DOI:
10.1039/D4QI00868E
(Research Article)
Inorg. Chem. Front., 2024,
11, 4158-4166
Light-induced splitting of P–C bonds in a lanthanum(III) hemiphosphinal complex†
Received
5th April 2024
, Accepted 21st May 2024
First published on 22nd May 2024
Abstract
Insertion of fluorenone into the lanthanum–phosphorus bond of terminal phosphido complexes (PN)2La(PHR) (R = Mes (1a), R = Ph (1b) and R = tBu (1c)) results in the formation of light-sensitive hemiphosphinal complexes 2a–c. Upon exposure to daylight, the P–C bonds in 2a–c break resulting in the formation of diphospines and a lanthanum bound fluorenonyl-radical. The scope and mechansim of this reaction are experimentally and theoretically evaluated.
Introduction
In the past decades, early transition metal and f-element phosphanido complexes have been found to adopt a rich and versatile insertion chemistry, which allows the construction of exotic and rare types of phosphorus functionalized molecules.1–4 From a thermodynamic point of view, the main driving force for this versatile insertion chemistry is the exchange of a rather weak M–P interaction for more ionic M–N and M–O bonds, alongside the formation of a new P–C bond.5 Thus, in the 1990s a large number of insertion reactions were reported by the groups of Hey-Hawkins and Stephan which included the build-up of phosphaguanidinates,6 phospha-(thio)ureates7 and phosphaamidinates8,9 at zirconium(IV). This chemistry also has been widely extended into the f-elements in the past decade.1–4,10–15 Furthermore, the insertion of carbodiimides, nitriles and iso(thio)cyanates, olefins or alkynes into M–P bonds (M = early transition metals or f-element) is also considered to be a fundamental step in metal-catalyzed hydrophosphin(yl)ation reactions.3,4,10,11,16–20 Despite this plethora of examples, the insertion chemistry of ketones, especially of fluorenone (and related diaryl ketones e.g. benzophenone) into M–P bonds, yielding hemiphosphinals, is still scarcely studied and so far only a handful of hemiphosphinal complexes have been reported (Fig. 1, left).13,21–26 This is surprising, since diarylketones hold a variety of promising properties such as their easy reduction to form (stable) ketyl-radicals27–40 and to undergo reductive coupling41,42 reactions or 1,2-migratory insertion reactions.13,43–46 This makes them very attractive synthons for organometallic chemists, which is further demonstrated by a recent example from Erker and co-workers.44,47 Using hemiphosphinal zirconium(IV) complexes, originally described by Stephan and co-workers26 in 1996, they promoted new reactivity in FLP chemistry.44,47 Furthermore, when electron accepting ligands, such as bipyridine, are introduced, the Arnold group has recently shown, that terminal thorium phosphanido complexes can undergo reductive coupling of the phosphanido-ligands.48
 |
| | Fig. 1 Left: Selected examples of hemiphosphinal complexes reported in the literature.13,25,26,44 Right: Selection of anilidophosphine supported lanthanum complexes showing unexpected and versatile chemistry recently reported by our group. | |
We have recently reported a versatile lanthanum anilidophosphine complex I49 (Fig. 1, right) which readily undergoes salt metathesis reactions leading to highly functionalized metal complexes, such as the first stable 2-phosphaethynthiolate ([SCP]−) complex50II or the first terminal, primary phosphanido complex 1a of the lanthanides.49 The latter was found to be a promising candidate for the design of lanthanum phosphinidenes51 and readily undergoes insertion chemistry with e.g. iso(thio)cyanates to give phospha(thio)ureates of the general structure III.52 Given the previous mentioned propensity of electropositive metal-phosphorus insertion chemistry, and the lack of follow-up chemistry explored on hemiphosphinals, here we expand the reactivity of 1a towards fluorenone. To our surprise and in contrast to a previously reported thorium hemiphosphinal complex by the Walensky group,13 the resulting hemiphosphinal is light sensitive leading to a P–C bond scission, forming diphosphine and a fluorenonyl radical stabilized on lanthanum(III).
Results and discussion
Reaction between fluorenone and 1a in the absence of light resulted in the clean formation of the desired hemiphosphinal complex 2a (Scheme 1). Successful formation of the complexes is evident by the shift of the resonance corresponding to the phosphanido ligand in the 31P{1H} NMR spectrum from δ = –36.4 ppm in 1a to δ = –32.1 ppm in 2a (Fig. S19†). The retention of the proton on the phosphorus atom is confirmed by 1H NMR spectroscopy showing a doublet at 5.17 ppm (1JPH = 210.6 Hz). Additionally, the observation of a new doublet in the 13C{1H} NMR of 2a at δ = 95.3 (1JCP = 12.3 Hz) corresponding to the former fluorenone carbonyl carbon atom is indicative for the successful insertion into the La–P bond. X-ray diffraction studies (Fig. 2) unambiguously proved the insertion of the fluorenone moiety into the former La–P bond, forming a new La–O bond (La1–O1 2.1867(13) Å) and a new P–C bond (P70–C80 1.9163(19) Å). The distance of 4.462(2) Å between P70 and La1 proves that no interaction remains between the phosphorus and the lanthanum atom after the insertion of the fluorenone moiety. To our surprise, and in contrast to all previous reported insertion reactions of ketones into polarized M–P bonds,13,26 samples of 2a were not stable towards ambient light (Fig. 3a). Keeping NMR samples exposed to daylight resulted in a colour change from orange to dark red (Fig. 3b). Along with the colour change, the broad hemiphosphinal 31P NMR resonance at −32.1 ppm vanishes and two new multiplets at −110.8 ppm and −118.2 ppm appear, which correspond to dimesityldiphosphine [PHMes]2 (Fig. 3b and c).48,53
 |
| | Scheme 1 Synthesis of the phosphanido complexes 1a–d and the corresponding hemiphosphinal complexes 2a–2c. | |
 |
| | Fig. 2 Molecular structure of the hemiphosphinal complex 2a. Thermal ellipsoids are shown at a probability level of 50%. Hydrogen atoms (except for H70 on P70) and lattice solvent molecules have been omitted for clarity. Selected bond parameters of the hemiphosphinal moiety: 2a: La1–O1 2.1867(13) Å, O1–C80 1.401(2) Å, C80–P70 1.9163(19) Å, La1–O1–C80 177.79(12)°, O1–C80–P70 103.22(12)°. | |
 |
| | Fig. 3 (a) Net reaction scheme of the photolysis of complex 2a. (The radical is highlighted in red) (b) 31P NMR spectra of 2a before and after irradiation with sunlight for 18 h together with the visual colour change observed. (c) Multiplet of [PHMes]2. | |
Intrigued by the distinct colour change from orange to intense red, the disappearance of the PN resonances in the 31P NMR spectra over time, and the fact that diarylketones, especially benzophenone, can be photo-reduced using visible light in the presence of tert-butanolate,54 we investigated if the reaction might also proceed via a fluorenonyl-radical 3. Indeed, when fresh samples of 2a were irradiated with light inside the EPR spectrometer, we saw the formation of radical species 3 (Fig. 4, top). Simulation of the radical forming is in good agreement with a fluorenyl radical being bound to a lanthanum center with giso = 2.0026 and hyperfine coupling to both the lanthanum center (aiso = 1.7 MHz) and the four distinct fluorenone hydrogens (aiso = 10.1, 8.11, 3.04, 2.18 MHz) (Fig. 4). Complete photolysis of a separate sample of 2a using a high-intensity photoreactor (5000 K cold-white LED, 36 min, Fig. S48†) and subsequent recrystallisation of the reaction mixture by vapour diffusion of hexane into a benzene solution resulted in the formation of red blocks of 3. EPR of these red blocks confirmed their identity to be the same radical as observed after in situ photolysis of complex 2a in the EPR tube (Fig. 4, middle). NMR Evans Method revealed a magnetic moment of 1.42 μB which agrees with the presence of one unpaired electron. X-ray diffraction analysis of the new complex 3 unambiguously confirmed the presence of a fluorenonyl radical being bound to the lanthanum center (Fig. 5, middle). This is clearly observable from three bond parameters: (I) The La1–O1 bond distances which increases from 2.1867(13) Å in 2a to 2.261(4) Å in 3; (II) the decrease of the O1–C80 bond distance from 1.401(2) Å in 2a to 1.303(6) Å in 3 and the change of the La1–O1–C80 bond angle, moving from 177.79(12)° to 180.0° in 2a and 3 respectively. All these parameters indicate a major change of the electronic situation within the fluorenone-moiety affecting the electron density within its carbonyl functionality and are in line with previously reported lanthanide fluorenonyl radicals,27–34e.g. supported by samarium9,28,40 or ytterbium.38
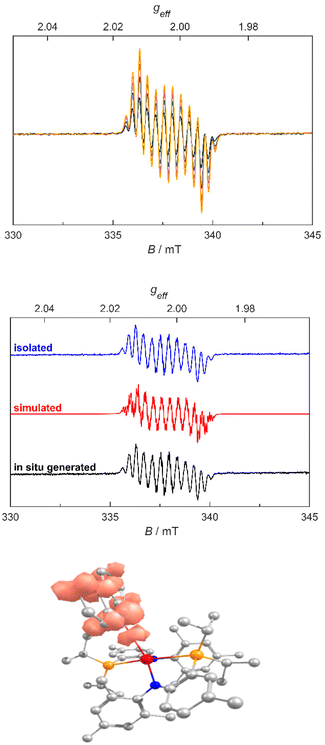 |
| | Fig. 4 Top: EPR spectra of the in situ irradiated sample of complex 2a, each shown step corresponds to a 40 s irradiation with 0.1% lamp power. Please note that the initial radical concentration at 0 minutes is not 0 as the sample already decomposes when being transferred to the EPR tube even in the dark. Middle: highly resolved EPR of complex 3 from in situ photolysis (black), isolated radical (blue) and simulation (red). The intensities of these spectra have been normalized for clarity. Bottom: calculated Spin density plot of the fluorenyl radical complex 3. | |
 |
| | Fig. 5 Molecular structures of the fluorenolate complex 4 (left), the fluorenonyl radical complex 3 (middle) and the dicationic fluorenone cluster 5′ (right). Thermal ellipsoids are shown at a probability level of 50%. Hydrogen atoms (except for the H at C80 in 4), counter ions and lattice solvent molecules have been omitted for clarity. In the structure of 5′ the PN ligands as well as the majority of the fluorenone ligands have been truncated for clarity. The La1–O1 and the O1–C80 bond distances have been depicted further for clarity and comparison. | |
To set these values into context (and to rule out the influence of potential steric and crystal packing effects) we synthesized the pure fluorenolate complex 4 and a corresponding cationic fluorenone complex 5. The “isostructural” series of complexes 3–5 allows the direct comparison between the bond metrics in the fluorenolate, the fluorenonyl-radical and the neutral fluorenone moiety. Synthesis of the fluorenolate complex 4 was achieved by salt metathesis between the chloride precursor I and lithium fluorenolate (Scheme 2). The product is isolated in 76% yield as a faint yellow powder. Characteristic NMR features that indicate the successful formation of the fluorenol complex are (I) the presence of the fluorenolate CH resonance at 6.10 ppm in its 1H NMR spectrum, together with the characteristic fluorenolate multiplets between 7.58–6.80 ppm (II) the shift of the characteristic aryl-H of the PN ligand from 5.65 ppm in complex I to 5.75 ppm in 4. Notably in this case the shift of the 31P NMR resonance of the PN ligand cannot be viewed as indicative for the formation of the new species 4 as it shifts from 9.20 ppm in I to 9.17 ppm in 4. X-ray quality crystals could be obtained from a concentrated toluene solution of 4 within 3 h at room temperature. Unfortunately, the synthesis of the desired cationic complex 5 proved to be more tricky and despite various attempts (halide abstraction was attempted using AgBArF20, AgBArF24, NaBArF24 and [(Et3Si)2H][BArF20] in the presence of complex I and fluorenone) no defined cationic mono-fluorenone complex 5 could be isolated. However, using [(Et3Si)2H][BArF20] as a chloride abstraction reagent we observed the formation of the dicationic tri-lanthanum fluorenone cluster 5′ after crystallisation from dichloromethane layered by pentane at −40 °C. Although this cluster is not the desired complex 5 it still holds valuable structural information concerning the interactions between lanthanum and neutral fluorenone. Comparison between the structures of 3, 4, and 5′ shows that (as expected) the La1–O1 distance increases from 2.193(3) Å in 4 to 2.261(4) Å in 3 culminating in the longest 2.469(9) Å in 5′. At the same time the O1-C80 distance decreases from 1.395(6) Å in 4 to 1.303(6) Å in 3 to 1.228(16) Å in 5′ which agrees with an increasing carbonyl double bond character between O1 and C80 (Fig. 5).
 |
| | Scheme 2 Synthesis of the fluorenolate complex 4 and attempted synthesis of the cationic fluorenone complex 5 as structural comparisons for the radical complex 3. Please note, that complex 5 has never been isolated and only complex 5′ (X-ray structure see Fig. 5, right) was accessible in single crystal yield. | |
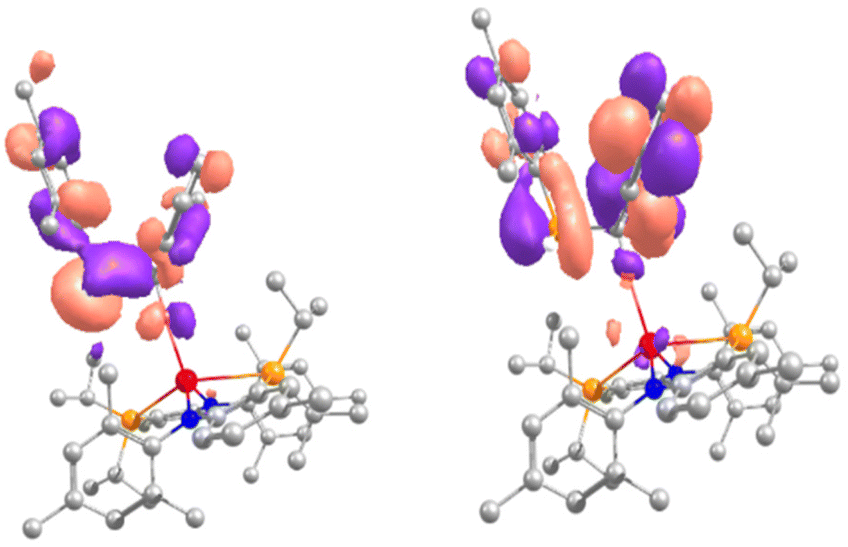
To gain further insights into the reaction (Scheme 1) its mechanism was further elucidated by computational methods (DFT, B3PW91, see ESI† for more information) for the hemiphosphinal complex 2a. For simplification, we modelled the insertion reaction the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond of benzophenone into the La–P bond, which is predicted to be kinetically facile (barrier of 1.7 kcal mol−1) and thermodynamically favourable (−21.6 kcal mol−1) (Fig. S74†).55 Therefore, the irradiation used experimentally is needed in order to break the P–C bond. This was investigated by computing the TDDFT spectrum of 2a (Fig. S69†), showing a transition at 602 nm that involves a HOMO−3 to LUMO transition. The HOMO−3 displays a bonding P–C interaction while the LUMO has primarily an antibonding P–C one so that populating the latter would imply breaking of the P–C bond (Fig. 6).
O double bond of benzophenone into the La–P bond, which is predicted to be kinetically facile (barrier of 1.7 kcal mol−1) and thermodynamically favourable (−21.6 kcal mol−1) (Fig. S74†).55 Therefore, the irradiation used experimentally is needed in order to break the P–C bond. This was investigated by computing the TDDFT spectrum of 2a (Fig. S69†), showing a transition at 602 nm that involves a HOMO−3 to LUMO transition. The HOMO−3 displays a bonding P–C interaction while the LUMO has primarily an antibonding P–C one so that populating the latter would imply breaking of the P–C bond (Fig. 6).
 |
| | Fig. 6 HOMO−3 and LUMO of complex 2a calculated by DFT, irradiation at 602 nm induces a transition from a binding P–C bond orbital (HOMO−3) into the antibonding P–C bond orbital (LUMO). | |
Intrigued by this result, we further investigated the possibility of a homolytic cleavage of the P–C bond, and if the second reaction product corresponds to a mesitylphosphinyl radical that quickly recombines to form the observed dimesityldiphosphine. However, performing the P–C bond photolysis in the presence of various radical scavengers (TEMPO, styrene, radical clocks etc.) gave no indication of the presence of free phosphinyl radicals. This was further confirmed by X-ray photolysis experiments of single crystals of 2a showing no signs of decomposition or changes along the C80–P70 bond (Table S2†). Furthermore, no EPR signals were observed if the EPR photolysis experiment were performed in a frozen solution of 2a. Since these experiments suggest that the reaction does not proceed via a monomolecular process, an alternative pathway involving a concerted mechanism for the P–P dimerization and the P–C bond splitting seems to be more likely (Scheme 3).
 |
| | Scheme 3 Postulated mechanism for the photoinduced P–C bond cleavage observed in complex 2avia a concerted (homolytic) C–P bond scission / P–P bond formation mechanism. | |
To figure out whether this decomposition reaction is unique to the primary mesitylphosphine moiety, we prepared the phenylphosphanido, the tert-butylphosphanido and the dimesitylphosphanido complexes 1b–d. The complexes of the primary phosphines 1b and 1c were readily accessible in good yields (60 and 82% respectively), following a salt metathesis protocol between complex I and the corresponding potassium phosphanides (KPHR = R = Ph or tBu). Formation of the desired complexes 1b and 1c is indicated by the 1H and 31P NMR spectra. The 1H NMR spectra show the characteristic doublets of the primary phosphanido ligand at 4.06 ppm (1JPH = 194.8 Hz) and 3.47 ppm (1JPH = 200.7 Hz) for 1b and 1c, respectively. These 1JPH coupling constants can also be extracted from the 31P NMR spectra showing distinct doublets at −10.2 ppm (1JPH = 194.8 Hz, Fig. S5†) and 97.5 ppm (1JPH = 199.3 Hz, Fig. S11†) for 1b and 1c respectively. Furthermore, the characteristic aryl proton of the PN ligand is shifted from 5.65 ppm in complex I to 5.67 ppm and 5.68 ppm in the 1H NMR spectra of 1b and 1c. Accordingly, the resonances of the PN-ligands phosphorus atom in the 31P{1H} NMR spectra are shifted from 9.2 ppm for complex I to 10.4 ppm and 12.3 ppm for 1b and 1c respectively.
On the other hand, synthesis of the dimesitylphosphanide complex 1d was found to be rather messy, producing multiple inseparable products. Apart from a few single crystals (yield less than 1%, structure see Fig. 7) no pure bulk material could be obtained, wherefore no further reactivity (insertion chemistry) with 1d could be investigated. Single crystals could be obtained by storing a concentrated hexane solution for 5 weeks at −40 °C for complex 1b or at room temperature overnight for complex 1c or by storing a concentrated diethyl ether solution at −40 °C over three days for complex 1d. Structural investigation on the phosphanido complexes 1b–d (Fig. 7, top row) revealed that the La–P bond distance is strongly depending on the substitution on the phosphorus donor and a La1–P70 distance of 3.009(1) Å was measured for 1b (3.053(1) Å in 1a), which shortened to 2.8364(6) Å in 1c. Interestingly, the steric congestion/bulk of the phosphanido ligand seems to play a minor role over the electronic properties of the phosphanido ligand towards the La1–P70 distance, since with 2.9216(14) Å this distance in 1d is shorter than in the primary phosphanido complexes 1a and 1b. Insertion reactions with fluorenone proceeded similarly to the insertion forming 2a and the complexes 2b and 2c were obtained as orange solids in yields of 86% and 68%. The shift of the PH-resonance in the 31P NMR spectra is again the most indicative feature for the formation of the complexes 2b and 2c, similar to 2a. Conversion from 1b to 2b shifts this phosphorus resonance from −10.2 ppm to 5.46 ppm (1JPH = 196.9 Hz) while 1c to 2c results in a shift from 97.5 ppm to 23.6 ppm (1JPH = 191.6 Hz). Similar to 2a, 2b and 2c were also highly sensitive towards light and a fast colour change was observed when samples where left exposed to daylight. Crystals suitable for X-ray diffraction studies were grown from concentrated toluene solutions of the complexes at room temperature (Fig. 7, bottom row). The structural features of the complexes are similar to 2a wherefore a detailed discussion is omitted at this place. Further structural information on the complexes can be extracted from the ESI (Tables S1 and S2†).
 |
| | Fig. 7 Molecular structure of the phosphanido complexes 1b–d (top from left to right). Bottom row shows the insertion complexes 2b and 2c after fluorenone insertion into the corresponding phosphanido complexes. Thermal ellipsoids are shown at a probability level of 50%. Hydrogen atoms (except for H70 on P70) and lattice solvent molecules have been omitted for clarity. Selected bond parameters: 1b: La1–P70 3.009(1) Å, 1c: La1–P70 2.8364(6) Å, 1d: La1–P70 2.9216(14) Å. 2b: La1–O1 2.191(3) Å, O1–C80 1.402(5) Å, C80–P70 1.924(4) Å, La1–O1–C80 167.8(3)°, O1–C80–P70 107.4(3)°. 2c: La1–O1 2.1776(14) Å, O1–C80 1.413(2) Å, C80–P70 1.895(2) Å, La1–O1–C80 176.90(15)°, O1–C80–P70 103.35(14)°. | |
To determine the influence of the phosphine, we monitored the reaction over time using triphenylphosphine (PPh3) as an internal standard (Fig. 8). Firstly, this reveals that the reaction is the fastest for complex 2a. Additionally it shows that the starting complexes are consumed at a much faster rate than the diphosphines are formed and also that the diphosphine concentration does not increase further once a threshold level is reached, which appears to be distinctive for the corresponding diphosphines. While in the case of 2a, about 60% conversion to dimesityldiphoshine can be reached after 16 minutes, for 2b and 2c only 16% and 10% conversions are reached within 120 and 36 min respectively. This further shows that reaction for 2b is much slower compared to the more electron rich phosphines in 2a and 2c. Notably, in the case of 2c, the concentration of the starting material is further decreasing even after the diphosphine concentration has reached its conversion. This indicates that another decomposition path for 2c might be possible, which is in agreement with the fact that the 31P NMRs of 2c show substantial formation of HPN and other phosphorous species (Fig. S50†). Thus, we deduce that aryl phosphines seem to react much more cleanly. To further elucidate if our postulated mechanism from Scheme 3 might be valid, we further performed scrambling experiments using equimolar mixtures of 2a and 2b. Indeed, during irradiation we saw the formation of three diphosphine species, being the two symmetrical dimesityldiphosphine ([PHMes]2) and diphenyldiphosphine ([PHPh]2), as well as the mixed phenyl-mesityl diphosphine (PhPH-PHMes) (Fig. S51 and S52†). Together with the fact, that no free phosphanyl radicals could be trapped and evidenced during the reaction (vide supra) the observation of the scrambling product PhPH-PHMes further supports our theory of a concerted reaction mechanism.
 |
| | Fig. 8 Time-conversion-plots of the light-promoted decomposition of 2a, 2b and 2c. The blue curves show the amount of diphosphine being formed, while the orange lines show the consumption of the starting hemiphosphinal complex. Conversions were determined via31P NMR spectroscopy using PPh3 as an internal standard. | |
Conclusion
We have reported the insertion reaction of fluorenone into labile La–P bonds in 1a–c, giving access to hemiphosphinal complexes 2a–c. These complexes are sensitive to light, decomposing to a fluorenonyl radical complex 3 and the corresponding diphosphines [PHMes]2, [PHPh]2 and [PHtBu]2. The former can be detected by EPR spectroscopy and can even be isolated in (single) crystalline form. On the phosphine side, although calculations suggest a direct P–C bond splitting no free phosphanyl radicals could be detected. Thus, we propose a concerted P-C bond scission / P-P bond formation mechanism which is also indicated experimentally via scrambling experiments (Scheme 3).
In summary we have reported a rare case of a light sensitive hemiphopshinal complex, which easily decomposes to give access to synthetically challenging diphosphines. Given the fact, that similar decomposition reactions have not been observed for transition metal-8,9,21,22,26 and actinide13 based hemiphosphinal complexes we believe that this reaction opens new venues in the use of lanthanides in (phosphor-)organic chemistry. Additionally, it gives facile access to multi-spin systems with fluorenone radicals attached to optically and magnetically active lanthanides.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to the Daimler and Benz Foundation, the Fonds der Chemischen Industrie, the Young Academy of the North-Rhine-Westphalian Academy of Sciences, Humanities and the Arts, the Paderborn University, and the University of Innsbruck for financial support. Dr Fabian Watt is kindly acknowledged for providing preliminary results on the insertion chemistry. Eva Burger and Roland Egger are kindly acknowledged for the measurements of elemental analysis. L. M. is a senior member of the Institut Universitaire de France. CalMip is acknowledged for a generous grant of computing time.
References
- S. P. Vilanova, I. Del Rosal, M. L. Tarlton, L. Maron and J. R. Walensky, Functionalization of Carbon Monoxide and tert-Butyl Nitrile by Intramolecular Proton Transfer in a Bis(Phosphido) Thorium Complex, Angew. Chem., Int. Ed., 2018, 57, 16748–16753 CrossRef CAS PubMed.
- M. L. Tarlton, I. Del Rosal, S. P. Vilanova, S. P. Kelley, L. Maron and J. R. Walensky, Comparative Insertion Reactivity of CO, CO2tBuCN, and tBuNC into Thorium– and Uranium–Phosphorus Bonds, Organometallics, 2020, 39, 2152–2161 CrossRef CAS.
- A. C. Behrle and J. R. Walensky, Insertion of (t)BuNC into thorium-phosphorus and thorium-arsenic bonds: phosphaazaallene and arsaazaallene moieties in f element chemistry, Dalton Trans., 2016, 45, 10042–10049 RSC.
- M. E. Garner, B. F. Parker, S. Hohloch, R. G. Bergman and J. Arnold, Thorium Metallacycle Facilitates Catalytic Alkyne Hydrophosphination, J. Am. Chem. Soc., 2017, 139, 12935–12938 CrossRef CAS PubMed.
- F. T. Edelmann, Lanthanide amidinates and guanidinates in catalysis and materials science: a continuing success story, Chem. Soc. Rev., 2012, 41, 7657–7672 RSC.
- F. Lindenberg, J. Sieler and E. Hey-Hawkins, Formation of novel P- and As-functionalized ligands by insertion reactions into the Zr·E bond of (η5-C5H4R)2ZrCl{E(SiMe3)2} (R = Me, E = P, As; R = H, E = P), Polyhedron, 1996, 15, 1459–1464 CrossRef CAS.
- U. Segerer, J. Sieler and E. Hey-Hawkins, Formation of Novel P - Functionalized Ligands by Insertion Reactions of RNCX (R = Ph, X = O, S; R = Pr i X = O) into the Zr–P Bond of [Cp° 2ZrCl(PHCy)] (Cp° = η5-C5EtMe4Cy = Cyclohexyl) and [Cp′ 2 ZrCl{PH(TRIP)}] (Cp′ = η5-C5MeH4 TRIP = 2,4,6-Pr i3C6H2), Organometallics, 2000, 19, 2445–2449 CrossRef CAS.
- Z. Hou and D. W. Stephan, Generation and reactivity of the first mononuclear early metal phosphinidene complex, Cp*2Zr:P(C6H2Me3-2,4,6), J. Am. Chem. Soc., 1992, 114, 10088–10089 CrossRef CAS.
- Z. Hou, T. L. Breen and D. W. Stephan, Formation and reactivity of the early metal phosphides and phosphinidenes Cp*2Zr:PR, Cp*2Zr(PR)2, and Cp*2Zr(PR)3, Organometallics, 1993, 12, 3158–3167 CrossRef CAS.
- A. C. Behrle and J. A. R. Schmidt, Insertion Reactions and Catalytic Hydrophosphination of Heterocumulenes using α-Metalated N,N-Dimethylbenzylamine Rare-Earth-Metal Complexes, Organometallics, 2013, 32, 1141–1149 CrossRef CAS.
- W.-X. Zhang, M. Nishiura, T. Mashiko and Z. Hou, Half-sandwich o-N,N-dimethylaminobenzyl complexes over the full size range of group 3 and lanthanide metals. synthesis, structural characterization, and catalysis of phosphine P–H bond addition to carbodiimides, Chem. – Eur. J., 2008, 14, 2167–2179 CrossRef CAS PubMed.
- W. Yi, J. Zhang, L. Hong, Z. Chen and X. Zhou, Insertion of Isocyanate and Isothiocyanate into the Ln–P σ-Bond of Organolanthanide Phosphides, Organometallics, 2011, 30, 5809–5814 CrossRef CAS.
- S. P. Vilanova, M. L. Tarlton, C. L. Barnes and J. R. Walensky, Double insertion of benzophenone into thorium-phosphorus bonds, J. Organomet. Chem., 2018, 857, 159–163 CrossRef CAS.
- C. Zhang, Y. Wang, G. Hou, W. Ding, G. Zi and M. D. Walter, Experimental and computational studies on a three-membered diphosphido thorium metallaheterocycle η5-1,3-(Me3C)2C5H32Thη2-P2(2,4,6-iPr3C6H2)2, Dalton Trans., 2019, 48, 6921–6930 RSC.
- R. J. Ward, P. Rungthanaphatsophon, I. Del Rosal, S. P. Kelley, L. Maron and J. R. Walensky, Divergent uranium- versus phosphorus-based reduction of Me3SiN3 with steric modification of phosphido ligands, Chem. Sci., 2020, 11, 5830–5835 RSC.
- I. V. Basalov, O. S. Yurova, A. V. Cherkasov, G. K. Fukin and A. A. Trifonov, Amido Ln(II) Complexes Coordinated by Bi- and Tridentate Amidinate Ligands: Nonconventional Coordination Modes of Amidinate Ligands and Catalytic Activity in Intermolecular Hydrophosphination of Styrenes and Tolane, Inorg. Chem., 2016, 55, 1236–1244 CrossRef CAS PubMed.
- M. M. I. Basiouny and J. A. R. Schmidt, Lanthanum-Catalyzed Double Hydrophosphinylation
of Nitriles, Organometallics, 2017, 36, 721–729 CrossRef CAS.
- Y. A. Rina and J. A. R. Schmidt, Lanthanum-Catalyzed Regioselective Anti-Markovnikov Hydrophosphinylation of Styrenes, Organometallics, 2019, 38, 4261–4270 CrossRef CAS.
- A. A. Trifonov, I. V. Basalov and A. A. Kissel, Use of organolanthanides in the catalytic intermolecular hydrophosphination and hydroamination of multiple C–C bonds, Dalton Trans., 2016, 45, 19172–19193 RSC.
- C. J. Weiss and T. J. Marks, Organo-f-element catalysts for efficient and highly selective hydroalkoxylation and hydrothiolation, Dalton Trans., 2010, 39, 6576–6588 RSC.
- T. L. Breen and D. W. Stephan, Synthesis and Reactivity of Phosphametallacycles: Sterically Induced Epimerizations and Retrocycloadditions, Organometallics, 1996, 15, 5729–5737 CrossRef CAS.
- T. L. Breen and D. W. Stephan, Synthesis and Reactivity of Phosphametallacyclobutenes: Sterically Induced [4 + 2] Retrocycloadditions, J. Am. Chem. Soc., 1996, 118, 4204–4205 CrossRef CAS.
- W. Mao, L. Xiang, C. A. Lamsfus, L. Maron, X. Leng and Y. Chen, Are Sc–C and Sc–P Bonds Reactive in Scandium Phosphinoalkylidene Complex? Insights on a Versatile Reactivity, Chin. J. Chem., 2018, 36, 904–908 CrossRef CAS.
- S. L. Marquard, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Stoichiometric CO bond oxidative addition of benzophenone by a discrete radical intermediate to form a cobalt(I) carbene, J. Am. Chem. Soc., 2013, 135, 6018–6021 CrossRef CAS PubMed.
- A. Ziółkowska, N. Szynkiewicz and Ł. Ponikiewski, Experimental and theoretical investigation of the reactivity of (BDI*)Ti(Cl){η2-P(SiMe3)-PiPr2} towards selected ketones, Dalton Trans., 2021, 50, 1390–1401 RSC.
- T. L. Breen and D. W. Stephan, Reactivity Studies of Methylzirconocene Phosphide Complexes, Organometallics, 1996, 15, 4509–4514 CrossRef CAS.
- Z. Hou, T. Koizumi, M. Nishiura and Y. Wakatsuki, Lanthanide(II) Complexes Bearing Linked Cyclopentadienyl−Anilido Ligands: Synthesis, Structures, and One-Electron-Transfer and Ethylene Polymerization Reactions, Organometallics, 2001, 20, 3323–3328 CrossRef CAS.
- Z. Hou, A. Fujita, H. Yamazaki and Y. Wakatsuki, First Isolation of a Tris(ketyl) Metal Complex, J. Am. Chem. Soc., 1996, 118, 7843–7844 CrossRef CAS.
- A. Domingos, I. Lopes, J. C. Waerenborgh, N. Marques, G. Y. Lin, X. W. Zhang, J. Takats, R. McDonald, A. C. Hillier, A. Sella, M. R. J. Elsegood and V. W. Day, Trapping of anionic organic radicals by (TpMe2)2Ln (Ln = Sm, Eu), Inorg. Chem., 2007, 46, 9415–9424 CrossRef CAS PubMed.
- N. J. C. van Velzen and S. Harder, Synthesis and reactivity of a β-diketiminate Sm, Aust. J. Chem., 2022, 75, 549–557 CrossRef CAS.
- F. Jaroschik, F. Nief and X.-F. Le Goff, Sterically hindered cyclopentadienyl and phospholyl ligands in dysprosium chemistry, Polyhedron, 2009, 28, 2744–2748 CrossRef CAS.
- Z. Hou, T. Miyano, H. Yamazaki and Y. Wakatsuki, Well-Defined Metal Ketyl Complex: Sm(ketyl)(OAr)2(THF)2 and Its Reversible Coupling to a Disamarium(III) Pinacolate, J. Am. Chem. Soc., 1995, 117, 4421–4422 CrossRef CAS.
- T. Yoshimura, Z. Hou and Y. Wakatsuki, Protonation at the Aromatic Ring vs. at the Carbonyl Group of Lanthanide-Diaryl Ketone Dianion Species by Aryl Alcohols. Formation, Structural Characterization, and Reactivity of Lanthanide Aryloxide, Mixed Aryloxide/Alkoxide, and Aryloxide/Enolate Complexes, Organometallics, 1995, 14, 5382–5392 CrossRef CAS.
- A. R. Crozier, K. W. Törnroos, C. Maichle-Mössmer and R. Anwander, Trivalent Cerium and Praseodymium Aromatic Ketone Adducts, Eur. J. Inorg. Chem., 2013, 2013, 409–414 CrossRef CAS.
- O. P. Lam, C. Anthon, F. W. Heinemann, J. M. O'Connor and K. Meyer, Structural and spectroscopic characterization of a charge-separated uranium benzophenone ketyl radical complex, J. Am. Chem. Soc., 2008, 130, 6567–6576 CrossRef CAS PubMed.
- K. C. Mullane, T. Cheisson, E. Nakamaru-Ogiso, B. C. Manor, P. J. Carroll and E. J. Schelter, Reduction of Carbonyl Groups by Uranium(III) and Formation of a Stable Amide Radical Anion, Chem. – Eur. J., 2018, 24, 826–837 CrossRef CAS PubMed.
- I. Nagl, M. Widenmeyer, S. Grasser, K. Köhler and R. Anwander, Surface Confined Ketyl Radicals via Samarium(II)-Grafted Mesoporous Silicas, J. Am. Chem. Soc., 2000, 122, 1544–1545 CrossRef CAS.
- D. Werner, X. Zhao, S. P. Best, L. Maron, P. C. Junk and G. B. Deacon, Bulky Ytterbium Formamidinates Stabilise Complexes with Radical Ligands, and Related Samarium “Tetracyclone” Chemistry, Chem. – Eur. J., 2017, 23, 2084–2102 CrossRef CAS PubMed.
-
K. A. Choquette and R. A. Flowers, in Comprehensive organic synthesis, ed. P. Knochel and G. A. Molander, Elsevier, Amsterdam, 2014, pp. 278–343 Search PubMed.
- Z. Hou, A. Fujita, Y. Zhang, T. Miyano, H. Yamazaki and Y. Wakatsuki, One-Electron Reduction of Aromatic Ketones by Low-Valent Lanthanides. Isolation, Structural Characterization, and Reactivity of Lanthanide Ketyl Complexes, J. Am. Chem. Soc., 1998, 120, 754–766 CrossRef CAS.
- M. H. Chisholm, K. Folting and J. A. Klang, Reaction between benzophenone and ditungsten hexaalkoxides. Molecular structure and reactivity of W(OCH2-tert-Bu)4(py)(.eta.2-OCPh2), Organometallics, 1990, 9, 607–613 CrossRef CAS.
- G. B. Deacon, P. C. Junk, J. Wang and D. Werner, Reactivity of bulky formamidinatosamarium(II or III) complexes with CO and CS bonds, Inorg. Chem., 2014, 53, 12553–12563 CrossRef CAS PubMed.
- A. J. Roering, S. E. Leshinski, S. M. Chan, T. Shalumova, S. N. MacMillan, J. M. Tanski and R. Waterman, Insertion Reactions and Catalytic Hydrophosphination by Triamidoamine-Supported Zirconium Complexes, Organometallics, 2010, 29, 2557–2565 CrossRef CAS.
- A. T. Normand, C. G. Daniliuc, B. Wibbeling, G. Kehr, P. Le Gendre and G. Erker, Insertion Reactions of Neutral Phosphidozirconocene Complexes as a Convenient Entry into Frustrated Lewis Pair Territory, Chem. – Eur. J., 2016, 22, 4285–4293 CrossRef CAS PubMed.
- J. E. Kim, A. V. Zabula, P. J. Carroll and E. J. Schelter, 1,2-Addition or Enolization? Variable Reactivity of a Cerium Acetylide Complex toward Carbonyl Compounds, Organometallics, 2016, 35, 2086–2091 CrossRef CAS.
- S. J. Kraft, P. E. Fanwick and S. C. Bart, Exploring the Insertion Chemistry of Tetrabenzyluranium Using Carbonyls and Organoazides, Organometallics, 2013, 32, 3279–3285 CrossRef CAS.
- Z. Jian, C. G. Daniliuc, G. Kehr and G. Erker, Frustrated Lewis Pair vs Metal–Carbon σ-Bond Insertion Chemistry at an o -Phenylene-Bridged Cp2 Zr+/PPh2 System, Organometallics, 2017, 36, 424–434 CrossRef CAS.
- M. E. Garner and J. Arnold, Reductive Elimination of Diphosphine from a Thorium–NHC–Bis(phosphido) Complex, Organometallics, 2017, 36, 4511–4514 CrossRef CAS.
- F. A. Watt, A. Krishna, G. Golovanov, H. Ott, R. Schoch, C. Wölper, A. G. Neuba and S. Hohloch, Monoanionic Anilidophosphine Ligand in Lanthanide Chemistry: Scope, Reactivity, and Electrochemistry, Inorg. Chem., 2020, 59, 2719–2732 CrossRef CAS PubMed.
- F. A. Watt, L. Burkhardt, R. Schoch, S. Mitzinger, M. Bauer, F. Weigend, J. M. Goicoechea, F. Tambornino and S. Hohloch, η3-Coordination and Functionalization of the 2-Phosphaethynthiolate Anion at Lanthanum(III)*, Angew. Chem., Int. Ed., 2021, 60, 9534–9539 CrossRef CAS PubMed.
- F. A. Watt, K. N. McCabe, R. Schoch, L. Maron and S. Hohloch, A transient lanthanum phosphinidene complex, Chem. Commun., 2020, 56, 15410–15413 RSC.
- F. A. Watt, N. Dickmann, R. Schoch and S. Hohloch, Isocyanate Insertion into a La–P Phosphide Bond: A Versatile Route to Phosphaureate-Bridged Heterobimetallic Lanthanide-Coinage-Metal Complexes, Inorg. Chem., 2020, 59, 13621–13631 CrossRef CAS PubMed.
- S. Kurz, H. Oesen, J. Sieler and E. Hey-Hawkins, Synthesis and molecular structure OF Mes(H)P-P(H)Mes (Mes = 2,4,6-Me3C6H2), Phosphorus Sulfur, 1996, 117, 189–196 CrossRef CAS.
- G. Nocera, A. Young, F. Palumbo, K. J. Emery, G. Coulthard, T. McGuire, T. Tuttle and J. A. Murphy, Electron Transfer Reactions: KO tBu (but not NaO tBu) Photoreduces Benzophenone under Activation by Visible Light, J. Am. Chem. Soc., 2018, 140, 9751–9757 CrossRef CAS PubMed.
- Indeed the experimental insertion reaction with benzophenone works equally well, giving access to the corresponding semiphosphinal complex, which is very light sensitive as well. Upon irradiation this complex also extrudes diphosphines and a benzophenone radical, which however cannot be crystallized.
|
| This journal is © the Partner Organisations 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
Stephan
Hohloch
*b and
Stephan
Hohloch