
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirected C–H activation with iron carbene complexes†
Zachary S.
Lincoln
 ,
Melissa R.
Hoffbauer
and
Vlad M.
Iluc
*
,
Melissa R.
Hoffbauer
and
Vlad M.
Iluc
*
Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556, USA. E-mail: viluc@nd.edu
First published on 18th July 2024
Abstract
The reactivity of PCcarbeneP iron carbenes, [{PC(sp2)P}Fe(L)(N2)] (1-L, L = NCtBu, PMe3), was investigated toward imines, ketones, diazenes, 2-vinylpyridine, and 8-methylquinoline, and revealed the directed activation of aryl, vinyl, or benzyl C–H bonds by 1,2-addition across the iron-carbene bond. Our results support a syn 1,2-addition of these bonds, suggesting a metal–ligand cooperative process for the activation of these substrates. These findings highlight the potential of iron-carbene complexes in C–H activation processes, and deviate from the traditional mechanisms employed for iron promoted C–H activations.
Introduction
Transition metal-catalyzed C–H activation has been of great interest as an econonomical and environmentally friendly alternative to traditional cross-coupling reactions.1–6 Usually, to achieve positional selectivity, directing groups within the substrate of interest are used,7 allowing for chelation-assisted C–H activation and resulting in cyclometalated species that can either be isolated and used for catalysis, or are transient species during functionalization.8 Though catalytic C–H activations have been highly desirable for many years, the use of iron complexes for C–H activation has been only recently reported.9–11 As such, mechanistic understanding of these systems is lacking, and good ligand scaffolds have yet to be realized. Pioneering work showed that low-valent iron(0/II) complexes can stoichiometrically activate certain C–H bonds via oxidative addition or σ-bond metathesis mechanisms (Fig. 1A and B).12 Modern reactions can be achieved catalytically, and typically employ iron(III) species alongside the use of directing groups within the substrate.13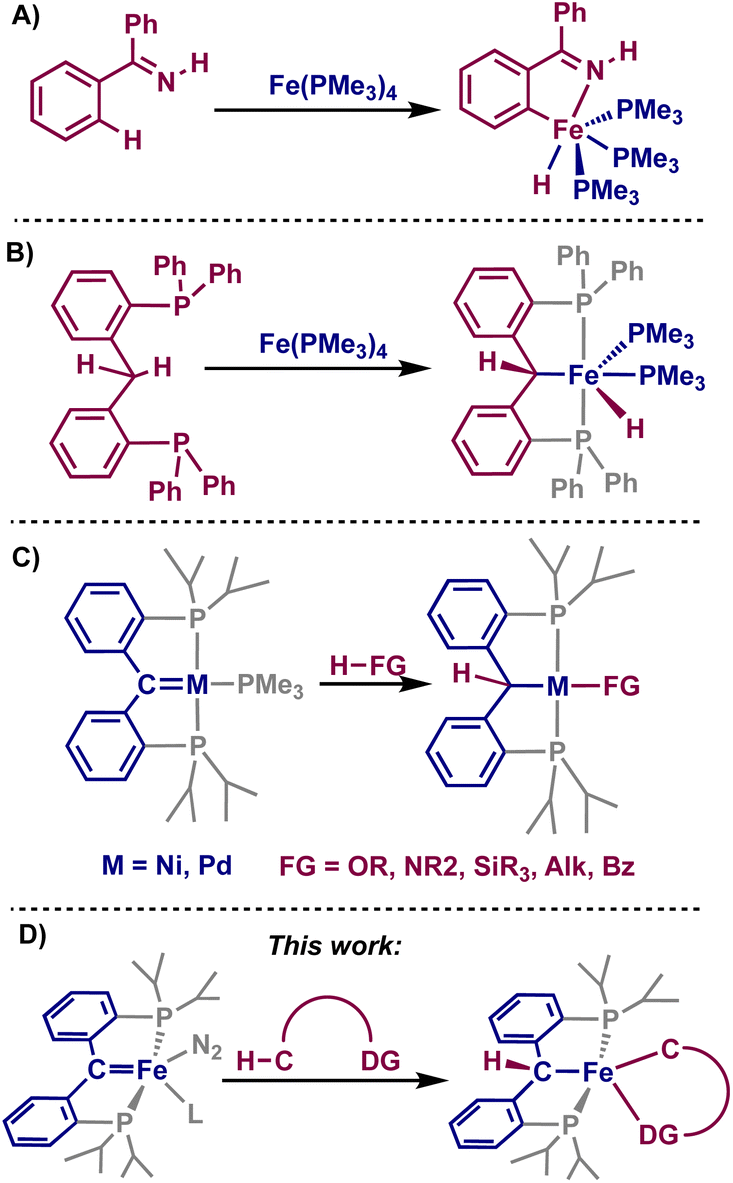 | ||
| Fig. 1 (A and B) Examples of C–H activation promoted by iron-complexes. (C) E–H activations with PCcarbeneP complexes. (D) This work: directed C–H activations across an iron–PCcarbeneP bond. | ||
Transition metal complexes employing pincer-type ligands have been shown to be catalytically proficient in a number of C–H activation reactions,14–16 as they closely control the environment of the transition metal and can even be involved in the reaction as the metal and ligand work cooperatively.17,18 Compounds containing PCcarbeneP, whereby the pincer complex contains a central-carbene moiety, have shown promise in E–H (E = H, B, C, N, O, Si, Ge) activations by accessibility of a 1,2-addition across the metal–carbene moiety (Fig. 1C).19,20 Despite the breadth of 1,2-addition reactions displayed by PCcarbeneP complexes, C–H activations have been limited to classical substrates such as acetonitrile, alkynes, and 9,10-dihydroanthracene.21–23
Considering the established reactivity of PCcarbeneP complexes toward small molecule activation, we sought whether the use of substrates incorporating directing groups could undergo directed C–H activation with an iron PCcarbeneP complex, denoted as [{PC(sp2)P}Fe(L)(N2)] (1-L, L = NCtBu, PMe3). Herein, we discuss the directed C–H activations of a variety of substrates using an iron carbene, resulting in cyclometalated species. These results showcase that a cooperative 1,2-addition occurs with the iron-carbene moiety, displaying unique reactivity from the typical oxidative addition and σ-bond metathesis mechanisms invoked for iron-promoted C–H bond activation.
Results and discussion
Imines have been extensively explored as directing groups for C–H activation owing to the strong coordination of the N-group to transition metals.24 As such, we explored the reaction of N-benzylideneaniline towards 1-NCtBu. Addition of one equivalent of N-benzylideneaniline to a diethyl ether solution of 1-NCtBu or 1-PMe3 results in the precipitation of a green solid immediately. NMR analysis of this material reveals a new paramagnetic compound in 29% yield, which was identified as the cyclometalated imine, [{PC(sp3)HP}Fe(–CAr–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NPh–)] (2) by X-ray crystallography (Scheme 1 and Fig. 2). C–H activation of the ortho-position of the carbon-aryl group is evident with a new Fe–C(52) bond of 1.990(2) Å, which is in the typical range of other iron(II)-aryl complexes.12,25 Loss of the carbene character is indicated by lengthening of the Fe–C distance from 1.898(2) Å in 1-NCtBu
NPh–)] (2) by X-ray crystallography (Scheme 1 and Fig. 2). C–H activation of the ortho-position of the carbon-aryl group is evident with a new Fe–C(52) bond of 1.990(2) Å, which is in the typical range of other iron(II)-aryl complexes.12,25 Loss of the carbene character is indicated by lengthening of the Fe–C distance from 1.898(2) Å in 1-NCtBu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26 to 2.127(1) Å – which is consistent with other reported [PC(sp3)HP] iron complexes,26–28 and iron(II)-alkyl complexes.29,30 The CN bond experiences only minimal elongation to 1.303(2) Å, and is in range for other isolated iron-imine chelates.12,31–33 Compound 2 is square-pyramidal, with the two carbon groups trans to one another and C–Fe–C(52) angle of 162.61(5)° and P(1)–Fe–P(2) angle of 156.23(2)°. The five-membered ring achieves the ideal value of 540° for a five-membered metallacycle, with the sum of the internal angles at 540.0°. The solution-state magnetic moment was measured, and corresponds to S = 2, pointing to a high-spin state.
26 to 2.127(1) Å – which is consistent with other reported [PC(sp3)HP] iron complexes,26–28 and iron(II)-alkyl complexes.29,30 The CN bond experiences only minimal elongation to 1.303(2) Å, and is in range for other isolated iron-imine chelates.12,31–33 Compound 2 is square-pyramidal, with the two carbon groups trans to one another and C–Fe–C(52) angle of 162.61(5)° and P(1)–Fe–P(2) angle of 156.23(2)°. The five-membered ring achieves the ideal value of 540° for a five-membered metallacycle, with the sum of the internal angles at 540.0°. The solution-state magnetic moment was measured, and corresponds to S = 2, pointing to a high-spin state.
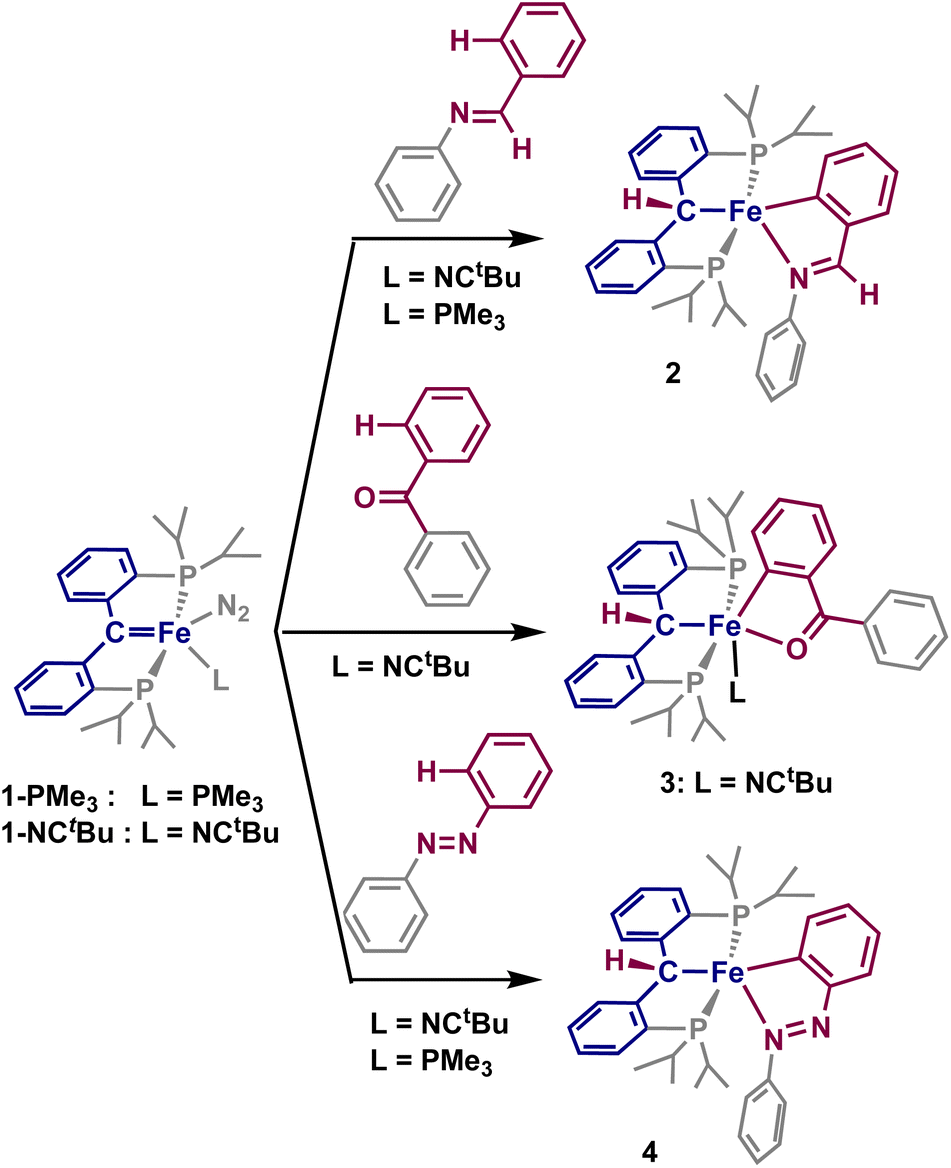 | ||
| Scheme 1 Synthetic routes to 2–4 from 1-L. | ||
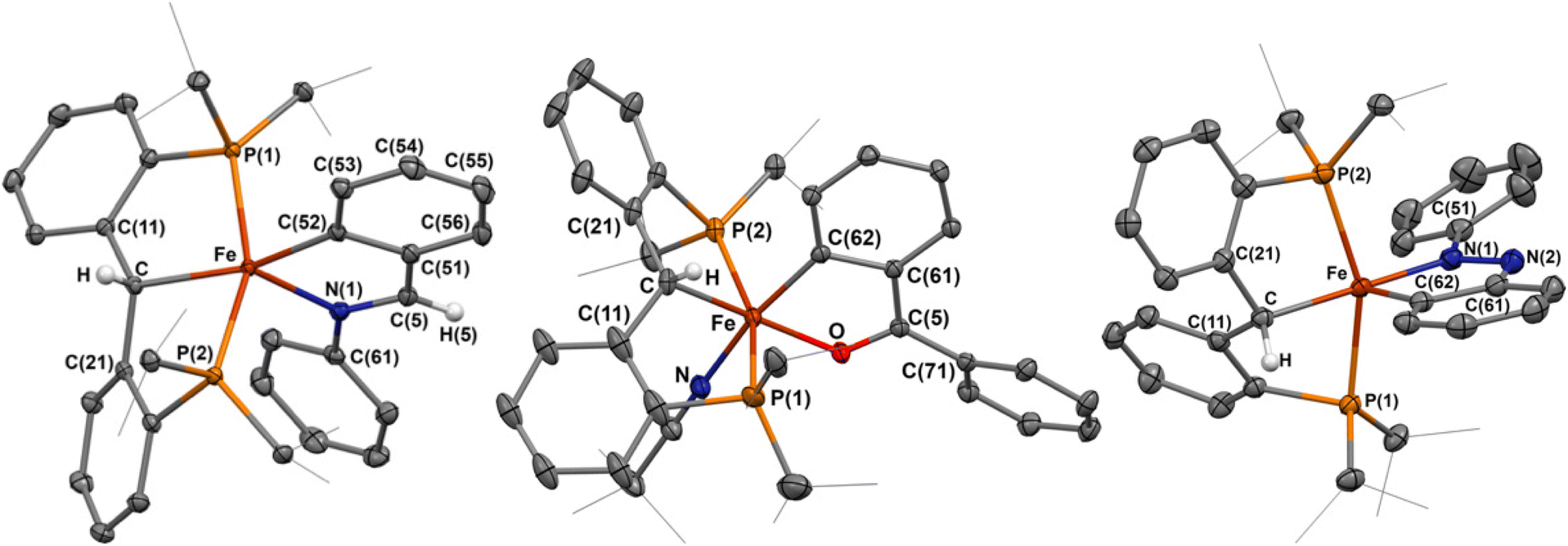 | ||
| Fig. 2 Molecular structures of 2, 3, and 4. Most H atoms have been omitted for clarity, atomic displacements are displayed at the 50% probability level. Selected distances (Å) and angles (°) for 2: Fe–C, 2.127(1); Fe–C(52), 1.990(2); Fe–N(1), 2.1189(9); N(1)–C(5), 1.303(2); Fe–P(1), 2.2160(4); Fe–P(2), 2.2599(4); C–Fe–C(52), 162.61(5); P(1)–Fe–P(2), 156.23(2). Selected distances (Å) and angles (°) for 3: Fe–C, 2.102(3); Fe–C(62), 1.927(2); Fe–O(1), 1.973(2); C(5)–O, 1.272(3); Fe–P(1), 2.288(1); Fe–P(2), 2.2272(8); Fe–N, 1.949(2); C–Fe–O(1), 171.3(1); P(1)–Fe–P(2), 159.3; N(1)–Fe–C(62), 169.1(1). Selected distances (Å) and angles (°) for 4: Fe–C, 2.091(1); Fe–C(62), 1.869(1); Fe–N(1), 1.901(1); N(1)–N(2), 1.312(1); Fe–P(1), 2.2590(5); Fe–P(2), 2.2398(5); C–Fe–N(1), 178.41(5); P(1)–Fe–P(2), 157.16(2). | ||
Having established that the strongly coordinating imine undergoes this directed ortho C–H activation of the aryl group, we also sought whether the significantly weaker O-director could also achieve this directed-activation. Though less extensively studied, carbonyl-containing directing groups have appealed as the more-abundant, easily-handled, and functionally useful alternative to imines.34 Young and coworkers described the 2π + 2π cycloaddition and subsequent β-hydride elimination of PCcarbeneP cobalt complexes with aldehydes,35 thus we sought ketones as directing groups for C–H activation, which have previously demonstrated directed C–H activation and insertion with a yttrium methandiide-derived carbene.36
Addition of one equivalent of benzophenone to a solution of 1-NCtBu led to the rapid formation of a new paramagnetic product as indicated by 1H and 31P NMR spectra. X-ray crystallography identified the product as the iron(II) cyclometalated complex 3, [{PC(sp3)HP}Fe(–CAr–CBzO–)(NCtBu)], isolated in 39% yield (Scheme 1 and Fig. 2). As with 2, C–H activation of the ortho-position of the ketone has occurred, and protonation of the carbene is evident in the crystal structure. The Fe–C distance 2.102(3) Å and Fe–C(62) distance of 1.927(2) Å are comparable to that of 2, though minor decrease in the distances can be attributed to the cis-arrangement of the two carbon groups, whereas they are trans in 2. Minimal elongation of the C(5)O bond to 1.272(3) Å occurs as a result of coordination to iron, with a Fe–O distance of 1.973(2) Å – consistent with other isolated iron cyclometalated ketones.37–39 The newly formed ortho-metalated ketone ligand has a bite angle of 81.6(1)°, and the sum of the internal angles (539.4°) is close to the ideal value of 540° for a planar five-member ring. The phosphines are comparable in orientation to 2 with a P(1)–Fe–P(2) angle of 159.3(3)°, and the ketone and alkyl groups lie essentially trans from one another with a C–Fe–O angle of 171.3(1)°. Unlike in 2, the final coordination site is occupied with a pivalonitrile ligand, likely as a result of the weak electron-donating capabilities of the ketone. Measurement of the solution-state magnetic moment is consistent with S = 2, and suggests a high-spin iron(II) complex.
Though imines and carbonyl compounds are extensively studied, directed C–H activation of non-traditional directing groups is also of interest for building complex molecular structures. Aryl-diazenes represent an interesting class of molecules in this regard, as they have a wide range of significant applications, particularly as azo dyes.35 In addition, directed C–H activation has been shown with diazenes,40 however these transformations have yet to be catalytically applied to iron-containing systems.
Addition of one equivalent of azobenzene to a sealed NMR tube of 1-NCtBu or 1-PMe3 led to the slow formation of a new paramagnetic product, alongside concomitant formation of trimethylphosphine and a color change to dark blue. Single crystals obtained from this reaction revealed the formation of the cyclometalated diazene, [{PC(sp3)HP}Fe(–CAr–NNPh–)] (4) in 55% yield (Scheme 1 and Fig. 2). As with 3, the activated arene lies cis to the erstwhile carbene. The Fe–C distance of 2.091(1) Å is shorter than in both 2 and 3, while the Fe–C(62) distance is considerably shorter at 1.869(1) Å than in 2, 3, and the parent carbene (1.9037(18) Å).26 Accordingly, an increase in the adjacent C–C bonds is observed with the C(62)–C(63) and C(61)–C(62) distances of 1.411(2) Å and 1.428(2) Å, respectively. The N(1)–N(2) bond experiences considerable elongation to 1.312(1) Å, which falls between a single and double bond, as supported by an intermediate single/double bond distance for N(2)–C(61) of 1.380(2) Å. Despite this, the Fe–N(1) interaction does not adopt an iron-imido configuration, as the Fe–N(1) distance of 1.901(1) Å is significantly longer than in reported iron-imido species.41 The internal angles of the five-membered metallacycle once again approaches the ideal value of 540° with a sum of 539.88°, supporting significant aromaticity in the five-membered metallacycle. Solution-state magnetic measurements were performed and a spin state of S = 2 was determined, which was consistent with the high-spin states of 2 and 3. Together, these structural parameters are consistent with other cyclometalated diazenes isolated by C–H activation of azobenzenes with first-row transition metals.42
We sought to understand the mechanism by which 2–4 are formed. The most common mode of C–H activation by iron is σ-bond metathesis, and the use of directing groups for C–H activation with iron is relatively rare: examples in the literature primarily rely on the use of Fe(CH3)2(PMe3)2, through a σ-bond metathesis mechanism, or transient iron(0) complexes through an oxidative addition mechanism (Fig. 1A and B).43 On the basis of several reports of metal–ligand cooperative C–H activation with PCcarbeneP complexes,19 we also propose that 2–4 can be formed by a cooperative 1,2-addition of the ortho C–H moiety.
To understand whether 2 and 4 could be formed through σ-bond metathesis, we investigated whether N-benzylideneaniline or azobenzene would form the respective products when added to [{PC(sp3)HP}Fe(CH3)]. No reaction was observed with either substrate, and the addition of benzophenone also did not elicit any reaction – implying that the iron-carbene bond of 1 is relevant to the addition reactions observed above (Scheme 2). Previous studies of palladium PCcarbeneP complexes with silanes and germanes implicated a concerted 1,2-addition mechanism by isolation of the syn-addition intermediate species;44 as such, we propose that 2–4 are formed through a similar 1,2-addition, as they are also isolated exclusively as the syn-addition products. However, the oxidative addition of the ortho C–H group and subsequent reductive elimination of the hydride and carbene cannot be directly ruled out (Scheme 2).
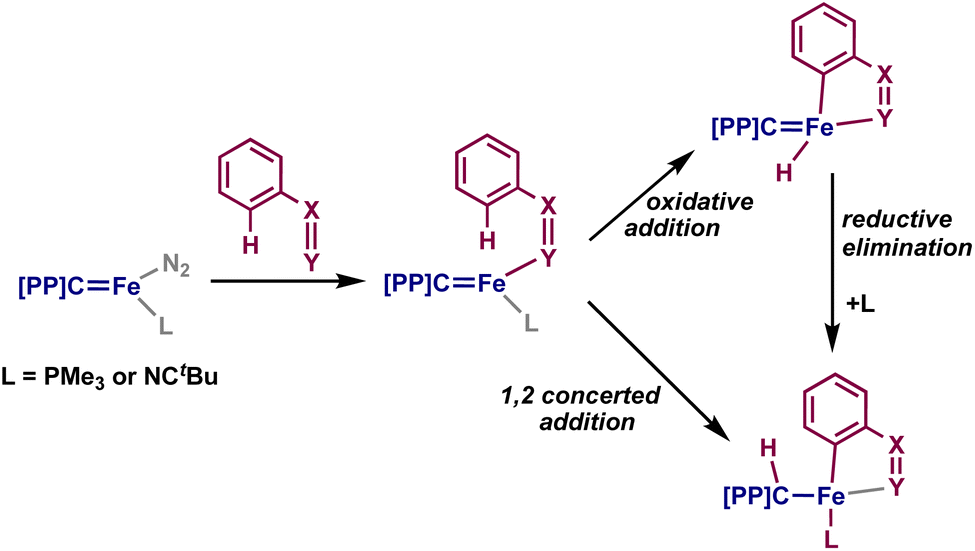 | ||
| Scheme 2 Two possible pathways for the formation of 2–4. | ||
Having established that directed ortho C–H activation can be achieved using cooperative action of an iron-carbene complex, we also sought to study other directed C–H activations. Another attractive candidate is 2-vinylpyridine, as it has shown similar reactivity to what was observed with 2–4.32,45,46 Addition of one equivalent of 2-vinylpyridine to a sealed tube of 1-PMe3 resulted in the slow formation of a new paramagnetic product by 1H and 31P NMR spectroscopy, alongside the concomitant release of trimethylphosphine. Interrogation of this product by X-ray crystallography reveals the formation of the vinyl C–H activated compound 5, [{PC(sp3)HP}Fe{–C(sp2)H–CH–Py–}] (Scheme 3 and Fig. 3). Loss of the carbene character is indicated by the Fe–C distance of 2.104(1) Å, while the Fe–C(5) distance of 1.965(1) Å is slightly shorter than other reported η1-vinyl iron complexes.47,48 The C(5)–C(6) distance 1.360(2) Å as well as the C(5)–C(6)–C(61) angle of 117.3(1)° and the Fe–C(5)–C(6) of 114.98(9)° are within range for other vinyl η1-vinyl complexes. The Fe–N(6) distance of 2.083(1) Å is within range of typical iron-pyridine distances, indicating that no increase in bond order occurs. The metallacycle once again approaches the ideally planar value of 540°, with the sum of internal angles at 539.97°. Complex 5 is a square-pyramidal structure, similar to 2, where the vinyl group lies cis to the protonated carbene, with a C(5)–Fe–C angle of 174.00(5)° and P(1)–Fe–P(2) angle of 152.52(2)°. Solution-state magnetic moment measurements were consistent with S = 1, pointing to an intermediate spin-state, which is different from the high-spin states of the square-pyramidal compounds 2 and 4.
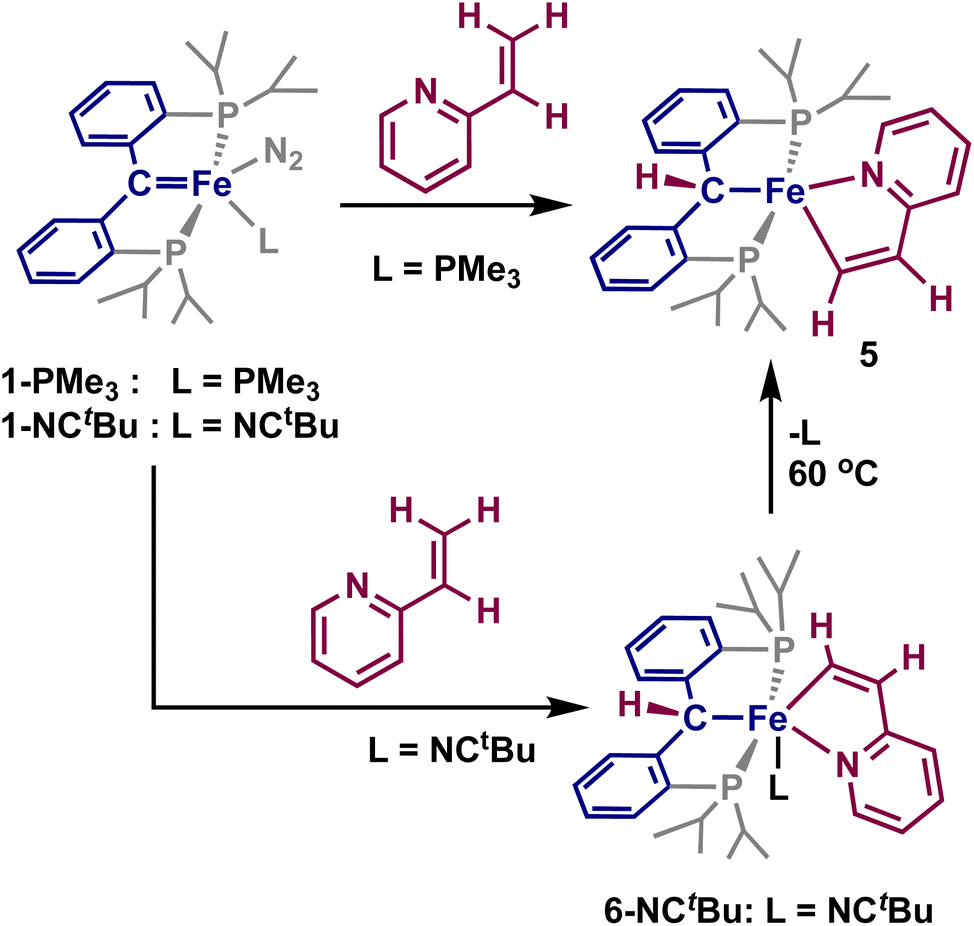 | ||
| Scheme 3 Synthesis of 5 and 6 by addition of 2-vinylpyridine to 1-PMe3 and 1-NCtBu, respectively. | ||
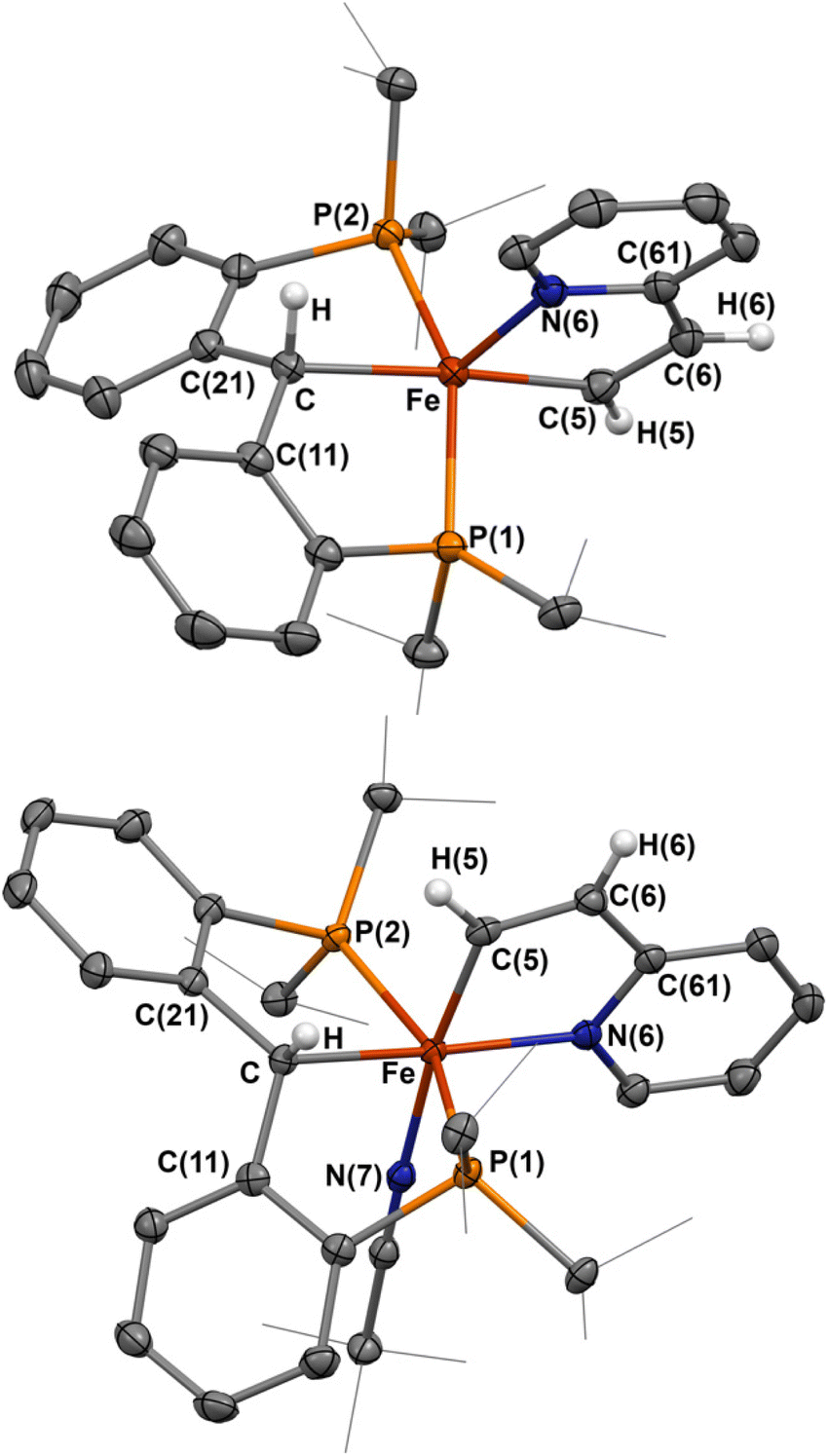 | ||
| Fig. 3 Molecular structures of 5 and 6-NCtBu. Most H atoms have been omitted for clarity, atomic displacements are displayed at the 50% probability level. Selected distances (Å) and angles (°) for 5: Fe–C, 2.104(1); Fe–C(5), 1.965(1); Fe–N(6), 2.083(1); C(5)–C(6), 1.360(2); Fe–P(1), 2.2306(4); Fe–P(2), 2.2567(5); C–Fe–C(5), 174.00(5); P(1)–Fe–P(2), 152.52(2). Selected distances (Å) and angles (°) for 6-NCtBu: Fe–C, 2.105(1); Fe–C(5), 1.905(1); Fe–N(6), 2.015(1); Fe–P(1), 2.2882(4); Fe–P(2), 2.2358(4); Fe–N(7), 1.946(1); C–Fe–N(6), 171.70(5); C(5)–Fe–N(7), 172.71(5); P(1)–Fe–P(2), 158.23(2). | ||
When the same reaction was attempted with 1-NCtBu, 5 was not immediately formed. Instead, two new diamagnetic products are observed in the 31P NMR spectrum at 65.4 and 64.5 ppm in a 95:5 ratio. Single crystals obtained from this reaction revealed the formation of [{PC(sp3)HP}Fe{–C(sp2)H–CH–Py–}(NCtBu)] (6-NCtBu) (Scheme 3 and Fig. 3). Due to the similar chemical shifts between the two products, we assign the other species to be [{PC(sp3)HP}Fe{–C(sp2)H–CH–Py–}(N2)] (6-N2), though we were unable to isolate this product.26,49
Compound 6-NCtBu is the six-coordinated analogue of 5, however, isomerization of the cyclometalated vinylpyridine occurs such that the vinyl moiety is cis to the erstwhile carbene. The Fe–C distance (2.105(1) Å) is comparable to the others discussed herein, while the Fe–C(5) distance is noticeably shorter at 1.905(1) Å. Despite this, C(5)–C(6) of 1.365(2) Å, the C(5)–C(6)–C(61) angle of 114.7(1)° and the Fe–C(5)–C(6) angle of 117.3(1)° are comparable to 5, suggesting that 6-NCtBu does not adopt a η1-vinyl carbene in the solid state. This is supported by the solution structure, whereby the vinyl proton resonates at 11.1 ppm as a triplet (J = 8.0 Hz), which is upfield relative to true vinyl carbene metal complexes.50,51 The 13C NMR spectrum paints a more complex picture: the vinyl carbon resonates as a triplet at 233.5 ppm (JCP = 28 Hz), which is within the wide range of reported η1-vinyl metal complexes (ca. 260–150 ppm),46,52 and in between the closely related vinyl PCP iron carbenes isolated by cycloaddition of alkynes to 1-PMe3 (242–228 ppm).26 Together, these results point toward 6 being an intermediate species between a true η1-vinyl carbene and an η1-vinyl metal complex.
Upon heating a solution of 6-NCtBu to 60° C, formation of 5 and free NCtBu is observed, demonstrating that isomerization of the vinyl adduct occurs upon loss of a ligand (Scheme 4). Knowing that 6 is an intermediate in the formation of 5, the mechanism seems to point toward the metal–ligand cooperative 1,2-addition across the carbene. The mechanism likely proceeds by initial coordination of the pyridine to the iron center, displacing the majority of the N2 ligand of 1-NCtBu. From here, a 1,2-addition of the vinyl group across the iron-carbene bond proceeds, leading to the syn-addition product 6-NCtBu. No observation of the anti-product is obvious in the solid state or by NMR spectroscopy, suggesting this is the only mode of C–H activation present with 2-vinylpyridine. Furthermore, because 6-NCtBu undergoes ligand loss and irreversible isomerization to 5, it is likely that 5 proceeds through this same mechanism – though the six-coordinate complex with trimethylphosphine is significantly less stable and it is never observed by NMR spectroscopy (Scheme 4).
 | ||
| Scheme 4 Proposed mechanism for the stepwise formation of 6 by 1,2-addition of 2-vinylpyridine to 1-NCtBu to yield 6-NCtBu, from which ligand loss and isomerization occurs to yield 5. The direct formation of 5 from 1-PMe3 likely proceeds by this same mechanism, with L = PMe3. | ||
Having established that directed C(sp2)–H activation can be accomplished by 1 with the use of directing groups, we turned our attention to whether C(sp3)–H bonds could be activated in a similar fashion. Several directed C(sp3)–H activation strategies are known,3,53–55 which have frequently utilized N-heteroarenes as the directing group. We opted to use 8-methylquinoline to explore the reactivity of 1, as a C(sp3) analogue of 2-vinylpyridine.
The addition of an equivalent of 8-methylquinoline to a solution of 1-NCtBu rapidly results in formation of a new paramagnetic product by 1H and 31P NMR spectroscopy. Single crystals obtained of this material revealed the formation of the five-coordinate complex, [{PC(sp3)HP}Fe{–C(sp3)H2–Nquin}] (7, Scheme 5 and Fig. 4) Once more, loss of the carbene is indicated by a Fe–C distance of 2.087(3) Å, which is comparable to the other C–H activated products 2–6. The methyl-group of the 8-methylquinoline has undergone C–H activation, indicated by a Fe–C(5) bond of 2.077(3) Å, which is notably longer than any of the activated Fe–C bonds of 2–6 but within the typical range for a Fe–C(sp3) single bond. Furthermore, the Fe–N(5) distance of 2.089(2) Å as well as the C–C distances of the quinoline moiety are consistent with a coordinated quinoline. As with 2–6, the sum of the internal angles for the five-membered metallacycle approaches ideally planar at 539.6°, and the phosphines experience a similar angle at 152.48(3)° providing 7 as a pseudo-square pyramidal structure (Fig. 4). The C–Fe–C(5) angle of 174.0(1)° is nearly identical to that of 5, establishing 7 as the C(sp3) activated analogue.
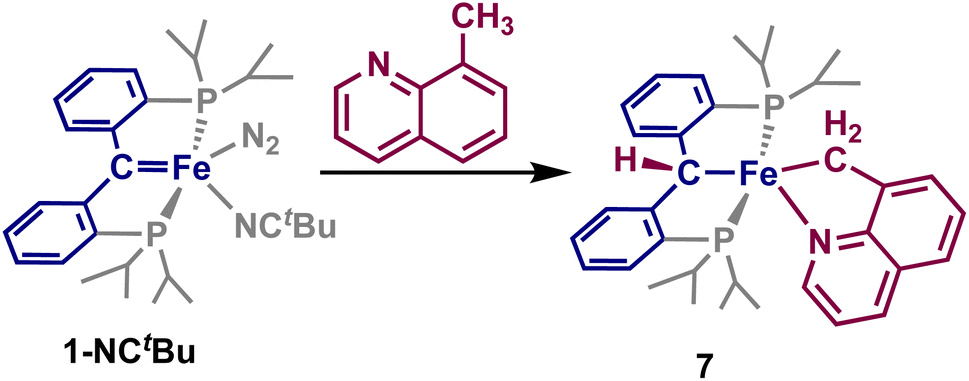 | ||
| Scheme 5 Synthesis of 7 by addition of 8-methylquinoline to 1-NCtBu. | ||
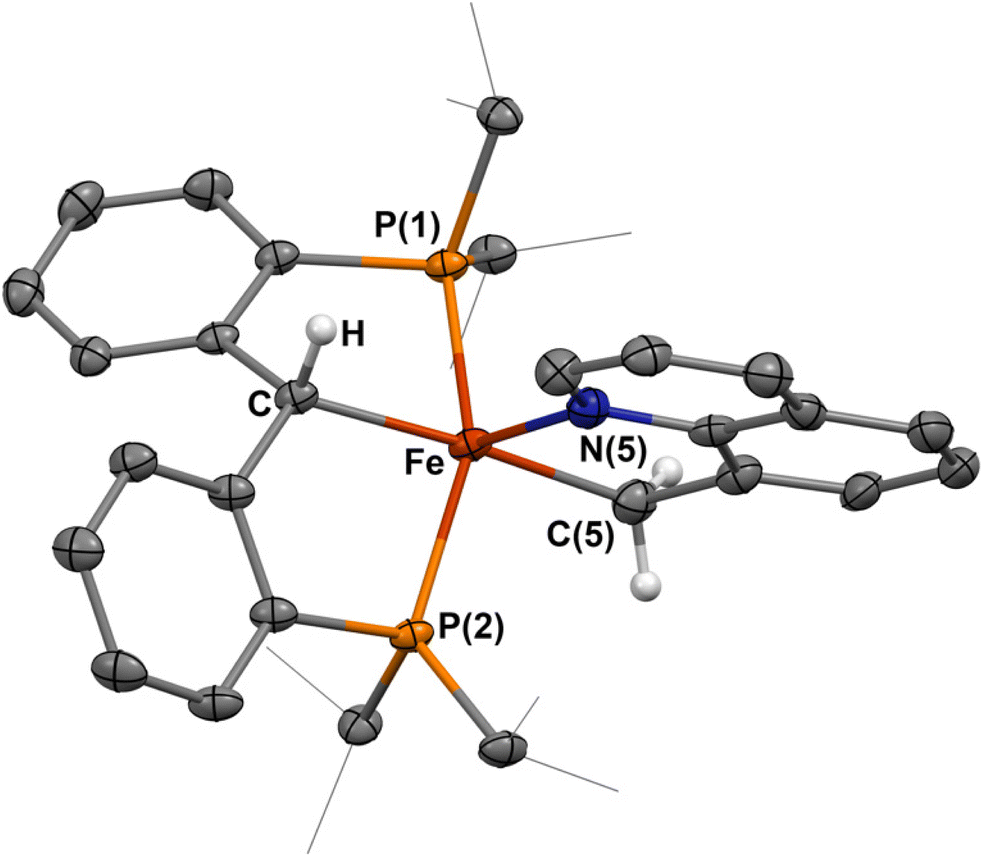 | ||
| Fig. 4 Molecular structure of 7. Most hydrogen atoms have been omitted for clarity; atomic displacements are displayed at the 50% probability level. Selected distances (Å) and angles (°) for 7: Fe–C, 2.087(3); Fe–C(5), 2.077(3); Fe–N(5), 2.089(2); Fe–P(1), 2.2625(8); Fe–P(2), 2.2331(8); C–Fe–C(5), 174.0(1); P(1)–Fe–P(2), 152.48(3). | ||
Notably, 7 appears to be the result of an anti-addition of C(sp3)H group, as the activated methyl group lies trans- to that of the carbene. We postulate that 7 follows a similar mechanism to that of 5, whereby generation of an intermediate species akin to 6-NCtBu occurs, before subsequent ligand loss and rearrangement. Attempts to observe this species by NMR were unsuccessful, highlighting that this rearrangement is more facile than that of 6-NCtBu. Attempts to generate 7 by addition of 8-methylquinoline to 1-PMe3 were unsuccessful, even at elevated temperatures, likely as a result of the quinoline not being able to outcompete the coordination of the trimethylphosphine ligand.
Conclusions
In conclusion, this study expands the toolbox of cooperative C–H activations by PCcarbeneP iron complexes, and shows that directed ortho C–H activation can be accomplished with imines, ketones, and diazenes as directing groups. Furthermore, 2-vinylpyridine leads to stepwise syn 1,2-addition of the vinyl moiety across the iron-carbon bond, from which ligand loss and isomerization eventually leads to the apparent anti-product 5. This reactivity was not limited to C(sp2)–H bonds, the iron carbene is also activating C(sp3)–H bonds, specifically the methyl group of 8-methylquinoline. These results display new reactivity for directed C–H activation by iron carbene complexes, and point toward the potential for exploitation of iron-carbene bonds as mediators of C–H activation processes in catalysis. These results also showcase that iron PCcarbeneP complexes can be utilized in C–H activation processes, much like previous examples of PCcarbeneP metal complexes, opening the door to new iron-ligand cooperative reactions.Data availability
All data supporting this article have been included as part of the ESI.†Crystallographic data for 2–7 has been deposited at the CCDC under 2352059–2352063, 2367770 and can be obtained from https://www.ccdc.cam.ac.uk.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation (award number: 2102517).References
- J. C. K. Chu and T. Rovis, Complementary Strategies for Directed C(sp3)−H Functionalization: A Comparison of Transition–Metal–Catalyzed Activation, Hydrogen Atom Transfer, and Carbene/Nitrene Transfer, Angew. Chem., Int. Ed., 2018, 57(1), 62–101, DOI:10.1002/anie.201703743
.
- Y. Yang, J. Lan and J. You, Oxidative C–H/C–H Coupling Reactions between Two (Hetero)arenes, Chem. Rev., 2017, 117(13), 8787–8863, DOI:10.1021/acs.chemrev.6b00567
- T. Rogge, N. Kaplaneris, N. Chatani, J. Kim, S. Chang, B. Punji, L. L. Schafer, D. G. Musaev, J. Wencel-Delord and C. A. Roberts,
et al., C–H activation, Nat. Rev. Methods Primers, 2021, 1(1), 43, DOI:10.1038/s43586-021-00041-2
- P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, 3d Transition Metals for C–H Activation, Chem. Rev., 2019, 119(4), 2192–2452, DOI:10.1021/acs.chemrev.8b00507
- J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Towards mild metal-catalyzed C–H bond activation, Chem. Soc. Rev., 2011, 40(9), 4740, 10.1039/c1cs15083a
- T. Dalton, T. Faber and F. Glorius, C–H Activation: Toward Sustainability and Applications, ACS Cent. Sci., 2021, 7(2), 245–261, DOI:10.1021/acscentsci.0c01413
- C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord and T. Besset,
et al., A comprehensive overview of directing groups applied in metal-catalysed C–H functionalisation chemistry, Chem. Soc. Rev., 2018, 47(17), 6603–6743, 10.1039/c8cs00201k
- J. M. Zakis, T. Smejkal and J. Wencel-Delord, Cyclometallated complexes as catalysts for C–H activation and functionalization, Chem. Commun., 2022, 58(4), 483–490, 10.1039/d1cc05195d
- T. Doba, R. Shang and E. Nakamura, Iron-Catalyzed C–H Activation for Heterocoupling and Copolymerization of Thiophenes with Enamines, J. Am. Chem. Soc., 2022, 144(47), 21692–21701, DOI:10.1021/jacs.2c09470
- N. Kimura, S. Katta, Y. Kitazawa, T. Kochi and F. Kakiuchi, Iron-Catalyzed Ortho C–H Homoallylation of Aromatic Ketones with Methylenecyclopropanes, J. Am. Chem. Soc., 2021, 143(12), 4543–4549, DOI:10.1021/jacs.1c00237
- J. Mo, A. M. Messinis, J. C. A. Oliveira, S. Demeshko, F. Meyer and L. Ackermann, Iron-Catalyzed Triazole-Enabled C–H Activation with Bicyclopropylidenes, ACS Catal., 2021, 11(3), 1053–1064, DOI:10.1021/acscatal.0c04748
- S. Camadanli, R. Beck, U. Flörke and H.-F. Klein, C−H Activation of Imines by Trimethylphosphine-Supported Iron Complexes and Their Reactivities, Organometallics, 2009, 28(7), 2300–2310, DOI:10.1021/om800828j
- N. Yoshikai, A. Matsumoto, J. Norinder and E. Nakamura, Iron–Catalyzed Chemoselective ortho Arylation of Aryl Imines by Directed C-H Bond Activation, Angew. Chem., Int. Ed., 2009, 48(16), 2925–2928, DOI:10.1002/anie.200900454
- M. Wilklow-Marnell, B. Li, T. Zhou, K. Krogh-Jespersen, W. W. Brennessel, T. J. Emge, A. S. Goldman and W. D. Jones, Catalytic Dehydrogenative C–C Coupling by a Pincer-Ligated Iridium Complex, J. Am. Chem. Soc., 2017, 139(26), 8977–8989, DOI:10.1021/jacs.7b03433
- J. V. Obligacion, S. P. Semproni and P. J. Chirik, Cobalt-Catalyzed C–H Borylation, J. Am. Chem. Soc., 2014, 136(11), 4133–4136, DOI:10.1021/ja500712z
- M. Kanzelberger, B. Singh, M. Czerw, K. Krogh-Jespersen and A. S. Goldman, Addition of C−H Bonds to the Catalytically Active Complex (PCP)Ir (PCP = η3-2,6-(tBu2PCH2)2C6H3), J. Am. Chem. Soc., 2000, 122(44), 11017–11018, DOI:10.1021/ja001626s
- J. R. Khusnutdinova and D. Milstein, Metal–Ligand Cooperation, Angew. Chem., Int. Ed., 2015, 54(42), 12236–12273, DOI:10.1002/anie.201503873
- M. R. Elsby and R. T. Baker, Strategies and mechanisms of metal–ligand cooperativity in first-row transition metal complex catalysts, Chem. Soc. Rev., 2020, 49(24), 8933–8987, 10.1039/d0cs00509f
- K.-S. Feichtner and V. H. Gessner, Cooperative bond activation reactions with carbene complexes, Chem. Commun., 2018, 54(50), 6540–6553, 10.1039/c8cc02198h
- R. A. Manzano and R. D. Young, Cataloguing a renaissance in late transition metal PCcarbeneP pincer complexes, Coord. Chem. Rev., 2021, 449, 214215, DOI:10.1016/j.ccr.2021.214215
- D. V. Gutsulyak, W. E. Piers, J. Borau-Garcia and M. Parvez, Activation of Water, Ammonia, and Other Small Molecules by PCcarbeneP Nickel Pincer Complexes, J. Am. Chem. Soc., 2013, 135(32), 11776–11779, DOI:10.1021/ja406742n
- C. C. Comanescu and V. M. Iluc, Synthesis and Reactivity of a Nucleophilic Palladium(II) Carbene, Organometallics, 2014, 33(21), 6059–6064, DOI:10.1021/om500682s
- C. C. Comanescu and V. M. Iluc, C–H Activation Reactions of a Nucleophilic Palladium Carbene, Organometallics, 2015, 34(19), 4684–4692, DOI:10.1021/acs.organomet.5b00414
- J. I. Higham and J. A. Bull, Transient imine directing groups for the C–H functionalisation of aldehydes, ketones and amines: an update 2018–2020, Org. Biomol. Chem., 2020, 18(37), 7291–7315, 10.1039/d0ob01587c
- P. Bhattacharya, J. A. Krause and H. Guan, Iron Hydride Complexes Bearing Phosphinite-Based Pincer Ligands: Synthesis, Reactivity, and Catalytic Application in Hydrosilylation Reactions, Organometallics, 2011, 30(17), 4720–4729, DOI:10.1021/om2005589
- M. R. Hoffbauer and V. M. Iluc, [2+2] Cycloadditions with an Iron Carbene: A Critical Step in Enyne Metathesis, J. Am. Chem. Soc., 2021, 143(15), 5592–5597, DOI:10.1021/jacs.0c12175
- Q. Wang, R. A. Manzano, H. Tinnermann, S. Sung, B. Leforestier, T. Krämer and R. D. Young, Access to and Reactivity of Fe0, Fe−I, FeI, and FeII PCcarbeneP Pincer Complexes, Angew. Chem., Int. Ed., 2021, 60(33), 18168–18177, DOI:10.1002/anie.202104130
- H. Zhao, H. Sun and X. Li, Synthesis and Catalytic Property of Iron Pincer Complexes Generated by Csp3−H Activation, Organometallics, 2014, 33(13), 3535–3539, DOI:10.1021/om500429r
- A. M. Tondreau, C. Milsmann, A. D. Patrick, H. M. Hoyt, E. Lobkovsky, K. Wieghardt and P. J. Chirik, Synthesis and Electronic Structure of Cationic, Neutral, and Anionic Bis(imino)pyridine Iron Alkyl Complexes: Evaluation of Redox Activity in Single-Component Ethylene Polymerization Catalysts, J. Am. Chem. Soc., 2010, 132(42), 15046–15059, DOI:10.1021/ja106575b
- C. Pranckevicius and D. W. Stephan, Three-Coordinate, Cyclic Bent Allene Iron Complexes, Organometallics, 2013, 32(9), 2693–2697, DOI:10.1021/om400166t
- Y. Shi, M. Li, Q. Hu, X. Li and H. Sun, C−Cl Bond Activation of ortho-Chlorinated Imine with Iron Complexes in Low Oxidation States, Organometallics, 2009, 28(7), 2206–2210, DOI:10.1021/om801162u
- H.-F. Klein, S. Camadanli, R. Beck and U. Flörke, Meridional bicyclometalation with iron: a novel way of forming dianionic [C,N,C]-ligands, Chem. Commun., 2005,(3), 381–382, 10.1039/b413409e
- A. M. Messinis, T. Von Münchow, M. Surke and L. Ackermann, Room temperature photo-promoted iron-catalysed arene C–H alkenylation without Grignard reagents, Nat. Catal., 2024, 7, 273–284, DOI:10.1038/s41929-023-01105-0
- Z. Huang, H. N. Lim, F. Mo, M. C. Young and G. Dong, Transition metal-catalyzed ketone-directed or mediated C–H functionalization, Chem. Soc. Rev., 2015, 44(21), 7764–7786, 10.1039/c5cs00272a
- S. Sung, Q. Wang, T. Krämer and R. D. Young, Synthesis and reactivity of a PCcarbeneP cobalt(I) complex: the missing link in the cobalt PXP pincer series (X = B, C, N), Chem. Sci., 2018, 9(43), 8234–8241, 10.1039/c8sc02782j
- D. P. Mills, L. Soutar, W. Lewis, A. J. Blake and S. T. Liddle, Regioselective C−H Activation and Sequential C−C and C−O Bond Formation Reactions of Aryl Ketones Promoted by an Yttrium Carbene, J. Am. Chem. Soc., 2010, 132(41), 14379–14381, DOI:10.1021/ja107958u
- X. Xu, J. Jia, H. Sun, Y. Liu, W. Xu, Y. Shi, D. Zhang and X. Li, Selective activation of C–F and C–H bonds with iron complexes, the relevant mechanism study by DFT calculations and study on the chemical properties of hydrido iron complex, Dalton Trans., 2013, 42(10), 3417–3428, 10.1039/c2dt31795h
- S. Garhwal, J. Panda, N. Fridman, A. Karton and G. De Ruiter, Formation, of distinct iron hydrides via mechanistic divergence in directed C–H Bond activation of aryl ketones, esters and amides, Chem. Commun., 2023, 59(4), 426–429, 10.1039/d2cc04394g
- A. M. Messinis, L. H. Finger, L. Hu and L. Ackermann, Allenes for Versatile Iron-Catalyzed C–H Activation by Weak O-Coordination: Mechanistic Insights by Kinetics, Intermediate Isolation, and Computation, J. Am. Chem. Soc., 2020, 142(30), 13102–13111, DOI:10.1021/jacs.0c04837
- N. Muniraj and K. R. Prabhu, Cobalt(III)-Catalyzed C–H Activation: Azo Directed Selective 1,4-Addition of Ortho C–H Bond to Maleimides, J. Org. Chem., 2017, 82(13), 6913–6921, DOI:10.1021/acs.joc.7b01094
- D. A. Iovan and T. A. Betley, Characterization of Iron-Imido Species Relevant for N-Group Transfer Chemistry, J. Am. Chem. Soc., 2016, 138(6), 1983–1993, DOI:10.1021/jacs.5b12582
- H.-F. Klein, M. Helwig, U. Koch, U. Flörke and H.-J. Haupt, Coordination and Reactions of Diazenes in Trimethylphosphinecobalt(I). Complexes - Syntheses and Structures of Complexes Containing μ2-(N,N′)-Benzo[c]cinnoline and η2-Azobenzene Ligands, Z. Naturforsch., B: J. Chem. Sci., 1993, 48(6), 778–784, DOI:10.1515/znb-1993-0612
- R. Shang, L. Ilies and E. Nakamura, Iron-Catalyzed C–H Bond Activation, Chem. Rev., 2017, 117(13), 9086–9139, DOI:10.1021/acs.chemrev.6b00772
- C. C. Comanescu and V. M. Iluc, E–H (E = B, Si, Ge) bond activation of pinacolborane, silanes, and germanes by nucleophilic palladium carbene complexes, Chem. Commun., 2016, 52(58), 9048–9051, 10.1039/c5cc09468b
- M. A. Esteruelas, F. J. Fernández-Alvarez, M. Oliván and E. Oñate, C−H Bond Activation and Subsequent C−C Bond Formation Promoted by Osmium: 2-Vinylpyridine−Acetylene Couplings, J. Am. Chem. Soc., 2006, 128(14), 4596–4597, DOI:10.1021/ja060901t
- L. Zhang, L. Dang, T. B. Wen, H. H. Y. Sung, I. D. Williams, Z. Lin and G. Jia, Cyclometalation of 2-Vinylpyridine with MCl2(PPh3)3 and MHCl(PPh3)3(M = Ru, Os), Organometallics, 2007, 26(11), 2849–2860, DOI:10.1021/om0611305
- Y. Yu, A. R. Sadique, J. M. Smith, T. R. Dugan, R. E. Cowley, W. W. Brennessel, C. J. Flaschenriem, E. Bill, T. R. Cundari and P. L. Holland, The Reactivity Patterns of Low-Coordinate Iron−Hydride Complexes, J. Am. Chem. Soc., 2008, 130(20), 6624–6638, DOI:10.1021/ja710669w
- J. M. Smith, R. J. Lachicotte and P. L. Holland, NN Bond Cleavage by a Low-Coordinate Iron(II) Hydride Complex, J. Am. Chem. Soc., 2003, 125(51), 15752–15753, DOI:10.1021/ja038152s
- Z. S. Lincoln and V. M. Iluc, Iron Olefin Metathesis: Unlocking Reactivity and Mechanistic Insights, J. Am. Chem. Soc., 2024, 146(26), 17595–17599, DOI:10.1021/jacs.4c04356
- P. Binger, P. Müller, R. Benn and R. Mynott, Vinylcarbene Complexes of Titanocene, Angew. Chem., Int. Ed. Engl., 1989, 28(5), 610–611, DOI:10.1002/anie.198906101
- S. T. Nguyen, L. K. Johnson, R. H. Grubbs and J. W. Ziller, Ring-opening metathesis polymerization (ROMP) of norbornene by a Group VIII carbene complex in protic media, J. Am. Chem. Soc., 1992, 114(10), 3974–3975, DOI:10.1021/ja00036a053
- M.-C. Chung, X. Gu, B. A. Etzenhouser, A. M. Spuches, P. T. Rye, S. K. Seetharaman, D. J. Rose, J. Zubieta and M. B. Sponsler, Intermetal Coupling in [(η5-C5R5)Fe(dppe)]2(μ-CHCHCHCH) and in Their Dicationic and Monocationic Mixed-Valence Forms, Organometallics, 2003, 22(17), 3485–3494, DOI:10.1021/om0300979
- V. G. Zaitsev, D. Shabashov and O. Daugulis, Highly Regioselective Arylation of sp3 C−H Bonds Catalyzed by Palladium Acetate, J. Am. Chem. Soc., 2005, 127(38), 13154–13155, DOI:10.1021/ja054549f
- K. M. Altus and J. A. Love, The continuum of carbon–hydrogen (C–H) activation mechanisms and terminology, Commun. Chem., 2021, 4(1), 173, DOI:10.1038/s42004-021-00611-1
- M. Sun, X.-X. Chen, J.-T. Ren, J.-L. Xu, H. Xie, W. Sun and Y.-M. Li, Cobalt(III)-Catalyzed 1,4-Addition of C(sp3)–H Bonds to Maleimides, Synlett, 2018, 29(12), 1601–1606, DOI:10.1055/s-0037-1609847
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, details f 1H/13C/31P NMR, spectra, and data for crystals 2–7. CCDC 2327758–2327763, 2367770. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qi01088d |
| This journal is © the Partner Organisations 2024 |