
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective syntheses of homoleptic Ir(III) complexes bearing di-CF3-functionalized benzoimidazol-2-ylidenes for generation of blue phosphorescence†
Jie
Yan‡
 a,
Yi
Pan‡
a,
I-Che
Peng‡
b,
Wen-Yi
Hung
*b,
Bingjie
Hu
a,
Guowei
Ni
a,
Shek-Man
Yiu
a,
Yun
Chi
*ac and
Kai Chung
Lau
*a
a,
Yi
Pan‡
a,
I-Che
Peng‡
b,
Wen-Yi
Hung
*b,
Bingjie
Hu
a,
Guowei
Ni
a,
Shek-Man
Yiu
a,
Yun
Chi
*ac and
Kai Chung
Lau
*a
aDepartment of Materials Science and Engineering and Department of Chemistry, City University of Hong Kong, Kowloon 999077, Hong Kong SAR. E-mail: yunchi@cityu.edu.hk; kaichung@cityu.edu.hk
bDepartment of Optoelectronics and Materials Technology, Taiwan Ocean University, Keelung 20224, Taiwan, China. E-mail: wenhung@mail.ntou.edu.tw
cCenter of Super-Diamond and Advanced Films (COSDAF), City University of Hong Kong, Kowloon 999077, Hong Kong SAR
First published on 12th March 2024
Abstract
Homoleptic Ir(III) based carbene complexes are known to be the most promising emitters of future blue OLED devices. To provide the proof-of-concept, we designed a series of functional di-CF3-functionalized benzo[d]imidazol-3-ium pro-chelates, which could afford single product emitters after proper modification. For benzoimidazol-2-ylidene with an N-methyl substituent, selective formation of the product can be achieved by introduction of t-butylphenyl for the phenyl group, as shown by shifting the product from mixed m-Ir(dfp)3 and f-Ir(dfp)3 to the single isomer f-Ir(dfpb)3. Alternatively, for di-N-aryl substituted carbene chelates, the steric encumbrance imposed between the ortho-CF3 group and the adjacent N-aryl substituent redirects the cyclometalation to the other N-aryl substituent, leading to the formation of one single product, e.g., f-Ir(tBpp)3 and f-Ir(ptBp)3. Moreover, the doped OLED based on f-Ir(tBpp)3 delivered true-blue emission centered at 457 nm and a maximum EQE of 15.6%. Furthermore, upon addition of terminal emitters ν-DABNA and t-DABNA, the respective hyper-OLEDs exhibited narrowband blue emission with a maximum EQE of 18.9% at 474 nm and 18.1% at 462 nm, respectively. These highlighted the potential of these Ir(III) emitters in the fabrication of blue OLEDs.
Introduction
Fluorination or incorporation of the perfluoroalkyl group to various functional materials has become an important methodology to enhance the performance of soft optoelectronic devices such as organic field-effect transistors (OFETs), light emitting transistors (OLETs) and light emitting diodes (OLEDs).1–5 The specific features of such a fluorinated entity, i.e., the trifluoromethyl (CF3) group, are related to its high electron withdrawing character, together with enhanced hydrophobicity, lipophobicity and volatility. It is also known that the CF3 group can lower the π-orbital energies of organic functional entities and materials.6,7 The lowered energy levels facilitate electron injection, thus giving balanced carrier transportation and, hence, improved device efficiencies.8–10 With this in mind, we decided to introduce the CF3 group to Ir(III) based phosphors bearing functional imidazolylidene cyclometalates, with an attempt to further improve their photo- and electro-luminescence characteristics.Similar manipulation has been documented in Ir(III) carbene complexes such as m-Ir(tfp_tz)3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11–13 and its analogues.14,15 Furthermore, Zysman-Colman and co-workers have also reported mer-arranged, homoleptic and blue Ir(III) phosphors comprising 1-methyl-3-(4-(trifluoromethyl)phenyl)-imidazol-2-ylidene chelates, cf., m-Ir(tfpmi)3 and its analogue.16 Next, they switched to similar Ir(III) complexes containing a sterically demanding benzyl (or 2,4,6-trimethylbenzyl) appendage on carbene chelates, cf., m-Ir(tfpi_Bn)3.17 One representative OLED device showed deep-blue electroluminescence with a peak maximum (λmax) of 429 nm and CIE color coordinates of (0.16, 0.08), illustrating excellent performance for those with CF3 substituted N-phenyl cyclometalating carbene chelates. However, their photoluminescence radiative rate constant (kr) is in the region of ∼105 s−1, suggesting the dominant ligand-centered ππ* transition characteristics in the excited state. These photophysical properties are inferior to their Ir(III) based benzoimidazol-2-ylidene counterparts with an improved metal-to-ligand charge transfer (MLCT) character, which exhibited a much faster kr of ∼106 s−1.18 This opens up one possible research direction for improving both the photochemical and physical properties by increasing the MLCT contribution in the excited states of relevant Ir(III) carbene complexes.18–20
11–13 and its analogues.14,15 Furthermore, Zysman-Colman and co-workers have also reported mer-arranged, homoleptic and blue Ir(III) phosphors comprising 1-methyl-3-(4-(trifluoromethyl)phenyl)-imidazol-2-ylidene chelates, cf., m-Ir(tfpmi)3 and its analogue.16 Next, they switched to similar Ir(III) complexes containing a sterically demanding benzyl (or 2,4,6-trimethylbenzyl) appendage on carbene chelates, cf., m-Ir(tfpi_Bn)3.17 One representative OLED device showed deep-blue electroluminescence with a peak maximum (λmax) of 429 nm and CIE color coordinates of (0.16, 0.08), illustrating excellent performance for those with CF3 substituted N-phenyl cyclometalating carbene chelates. However, their photoluminescence radiative rate constant (kr) is in the region of ∼105 s−1, suggesting the dominant ligand-centered ππ* transition characteristics in the excited state. These photophysical properties are inferior to their Ir(III) based benzoimidazol-2-ylidene counterparts with an improved metal-to-ligand charge transfer (MLCT) character, which exhibited a much faster kr of ∼106 s−1.18 This opens up one possible research direction for improving both the photochemical and physical properties by increasing the MLCT contribution in the excited states of relevant Ir(III) carbene complexes.18–20
Accordingly, these desired properties can be achieved by expanding π-conjugation on the imidazol-2-ylidene (carbene) fragment and incorporating electron deficient entities at the same time.18 One practical approach involved the replacement of the parent imidazolylidene with the benzo[d]imidazol-2-ylidene fragment,21,22 followed by further conversion to imidazo[4,5-b]pyridin-2-ylidene,23,24 imidazo[4,5-b]pyrazin-2-ylidene,25–27 and even the purin-8-ylidene entity,28,29 by addition of electro-negative nitrogen atom(s) at the designated skeletal position(s). Unlike the previously mentioned chelates and Ir(III) derivatives as shown in Scheme 1, these newly employed electron deficient carbene cyclometalates tended to afford two configurational isomers, e.g., m- and f-isomers under all conditions. Importantly, they showed structureless emission with a relatively fast radiative rate constant in solution at RT, confirming the dominant MLCT character in the excited state manifold.30,31
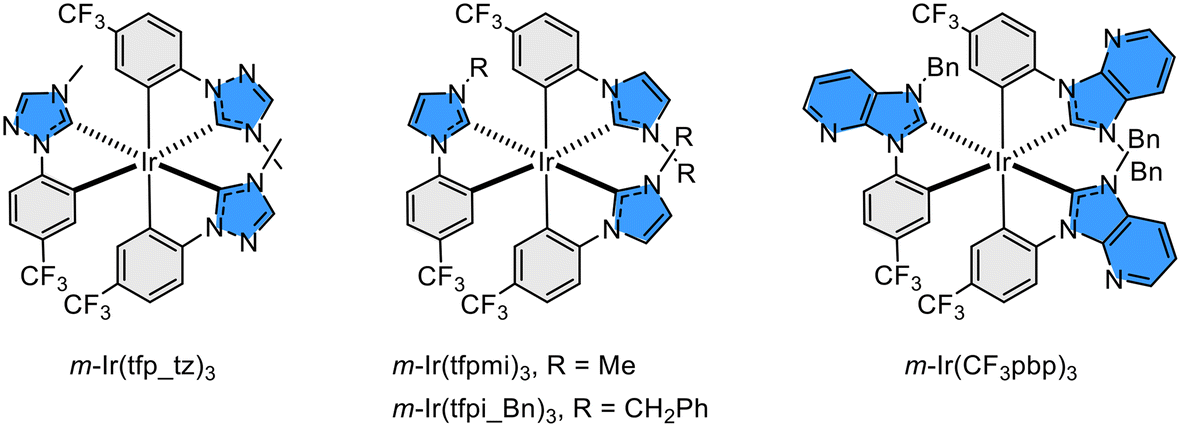 | ||
| Scheme 1 Drawings of homoleptic Ir(III) carbene complexes bearing CF3 substituted N-phenyl cyclometalates. | ||
Herein, we report the design and preparation of such Ir(III) emitters bearing 4,6-bis(trifluoromethyl)-benzo[d]imidazol-2-ylidene cyclometalates. Their molecular structures are depicted in Scheme 2, in which m-/f-Ir(dfp)3, f-Ir(dfpb)3, f-Ir(tBpp)3 and f-Ir(ptBp)3 are associated with (i) either a phenyl or p-t-butylphenyl cyclometalating unit and (ii) a methyl, phenyl or p-t-butylphenyl peripheral appendage. To these designs, the trifluoromethyl (CF3) group is expected to be inert to environmental perturbation and, thus, may demonstrate an increased chemical stability toward those offered by skeletal nitrogen atoms, which is known to be basic due to the possession of a lone pair of electrons. Not to mention that its ortho-CF3 group can provide critical steric encumbrance to allow the selective formation of a certain favorable geometrical isomer, i.e., achieving selective synthesis of one isomer out of four statistically distributed products. Hence, tedious separation of isomers was no longer required, which is particularly important for the scale-up operation and marking an essential milestone in syntheses. Moreover, this site-selectivity is of uttermost importance to the widespread employment of di-aryl substituted benzoimidazol-2-ylidene and its analogues,32–34 as they are deemed superior to their mono-N-aryl counterparts in serving as blue OLED emitters. However, the introduction of either 2,6- or 3,5-dialkylaryl substituents gave only decomposition, while using a 4-substituted aryl group afforded a mixture of up to four isomers,35–37 confirming the success of the current strategy. Finally, fabrication of OLED devices was executed using three Ir(III) emitters f-Ir(dfp)3, f-Ir(tBpp)3 and f-Ir(ptBp)3. These emitters can serve as both the sole dopant and Förster Resonance Energy Transfer (FRET) sensitizer to both the terminal emitters ν-DABNA and t-DABNA, in achieving narrow band true blue emission, as demanded by emerging industrial applications.
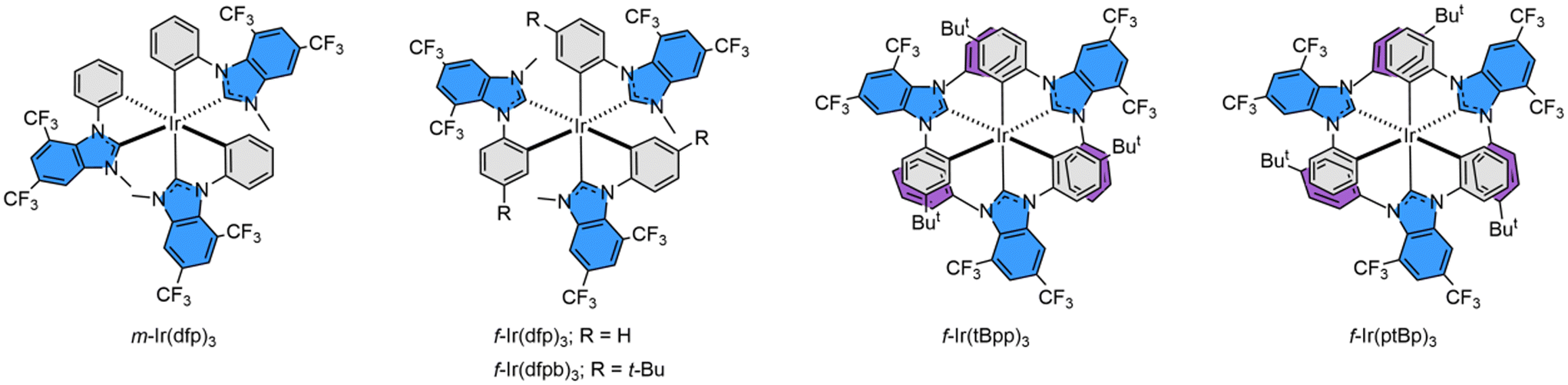 | ||
| Scheme 2 Structural drawings of Ir(III) complexes bearing dual CF3 substituted benzoimidazol-2-ylidene, confirming the selective cyclometalation of the N-aryl substituent. | ||
Results and discussion
Preparation and characterization
The functional 1-aryl-1H-benzo[d]imidazole with dual trifluoromethyl (CF3) substituents was best synthesized using a multi-step protocol as depicted in Scheme 3. First, the key starting material 2-bromo-3,5-bis(trifluoromethyl)aniline (A) was prepared by bromination of commercially available 3,5-bis(trifluoromethyl)aniline using N-bromosuccinimide in CH2Cl2 at ∼5 °C. After that, it was treated with a stoichiometric amount of triethyl orthoformate in the presence of glacial acetic acid as a catalyst at 120 °C, followed by the addition of aniline or 4-t-butylaniline at 140 °C. This two-step operation procedure is aimed at in situ preparation of functional ethyl N-arylformimidate as an intermediate,38 which is expected to undergo further coupling with aniline to afford asymmetric N,N′-diarylformamidines with a 2-bromo-3,5-bis(trifluoromethyl)phenyl substituent.39 After that, a copper-catalyzed intramolecular C–N bond coupling was executed to obtain the imidazole derivatives B1 and B2 in moderate yields,40 following which N-alkylation with methyl trifluoromethanesulfonate has successfully afforded the anticipated 3-methyl-1-aryl-5,7-bis(trifluoromethyl)-1H-benzo[d]imidazol-3-ium pro-chelates C1 and C2 in high yields. Concurrently, according to the literature,41 the reaction of B1 and B2 with diaryliodonium salt in DMF and with Cu(OAc)2 as the catalyst afforded the respective di-N-aryl substituted pro-chelates C3 and C4 in high yields.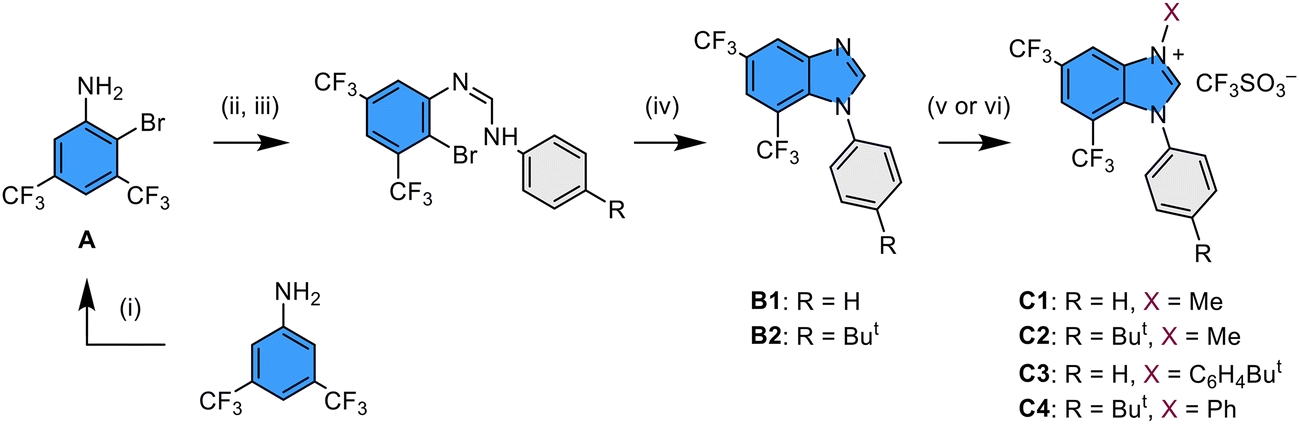 | ||
| Scheme 3 Synthetic protocols to the functional benzo[d]imidazol-3-ium pro-chelates: (i) NBS, DCM, 0 °C; (ii) triethyl orthoformate, glacial acetic acid, 120 °C; (iii) arylamine, 140 °C; (iv) DBU, CuI, DMSO, 150 °C; (v) CF3SO3Me, toluene, RT; (vi) diaryliodonium salt, Cu(OAc)2·H2O, DMF, 110 °C. | ||
Isolation of these carbene pro-chelates made the preparation of all homoleptic Ir(III) carbene complexes an easy task. The iridium reagent [IrCl3(tht)3]42 was selected as the metal source due to its enhanced solubility in nonpolar aromatic solvents which is essential for the employment of sodium acetate as a catalyst.43 Accordingly, treatment of pro-chelate C1 with IrCl3(tht)3 and sodium acetate in refluxing t-butylbenzene (bp = 169 °C) afforded a mixture of both m- and f-isomers, namely m-Ir(dfp)3 and f-Ir(dfp)3. In sharp contrast, treatment of respective pro-chelate C2 with IrCl3(tht)3 afforded only the symmetric derivative f-Ir(dfpb)3 in refluxing t-butylbenzene. This could be attributed to the facile m- to f-isomerization process and greater thermodynamic stability for the f-isomer according to the literature.44 Similarly, extensive heating of C3 and C4 with IrCl3(tht)3 in a higher boiling solvent 1,2,4-trichlorobenzene (bp = 214 °C) also afforded a single product f-Ir(tBpp)3 and f-Ir(ptBp)3, respectively. The gross arrangement of chelates can be further confirmed by 1H and 19F NMR spectroscopy, which revealed a single set and three distinctive sets of resonance signals for the f- and m-isomers, respectively. Their structural drawings are depicted in Scheme 2 for scrutiny. For f-Ir(ptBp)3, its structural assignment is confirmed by the observation of a total of four sets of doublet of doublets at δ 7.53 (dd, J = 8.4, 2.4 Hz), 7.36 (dd, J = 8.4, 2.4 Hz), 6.68 (dd, J = 8.0, 2.0 Hz) and 6.54 (dd, J = 8.0, 2.0 Hz) at room temperature, and the distinctive JHH coupling constants of 8.4 and 2.4 Hz, 8.0 and 2.0 Hz were assigned for the ABXY spin coupling pattern, due to the hindered rotation of these t-butylphenyl appendages. Notably, in addition to the formation of solely the f-isomers for both f-Ir(tBpp)3 and f-Ir(ptBp)3, the pro-chelates C3 and C4 possess two distinctive aryl groups which, in principle, have equal probability of undergoing metal cyclometalation in affording isomeric products. However, only the aryl group that is far away from the ortho-substituted CF3 group underwent selective cyclometalation, leaving the adjacent aryl group as a free appendage, as shown in f-Ir(tBpp)3 and f-Ir(ptBp)3. This enhanced selectivity is most probably caused by the steric encumbrance exerted between the CF3 group at the 7-position and the adjacent 1-aryl group of the benzo[d]imidazol-2-ylidene entity, which is best explained by the X-ray structural data of m-Ir(dfp)3 (vide infra). Furthermore, the reversible C–H activation and cyclometalation of aryl appendages is also responsible for the selective formation of a single product in the presence of sodium acetate at elevated temperature.45,46
A single crystal X-ray diffraction study on m-Ir(dfp)3 and f-Ir(tBpp)3 was executed to confirm the coordination arrangement of chelates. Fig. 1 depicts a distorted octahedral structure of m-Ir(dfp)3, as expected for the mer-configuration. Six Ir–C distances were found to be different, among which all Ir–C(carbene) distances (Ir–C7 = 2.006(3), Ir–C23 = 2.047(3) and Ir–C39 = 2.021(3) Å) are notably shorter than the corresponding Ir–C(aryl) distances (Ir–C1 = 2.076(3), Ir–C17 = 2.112(3), and Ir–C33 = 2.104(3) Å). Particularly, the unique Ir–C1 vector, that is uniquely located trans to the Ir–C(carbene) vector, is the shortest among all Ir–C(aryl) vectors, showing the reduced trans-effect of carbene in reference to the aryl entities. Concurrently, the Ir–C23 distance of the same carbene chelate is also longer than the other two, trans-arranged Ir–C(carbene) fragments, showing the relatively strengthened trans-effect exerted by its opposite phenyl cyclometalate. Furthermore, the distances of this trans-arranged C(aryl)–Ir–C(carbene) vector of m-Ir(dfp)3 are comparable to the respective C(aryl)–Ir–C(carbene) distances observed in f-coordinated f-Ir(tBpp)3, i.e., with Ir–C19 = 2.0903(19) and Ir–C13 = 2.0428(17) Å, as depicted in Fig. 2. These results confirmed the variation of Ir–C distances in response to their local coordinative environment.
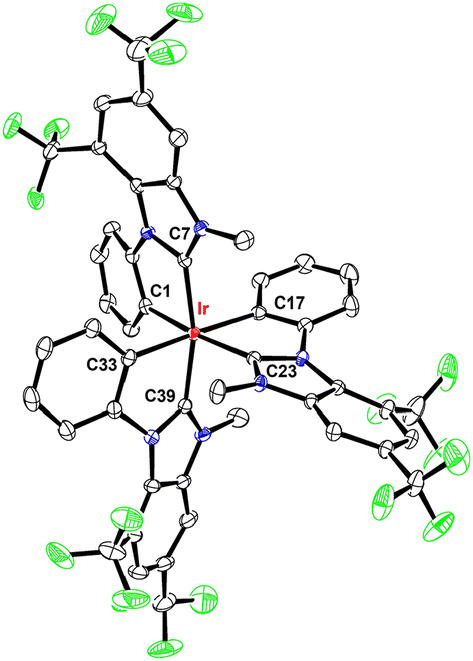 | ||
| Fig. 1 Structure drawing of m-Ir(dfp)3 with thermal ellipsoids shown at the 30% probability level. Selected bond length (Å): Ir–C1 = 2.076(3), Ir–C7 = 2.006(3), Ir–C17 = 2.112(3), Ir–C23 = 2.047(3), Ir–C33 = 2.104(3), and Ir–C39 = 2.021(3). Selected bond angle (°): C1–Ir–C23 = 169.91(10), C7–Ir–C39 = 166.42(10), and C17–Ir–C33 = 177.97(11). | ||
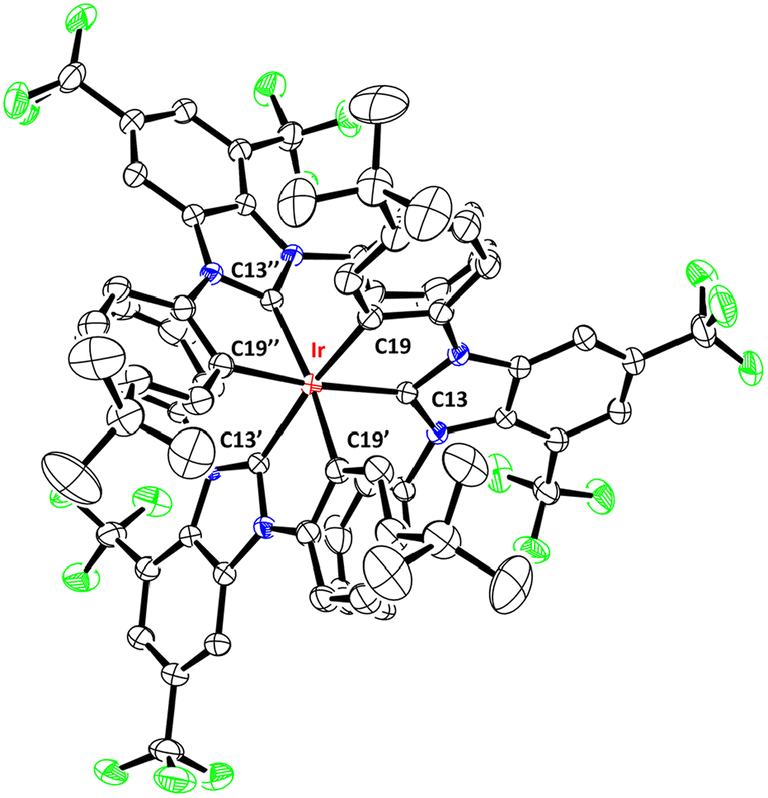 | ||
| Fig. 2 Structure drawing of f-Ir(tBpp)3 with thermal ellipsoids shown at the 30% probability level. Selected bond length (Å): Ir–C13 = 2.0428(17) and Ir–C19 = 2.0903(19). Selected bond angles (°): C13–Ir–C19′ = 168.52(8). | ||
Notably, there existed a large angular distortion between the imidazolylidene entity and phenyl cyclometalate of each individual carbene chelates of m-Ir(dfp)3, which is obviously caused by the steric hindrance exerted by the CF3 group at the 7-position and adjacent 1-phenyl cyclometalate. The averaged torsional angle is calculated to be around 23°, which is notably larger than the 14° distortion observed in f-Ir(tBpp)3, and the 7-position is occupied by a smaller hydrogen atom instead. Hence, this angular distortion will be a major driving force in giving the structural selectivity as observed in Ir(III) complexes f-Ir(tBpp)3 and f-Ir(ptBp)3. Furthermore, the closest contacts between the CF3 group and the ortho C–H entity of the adjacent phenyl cyclometalate in m-Ir(dfp)3 were found to be in the range of 2.306–2.355 Å, which are shorter than the sum of the van der Waals radii of hydrogen (1.2 Å) and fluorine (1.47 Å) and validated the repulsive interaction.
Photophysical measurement
Fig. 3 depicts the UV-Vis absorption and emission spectra of the studied Ir(III) complexes in toluene and degassed toluene, and the respective numerical data are summarized in Table 1. Generally speaking, all Ir(III) complexes exhibit multiple high energy bands beyond 320 nm, which can be assigned to ligand-centered ππ* and/or ligand-to-ligand charge transfer (LLCT) transitions.47 Notably, f-isomers show an additional, strong absorption band centered at ∼370 nm, and the pattern has been observed for many f-arranged Ir(III) carbene complexes documented in the literature, while m-Ir(dfp)3 exhibits a much weaker absorption band within this region; however, its peak onset was further extended to a longer wavelength of ∼440 nm. This was not observed for their f-counterpart, manifesting a relatively lower energy MLCT transition character.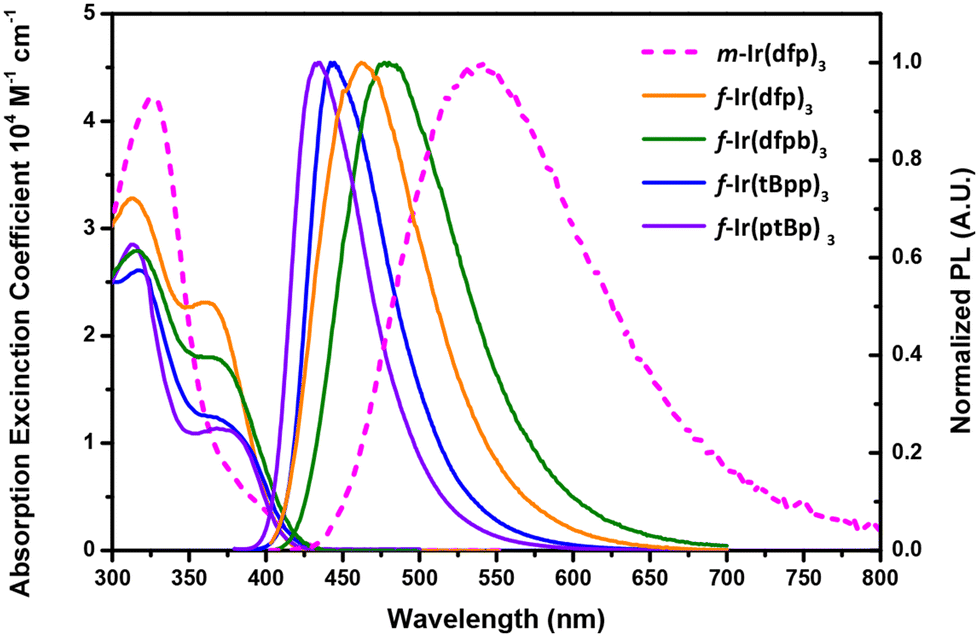 | ||
| Fig. 3 Absorption and emission spectra of the studied Ir(III) carbene complexes in degassed toluene at RT. | ||
| Abs λmax/nm; ε (104 M−1 cm−1)a | Em λmaxb (nm) | FWHMc (nm eV−1) | Φ , (%) | τ obs (μs) | τ rad (μs) | k r (105 s−1) | k nr (105 s−1) | |
|---|---|---|---|---|---|---|---|---|
| a Those were recorded in toluene at a concentration of 10−5 M at RT. b Those were recorded in degassed toluene at a concentration of 10−5 M at RT. c Full width at half maximum. d Coumarin 153 (C153) in ethanol (Q.Y. = 58% and λmax = 530 nm) and Coumarin 102 (C102) in methanol (Q.Y. = 80% and λmax = 480 nm) were employed as standards. | ||||||||
| m-Ir(dfp)3 | 327 (4.22), 381 (0.55) | 541 | 149/0.59 | 11.1 | 0.18 | 1.64 | 6.1 | 48.5 |
| f-Ir(dfp)3 | 314 (3.28), 362 (2.31) | 462 | 84/0.46 | 64.3 | 0.85 | 1.33 | 7.5 | 4.2 |
| f-Ir(dfpb)3 | 316 (2.79), 369 (1.78) | 472 | 83/0.47 | 62.0 | 0.9 | 1.45 | 6.8 | 4.2 |
| f-Ir(tBpp)3 | 318 (2.61), 372 (1.21) | 444 | 60/0.36 | 78.9 | 0.98 | 1.24 | 8.1 | 2.2 |
| f-Ir(ptBp)3 | 314 (2.85), 372 (1.13) | 435 | 55/0.35 | 60.7 | 0.85 | 1.40 | 7.1 | 4.6 |
For photoluminescence, f-Ir(dfb)3 and f-Ir(dfpb)3 exhibit true blue emission with a peak maximum located at 462 and 472 nm, and this difference in peak wavelength is obviously induced by the t-butyl substituent in the phenyl cyclometalates.27,28 The same trend was observed by comparison between f-Ir(tBpp)3 (444 nm) and f-Ir(ptBp)3 (435 nm) as the former possesses a t-butyl substituent at the cyclometalating sites. Interestingly, these two complexes with di-N-aryl substituted chelates display both a more blue-shifted emission peak wavelength and a narrowed emission band with a FWHM of <60 nm, showing the reduced influence from solvation. Moreover, the observed peak wavelength between f-Ir(dfp)3 and f-Ir(ptBp)3 is related to both the dislocated CF3 groups on the benzoimidazol-2-ylidene chelate and the replacement of N-methyl with an N-aryl appendage. In fact, our DFT data are also in agreement with this hypothesis (vide infra). Most importantly, complexes with facial configurations exhibit a high quantum yield of 60.3–78.9% and a short radiative lifetime between 1.24 μs and 1.45 μs under the same measurement conditions, confirming their potential in fabricating efficient OLED devices. In contrast, as the only mer-derivative of this family, m-Ir(dfp)3 exhibits broader and red-shifted emission and a lower photoluminescence quantum yield (PLQY) of 11.1%, which can be rationalized by much enhanced solvation.24 Furthermore, the emission of all studied Ir(III) complexes with a concentration of 2 wt% doped in PMMA was measured and is summarized in Fig. S1† and Table 2. The emission peak in PMMA of m-Ir(dfp)3 is blue shifted by 42 nm compared to that recorded in toluene, confirming the reduction of solvation. In contrast, all f-complexes showed red-shifted emission and no notable change in bandwidth at the same time, confirming their good photophysical properties in both the solution and thin film states.
| Em λmaxa (nm) | Φ , (%) | τ obs , (μs) | τ rad , (μs) | FWHMc (nm eV−1) | |
|---|---|---|---|---|---|
| a PL spectrum, quantum yield and lifetime were recorded in the doped PMMA thin film at RT (2 wt%) using an integration sphere. b Data in square brackets were measured in the co-doped PPT host (20 wt%). c Full width at half maximum. | |||||
| m-Ir(dfp)3 | 499 | 41 | 0.68 | 1.66 | 112/0.54 |
| f-Ir(dfp)3 | 466 | 59 | 1.14 | 1.94 | 86/0.48 |
| f-Ir(dfpb)3 | 474 [468] | 68 [78] | 1.20 [1.53] | 1.77 [1.96] | 88/0.47 |
| f-Ir(tBpp)3 | 452 [461] | 80 [85] | 1.08 [1.30] | 1.35 [1.53] | 62/0.37 |
| f-Ir(ptBp)3 | 438 [449] | 65 [78] | 0.94 [1.33] | 1.45 [1.86] | 52/0.33 |
Electrochemical and thermal characterization
Cyclic voltammetry was measured in acetonitrile solution at RT (Fig. S2†). As expected, oxidation mainly occurred at the Ir(III) metal center, with the oxidation onset potentials of f-isomers occurring in the region of 0.52–0.64 V (vs. Fc/Fc+), while that of m-Ir(dfp)3 exhibited a cathodic shift to 0.30 V, which was consistent with its red shifted emission spectrum. Complexes m-Ir(dfp)3, f-Ir(dfp)3 and f-Ir(dfpb)3 show irreversible oxidation processes with the anodic onset potentials at 0.30, 0.64, and 0.58 V, respectively, while complexes f-Ir(tBpp)3 and f-Ir(ptBp)3 showed quasi-reversible oxidation peaks with the anodic onset potentials at 0.52 and 0.64 V, respectively. It seems that the pendent N-aryl group had also improved the reversibility to the oxidized complexes. Alternatively, all these Ir(III) complexes presented irreversible reduction peaks spanning from −2.44 to −2.33 V. Thermogravimetric data were then recorded and are given in Fig. S3 and Table S1.† As can be seen, f-Ir(tBpp)3 appeared to exhibit the lowest decomposition temperature (Td) of 292 °C, while others displayed excellent thermal stability with a Td well above 345 °C. Interestingly, all these emitters can be purified by vacuum sublimation with the operational temperature being ∼60 °C lower than their Td, indicating that the CF3 group has greatly improved their volatility and enables the fabrication of OLED devices via more convenient vacuum deposition processes.Theoretical investigation
The lowest singlet (S1) and triplet (T1) excited states for the studied Ir(III) complexes were investigated by TD-DFT calculations48,49 to gain an understanding of the nature of the absorption/emission bands and the effect of chelate modification on their photophysical properties. The details of computational methods are described in the ESI.†The vertical excitation energies of the S0 → S1 transition were calculated to be 438, 389, 395, 401 and 394 nm for m-Ir(dfp)3, f-Ir(dfp)3, f-Ir(dfpb)3, f-Ir(tBpp)3 and f-Ir(ptBp)3, respectively (Table 3), which are correlated with their experimental lowest-energy absorption tails around 400 nm (Fig. 3). The calculated vertical excitation energies for the S0 → T1 transition, which is usually used to represent the T1 → S0 emission in the equilibrium structure of S0, were 451, 419, 424, 431 and 416 nm for m-Ir(dfp)3, f-Ir(dfp)3, f-Ir(dfpb)3, f-Ir(tBpp)3 and f-Ir(ptBp)3, respectively (Table 3). These values are comparable to the experimental phosphorescence (λPLmax = 541, 462, 472, 444 and 435 nm in toluene; λmax = 499, 466, 474, 452 and 438 nm in the doped PMMA thin film, respectively; see Fig. 3, Tables 1 and 2) with a MAD (mean absolute deviation) of about ≈0.25 eV (≈5.7 kcal mol−1) in toluene. Moreover, the calculated adiabatic emission energy (565, 456, 459, 441 and 424 nm, Table 4) is much closer to the experimental wavelengths in toluene with a smaller MAD (0.06 eV or 1.4 kcal mol−1).
| E HOMO (eV) | H–L gapa (eV) | Excitation | λ [nm eV−1] | f | MO contribution (>20%)b | Assignmentc | Sum contributiond | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MLCT | ILCT | LC | LMCT | MC | ||||||||
| a The EHOMO and H–L gap are computed at optimized S0 structures at the B3LYP-D3(BJ)/def2-SVP level with the polarizable continuum model (PCM) for toluene. b The results were calculated using TD-DFT using the B3LYP functional with PCM for toluene (cf., ESI†). c The percentages of all S0 → T1 excitation character were calculated using the IFCT(Hirshfeld) method. d The sum contribution is MLCT + ILCT + LC − LMCT. | ||||||||||||
| m-Ir(dfp)3 | −5.45 | 3.44 | S0 → T1 | 451/2.75 | 0 | HOMO → LUMO (72.3%) | 35.8% | 37.9% | 18.4% | 4.9% | 3.1% | 87.2% |
| S0 → S1 | 438/2.83 | 0.0010 | HOMO → LUMO (84.7%) | |||||||||
| f-Ir(dfp)3 | −5.67 | 3.80 | S0 → T1 | 419/2.96 | 0 | HOMO → LUMO (77.6%) | 30.7% | 38.0% | 26.0% | 3.6% | 1.7% | 90.9% |
| S0 → S1 | 389/3.18 | 0.0182 | HOMO → LUMO (86.5%) | |||||||||
| f-Ir(dfpb)3 | −5.57 | 3.70 | S0 → T1 | 424/2.92 | 0 | HOMO → LUMO (77.7%) | 30.0% | 38.8% | 25.7% | 3.8% | 1.7% | 90.7% |
| S0 → S1 | 395/3.14 | 0.0927 | HOMO → LUMO+1 (91.8%) | |||||||||
| f-Ir(tBpp)3 | −5.50 | 3.67 | S0 → T1 | 431/2.98 | 0 | HOMO → LUMO (79.0%) | 28.1% | 45.1% | 22.6% | 3.0% | 1.2% | 92.8% |
| S0 → S1 | 401/3.10 | 0.0053 | HOMO → LUMO (94.0%) | |||||||||
| f-Ir(ptBp)3 | −5.58 | 3.77 | S0 → T1 | 416/2.98 | 0 | HOMO → LUMO (81.0%) | 32.6% | 36.5% | 25.3% | 3.7% | 1.9% | 90.7% |
| S0 → S1 | 394/3.14 | 0.0552 | HOMO → LUMO+1 (95.9%) | |||||||||
| Emission (T1 → S0) | λ [nm eV−1] | λ [nm eV−1] | τ rad (μs) | k r (μs−1) |
|---|---|---|---|---|
| a The adiabatic emission energy was obtained from the difference between the optimized structures of S0 and T1 states using the B3LYP functional with a polarizable continuum model (PCM) for toluene (cf. ESI†). b The vertical emission energy between S0 and T1 states was obtained using the SOC-TDDFT method in ORCA at optimized structures of S0 (in normal font) and T1 (in italic and bold fonts in parentheses). c The τrad and kr are calculated by the arithmetic average/Boltzmann average (at 298 K) of the SOC substates of T1, at the optimized structures of S0 (in normal font) and T1 (in italic and bold fonts in parentheses). | ||||
| m-Ir(dfp)3 | 565/2.20 | 447/2.77 (822/1.51) | 0.23/0.38 (1.55/1.94) | 4.30/2.64 (0.65/0.52) |
| f-Ir(dfp)3 | 456/2.72 | 407/3.05 (830/1.50) | 1.02/1.35 (6.18/6.87) | 0.98/0.74 (0.16/0.15) |
| f-Ir(dfpb)3 | 459/2.70 | 417/2.97 (772/1.61) | 0.84/1.11 (5.89/5.99) | 1.20/0.90 (0.17/0.16) |
| f-Ir(tBpp)3 | 441/2.81 | 428/2.90 (520/2.38) | 0.44/0.45 (1.37/1.52) | 2.27/2.24 (0.73/0.66) |
| f-Ir(ptBp)3 | 424/2.92 | 415/2.99 (489/2.54) | 1.11/1.23 (1.09/1.16) | 0.90/0.82 (0.91/0.86) |
To further understand the emission properties of the studied Ir(III) complexes, the natural transition orbital (NTO) analysis50 to express the S0 → T1 transition as a single pair of orbitals was applied. The predominant NTO pairs found for the S0 → T1 transition are present in Fig. 4. For all the four facial Ir(III) complexes, namely f-Ir(dfp)3, f-Ir(dfpb)3, f-Ir(tBpp)3 and f-Ir(ptBp)3, the occupied NTOs are delocalized at both the Ir(III) metal center and cyclometalating carbene chelates (in π orbitals); while the virtual NTOs are mainly localized at the carbene donor fragments (in π* orbitals). Only for the meridional complex (m-Ir(dfp)3), the occupied NTO is delocalized at the Ir(III) metal center and carbene chelates whereas the virtual NTO is localized at the N-aryl cyclometalates. It indicates that for m-Ir(dfp)3, the overlaps between the occupied and virtual NTOs are greatly reduced and lead to a diminished ligand-centered (LC) character in the S0 → T1 transition.
 | ||
| Fig. 4 Natural transition orbital (NTO) pairs expressing the S0 → T1 excitation of the studied Ir(III) complexes at their geometries optimized for the ground state, with the contribution of dominant molecular orbitals (MOs) to NTOs provided. | ||
Although the NTO analysis makes use of the dominant molecular orbital pairs, the contribution of the NTO pairs is unable to reveal the full properties of S0 → T1 excitation. In fact, no obvious difference is found between the NTO analysis and TD-DFT results, for example, the calculated eigenvalues of the NTOs for the S0 → T1 transition of f-Ir(dfp)3 is 0.824 (Fig. 4), representing that 82.4% of S0 → T1 excitation is contributed by the “HOMO → LUMO” transition, and the results are similar to the TD-DFT value of 77.6% (Table 3).
The inter-fragment charge transfer (IFCT) method, available from the Multiwfn software package,51 was employed to quantify the contributions of relevant molecular orbitals (MOs) to the selected electronic transition. This manipulation is to provide a better picture of the S0 → T1 transition, and the percentage of MLCT, intra-ligand charge transfer (ILCT), ligand-centered (LC), ligand-to-metal charge transfer (LMCT) and metal-centered (MC) dd contributions are depicted in Table 4. Based on the IFCT analysis, the effect of ligand modification on kr is summarized as follows: (i) m-Ir(dfp)3 has the highest percentage of MCLT (35.8%), LMCT (4.9%) and MC (3.1%) among all studied Ir(III) complexes. It also features the lowest percentage of LC (18.4%), which is consistent with the lowest orbital overlaps between the occupied and virtual NTOs of m-Ir(dfp)3 (Fig. 4). (ii) When the meridional m-Ir(dfp)3 is changed to its facial isomer f-Ir(dfp)3 (Scheme 2), the MLCT character (30.7%) is reduced by 5.1%, the LMCT and MC characters are also slightly reduced whereas the LC character is greatly increased from 26.0% to 33.6%. The increased LC character also manifests on the orbital overlaps of NTO analysis on f-Ir(dfp)3 (Fig. 4). (iii) Replacing the N-phenyl cyclometalate of carbene chelate with the 4-(t-butyl)phenyl group of f-Ir(dfpb)3 does not impose any significant effect on the MLCT, ILCT, LC and LMCT character. This suggests that both f-Ir(dfpb)3 and f-Ir(dfp)3 should have similar photophysical properties. (iv) Upon changing the dual CF3 substituents (from the 1,3-position to the 2,4-position) and changing the N-methyl appendages to phenyl groups (Scheme 2, from f-Ir(dfpb)3 to f-Ir(tBpp)3), the ILCT character is greatly increased to 45.1%, being the highest ILCT percentage observed among all complexes. The modification of chelates in this step may reduce the steric hindrance and result in more effective ILCT character of f-Ir(tBpp)3. The LMCT character (3.0%) of f-Ir(tBpp)3 is also the smallest one among all complexes. (v) Alternately, simultaneous replacement of both the 4-(t-butyl)phenyl cyclometalate and the N-phenyl appendage on f-Ir(tBpp)3 with the phenyl cyclometalate and 4-(t-butyl)phenyl appendage gives f-Ir(ptBp)3 (Scheme 2). The ILCT character of f-Ir(ptBp)3 becomes lower (36.5%), but its MLCT and LC characters are increased slightly. The percentages of MLCT (32.6%), ILCT (36.5%) and LC (25.3%) characters of f-Ir(ptBp)3 are akin to those of f-Ir(dfp)3 and f-Ir(dfpb)3.
The IFCT method suggests that the S0 → T1 excitation exhibits a dominant mixture of MLCT, ILCT and LC characters for all the four facial Ir(III) complexes. For the solely meridional complex (m-Ir(dfp)3), the S0 → T1 excitation mainly consists of mixed MLCT and ILCT character, with a relatively smaller LC character. The respective sum contribution (Table 3) of “MLCT + ILCT + LC–LMCT” of 87.2%, 90.9%, 90.7%, 92.8% and 90.7% for m-Ir(dfp)3, f-Ir(dfp)3, f-Ir(dfpb)3, f-Ir(tBpp)3 and f-Ir(ptBp)3 correlates excellently with the respective measured kr values (in μs−1, Table 1) of 0.61, 0.75, 0.68, 0.81 and 0.71 from experimental measurements. Based on the IFCT method, it appears that the meridional complex (m-Ir(dfp)3, 87.2%) has the smallest radiative decay rate while the facial complex of f-Ir(tBpp)3 has the fastest radiative decay rate (92.8%).
Following the IFCT prediction, we perform calculations to investigate the theoretical τrad and kr. The emission energy calculated for the optimized structure (T1) might have a physical meaning, since phosphorescence is a long-lived process such that it may give the emitting species enough time to relax from the triplet manifold to a lower energy geometry. However, it has been pointed out that although a lot of calculations using the optimized structure (T1) can give good prediction to the experimental τrad, many other works of τrad using the ground state geometry (S0) also present a better correlation with the experimental data.52–54 As the “actual” emitting species sit between the ground state (S0) and excited state (T1) levels,55,56 we here apply the spin–orbit coupling (SOC)-TDDFT method on both the optimized S0 and T1 structures for the estimation of τrad and kr.
The calculated emission energies, τrad and kr at both optimized S0 and T1 geometries, are summarized in Table 4, and the results obtained using the arithmetic average or Boltzmann average of the SOC substates are similar. Here, we focus on the arithmetic average results. (i) For the T1 → S0 emission of m-Ir(dfp)3, the calculated τrad is 0.23 μs (at the S0 structure) and 1.55 μs (at the T1 structure). In comparison with the experimental τrad (1.64 μs, Table 1), the true emitting intermediate of m-Ir(dfp)3 is suggested to be closer to the T1 structure. (ii) Unlike the emission of m-Ir(dfp)3, the experimental τrad (1.33 μs) of its facial isomer f-Ir(dfp)3 is closer to the calculated τrad of 1.02 μs (in the S0 geometry), suggesting that the true emitting intermediate of f-Ir(dfp)3 is near to the S0 structure. (iii) A similar conclusion can also be drawn for f-Ir(dfpb)3, in which the true emitting intermediate is near to the S0 structure. The calculated τrad of 0.84 μs (in the S0 structure) is much closer to the experimental τrad (1.45 μs) than the calculated τrad (5.89 μs) based on the T1 structure. (iv) For f-Ir(tBpp)3, the experimental τrad of 1.24 μs deviates from the calculated τrad of 0.44 μs (in the S0 structure) and 1.37 μs (in the T1 structure) by +0.80 and −0.13 μs, respectively. We suggested that the actual emitting intermediate of f-Ir(tBpp)3 resembles both S0 and T1 structures. (v) The calculated τrad values of 1.11 μs (in the S0 geometry) and 1.09 μs (in the T1 geometry) of f-Ir(ptBp)3 are almost the same, hinting that its emitting intermediate resembles both the S0 and T1 structures. It is worth noting here that the uncertainty (∼1.7 μs) of τrad for other Ir(III) complexes predicted with the SOC-TDDFT method is relatively large.57 From the above analysis, we conclude that the phosphorescence process of all the four facial Ir(III) complexes likely occurred near the S0 structure while that of the meridional complex (m-Ir(dfp)3) is initiated in the T1 structure.
Device performance
To examine the electroluminescence (EL) characteristics, we chose f-Ir(dfpb)3, f-Ir(tBpp)3 and f-Ir(ptBp)3 as sensitizers for constructing both phosphorescent and hyper-OLED devices. Phosphorescent OLEDs are known to offer the full utilization of all excitons generated during operation, while hyper-OLEDs exhibit another capacity to further improve efficiency by transferring energy from sensitizers to fluorescent emitters.58–62 The adjustable emission energy, enhanced spin–orbit coupling, and a greater MLCT fraction designate these Ir(III) carbene emitters as promising candidates for realizing effective FRET.37,63–68 The respective phosphorescent and hyper-OLEDs were constructed using the device structure: indium tin oxide (ITO)/4 wt% rhenium oxide (ReO3): N,N-dicarbazolyl-3,5-benzene (mCP) (60 nm)/mCP (15 nm)/EML (20 nm)/tris-[3-(3-pyridyl)mesityl]borane (3TPYMB) (50 nm)/lithium 8-hydroxyquinolinolate (Liq) (0.5 nm)/Al (100 nm). This configuration, previously detailed in our earlier study, is illustrated in Fig. 5a.69 The hole-transport layer (HTL) and electron-transport layer (ETL) consisted of mCP and 3TPYMB, respectively. Thin layers of 4 wt% ReO3 doped mCP and Liq were also applied to enhance the carrier and charge injection from the respective electrodes.70 The emissive layer (EML) contained 20 wt% Ir(III) metal phosphors (f-Ir(dfpb)3, f-Ir(tBpp)3 and f-Ir(ptBp)3) and additional 1 wt% terminal emitters (ν-DABNA and t-DABNA) for hyper-OLEDs incorporated into the host material, 2,8-bis(diphenylphosphoryl)dibenzo[b,d]thiophene (PPT).71,72 Their EL characteristics are presented in Fig. 5 (and Fig. S4 and S5 of the ESI†), together with the pertinent parameters that are consolidated in Table 5.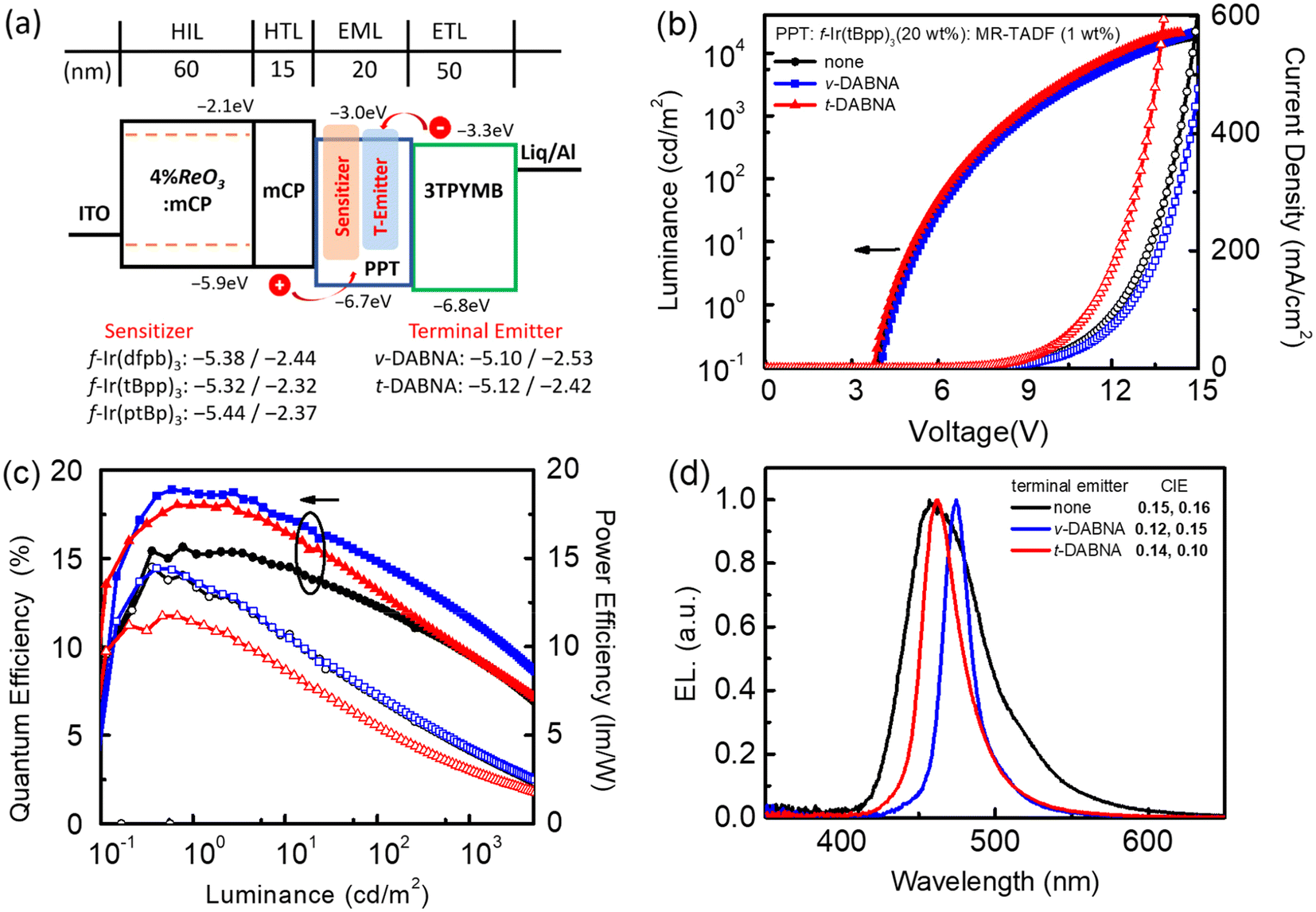 | ||
| Fig. 5 (a) The energy diagram of all OLED and hyper-OLED devices fabricated in this study, (b) current density–voltage–luminance (J–V–L) characteristics of devices fabricated using f-Ir(tBpp)3 as a dopant, and devices with Ir(tBpp)3 as a sensitizer, together with ν-DABNA and t-DABNA as terminal emitters, and (c) respective EQE and PE diagrams as a function of luminance, and (d) EL spectra. | ||
| Sensitizer | 1 wt% T-emitter | V on [V] | EQE/CE/PE at max. [%/cd A−1/lm W−1] | EQE/CE/PE at 103 cd m−2 [%/cd A−1/lm W−1/V] | λ max [nm] | FWHM [nm] | CIE [x,y] |
|---|---|---|---|---|---|---|---|
| f-Ir(dfpb)3 | None | 4.5 | 10.6/20.4/12.0 | 5.7/10.9/3.4/10.0 | 481 | 81 | 0.17, 0.29 |
| v-DABNA | 4.7 | 14.3/17.7/10.3 | 7.2/8.9/2.7/10.5 | 475 | 21 | 0.12, 0.19 | |
| t-DABNA | 4.7 | 14.3/14.2/9.2 | 4.5/4.5/1.4/10.1 | 466 | 30 | 0.13, 0.12 | |
| f-Ir(tBpp)3 | None | 3.9 | 15.6/19.2/14.5 | 9.6/11.7/4.2/8.8 | 457 | 58 | 0.15, 0.16 |
| v-DABNA | 3.9 | 18.9/20.1/14.4 | 11.6/12.4/4.4/9.0 | 474 | 19 | 0.12, 0.15 | |
| t-DABNA | 3.8 | 18.1/15.7/11.7 | 9.7/8.4/3.1/8.6 | 462 | 29 | 0.14, 0.10 | |
| f-Ir(ptBp)3 | None | 3.9 | 12.9/11.5/8.6 | 6.5/5.8/2.1/8.7 | 453 | 57 | 0.15, 0.10 |
| v-DABNA | 3.9 | 16.3/15.1/11.3 | 9.4/8.7/3.3/8.4 | 473 | 20 | 0.12, 0.13 | |
| t-DABNA | 3.9 | 14.1/11.9/8.1 | 5.19/4.39/1.31/9.5 | 461 | 33 | 0.14, 0.10 |
To confirm our device design, the EL characteristics were explored without the addition of an MR-TADF terminal emitter. These devices displayed a progressive hypsochromic shift from sky blue to blue with peak maxima at 481, 457, and 453 nm for f-Ir(dfpb)3, f-Ir(tBpp)3 and f-Ir(ptBp)3, respectively. They were also accompanied by a decrease of full width at half maximum (FWHM) from 81 nm, 58 nm and to 51 nm, aligning well with their photoluminescence (PL) spectra (Table 1). In comparison to the devices with f-Ir(dfpb)3 and f-Ir(tBpp)3, the third device with f-Ir(ptBp)3 demonstrated slightly superior chromaticity coordinates CIEx,y of 0.15 and 0.10 for deep-blue emission. On the other hand, the device based on f-Ir(tBpp)3 demonstrated the highest efficiency, displaying a maximum external quantum efficiency (EQEmax) of 15.6%, a maximum current efficiency (CEmax) of 19.2 cd A−1, and a maximum power efficiency (PEmax) of 14.5 lm W−1. Conversely, relatively inferior efficiencies, i.e., EQEmax, CEmax, and PEmax values of 10.6%, 20.4 cd A−1, and 12.0 lm W−1 and 12.9%, 11.5 cd A−1, and 8.6 lm W−1 were observed for f-Ir(dfpb)3 and f-Ir(ptBp)3, respectively. The efficiencies seemed to follow the photoluminescence quantum yield (ΦPL) recorded in toluene solution (Table 1) and both the PMMA polymer matrix and PPT host material (Table 2), while f-Ir(tBpp)3 exhibits a faster radiative lifetime among the three studied Ir(III) complexes.
With the objective of achieving deep-blue hyper-OLEDs, a low concentration of MR-TADF terminal emitters (ν-DABNA and t-DABNA) was next incorporated into the EML. Fig. 5a illustrates that all Ir(III) dopants possess a fitted energy level arrangement with respect to that of ν-DABNA and t-DABNA. Consequently, the sensitizers could suppress electron trapping in the EML. The concentration is optimized to 1 wt% to prevent undesired concentration quenching. As depicted in Table 5, as well as other literature precedents,71–75 the incorporation of both ν-DABNA and t-DABNA has given substantial enhancements in efficiencies and reduction in FWHM. Moreover, the PLQYs of the blended thin film of ν-DABNA and t-DABNA with the same blending ratios of Ir(III) phosphors and the PPT host are provided in Table S2 of the ESI.† The high EQEmax values of devices are in accordance with the results of the high PLQYs of blend-films. Notably, the EL performance of the f-Ir(tBpp)3 sensitized device is generally better than those of f-Ir(dfpb)3 and f-Ir(ptBp)3 based devices. The deep blue hyper-OLED devices with f-Ir(tBpp)3 have achieved remarkable EQEmax values of 18.9% and 18.1% for terminal emitters ν-DABNA and t-DABNA, representing a substantial increase of EQEmax with reference to that of the parent device (15.6%). Hence, these hyper-OLEDs also exhibited lower turn-on voltages and higher maximum EQE and EQE at a brightness of 1000 cd m−2 in comparison to the pure MR-TADF devices recorded using ν-DABNA under identical device architecture.69 Furthermore, the EL spectrum also exhibited a large reduction in FWHM from 58 nm for the parent device to 19 nm and 29 nm for terminal emitters ν-DABNA and t-DABNA, respectively.
Conclusion
In this work, we demonstrated the conceptual strategy of designing asymmetric carbene chelates for obtaining single Ir(III) carbene emitters through intra-chelate steric interactions. First, upon replacement of the phenyl substituent in carbene pro-chelate C1 with a 4-t-butylphenyl substituent as shown in C2, the distribution of products changed from a mixed product (i.e., both m-Ir(dfp)3 and f-Ir(dfp)3) to a single isomer, i.e., f-Ir(dfpb). This selectivity is probably attributed to the electron donating t-butyl group, which can increase the electron density at the Ir(III) metal center and, in turn, facilitate mer-to-fac isomerization under the applied synthetic conditions. On the other hand, this isomerization is not governed by the steric effect among three t-butyl groups, because this fac-orientation should exert a greater steric interaction than the mer-counterpart. Moreover, upon switching the carbene pro-chelates from C1 and C2 to C3 and C4, both with dual N-aryl substituents, it is expected that both N-aryl groups can undergo competitive C–H activation and cyclometalation with the Ir(III) metal center affording four isomeric products as shown in our previous studies.35–37 However, the ortho-substituted CF3 group of C3 and C4 provides a second steric encumbrance, leading to the selective formation of only one product, namely f-Ir(tBpp)3 and f-Ir(ptBp)3, independent of the employed t-butylphenyl and phenyl appendages. Apparently, the imposed steric effect has forced this N-aryl group to rotate, which then reduced its chance to react with the Ir(III) metal center leading to direct cyclometalation. This selectivity is important for the possible mass production of relevant Ir(III) complexes if they are required in the future.Moreover, all isolated fac-isomers exhibited high PLQYs and narrowed FWHMs. Among them, f-Ir(tBpp)3 with aryl appendages showed narrow photoluminescence with FWHM = 60 nm and a high PLQY approaching 80%. The fabricated OLED device using f-Ir(tBpp)3 as an emitter delivered a maximum EQE of 15.6% with CIE(x,y) coordinates of (0.15, 0.16). Furthermore, these Ir(III) complexes can also serve as sensitizers to convey their energy to the MR-TADF terminal emitters ν-DABNA and t-DABNA to achieve a maximum EQE of 18.9% and 18.1% with CIE(x,y) coordinates of (0.12, 0.15) and (0.14, 0.10), respectively. The findings elaborated a unique strategy for the advancement of OLED technologies in displays and lighting luminaries. Further research in this direction holds great promise in unlocking energy-efficient and high-performance OLEDs.
Experimental section
General information and materials
Commercially available reagents were used without further purification. 1H and 19F NMR spectra were obtained with an NMR 400 MHz instrument (Bruker AVANCE III, BBO probe). Mass spectra were recorded on an Applied Biosystems 4800 Plus MALDI TOF/TOF analyzer using 2,5-dihydroxybenzoic acid as the matrix substance. TGA measurements were performed on a TA Instruments TGAQ50, at a heating rate of 10 °C min−1 under a nitrogen atmosphere.Photophysical measurements
UV-Vis spectra were recorded with a UV-Visible NIR spectrophotometer system (HITACHI UH4150). The steady-state emission spectra were recorded with a spectrofluorometer (Fluormax-4) and the lifetime decay spectra were recorded with a photon-counting system (Edinburgh FLS980). All solution samples were degassed using at least three freeze–pump–thaw cycles. Photoluminescence quantum yields in solution were calculated using the standard sample which has a known quantum yield, while quantum yields in PMMA thin film were measured using an integrated sphere. Lifetimes were obtained using an Edinburgh FLS980 time-correlated single photon counting (TCSPC) system with an EPL-375 diode laser as the excitation source.Electrochemistry
Cyclic voltammetry was measured with an electrochemical analyzer (CHI660) equipped with a three-electrode system (glassy carbon: working electrode, platinum wire: auxiliary electrode, and Ag/AgCl: reference electrode). Nitrogen-purged acetonitrile was used as a solvent and NBu4PF6 (0.1 M) was used as a supporting electrolyte. The potentials were referenced externally to the ferrocenium/ferrocene (Fc+/Fc) couple.Selected spectroscopic data for m-Ir(dfp)3. HRMS (ESI, 193Ir): m/z 1223.1620 [M + H+], calcd for C48H28F18IrN6: 1223.1717; 1H NMR (400 MHz, acetone-d6) δ 8.20 (s, 2H), 8.13 (s, 1H), 8.03 (s, 1H), 7.97 (s, 1H), 7.94 (s, 1H), 7.64 (d, J = 8.1 Hz, 1H), 7.61–7.53 (m, 2H), 6.95–6.79 (m, 5H), 6.62 (m, 4H), 3.72 (s, 3H), 3.70 (s, 3H), 3.57 (s, 3H); 19F NMR (376 MHz, acetone-d6) δ −53.00 (s, 3F), −53.07 (s, 3F), −53.21 (s, 3F), −61.71 (s, 3F), −61.73 (s, 3F), −61.74 (s, 3F). Anal. calcd for C48H27F18IrN6: C, 47.18; H, 2.23; N, 6.88. Found: C, 47.20; H, 2.58; N, 6.79.
Selected crystal data for m-Ir(dfp)3. CCDC deposition number: 2110266.† C48H27F18IrN6; M = 1221.95; monoclinic; space group = P21/n; a = 9.3558(2) Å, b = 34.9470(8) Å, c = 14.2098(3) Å; β = 91.9000(10)°; V = 4643.44(18) Å3; Z = 4; ρcalcd = 1.748 g cm−3; F(000) = 2384.0, crystal size = 0.36 × 0.14 × 0.03 mm3; λ(Mo-Kα) = 0.71073 Å; T = 223 (2) K; μ = 2.990 mm−1; 43
608 reflections collected, 9485 independent reflections (Rint = 0.0363), max. and min. transmission = 0.605 and 0.745, data/restraints/parameters = 9485/186/697, GOF = 1.021, final R1[I > 2σ(I)] = 0.0241 and wR2(all data) = 0.0501.
Selected spectroscopic data for f-Ir(dfp)3. HRMS (ESI, 193Ir): m/z 1223.1585 [M + H+], calcd for C48H28F18IrN6: 1223.1717; 1H NMR (400 MHz, acetone-d6) δ 8.15 (s, 3H), 8.00 (s, 3H), 7.64 (d, J = 8.0 Hz, 3H), 6.97–6.90 (m, 3H), 6.62–6.61 (m, 6H), 3.62 (s, 9H); 19F NMR (376 MHz, acetone-d6) δ −53.08 (s, 9F), −61.74 (s, 9F). Anal. calcd for C48H27F18IrN6: C, 47.18; H, 2.23; N, 6.88. Found: C, 47.25; H, 2.26; N, 6.91.
Selected spectroscopic data for f-Ir(dfpb)3. HRMS (ESI, 193Ir): m/z 1391.3503 [M + H+], calcd for C60H52F18IrN6: 1391.3595; 1H NMR (400 MHz, acetone-d6) δ 8.13 (s, 3H), 7.98 (s, 3H), 7.57 (d, J = 8.5 Hz, 3H), 6.99 (dd, J = 8.5, 2.3 Hz, 3H), 6.48 (d, J = 2.3 Hz, 3H), 3.57 (s, 9H), 1.01 (s, 27H); 19F NMR (376 MHz, acetone-d6) δ −52.74 (s, 9F), −61.71 (s, 9F). Anal. calcd for C60H51F18IrN6: C, 51.83; H, 3.70; N, 6.04. Found: C, 51.78; H, 3.69; N, 5.93.
Selected spectroscopic data for f-Ir(tBpp)3. HRMS (ESI, 193Ir): m/z 1577.3892 [M + H+], calcd for C75H58F18IrN6: 1577.4065; 1H NMR (400 MHz, CDCl3) δ 8.46 (s, 3H), 7.79 (d, J = 8.0 Hz, 3H), 7.71 (s, 3H), 7.62 (d, J = 8.4 Hz, 3H), 7.19 (td, J = 7.6, 1.2 Hz, 3H), 7.10 (dd, J = 8.4, 2.4 Hz, 3H), 6.59 (td, J = 8.0, 1.6 Hz, 3H), 6.49 (tt, J = 7.6, 1.2 Hz, 3H), 6.42 (d, J = 2.4 Hz, 3H), 6.08 (dt, J = 8.0, 1.6 Hz, 3H), 0.98 (s, 27H); 19F NMR (376 MHz, CDCl3) δ −56.25 (s, 9F), −61.03 (s, 9F). Anal. calcd for C75H57F18IrN6: C, 57.14; H, 3.64; N, 5.33. Found: C, 57.07; H, 3.60; N, 5.37.
Selected crystal data for f-Ir(tBpp)3. CCDC deposition number: 2242122.† C75H57F18IrN6; M = 1576.46; trigonal; space group = P
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) ; a = 16.3890(5) Å, b = 16.3890(5) Å, c = 17.8634(5) Å; γ = 120°; V = 4155.3(3) Å3; Z = 2; ρcalcd = 1.260 g cm−3; F(000) = 1576.0, crystal size = 0.29 × 0.14 × 0.04 mm3; λ(Cu–Kα) = 1.54178 Å; T = 213 (2) K; μ = 3.811 mm−1; 74392 reflections collected, 5712 independent reflections (Rint = 0.0498), max. and min. transmission = 0.754 and 0.520, data/restraints/parameters = 5712/901/424, GOF = 1.068, final R1[I > 2σ(I)] = 0.0239 and wR2(all data) = 0.0651.
; a = 16.3890(5) Å, b = 16.3890(5) Å, c = 17.8634(5) Å; γ = 120°; V = 4155.3(3) Å3; Z = 2; ρcalcd = 1.260 g cm−3; F(000) = 1576.0, crystal size = 0.29 × 0.14 × 0.04 mm3; λ(Cu–Kα) = 1.54178 Å; T = 213 (2) K; μ = 3.811 mm−1; 74392 reflections collected, 5712 independent reflections (Rint = 0.0498), max. and min. transmission = 0.754 and 0.520, data/restraints/parameters = 5712/901/424, GOF = 1.068, final R1[I > 2σ(I)] = 0.0239 and wR2(all data) = 0.0651.
Selected spectroscopic data for f-Ir(ptBp)3. HRMS (ESI, 193Ir): m/z 1577.3932 [M + H+], calcd for C75H58F18IrN6: 1577.4065; 1H NMR (400 MHz, CDCl3) δ 8.57 (s, 3H), 7.80 (dd, J = 8.0, 1.2 Hz, 3H), 7.66 (s, 3H), 7.53 (dd, J = 8.4, 2.4 Hz, 3H), 7.36 (dd, J = 8.4, 2.4 Hz, 3H), 7.17 (td, J = 8.0, 1.6 Hz, 3H), 6.71 (td, J = 8.0, 1.2 Hz, 3H), 6.68 (dd, J = 8.0, 2.0 Hz, 3H), 6.54 (dd, J = 8.0, 2.0 Hz, 3H), 5.84 (dd, J = 8.0, 1.6 Hz, 3H), 0.84 (s, 27H).; 19F NMR (376 MHz, CDCl3) δ −56.09 (s, 9F), −61.18 (s, 9F). Anal. calcd for C75H57F18IrN6: C, 57.14; H, 3.64; N, 5.33. Found: C, 57.16; H, 3.66; N, 5.29.
Author contributions
J. Y., Y. P. and I. C. P.: investigation, experiments, data curation, writing – original draft preparation, and writing – review and editing. B. H., G. N. and S. M. Y.: experiments. W. Y. H., Y. C. and K. C. L.: funding acquisition, writing – review and editing, supervision, resources, funding acquisition, project administration, writing – original draft preparation, and writing – review and editing.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by funding from the Research Grant Council (CityU 11304221 and CityU 11312722) and the City University of Hong Kong, Hong Kong SAR. The computational studies were carried out using the High-Performance Computing facility, CityU Burgundy at the City University of Hong Kong.References
- M. L. Tang and Z. Bao, Halogenated Materials as Organic Semiconductors, Chem. Mater., 2011, 23, 446–455 CrossRef CAS.
- M. Stolar and T. Baumgartner, Organic n-type materials for charge transport and charge storage applications, Phys. Chem. Chem. Phys., 2013, 15, 9007–9024 RSC.
- X. Zhou, P. L. Burn and B. J. Powell, Bond Fission and Non-Radiative Decay in Iridium(III) Complexes, Inorg. Chem., 2016, 55, 5266–5273 CrossRef CAS PubMed.
- R. Ragni, A. Punzi, F. Babudri and G. M. Farinola, Organic and Organometallic Fluorinated Materials for Electronics and Optoelectronics: A Survey on Recent Research, Eur. J. Org. Chem., 2018, 3500–3519 CrossRef CAS.
- S. Dolui, D. Kumar, S. Banerjee and B. Ameduri, Well-Defined Fluorinated Copolymers: Current Status and Future Perspectives, Acc. Mater. Res., 2021, 2, 242–251 CrossRef CAS.
- F. Babudri, G. M. Farinola, F. Naso and R. Ragni, Fluorinated organic materials for electronic and optoelectronic applications: the role of the fluorine atom, Chem. Commun., 2007, 1003–1022, 10.1039/B611336B.
- A. Y. Sosorev, M. K. Nuraliev, E. V. Feldman, D. R. Maslennikov, O. V. Borshchev, M. S. Skorotetcky, N. M. Surin, M. S. Kazantsev, S. A. Ponomarenko and D. Y. Paraschuk, Impact of terminal substituents on the electronic, vibrational and optical properties of thiophene–phenylene co-oligomers, Phys. Chem. Chem. Phys., 2019, 21, 11578–11588 RSC.
- H.-B. Han, R.-Z. Cui, Y.-M. Jing, G.-Z. Lu, Y.-X. Zheng, L. Zhou, J.-L. Zuo and H. Zhang, Highly efficient orange-red electroluminescence of iridium complexes with good electron mobility, J. Mater. Chem. C, 2017, 5, 8150–8159 RSC.
- G.-Z. Lu, L. Liu, Z.-L. Tu, Y.-H. Zhou and Y. Zheng, Two green iridium(III) complexes containing the electron-transporting group of 4-phenyl-4H-1,2,4-triazole for highly efficient OLEDs, J. Mater. Chem. C, 2019, 7, 2022–2028 RSC.
- L. Zhang, Z.-P. Yan, Z.-L. Tu, Z.-G. Wu and Y.-X. Zheng, Green-emitting iridium(III) complexes containing pyridine sulfonic acid as ancillary ligands for efficient OLEDs with extremely low efficiency roll-off, J. Mater. Chem. C, 2019, 7, 11606–11611 RSC.
- Z. Chen, L. Wang, S. Su, X. Zheng, N. Zhu, C.-L. Ho, S. Chen and W.-Y. Wong, Cyclometalated Iridium(III) Carbene Phosphors for Highly Efficient Blue Organic Light-Emitting Diodes, ACS Appl. Mater. Interfaces, 2017, 9, 40497–40502 CrossRef CAS PubMed.
- C. You, X.-Q. Wang, X. Zhou, Y. Yuan, L.-S. Liao, Y.-C. Liao, P.-T. Chou and Y. Chi, Homoleptic Ir(III) Phosphors with 2-Phenyl-1,2,4-triazol-3-ylidene Chelates for Efficient Blue Organic Light-Emitting Diodes, ACS Appl. Mater. Interfaces, 2021, 13, 59023–59034 CrossRef CAS PubMed.
- R. Kumaresan, H.-Y. Park, A. Maheshwaran, H. Park, Y. Do, M. Song, J. Yoon, S. I. Ahn and S.-H. Jin, High Performance Solution-Processed Deep-Blue Phosphorescence Organic Light-Emitting Diodes with EQE Over 24% by Employing New Carbenic Ir(III) Complexes, Adv. Opt. Mater., 2022, 10, 2101686 CrossRef CAS.
- X.-K. Zheng, F.-Q. Zhao, M.-N. Yin, C. Qian, S.-H. Bi, P. Tao, Y.-Q. Miao, S.-J. Liu and Q. Zhao, New trifluoromethyl modified iridium(III) complex for high-efficiency sky-blue phosphorescent organic light-emitting diode, Tetrahedron Lett., 2021, 75, 153181 CrossRef CAS.
- J. Yan, Z.-H. Qu, D.-Y. Zhou, S.-M. Yiu, Y. Qin, X. Zhou, L.-S. Liao and Y. Chi, Bis-tridentate Ir(III) Phosphors and Blue Hyperphosphorescence with Suppressed Efficiency Roll-Off at High Brightness, ACS Appl. Mater. Interfaces, 2024, 16, 3809–3818 CrossRef CAS PubMed.
- A. K. Pal, S. Krotkus, M. Fontani, C. F. R. Mackenzie, D. B. Cordes, A. M. Z. Slawin, I. D. W. Samuel and E. Zysman-Colman, High-Efficiency Deep-Blue-Emitting Organic Light-Emitting Diodes Based on Iridium(III) Carbene Complexes, Adv. Mater., 2018, 30, 1804231 CrossRef PubMed.
- C. F. R. Mackenzie, L. Zhang, D. B. Cordes, A. M. Z. Slawin, I. D. W. Samuel and E. Zysman-Colman, Bulky Iridium NHC Complexes for Bright, Efficient Deep-Blue OLEDs, Adv. Opt. Mater., 2023, 11, 2201495 CrossRef CAS.
- G. Ni, J. Yan, Y. Wu, F. Zhou, P.-T. Chou and Y. Chi, Transition-metal phosphors with emission peak maximum on and beyond the visible spectral boundaries, Inorg. Chem. Front., 2023, 10, 1395–1401 RSC.
- S. Lee and W.-S. Han, Cyclometalated Ir(iii) Complexes Towards Blue-Emissive Dopant for Organic Light-Emitting Diodes: Fundamentals of Photophysics and Designing Strategies, Inorg. Chem. Front., 2020, 7, 2396–2422 RSC.
- A. Bonfiglio and M. Mauro, Phosphorescent Tris-Bidentate IrIII Complexes with N-Heterocyclic Carbene Scaffolds: Structural Diversity and Optical Properties, Eur. J. Inorg. Chem., 2020, 2020, 3427–3442 CrossRef CAS.
- T. Sajoto, P. I. Djurovich, A. B. Tamayo, J. Oxgaard, W. A. Goddard and M. E. Thompson, Temperature Dependence of Blue Phosphorescent Cyclometalated Ir(III) Complexes, J. Am. Chem. Soc., 2009, 131, 9813–9822 CrossRef CAS PubMed.
- K. Tsuchiya, S. Yagai, A. Kitamura, T. Karatsu, K. Endo, J. Mizukami, S. Akiyama and M. Yabe, Synthesis and Photophysical Properties of Substituted Tris(phenylbenzimidazolinato) IrIII Carbene Complexes as a Blue Phosphorescent Material, Eur. J. Inorg. Chem., 2010, 2010, 926–933 CrossRef.
- J. Lee, H.-F. Chen, T. Batagoda, C. Coburn, P. I. Djurovich, M. E. Thompson and S. R. Forrest, Deep Blue Phosphorescent Organic Light-Emitting Diodes with Very High Brightness and Efficiency, Nat. Mater., 2016, 15, 92–98 CrossRef CAS PubMed.
- X. Yang, X. Zhou, Y.-X. Zhang, D. Li, C. Li, C. You, T.-C. Chou, S.-J. Su, P.-T. Chou and Y. Chi, Blue Phosphorescence and Hyperluminescence Generated from Imidazo[4,5-b]pyridin-2-ylidene Based Iridium(III) Phosphors, Adv. Sci., 2022, 9, 2201150 CrossRef CAS PubMed.
- A. Maheshwaran, V. G. Sree, H.-Y. Park, H. Kim, S. H. Han, J. Y. Lee and S.-H. Jin, High Efficiency Deep-Blue Phosphorescent Organic Light-Emitting Diodes with CIE x, y (<0.15) and Low Efficiency Roll-Off by Employing a High Triplet Energy Bipolar Host Material, Adv. Funct. Mater., 2018, 28, 1802945 CrossRef.
- M. Idris, S. C. Kapper, A. C. Tadle, T. Batagoda, D. S. M. Ravinson, O. Abimbola, P. I. Djurovich, J. Kim, C. Coburn, S. R. Forrest and M. E. Thompson, Blue Emissive fac/mer-Iridium(III) NHC Carbene Complexes and their Application in OLEDs, Adv. Opt. Mater., 2021, 9, 2001994 CrossRef CAS.
- J. Yan, Q. Xue, H. Yang, S.-M. Yiu, Y.-X. Zhang, G. Xie and Y. Chi, Regioselective Syntheses of Imidazo[4,5-b]pyrazin-2-ylidene Based Chelates and Blue Emissive Iridium(III) Phosphors for Solution Processed OLEDs, Inorg. Chem., 2022, 61, 8797–8805 CrossRef CAS PubMed.
- J. Jin, Z. Zhu, J. Yan, X. Zhou, C. Cao, P.-T. Chou, Y.-X. Zhang, Z. Zheng, C.-S. Lee and Y. Chi, Iridium(III) Phosphors–Bearing Functional 9-Phenyl-7,9-dihydro-8H-purin-8-ylidene Chelates and Blue Hyperphosphorescent OLED Devices, Adv. Photonics Res., 2022, 3, 2100381 CrossRef CAS.
- Y. Qin, X. Yang, J. Jin, D. Li, X. Zhou, Z. Zheng, Y. Sun, W.-Y. Wong, Y. Chi and S.-J. Su, Facially Coordinated, Tris-bidentate Purin-8-ylidene Ir(III) Complexes for Blue Electrophosphorescence and Hyperluminescence, Adv. Opt. Mater., 2022, 10, 2201633 CrossRef CAS.
- Y. Chi and P.-T. Chou, Transition Metal Phosphors with Cyclometalating Ligands; Fundamental and Applications, Chem. Soc. Rev., 2010, 39, 638–655 RSC.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, The Triplet State of Organo-Transition Metal Compounds. Triplet Harvesting and Singlet Harvesting for Efficient OLEDs, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS.
- T. Strassner, Phosphorescent Platinum(II) Complexes with C^C* Cyclometalated NHC Ligands, Acc. Chem. Res., 2016, 49, 2680–2689 CrossRef CAS PubMed.
- Z. Zheng, L. Wang, Y. Xin, Q. Wang, X. Hong, Y. Zhang, S.-M. Yiu, F. Zhou, J. Yan, D. Zhang, L. Duan and Y. Chi, Iridium(III) Carbene Phosphors with Fast Radiative Transitions for Blue Organic Light Emitting Diodes and Hyperphosphorescence, Adv. Funct. Mater., 2024, 34, 2311692 CrossRef.
- J. Yan, C. Wu, K.-N. Tong, F. Zhou, Y. Chen, Y. Pan, G. Xie, Y. Chi, K.-C. Lau and G. Wei, Structural Engineering of Iridium(III) Phosphors with Imidazo[4,5-b]pyrazin-2-ylidene Cyclometalates for Efficient Blue Electroluminescence, Small Methods, 2024, 8, 2301555 CrossRef PubMed.
- J. Yan, Y. Wu, I.-C. Peng, Y. Pan, S.-M. Yiu, K.-T. Wong, W.-Y. Hung, Y. Chi and K.-C. Lau, Peripheral engineering of Ir(III) emitters with imidazo[4,5-b]pyrazin-2-ylidene cyclometalates for blue organic light emitting diodes, J. Mater. Chem. C, 2023, 11, 12270–12279 RSC.
- J. Yan, D.-Y. Zhou, L.-S. Liao, M. Kuhn, X. Zhou, S.-M. Yiu and Y. Chi, Electroluminescence and hyperphosphorescence from stable blue Ir(III) carbene complexes with suppressed efficiency roll-off, Nat. Commun., 2023, 14, 6419 CrossRef CAS PubMed.
- J. Yan, Z.-Q. Feng, Y. Wu, D.-Y. Zhou, S.-M. Yiu, C.-Y. Chan, Y. Pan, K. C. Lau, L.-S. Liao and Y. Chi, Blue Electrophosphorescence from Iridium(III) Phosphors Bearing Asymmetric Di-N-Aryl 6-(trifluoromethyl)-2H-imidazo[4,5-b]pyridin-2-ylidene Chelates, Adv. Mater., 2024, 36, 2305273 Search PubMed.
- R. M. Roberts and P. J. Vogt, Ethyl N-phenylformimidate, Org. Synth., 1955, 35, 65–66 CrossRef CAS.
- E. B. Knott and R. A. Jeffreys, The structure of the solid reaction product of anilines and ethyl orthoformate, J. Org. Chem., 1949, 14, 879–885 CrossRef CAS.
- K. Hirano, A. T. Biju and F. Glorius, Copper-Catalyzed Synthesis of 2-Unsubstituted, N-Substituted Benzimidazoles, J. Org. Chem., 2009, 74, 9570–9572 CrossRef CAS PubMed.
- T. Lv, Z. Wang, J. You, J. Lan and G. Gao, Copper-Catalyzed Direct Aryl Quaternization of N-Substituted Imidazoles to Form Imidazolium Salts, J. Org. Chem., 2013, 78, 5723–5730 CrossRef CAS PubMed.
- Y.-C. Chiu, C.-H. Lin, J.-Y. Hung, Y. Chi, Y.-M. Cheng, K.-W. Wang, M.-W. Chung, G.-H. Lee and P.-T. Chou, Authentic-Blue Phosphorescent Iridium(III) Complexes Bearing Both Hydride and Benzyl Diphenylphosphine; Control of the Emission Efficiency by Ligand Coordination Geometry, Inorg. Chem., 2009, 48, 8164–8172 CrossRef CAS PubMed.
- J.-Y. Hung, C.-H. Lin, Y. Chi, M.-W. Chung, Y.-J. Chen, G.-H. Lee, P.-T. Chou, C.-C. Chen and C.-C. Wu, Phosphorescent Ir(III) Complexes Bearing Double Benzyldiphenylphosphine Cyclometalates; Strategic Synthesis, Fundamental and Integration for White OLED Fabrication, J. Mater. Chem., 2010, 20, 7682–7693 RSC.
- J. G. Osiak, T. Setzer, P. G. Jones, C. Lennartz, A. Dreuw, W. Kowalsky and H.-H. Johannes, Twist it! The Acid-dependent Isomerization of Homoleptic Carbenic Iridium(III) Complexes, Chem. Commun., 2017, 53, 3295–3298 RSC.
- N. Tamosiunaite, L. C. Logie, S. E. Neale, K. Singh, D. L. Davies and S. A. Macgregor, Experimental and Computational Studies on the Acetate-Assisted C–H Activation of N-Aryl Imidazolium Salts at Rhodium and Iridium: A Chloride Additive Changes the Selectivity of C–H Activation, J. Org. Chem., 2022, 87, 1445–1456 CrossRef CAS PubMed.
- A. Y. Gitlina, F. Fadaei-Tirani and K. Severin, The acid-mediated isomerization of iridium(III) complexes with cyclometalated NHC ligands: kinetic vs. thermodynamic control, Dalton Trans., 2023, 52, 2833–2837 RSC.
- C. Wu, K. Shi, S. Li, J. Yan, Z.-Q. Feng, K.-N. Tong, S.-W. Zhang, Y. Zhang, D. Zhang, L.-S. Liao, Y. Chi, G. Wei and F. Kang, Design Strategies of Iridium(III) Complexes for Highly Efficient Saturated Blue Phosphorescent OLEDs with Improved Lifetime, EnergyChem, 2024, 6, 100120 CrossRef CAS.
- C. Adamo and D. Jacquemin, The calculations of excited-state properties with Time-Dependent Density Functional Theory, Chem. Soc. Rev., 2013, 42, 845–856 RSC.
- A. D. Laurent, C. Adamo and D. Jacquemin, Dye chemistry with time-dependent density functional theory, Phys. Chem. Chem. Phys., 2014, 16, 14334–14356 RSC.
- R. L. Martin, Natural Transition Orbitals, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- J. M. Younker and K. D. Dobbs, Correlating Experimental Photophysical Properties of Iridium(III) Complexes to Spin–Orbit Coupled TDDFT Predictions, J. Phys. Chem. C, 2013, 117, 25714–25723 CrossRef CAS.
- P. Pander, A. V. Zaytsev, A. Sil, J. A. G. Williams, P.-H. Lanoe, V. N. Kozhevnikov and F. B. Dias, The role of dinuclearity in promoting thermally activated delayed fluorescence (TADF) in cyclometallated, N^C^N-coordinated platinum(II) complexes, J. Mater. Chem. C, 2021, 9, 10276–10287 RSC.
- P. Pander, L. Gomes Franca, F. B. Dias and V. N. Kozhevnikov, Electroluminescence of Tetradentate Pt(II) Complexes: O^N^N^O versus C^N^N^O Coordination, Inorg. Chem., 2023, 62, 5772–5779 CrossRef CAS PubMed.
- E. Jansson, P. Norman, B. Minaev and H. Ågren, Evaluation of low-scaling methods for calculation of phosphorescence parameters, J. Chem. Phys., 2006, 124, 114106 CrossRef PubMed.
- E. Jansson, B. Minaev, S. Schrader and H. Ågren, Time-dependent density functional calculations of phosphorescence parameters for fac-tris(2-phenylpyridine) iridium, Chem. Phys., 2007, 333, 157–167 CrossRef CAS.
- K. Mori, T. P. M. Goumans, E. van Lenthe and F. Wang, Predicting phosphorescent lifetimes and zero-field splitting of organometallic complexes with time-dependent density functional theory including spin–orbit coupling, Phys. Chem. Chem. Phys., 2014, 16, 14523–14530 RSC.
- M. A. Baldo, M. E. Thompson and S. R. Forrest, High-efficiency Fluorescent Organic Light-emitting Devices using a Phosphorescent Sensitizer, Nature, 2000, 403, 750–753 CrossRef CAS PubMed.
- K. H. Lee and J. Y. Lee, Phosphor Sensitized Thermally Activated Delayed Fluorescence Organic Light-emitting Diodes with Ideal Deep Blue Device Performances, J. Mater. Chem. C, 2019, 7, 8562–8568 RSC.
- S. Nam, J. W. Kim, H. J. Bae, Y. M. Maruyama, D. Jeong, J. Kim, J. S. Kim, W.-J. Son, H. Jeong, J. Lee, S.-G. Ihn and H. Choi, Improved Efficiency and Lifetime of Deep-Blue Hyperfluorescent Organic Light-Emitting Diode using Pt(II) Complex as Phosphorescent Sensitizer, Adv. Sci., 2021, 8, 2100586 CrossRef CAS PubMed.
- A. Monkman, Why Do We Still Need a Stable Long Lifetime Deep Blue OLED Emitter?, ACS Appl. Mater. Interfaces, 2022, 14, 20463–20467 CrossRef CAS PubMed.
- P. Zuo, Y.-K. Qu, Q. Zheng, L.-S. Liao and Z.-Q. Jiang, Sensitized organic light-emitting diodes: towards high efficiency and long lifetimes, Mater. Chem. Front., 2023, 7, 1760–1780 RSC.
- D. Zhang, X. Song, A. J. Gillett, B. H. Drummond, S. T. E. Jones, G. Li, H. He, M. Cai, D. Credgington and L. Duan, Efficient and Stable Deep-Blue Fluorescent Organic Light-Emitting Diodes Employing a Sensitizer with Fast Triplet Upconversion, Adv. Mater., 2020, 32, 1908355 CrossRef CAS PubMed.
- C.-Y. Chan, M. Tanaka, Y.-T. Lee, Y.-W. Wong, H. Nakanotani, T. Hatakeyama and C. Adachi, Stable pure-blue hyperfluorescence organic light-emitting diodes with high-efficiency and narrow emission, Nat. Photonics, 2021, 15, 203–207 CrossRef CAS.
- C. Duan, Y. Xin, Z. Wang, J. Zhang, C. Han and H. Xu, High-efficiency hyperfluorescent white light-emitting diodes based on high-concentration-doped TADF sensitizer matrices via spatial and energy gap effects, Chem. Sci., 2022, 13, 159–169 RSC.
- K.-W. Lo, G. S. M. Tong, G. Cheng, K.-H. Low and C.-M. Che, Dinuclear PtII Complexes with Strong Blue Phosphorescence for Operationally Stable Organic Light-Emitting Diodes with EQE up to 23% at 1000 cd m−2, Angew. Chem., Int. Ed., 2022, 61, e202115515 CrossRef CAS PubMed.
- C. Wu, M. Wang, K.-N. Tong, M. Zhang, W. Li, Z. Xu, W.-L. Zhang, Y. Wu, C. Yang, H.-Y. Fu, S. S. Chen, M. Ng, M.-C. Tang and G. Wei, Blue Iridium(III) Phosphorescent OLEDs with High Brightness Over 10 000 cd m−2 and Ultralow Efficiency Roll-Off, Adv. Opt. Mater., 2023, 11, 2201998 CrossRef CAS.
- H. Liu, Y. Fu, J. Chen, B. Z. Tang and Z. Zhao, Energy-Efficient Stable Hyperfluorescence Organic Light-Emitting Diodes with Improved Color Purities and Ultrahigh Power Efficiencies Based on Low-Polar Sensitizing Systems, Adv. Mater., 2023, 35, 2212237 CrossRef CAS PubMed.
- J. Yan, S. F. Wang, C.-H. Hsu, E. H.-C. Shi, C.-C. Wu, P.-T. Chou, S.-M. Yiu, Y. Chi, C. You, I. C. Peng and W.-Y. Hung, Engineering of Cyano Functionalized Benzo[d]imidazol-2-ylidene Ir(III) Phosphors for Blue Organic Light-Emitting Diodes, ACS Appl. Mater. Interfaces, 2023, 15, 21333–21343 CrossRef CAS PubMed.
- H. Shin, S. Lee, K.-H. Kim, C.-K. Moon, S.-J. Yoo, J.-H. Lee and J.-J. Kim, Blue Phosphorescent Organic Light-Emitting Diodes Using an Exciplex Forming Co-Host with the External Quantum Efficiency of Theoretical Limit, Adv. Mater., 2014, 26, 4730–4734 CrossRef CAS PubMed.
- Y. Kondo, K. Yoshiura, S. Kitera, H. Nishi, S. Oda, H. Gotoh, Y. Sasada, M. Yanai and T. Hatakeyama, Narrowband deep-blue organic light-emitting diode featuring an organoboron-based emitter, Nat. Photonics, 2019, 13, 678–682 CrossRef CAS.
- S. H. Han, J. H. Jeong, J. W. Yoo and J. Y. Lee, Ideal Blue Thermally Activated Delayed Fluorescence Emission Assisted by a Thermally Activated Delayed Fluorescence Assistant Dopant Through a Fast Reverse Intersystem Crossing Mediated Cascade Energy Transfer Process, J. Mater. Chem. C, 2019, 7, 3082–3089 RSC.
- S. O. Jeon, K. H. Lee, J. S. Kim, S.-G. Ihn, Y. S. Chung, J. W. Kim, H. Lee, S. Kim, H. Choi and J. Y. Lee, High-efficiency, long-lifetime deep-blue organic light-emitting diodes, Nat. Photonics, 2021, 15, 208–215 CrossRef CAS.
- W. J. Chung, K. H. Lee, M. Jung, K. M. Lee, H. C. Park, M.-S. Eum and J. Y. Lee, Over 30 000 h Device Lifetime in Deep Blue Organic Light-Emitting Diodes with y Color Coordinate of 0.086 and Current Efficiency of 37.0 cd A−1, Adv. Opt. Mater., 2021, 9, 2100203 CrossRef CAS.
- Z.-L. Zhu, J. Yan, L.-W. Fu, C. Cao, J.-H. Tan, S.-F. Wang, Y. Chi and C.-S. Lee, Peripheral engineering of platinum(II) dicarbene pincer complexes for efficient blue hyperphosphorescent organic light-emitting diodes, Mater. Chem. Front., 2023, 7, 3398–3405 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: General synthetic protocol of chelates, supportive photophysical and electrochemical data, procedures for a computational study, detailed TD-DFT results, and supportive OLED performance data of all studied Ir(III) metal complexes. CCDC 2110266 and 2242122. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qi00454j |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2024 |