodic to cathodic peak separations for the first two reduction waves (ΔEp,2 = 71–95 mV and ΔEp,3 = 71–93 mV) are comparable to that of the Fc+/Fc redox couple (ΔEp = 80 mV) under the same conditions. However, those for the oxidation wave are somewhat larger (ΔEp,1 = 78–260 mV), with 5 even revealing a two-step oxidation process. Yet, scan rate-dependence studies for this oxidation wave in the range 20–250 mV s−1 (insets of Fig. S25‡) show a linear dependence of the difference between the cathodic and anodic peak intensities with the square root of the scan rate in all compounds (data not shown), indicating reversibility on the voltametric time-scale.
The assignment of the observed redox processes for 1–9 is illustrated in Scheme 2.26 The reversible oxidation wave corresponds to the one-electron metal oxidation to afford the cobalt(III) species (Scheme 2a). In contrast, the first pair of well-separated reversible reduction waves conform to the ligand's stepwise one-electron reduction to give the single and double imide-type π-radical anion cobalt(II) species (Scheme 2b). That being so, the second pair of irreversible reduction waves is likely attributed to the further stepwise one-electron reduction of the ligands to give the single and double imide-type (non-radical) dianion cobalt(II) species (Scheme 2c).
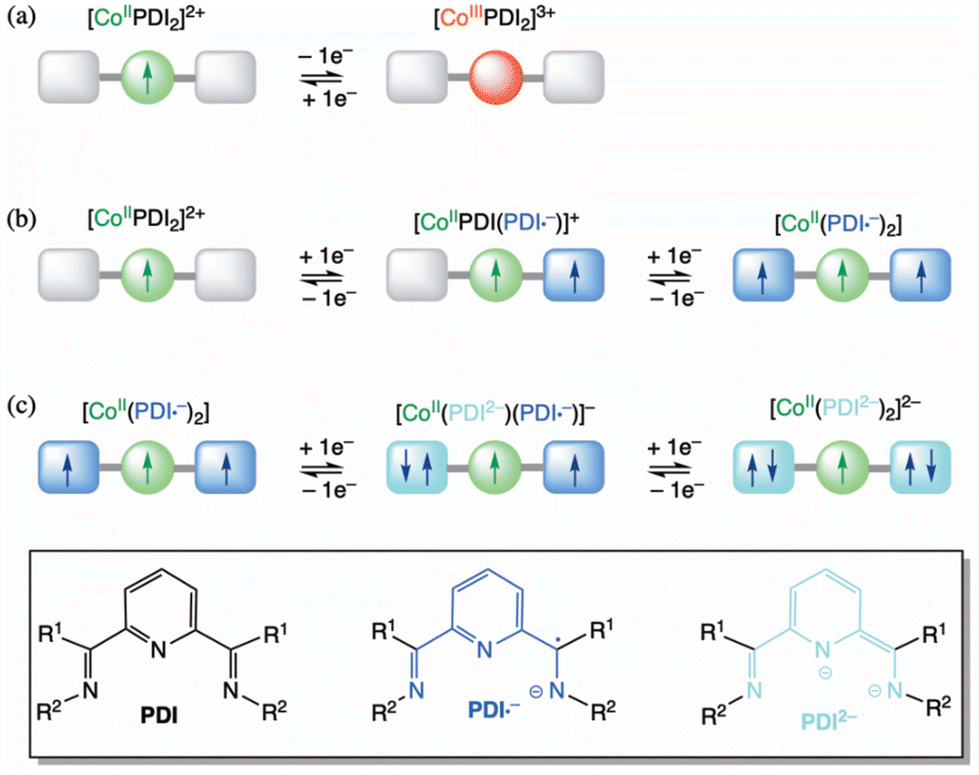 | ||
| Scheme 2 Proposed redox model for the multielectron transfer series in mononuclear six-coordinate cobalt(II)-PDI complexes as prototypes of molecular spin quantum transistors and capacitors. The boxed structure shows redox isomers for the non-innocent PDI ligands. | ||
The σ-donor capacity and π-acceptor ability of the PDI ligands are the leading causes of the dual metal- and ligand-centred multiredox behaviour of this unique series of complexes. Moreover, they are eventually responsible for the observed thermodynamic and kinetic stability of the corresponding cobalt(III) and the single or double imide-type π-radical anion cobalt(II) species. Hence, the significant separation between the first pair of reversible reduction waves along this series (ΔE23) points out high thermodynamic stability of the mixed-valent, one-electron reduced cobalt(II) π-radical species resulting from a substantial electronic delocalisation, as evidenced by the estimated values of the comproportionation constant (Kc,23 = 2.6–8.2 × 1014).
There is a linear correlation between the formal redox potentials for each of the three reversible redox potentials along 1–9 and the electron-withdrawing nature of the ligand substituents expressed by the Hammett constant (Fig. 11).85E1–3 moderately increase with σH, favouring thus the metal oxidation and disfavouring the ligand reduction. The deviation of 5 from this correlation could be attributed to several reasons, with the most probable cause being the significant distortion from planarity underwent by the PDI ligand due to steric hindrances between imine hydrogen atoms and the ortho-methyl groups, leading to a loss of ligand aromaticity. Notably, 5 systematically deviates from all correlations established in this study between a physical property and σH. This discrepancy is particularly evident in the correlation for the 1H NMR chemical shift, carried out only on the ligands, supporting the hypothesis that the ligand distortion is the underlying cause, as confirmed by theoretical calculations (see discussion above). It must be emphasised that the Hammett parameter is applied without considering other factors, such as geometric changes, which may lead to an inaccurate estimation of the electron-withdrawing effects of the substituents. Thus, this electronic feature can be in 5 less than in 1, despite the former containing an additional electron-donating methyl group, both groups being not very efficient in the geometry shown.
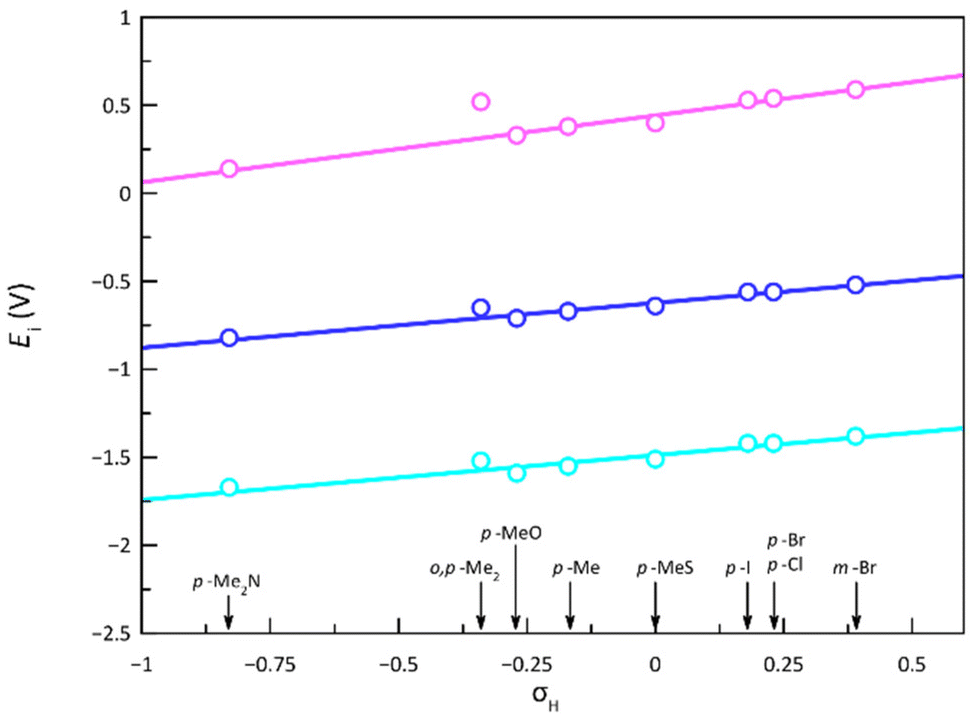 | ||
| Fig. 11 Variation of the formal redox potentials [Ei, i = 1 (pink), 2 (blue), and 3 (cyan)] for the cobalt(II)-PDI complexes 1–9 with the Hammett constants (σH) of the ligand substituents. The identity of the ligand substituents is included for clarity. The solid lines are the best linear fits (see text). | ||
Thus, our DFT calculations on the unsubstituted [CoII(PDI)2]2+ model complex support that the electronic reductions occur in the ligands instead of the metal ion. Geometry optimisation on the double reduced species in both ferromagnetic (F) and antiferromagnetic (AF) states confirm that the [CoII(PDI˙−)2] electronic nature is the most stable,95,96 exhibiting partial molecular asymmetry that distinguishes both radicals. For the optimised geometries of the most stable spin configurations with LS and HS CoII ions, the magnetic couplings between the PDI˙− radicals and the CoII ion are F (J ≈ +299/+309 cm−1, Ĥ = −JŜ1·Ŝ2) for the LS metal ion and AF (J ≈ −590/−579 cm−1) for the HS one (Fig. 12).
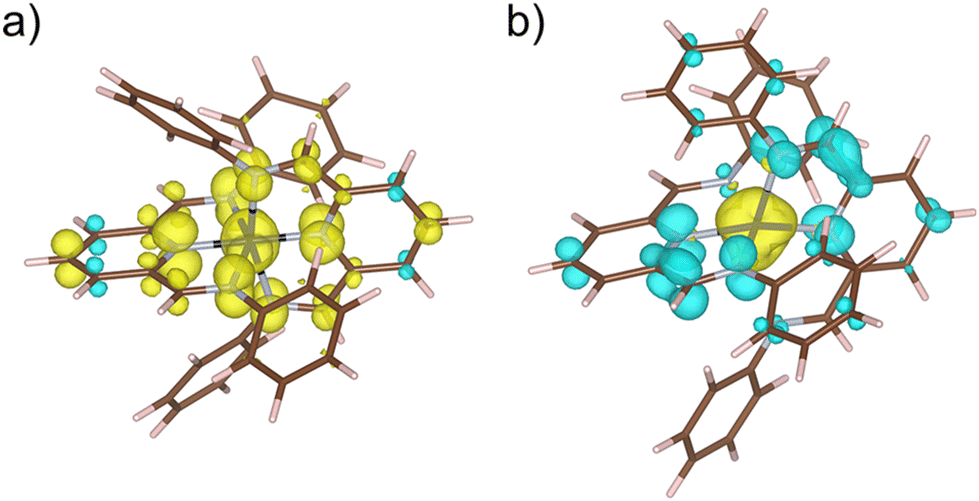 | ||
| Fig. 12 Spin density maps for the optimised geometries of the most stable spin configurations of the [CoII(PDI˙−)2] species incorporating (a) LS and (b) HS CoII ions. Isosurfaces are shown setting cut-off at 0.006 e per bohr3. Positive and negative spin densities are displayed in yellow and blue colours. | ||
For a LS CoII ion, there is only one contribution, which is ferromagnetic, as it involves two orthogonal magnetic orbitals, one π* from the radical and one eg from the metal ion. Conversely, for a HS CoII ion, although a new F contribution appears with a second eg orbital from the metal ion, another AF contribution also arises, involving a t2g orbital that intensely overlaps with that describing the radical and, therefore, prevails. Since the PDI˙− radical places a large part of its spin density on the nitrogen donor atoms (Fig. 12), metal ion and radicals are strongly coupled, as evidenced by the calculated atomic spin densities (Table S8‡) and magnetic couplings.
For the same reason, and having a single atom in the exchange path between the two radicals, their magnetic coupling is non-negligible, being AF or F for an HS or LS configuration of the CoII ion, respectively. While a F coupling is initially expected due to the accidental orthogonality between the magnetic orbitals of the radicals, notable electronic transfers occur with HS CoII ion favouring the AF configuration (Scheme 3), which is overstabilised.97 This fact can partially distort results for JM–R and JR–R, requiring careful consideration within its context.
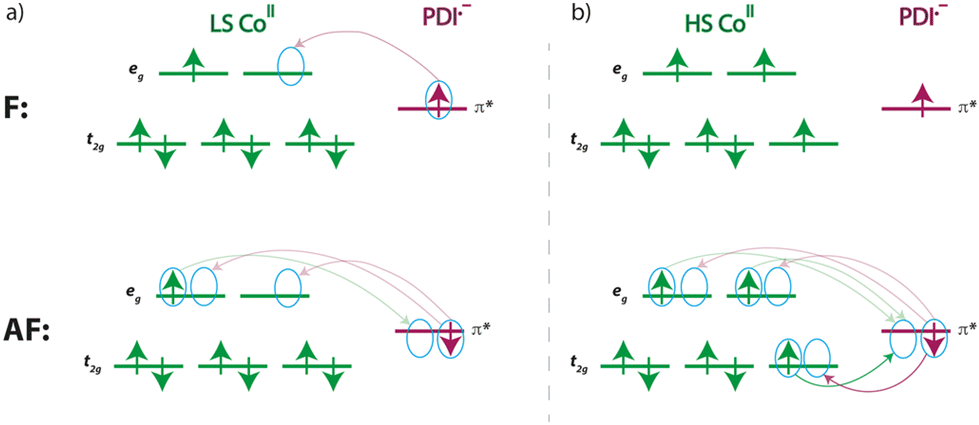 | ||
| Scheme 3 Simplified visualisation of the most significant partial charge transfers occurring in the F (top) and AF (down) configurations of the [CoII(PDI˙−)2] species incorporating (a) LS and (b) HS CoII ions. Only contributions that differentiate both states are shown. Fainter arrows are used for less notable charge transfers. | ||
The calculated charge-induced S = 1/2 ↔ S = 3/2 (F) [(LS)-CoII(PDI)2]2+/[(LS)-CoII(PDI˙−)2] or, alternatively, S = 3/2 ↔ S = 1/2 (AF) [(HS)-CoII(PDI)2]2+/[(HS)-CoII(PDI˙−)2] magnetic switching behaviour makes of these opto-electroactive spin crossover complexes potential prototypes of electric field-effect spin quantum transistors (EFE-SQTs) for molecular spintronics. Although SCO and SMM properties do not converge in the same temperature range, which will hardly happen in other systems since only a few SMMs based on organometallic lanthanide complexes reaching magnetisation-blocking temperatures up to that of N2 liquefaction (Tbca. 80 K) are known, another stimulus, the electronic reduction or oxidation of the system, interacts with these two properties. Depending on the applied electric gate voltage, these compounds can demonstrate significant changes in electrical conductance when transitioning from the HS or LS [CoII(PDI)2]2+ complexes to the magnetically coupled metal-radical species, [CoII(PDI˙−)2] (Scheme 2b).
In particular, the tailor-made mononuclear cobalt(II)-4-SMePhPDI complex is a very appealing candidate for the physical implementation of EFE-SQTs when integrated into gold-based molecular junctions because of the presumed high affinity for gold electrodes through the free sulphur atoms of the para-substituted thiomethyl groups. Thus, in a related cobalt(II) complex with a redox-active mercapto-functionalised TERPY ligand, the single Kondo peak in the current–voltage (I–V) plots splits after mechanical stretching of the attached individual molecules anchored between two mobile gold electrodes.46 The authors argued that this unique mechanical control of the EFE-SQT behaviour eventually originates from the reduced octahedral cobalt(I) species resulting upon current flow through the molecule, which would possess a non-negligible uniaxial magnetic anisotropy (D < 0) of the triplet spin state of the CoI ion.
Yet, the occurrence of a spin transition between HS and LS CoII ions typical of octahedral bis(terpyridine)cobalt(II) SCO complexes cannot be discarded, as previously found in the related iron(II) SCO complexes with the redox-active 2,6-bis(pyrazol-1-yl)pyridine (dpzpy) and 3,5-bis(pyrazol-1-yl)-4,4′-bipyridine (dpzbpy) ligands.47 In this other case, the authors proposed that, upon electrical current flow, the EFE-SQT behaviour occurs by reduction of the two ancillary ligands, which indirectly alters the spin state of the FeII ion, as supported by theoretical calculations on a model.47 This charge-induced SCO is associated to the appearance of a split zero bias resistance Kondo peak in the electrical conductance for the diamagnetic LS [FeII(dpzbpy)2]2+ complex upon increasing the gate voltage, which would be explained by the zero-field splitting of the triplet state in [FeII(dpzbpy˙−)2] resulting from an antiferromagnetic coupling between the quintet HS FeII ion and the two π-radical ligands arising from a double reduction.
Conclusions
Ligand tuning has led to the preparation of a unique family of mononuclear spin crossover cobalt(II) complexes featuring PDI ligands with electron-donating and withdrawing substituents at the para, meta, or ortho positions of the terminal N-substituted phenyl rings. Depending on the electronic nature of the substituent, this novel series exhibits a range of thermally induced spin transitions, including complete, partial, or no transition, but at the same time also field-induced slow magnetic relaxation at low temperatures. Key conclusions regarding their magnetic relaxation dynamics are as follows: (i) the SMM behaviour is governed by two distinct regimes, with slower- and faster-relaxing centres depending on their HS and LS nature; (ii) a strong dual field-dependence of the SMM behaviour is observed in the LS form, with spin reversal slowing down as the applied magnetic field increases; and (iii) the slow magnetic relaxation of LS and HS complexes involves distinct optical and acoustic two-phonon Raman mechanistic pathways, occasionally competing with an additional temperature-independent IK mechanism at the lowest temperatures.This unique family of redox-active complexes with electron-donating and withdrawing substituents possesses both metal- and ligand-centred electrochemical activity because of the strong σ-donor capacity and the π-acceptor character of the PDI ligand. Throughout this family, an overall correlation between the redox behaviour and the electron-donating/withdrawing nature of the ligand's para-substituent is established. Otherwise, the distinct redox behaviour of the ortho, para-dimethyl derivative is likely due to the steric effects of the ortho-methyl group, so the larger deviations from planarity in the ligand result in a significant reduction of the π-type inductive donor effects of the para-methyl group.
This new class of triply thermal-, magneto-, and electro-switchable SCO cobalt(II)-PDI compounds with a field-induced SMM behaviour possesses multiple metal- or ligand-based redox and spin states, capable of reversible interconversion under various external stimuli, including temperature, magnetic field, and redox potential. The reported electronic and magnetic multistabilities underscore the potential of multiresponsive and multifunctional SCO/SMMs in the emerging fields of molecular spintronics and quantum computation.
Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Data availability
Crystallographic data (excluding structure factors) of 1–5 have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 2333341 (1), 1989325 (2), 2333343 (3), 2333342 (4), and 2333340 (5). Experimental (EPR spectroscopy, …) and theoretical (optimized geometries, NMR shields, …) data: https://doi.org/10.5281/zenodo.12739876.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Spanish MINECO (Projects PID2019-109735GB-I00 and Unidad de Excelencia María de Maeztu CEX2019-000919-M) and the Generalitat Valenciana (AICO/2020/183 and AICO/2021/295). This work has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no 748921-SIMOF. R. R. thanks the Generalitat Valenciana and CAPES/BRASIL for the doctoral (GRISOLIAP/2017/057) and postdoctoral (88887.798611/2022-00) grants. We are indebted to Dr Nicolás Moliner, Dr Gloria Agustí, and José María Martínez (Universitat de València) for their assistance with the magnetic and EPR measurements.References
- J. M. Savéant and C. Costentin, Elements of Molecular and Biomolecular Electrochemistry: An Electrochemical Approach to Electron Transfer Chemistry, John Wiley & Sons, Inc., Hoboken, USA, 2006 Search PubMed.
- S. Lee, J. Hong and K. Kang, Redox–Active Organic Compounds for Future Sustainable Energy Storage System, Adv. Energy Mater., 2020, 10, 2001445 CrossRef CAS.
- G. Kwon, Y. Ko, Y. Kim, K. Kim and K. Kang, Versatile Redox-Active Organic Materials for Rechargeable Energy Storage, Acc. Chem. Res., 2021, 54, 4423–4433 CrossRef CAS PubMed.
- J. Stubbe and W. A. van der Donk, Protein Radicals in Enzyme Catalysis, Chem. Rev., 1998, 98, 705–762 CrossRef CAS PubMed.
- L. Que and W. B. Tolman, Biologically inspired oxidation catalysis, Nature, 2008, 455, 333–340 CrossRef CAS PubMed.
- Y. Yuan and A. Lei, Electrochemical Oxidative Cross-Coupling with Hydrogen Evolution Reactions, Acc. Chem. Res., 2019, 52, 3309–3324 CrossRef CAS PubMed.
- P. Gotico, D. Moonshiram, C. Liu, X. Zhang, R. Guillot, A. Quaranta, Z. Halime, W. Leibl and A. Aukauloo, Spectroscopic Characterisation of a Bio–Inspired Ni–Based Proton Reduction Catalyst Bearing a Pentadentate N2S3 Ligand with Improved Photocatalytic Activity, Chem. – Eur. J., 2020, 26, 2859–2868 CrossRef CAS PubMed.
- P. Gotico, L. Roupnel, R. Guillot, M. Sircoglou, W. Leibl, Z. Halime and A. Aukauloo, Atropisomeric Hydrogen Bonding Control for CO2 Binding and Enhancement of Electrocatalytic Reduction at Iron Porphyrins, Angew. Chem., Int. Ed., 2020, 59, 22451–22455 CrossRef CAS PubMed.
- G. Ruan, L. Engelberg, P. Ghosh and G. Maayan, A unique Co(III)-peptoid as a fast electrocatalyst for homogeneous water oxidation with low overpotential, Chem. Commun., 2021, 57, 939–942 RSC.
- G. L. Soloveichik, Flow Batteries: Current Status and Trends, Chem. Rev., 2015, 115, 11533–11558 CrossRef CAS PubMed.
- M. Park, J. Ryu, W. Wang and J. Cho, Material design and engineering of next-generation flow-battery technologies, Nat. Rev. Mater., 2016, 2, 16080 CrossRef.
- J. Winsberg, T. Hagemann, T. Janoschka, M. D. Hager and U. S. Schubert, Redox–Flow Batteries: From Metals to Organic Redox–Active Materials, Angew. Chem., Int. Ed., 2017, 56, 686–711 CrossRef CAS PubMed.
- S. Mukhopadhyay, S. K. Mandal, S. Bhaduri and W. H. Armstrong, Manganese Clusters with Relevance to Photosystem II, Chem. Rev., 2004, 104, 3981–4026 CrossRef CAS PubMed.
- C. Herrero, B. Lassallekaiser, W. Leibl, A. Rutherford and A. Aukauloo, Artificial systems related to light driven electron transfer processes in PSII, Coord. Chem. Rev., 2008, 252, 456–468 CrossRef CAS.
- T. J. Wydrzynski and W. Hillier, Molecular Solar Fuels, RSC Publishing, Cambridge, UK, 2011 Search PubMed.
- M. Desage-El Murr, Redox-Active Ligands: Concepts and Catalysis, Wiley-VCH, Weinheim, Germany, 2024 Search PubMed.
- M. D. Ward and J. A. McCleverty, Non-innocent behaviour in mononuclear and polynuclear complexes: consequences for redox and electronic spectroscopic properties, J. Chem. Soc., Dalton Trans., 2002, 275–288 RSC.
- K. Ray, T. Petrenko, K. Wieghardt and F. Neese, Joint spectroscopic and theoretical investigations of transition metal complexes involving non-innocent ligands, Dalton Trans., 2007, 1552–1566 RSC.
- R. Eisenberg and H. B. Gray, Noninnocence in Metal Complexes: A Dithiolene Dawn, Inorg. Chem., 2011, 50, 9741–9751 CrossRef CAS PubMed.
- W. Kaim, Manifestations of Noninnocent Ligand Behavior, Inorg. Chem., 2011, 50, 9752–9765 CrossRef CAS PubMed.
- C. G. Pierpont, Ligand Redox Activity and Mixed Valency in First-Row Transition-Metal Complexes Containing Tetrachlorocatecholate and Radical Tetrachlorosemiquinonate Ligands, Inorg. Chem., 2011, 50, 9766–9772 CrossRef CAS PubMed.
- J. Ferrando-Soria, M. Castellano, R. Ruiz-García, J. Cano, M. Julve, F. Lloret, J. Pasán, C. Ruiz-Pérez, L. Cañadillas-Delgado, Y. Li, Y. Journaux and E. Pardo, Redox switching of the antiferromagnetic coupling in permethylated dicopper(II) paracyclophanes, Chem. Commun., 2012, 48, 8401–8403 RSC.
- J. Ferrando-Soria, M. Castellano, R. Ruiz-García, J. Cano, M. Julve, F. Lloret, C. Ruiz-Pérez, J. Pasán, L. Cañadillas-Delgado, D. Armentano, Y. Journaux and E. Pardo, Dicopper(II) Metallacyclophanes with Electroswitchable Polymethyl–Substituted para –Phenylene Spacers, Chem. – Eur. J., 2013, 19, 12124–12137 CrossRef CAS PubMed.
- M. Castellano, R. Ruiz-García, J. Cano, M. Julve, F. Lloret, Y. Journaux, G. De Munno and D. Armentano, Multielectron transfer in a dicopper(II) anthraquinophane, Chem. Commun., 2013, 49, 3534–3536 RSC.
- M. Castellano, W. P. Barros, A. Acosta, M. Julve, F. Lloret, Y. Li, Y. Journaux, G. De Munno, D. Armentano, R. Ruiz-García and J. Cano, Dicopper(II) Anthraquinophanes as Multielectron Reservoirs for Oxidation and Reduction: A Joint Experimental and Theoretical Study, Chem. – Eur. J., 2014, 20, 13965–13975 CrossRef CAS PubMed.
- N. El Alouani Dahmouni, A. Switlicka, B. Machura, N. Moliner, R. Rabelo, R. Ruiz-García, S.-E. Stiriba, J. Cano and M. Julve, Bulky countercation effects on the crystal packing of anionic dithiooxalato-containing Ni(II), Pd(II) and Pt(II) complexes: spectroscopic–redox correlations, CrystEngComm, 2022, 24, 4787–4799 RSC.
- J. McGuire, B. Wilson, J. McAllister, H. N. Miras, C. Wilson, S. Sproules and J. H. Farnaby, Molecular and electronic structure of the dithiooxalato radical ligand stabilised by rare earth coordination, Dalton Trans., 2019, 48, 5491–5495 RSC.
- W. I. Dzik, J. I. van der Vlugt, J. N. H. Reek and B. de Bruin, Ligands that Store and Release Electrons during Catalysis, Angew. Chem., Int. Ed., 2011, 50, 3356–3358 CrossRef CAS PubMed.
- V. K. K. Praneeth, M. R. Ringenberg and T. R. Ward, Redox–Active Ligands in Catalysis, Angew. Chem., Int. Ed., 2012, 51, 10228–10234 CrossRef CAS PubMed.
- V. Lyaskovskyy and B. de Bruin, Redox Non-Innocent Ligands: Versatile New Tools to Control Catalytic Reactions, ACS Catal., 2012, 2, 270–279 CrossRef CAS.
- A. I. O. Suarez, V. Lyaskovskyy, J. N. H. Reek, J. I. van der Vlugt and B. de Bruin, Complexes with Nitrogen–Centered Radical Ligands: Classification, Spectroscopic Features, Reactivity, and Catalytic Applications, Angew. Chem., Int. Ed., 2013, 52, 12510–12529 CrossRef CAS PubMed.
- O. R. Luca and R. H. Crabtree, Redox-active ligands in catalysis, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC.
- D. L. J. Broere, R. Plessius and J. I. van der Vlugt, New avenues for ligand-mediated processes – expanding metal reactivity by the use of redox-active catechol, o-aminophenol and o-phenylenediamine ligands, Chem. Soc. Rev., 2015, 44, 6886–6915 RSC.
- J. Jacquet, M. Desage-El Murr and L. Fensterbank, Metal–Promoted Coupling Reactions Implying Ligand–Based Redox Changes, ChemCatChem, 2016, 8, 3310–3316 CrossRef CAS.
- J. I. van der Vlugt, Radical–Type Reactivity and Catalysis by Single–Electron Transfer to or from Redox–Active Ligands, Chem. – Eur. J., 2019, 25, 2651–2662 CrossRef CAS PubMed.
- A. Nakada, T. Matsumoto and H.-C. Chang, Redox-active ligands for chemical, electrochemical, and photochemical molecular conversions, Coord. Chem. Rev., 2022, 473, 214804 CrossRef CAS.
- H. Tang, E. N. Brothers and M. B. Hall, The Distinctive Electronic Structures of Rhenium Tris(thiolate) Complexes, an Unexpected Contrast to the Valence Isoelectronic Ruthenium Tris(thiolate) Complexes, Inorg. Chem., 2017, 56, 583–593 CrossRef CAS PubMed.
- I. F. Mansoor, D. I. Wozniak, Y. Wu and M. C. Lipke, A delocalized cobaltoviologen with seven reversibly accessible redox states and highly tunable electrochromic behaviour, Chem. Commun., 2020, 56, 13864–13867 RSC.
- K. A. Maxwell, M. Sykora, J. M. DeSimone and T. J. Meyer, One-Pot Synthesis and Characterization of a Chromophore−Donor−Acceptor Assembly, Inorg. Chem., 2000, 39, 71–75 CrossRef CAS PubMed.
- R. Konduri, H. Ye, F. M. MacDonnell, S. Serroni, S. Campagna and K. Rajeshwar, Ruthenium Photocatalysts Capable of Reversibly Storing up to Four Electrons in a Single Acceptor Ligand: A Step Closer to Artificial Photosynthesis, Angew. Chem., Int. Ed., 2002, 41, 3185–3187 CrossRef CAS PubMed.
- M.-J. Kim, R. Konduri, H. Ye, F. M. MacDonnell, F. Puntoriero, S. Serroni, S. Campagna, T. Holder, G. Kinsel and K. Rajeshwar, Dinuclear Ruthenium(II) Polypyridyl Complexes Containing Large, Redox-Active, Aromatic Bridging Ligands: Synthesis, Characterization, and Intramolecular Quenching of MLCT Excited States, Inorg. Chem., 2002, 41, 2471–2476 CrossRef CAS PubMed.
- F. Lachaud, A. Quaranta, Y. Pellegrin, P. Dorlet, M. F. Charlot, S. Un, W. Leibl and A. Aukauloo, A Biomimetic Model of the Electron Transfer between P680 and the TyrZ–His190 Pair of PSII, Angew. Chem., Int. Ed., 2005, 44, 1560–1564 CrossRef.
- J. Jortner and M. A. Ratner, Molecular Electronics, Blackwell Science, Oxford, UK, 1997 Search PubMed.
- M. A. Reed and T. Lee, Molecular Nanoelectronics, Amer Scientific Pub, Stevenson Ranch, CA, 2003 Search PubMed.
- J. Ferrando-Soria, J. Vallejo, M. Castellano, J. Martínez-Lillo, E. Pardo, J. Cano, I. Castro, F. Lloret, R. Ruiz-García and M. Julve, Molecular magnetism, quo vadis? A historical perspective from a coordination chemist viewpoint, Coord. Chem. Rev., 2017, 339, 17–103 CrossRef CAS.
- J. J. Parks, A. R. Champagne, T. A. Costi, W. W. Shum, A. N. Pasupathy, E. Neuscamman, S. Flores-Torres, P. S. Cornaglia, A. A. Aligia, C. A. Balseiro, G. K.-L. Chan, H. D. Abruña and D. C. Ralph, Mechanical Control of Spin States in Spin-1 Molecules and the Underscreened Kondo Effect, Science, 2010, 328, 1370–1373 CrossRef CAS PubMed.
- V. Meded, A. Bagrets, K. Fink, R. Chandrasekar, M. Ruben, F. Evers, A. Bernand-Mantel, J. S. Seldenthuis, A. Beukman and H. S. J. van der Zant, Electrical control over the Fe(II) spin crossover in a single molecule: Theory and experiment, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 245415 CrossRef.
- T. Mahfoud, G. Molnár, S. Cobo, L. Salmon, C. Thibault, C. Vieu, P. Demont and A. Bousseksou, Electrical properties and non-volatile memory effect of the [Fe(HB(pz)3)2] spin crossover complex integrated in a microelectrode device, Appl. Phys. Lett., 2011, 99, 053307 CrossRef.
- M. G. Cowan, J. Olguín, S. Narayanaswamy, J. L. Tallon and S. Brooker, Reversible Switching of a Cobalt Complex by Thermal, Pressure, and Electrochemical Stimuli: Abrupt, Complete, Hysteretic Spin Crossover, J. Am. Chem. Soc., 2012, 134, 2892–2894 CrossRef CAS PubMed.
- D. Aravena and E. Ruiz, Coherent Transport through Spin-Crossover Single Molecules, J. Am. Chem. Soc., 2012, 134, 777–779 CrossRef CAS PubMed.
- E. Ruiz, Charge transport properties of spin crossover systems, Phys. Chem. Chem. Phys., 2014, 16, 14–22 RSC.
- G. D. Harzmann, R. Frisenda, H. S. J. van der Zant and M. Mayor, Single–Molecule Spin Switch Based on Voltage–Triggered Distortion of the Coordination Sphere, Angew. Chem., Int. Ed., 2015, 54, 13425–13430 CrossRef CAS PubMed.
- E. J. Devid, P. N. Martinho, M. V. Kamalakar, I. Šalitroš, Ú. Prendergast, J.-F. Dayen, V. Meded, T. Lemma, R. González-Prieto, F. Evers, T. E. Keyes, M. Ruben, B. Doudin and S. J. van der Molen, Spin Transition in Arrays of Gold Nanoparticles and Spin Crossover Molecules, ACS Nano, 2015, 9, 4496–4507 CrossRef CAS PubMed.
- A. C. Aragonès, D. Aravena, J. I. Cerdá, Z. Acís-Castillo, H. Li, J. A. Real, F. Sanz, J. Hihath, E. Ruiz and I. Díez-Pérez, Large Conductance Switching in a Single-Molecule Device through Room Temperature Spin-Dependent Transport, Nano Lett., 2016, 16, 218–226 CrossRef PubMed.
- L. D. Wickramasinghe, S. Mazumder, K. K. Kpogo, R. J. Staples, H. B. Schlegel and C. N. Verani, Electronic Modulation of the SOMO–HOMO Energy Gap in Iron(III) Complexes towards Unimolecular Current Rectification, Chem. – Eur. J., 2016, 22, 10786–10790 CrossRef CAS PubMed.
- A. C. Aragonès, D. Aravena, F. J. Valverde-Muñoz, J. A. Real, F. Sanz, I. Díez-Pérez and E. Ruiz, Metal-Controlled Magnetoresistance at Room Temperature in Single-Molecule Devices, J. Am. Chem. Soc., 2017, 139, 5768–5778 CrossRef PubMed.
- T. Jasper-Toennies, M. Gruber, S. Karan, H. Jacob, F. Tuczek and R. Berndt, Robust and Selective Switching of an FeIII Spin-Crossover Compound on Cu2N/Cu(100) with Memristance Behavior, Nano Lett., 2017, 17, 6613–6619 CrossRef CAS PubMed.
- M. M. Paquette, D. Plaul, A. Kurimoto, B. O. Patrick and N. L. Frank, Opto-Spintronics: Photoisomerization-Induced Spin State Switching at 300 K in Photochrome Cobalt–Dioxolene Thin Films, J. Am. Chem. Soc., 2018, 140, 14990–15000 CrossRef CAS PubMed.
- S. K. Karuppannan, A. Martín-Rodríguez, E. Ruiz, P. Harding, D. J. Harding, X. Yu, A. Tadich, B. Cowie, D. Qi and C. A. Nijhuis, Room temperature conductance switching in a molecular iron(III) spin crossover junction, Chem. Sci., 2021, 12, 2381–2388 RSC.
- X. Feng, C. Mathonière, I.-R. Jeon, M. Rouzières, A. Ozarowski, M. L. Aubrey, M. I. Gonzalez, R. Clérac and J. R. Long, Tristability in a Light-Actuated Single-Molecule Magnet, J. Am. Chem. Soc., 2013, 135, 15880–15884 CrossRef CAS PubMed.
- C. Mathonière, H.-J. Lin, D. Siretanu, R. Clérac and J. M. Smith, Photoinduced Single-Molecule Magnet Properties in a Four-Coordinate Iron(II) Spin Crossover Complex, J. Am. Chem. Soc., 2013, 135, 19083–19086 CrossRef PubMed.
- A. Urtizberea and O. Roubeau, Switchable slow relaxation of magnetization in the native low temperature phase of a cooperative spin-crossover compound, Chem. Sci., 2017, 8, 2290–2295 RSC.
- H.-H. Cui, J. Wang, X.-T. Chen and Z.-L. Xue, Slow magnetic relaxation in five-coordinate spin-crossover cobalt(II) complexes, Chem. Commun., 2017, 53, 9304–9307 RSC.
- L. Chen, J. Song, W. Zhao, G. Yi, Z. Zhou, A. Yuan, Y. Song, Z. Wang and Z.-W. Ouyang, A mononuclear five-coordinate Co(II) single molecule magnet with a spin crossover between the S = 1/2 and 3/2 states, Dalton Trans., 2018, 47, 16596–16602 RSC.
- M.-X. Xu, Z. Liu, B.-W. Dong, H.-H. Cui, Y.-X. Wang, J. Su, Z. Wang, Y. Song, X.-T. Chen, S.-D. Jiang and S. Gao, Single-Crystal Study of a Low Spin Co(II) Molecular Qubit: Observation of Anisotropic Rabi Cycles, Inorg. Chem., 2019, 58, 2330–2335 CrossRef CAS PubMed.
- H. Xie, K. R. Vignesh, X. Zhang and K. R. Dunbar, From spin-crossover to single molecule magnetism: tuning magnetic properties of Co(II) bis-ferrocenylterpy cations via supramolecular interactions with organocyanide radical anions, J. Mater. Chem. C, 2020, 8, 8135–8144 RSC.
- D. Shao, L. Shi, L. Yin, B.-L. Wang, Z.-X. Wang, Y.-Q. Zhang and X.-Y. Wang, Reversible on–off switching of both spin crossover and single-molecule magnet behaviours via a crystal-to-crystal transformation, Chem. Sci., 2018, 9, 7986–7991 RSC.
- R. Rabelo, L. Toma, N. Moliner, M. Julve, F. Lloret, J. Pasán, C. Ruiz-Pérez, R. Ruiz-García and J. Cano, Electroswitching of the single-molecule magnet behaviour in an octahedral spin crossover cobalt(II) complex with a redox-active pyridinediimine ligand, Chem. Commun., 2020, 56, 12242–12245 RSC.
- R. Rabelo, L. Toma, N. Moliner, M. Julve, F. Lloret, M. Inclán, E. García-España, J. Pasán, R. Ruiz-García and J. Cano, pH-Switching of the luminescent, redox, and magnetic properties in a spin crossover cobalt(II) molecular nanomagnet, Chem. Sci., 2023, 14, 8850–8859 RSC.
- R. Rabelo, L. Toma, M. Julve, F. Lloret, J. Pasán, D. Cangussu, R. Ruiz-García and J. Cano, How the spin state tunes the slow magnetic relaxation field dependence in spin crossover cobalt(II) complexes, Dalton Trans., 2024, 53, 5507–5520 RSC.
- F. Kobayashi, Y. Komatsumaru, R. Akiyoshi, M. Nakamura, Y. Zhang, L. F. Lindoy and S. Hayami, Water Molecule-Induced Reversible Magnetic Switching in a Bis-Terpyridine Cobalt(II) Complex Exhibiting Coexistence of Spin Crossover and Orbital Transition Behaviors, Inorg. Chem., 2020, 59, 16843–16852 CrossRef CAS PubMed.
- S. E. Canton, M. Biednov, M. Pápai, F. A. Lima, T. K. Choi, F. Otte, Y. Jiang, P. Frankenberger, M. Knoll, P. Zalden, W. Gawelda, A. Rahaman, K. B. Møller, C. Milne, D. J. Gosztola, K. Zheng, M. Retegan and D. Khakhulin, Ultrafast Jahn–Teller Photoswitching in Cobalt Single–Ion Magnets, Adv. Sci., 2023, 10, 2206880 CrossRef CAS PubMed.
- P. E. Figgins and D. H. Busch, Complexes of Iron(II), Cobalt(II) and Nickel(II) with Biacetyl-bis-methylimine, 2-Pyridinal-methylimine and 2,6-Pyridindial-bis-methylimine, J. Am. Chem. Soc., 1960, 82, 820–824 CrossRef CAS.
- C. Römelt, T. Weyhermüller and K. Wieghardt, Structural characteristics of redox-active pyridine-1,6-diimine complexes: Electronic structures and ligand oxidation levels, Coord. Chem. Rev., 2019, 380, 287–317 CrossRef.
- B. de Bruin, E. Bill, E. Bothe, T. Weyhermüller and K. Wieghardt, Molecular and Electronic Structures of Bis(pyridine-2,6-diimine)metal Complexes [ML2](PF6)n (n = 0, 1, 2, 3; M = Mn, Fe, Co, Ni, Cu, Zn), Inorg. Chem., 2000, 39, 2936–2947 CrossRef CAS PubMed.
- A. Arnáiz, J. V. Cuevas, G. García-Herbosa, A. Carbayo, J. A. Casares and E. Gutierrez-Puebla, Revealing the diastereomeric nature of pincer terdentate nitrogen ligands 2,6-bis(arylaminomethyl)pyridine through coordination to palladium, J. Chem. Soc., Dalton Trans., 2002, 2581–2586 RSC.
- G. Juhász, S. Hayami, K. Inoue and Y. Maeda, [CoII(phimpy)2](ClO4)2 and [CoII(ipimpy)2](ClO4)2: New Cobalt(II) Spin Crossover Compounds, and the Role of the Ligand Flexibility in Spin Transition Behavior, Chem. Lett., 2003, 32, 882–883 CrossRef.
- F. Dumitru, Y.-M. Legrand, A. Van der Lee and M. Barboiu, Constitutional self-sorting of homochiral supramolecular helical single crystals from achiral components, Chem. Commun., 2009, 2667–2669 RSC.
- M. L. Saha and M. Schmittel, From 3-Fold Completive Self-Sorting of a Nine-Component Library to a Seven-Component Scalene Quadrilateral, J. Am. Chem. Soc., 2013, 135, 17743–17746 CrossRef CAS PubMed.
- F. Dumitru, Y.-M. Legrand, M. Barboiu and A. van der Lee, Weak intermolecular hydrogen and halogen interactions in an isomorphous halogen series of pseudoterpyridine ZnII complexes, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, 69, 43–54 CrossRef CAS.
- S. W. Li, Y. F. Wang, X. P. Li, J. S. Zhao and J. L. Wang, Synthesis, characterization, and mechanism studies of bis(imino)pyridine ligands and their Cr(III) compounds, Russ. J. Coord. Chem., 2015, 41, 129–134 CrossRef CAS.
- J. Zhang, D. Campolo, F. Dumur, P. Xiao, J. P. Fouassier, D. Gigmes and J. Lalevée, Visible–light–sensitive photoredox catalysis by iron complexes: Applications in cationic and radical polymerization reactions, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2247–2253 CrossRef CAS.
- B. R. Reed, M. Yousif, R. L. Lord, M. McKinnon, J. Rochford and S. Groysman, Coordination Chemistry and Reactivity of Bis(aldimino)pyridine Nickel Complexes in Four Different Oxidation States, Organometallics, 2017, 36, 582–593 CrossRef CAS.
- R. L. Hollingsworth, J. W. Beattie, A. Grass, P. D. Martin, S. Groysman and R. L. Lord, Reactions of dicobalt octacarbonyl with dinucleating and mononucleating bis(imino)pyridine ligands, Dalton Trans., 2018, 47, 15353–15363 RSC.
- C. Hansch, A. Leo and R. W. Taft, A survey of Hammett substituent constants and resonance and field parameters, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- S. Sun, S. Liang, W.-C. Xu, G. Xu and S. Wu, Photoresponsive polymers with multi-azobenzene groups, Polym. Chem., 2019, 10, 4389–4401 RSC.
- H. M. D. Bandara and S. C. Burdette, Photoisomerization in different classes of azobenzene, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- J. A. Gareth Willimas, Photochemistry and Photophysics of Coordination Compounds: Platinum in Photochemistry and Photophysics of Coordination Compounds II, ed. V. Balzani and S. Campagna, Springer Berlin Heidelberg, Berlin, Heidelberg, 2007, vol. 281 Search PubMed.
- H. Knoll, Photoisomerism of Azobenzene, in CRC Handbook of Organic Photochemistry and Photobiology, Volumes 1 & 2, ed. W. M. Horspool and F. Lenci, CRC Press, Boca Raton, 2003 Search PubMed.
- S. Decurtins, P. Gütlich, C. P. Köhler, H. Spiering and A. Hauser, Light-induced excited spin state trapping in a transition-metal complex: The hexa-1-propyltetrazole-iron(II) tetrafluoroborate spin-crossover system, Chem. Phys. Lett., 1984, 105, 1–4 CrossRef CAS.
- S. Decurtins, P. Gutlich, K. M. Hasselbach, A. Hauser and H. Spiering, Light-induced excited-spin-state trapping in iron(II) spin-crossover systems. Optical spectroscopic and magnetic susceptibility study, Inorg. Chem., 1985, 24, 2174–2178 CrossRef CAS.
- A. Cui, K. Takahashi, A. Fujishima and O. Sato, Mechanism and relaxation kinetics of photo-induced valence tautomerism of [Co(phen)(3,5-DBSQ)2]·C6H5Cl, J. Photochem. Photobiol., A, 2004, 167, 69–73 CrossRef CAS.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, PHI: A powerful new program for the analysis of anisotropic monomeric and exchange–coupled polynuclear d – and f –block complexes, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
- K. S. Cole and R. H. Cole, Dispersion and Absorption in Dielectrics I. Alternating Current Characteristics, J. Chem. Phys., 1941, 9, 341–351 CrossRef CAS.
- E. Ruiz, J. Cano, S. Alvarez and P. Alemany, Broken symmetry approach to calculation of exchange coupling constants for homobinuclear and heterobinuclear transition metal complexes, J. Comput. Chem., 1999, 20, 1391–1400 CrossRef CAS.
- E. Ruiz, A. Rodríguez-Fortea, J. Cano, S. Alvarez and P. Alemany, About the calculation of exchange coupling constants in polynuclear transition metal complexes, J. Comput. Chem., 2003, 24, 982–989 CrossRef CAS PubMed.
- R. Carrasco, I. Morgenstern-Badarau and J. Cano, Charge transfers influence on the spin ground state of manganese and iron superoxide dismutases: a DFT study on a model of the reduced active site interacting with O2−, Chem. Commun., 2003, 436–437 RSC.
Footnotes |
| † In memory of Prof. Miguel Julve, a fine researcher, excellent professor, and better friend and colleague who passed away in July 2024. |
| ‡ Electronic supplementary information (ESI) available: Experimental section and computational details. Details of powder X-Ray diffraction (Fig. S1) and crystallographic data (Fig. S2–S8 and Tables S1 and S2), structural and spectroscopic correlations (Fig. S9–S12), and theoretical calculations (Scheme S1 and Tables S3 and S4), EPR spectroscopic (Table S5), magnetic (Fig. S13–S25 and Table S6), and electrochemical data (Fig. S26 and Table S7). CCDC 1989325, 2333340–2333343. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qi01218f |
| This journal is © the Partner Organisations 2024 |