
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Straightforward construction of a rare-earth diphosphanato complex from white phosphorus: synthesis and reactivity†
Fangjun
Zhang
a,
Iker
del Rosal
 b,
Keyu
Han
a,
Jie
Zhang
*a,
Laurent
Maron
*b and
Xigeng
Zhou
*a
b,
Keyu
Han
a,
Jie
Zhang
*a,
Laurent
Maron
*b and
Xigeng
Zhou
*a
aDepartment of Chemistry, Fudan University, No. 2005, Songhu Road, Shanghai 200438, China. E-mail: zhangjie@fudan.edu.cn; xgzhou@fudan.edu.cn
bLPCNO, CNRS, and INSA, Université Paul Sabatier, 135 Avenue de Rangueil, Toulouse 31077, France. E-mail: laurent.maron@irsamc.ups-tlse.fr
First published on 20th February 2024
Abstract
The direct construction of functionalized diphosphine ligands from P4 is a highly valuable but challenging transformation. In this work, a simple method has been developed for the direct formation of an amido-functionalized diphosphanato ligand from a silyl-bridged amido/methylene yttrium complex and white phosphorus. The resulting amido-functionalized diphosphanato yttrium complex 2 has proved to be a useful intermediate for further diversification of the diphosphine ligand through the metal template procedure, for which no evident limitations are enforced by initially present substituents and ancillary ligands, and some unprecedented reactivity patterns of the RPPR unit are revealed. The results described here demonstrate that appending a strongly coordinative substituent to the alkyl ligand together with a coordination to rare earth ions is an efficient strategy for controlling the cleavage modes of P4 during alkylation and the reactivity of the resulting diphosphanato complex.
Introduction
Diphosphine-based ligands play an important role in catalysis, coordination and organometallic chemistry.1–3 To tune their properties for potential applications and improve the overall synthetic efficiency, significant effort continues to be devoted to the development of new approaches for their synthesis and new diphosphine-based structures.4–6 Among these approaches, the introduction of one or more heteroatoms (e.g. O, N, S, Se, Si, and F) into diphosphine backbones is especially attractive, as the presence of heteroatoms will have a much greater influence on the coordination modes and donor ability than the variation of organic substituents at the P atoms, and thus offers new opportunities for controlling the reactivity trends, catalytic behaviour and physical properties of metal complexes.7–12 For example, the pendant amines on diphosphine ligands facilitate the binding and heterolytic cleavage of H2 or serve as proton and hydride relays for metal-mediated hydrogen transfers.9c,dTraditionally, multifunctionalized diphosphine ligands are prefabricated by stepwise processes starting from PCl3 or its equivalents before application in organic and organometallic syntheses. Alternatively, in a few cases they can be generated in situ by the reaction of a metal phosphinido or phosphinidene complex intermediate (Scheme 1A).13 Obviously, these synthetic approaches to metal complexes containing polyfunctional diphosphine ligands often require expensive and/or toxic reagents and generate a large amount of waste, further compounded by limitations of substituent scope due to difficulties in preparing related P-based reactant precursors. If white phosphorus (P4) could be directly used in the construction of polyfunctional diphosphine ligands, it would provide a simpler, more environmentally benign and higher atom-economical process compared to the use of classical methods involving pollutants, such as PCl3, as starting materials. Although a great deal of effort has been devoted to the activation of P4 over the last four decades, reactions that can form diphosphine skeletons either as free species or as complexed ligands remain scarce.6,14–17 In fact, there are currently no generally satisfactory routes to the formation of polyfunctional diphosphine moieties from P4 without the formation of byproducts. The functionalization of P4 using alkyl/aryl metal complexes is relatively uncontrollable and tends to form monophosphanes or partly P-alkylated (arylated) polyphosphorus species (Scheme 1B).16,18,19 The difficulty of forming the diphosphanato or diphosphenyl complexes may reflect the challenge of controlling the cleavage pattern of P4 during alkylation/arylation, requiring a larger energy input for the simultaneous cleavage of four P–P bonds compared with that for the relatively small number of P–P bonds in the formation of [P4Rm]n− (m = 1–3; n = 0–4; R = alkyl, aryl) moieties.
 | ||
| Scheme 1 Synthetic strategies to access functionalized diphosphanato and diphosphine metal complexes and main reactivity patterns of metal alkyl/aryl complexes toward P4. | ||
On the other hand, although metal-based reductive cleavage of P4 to give a diphosphorus ligand is a well-established process, the exploration of the resulting P2 fragment as a synthon for diphosphines remains limited.20 Pioneering works reported by West, Bertrand, and Cummins et al. showed that the activation of P4 by disilenes, carbenes or photochemical alkylation could lead to the formation of the corresponding silylene-, alkylidenyl- and alkyl-substituted diphosphanes, respectively, and these findings open new avenues for the generation of reactive intermediates in P2 chemistry.4a,e,5b,6
Metal diphosphanato complexes could be considered as attractive intermediates for the synthesis of a variety of neutral or anionic diphosphine ligands as evidenced by their participation in the formation of carbon–phosphorus and heteroatom–phosphorus bonds.21,22 However, this class of compounds are still rare due to challenges in their synthesis.23 Given the pronounced ability of rare earth metals to influence the reactivity of small molecule substrates and high activity of lanthanide–alkyl bonds,24,25 we reasoned that the obstacles encountered in the direct conversion of P4 to dialkyldiphosphanato complexes via reductive insertion processes might be overcome by appending the strong chelating coordination moiety to the alkyl ligand together with a coordination to lanthanide ions, because this not only offers a sterically forced preferred orientation of the nucleophilic attack at P4 in an expected position for regioselective cleavage of P–P bonds, but also may inhibit the alkyl migration between P atoms (Scheme 1C). Here, we report a simple method for the direct and highly atom-economical construction and late-stage diversification of amido-functionalized diphosphanato ligands from white phosphorus enabled by rare earth metals, for which some unprecedented reactivity patterns of the RPPR moiety toward small molecule substrates are revealed.
Results and discussion
A new silyl-bridged amido/methylene yttrium complex 1 was prepared by the reaction of TpMe2YBn2(THF) (Bn = CH2Ph) with HN(SiMe3)2. Surprisingly, only a low yield of 1 was obtained when 1 equiv. of HN(SiMe3)2 was used, and complete transformation of TpMe2YBn2(THF) into 1 would require the use of 2 equiv. of HN(SiMe3)2 (Scheme 2). These results indicate that 1 probably results from amine elimination of intermediate B rather than the toluene elimination of A, and steric crowding around the metal center might play a more important role than ligand basicity in driving the methyl deprotonation. Complex 1 has been characterized by elemental analysis, 1H and 13C {1H} NMR spectra, and its solid-state structure has also been determined by X-ray diffraction analysis (Fig. S26†). | ||
| Scheme 2 Synthesis of 1,2-dialkyldiphosphanato yttrium complex 2. | ||
Treatment of 1 with 0.25 equiv. of P4 in toluene at ambient temperature afforded the Y–C bond insertion/coupling product 2 in 60% isolated yield (Scheme 2), which represents the first example of the efficient formation of amido-functionalized diphosphanato metal complexes directly from P4. Complex 2 is readily soluble in THF but sparingly soluble in toluene. The 31P NMR spectrum of 2 displayed a triplet peak at δ = 97.10 ppm (1JYP = 30 Hz), indicating that its binuclear structure is symmetric. Consistent with this, only one set of signals attributable to the TpMe2 ligand was observed in the 1H and 13C {1H} NMR spectra of 2. Crystallization of 2 in THF gave the THF adduct 2·THF. Significantly, the THF coordination triggers the difunctionalized diphosphanato ligand to isomerize from trans to cis (Fig. 1). Furthermore, in contrast to 2, 2·THF is insoluble in THF. The P–P distance in 2 (2.213(2) Å) is slightly longer than the values observed in Cp2NbH(P2Ph2) (2.136(5) Å) and Cp2TaH(P2Cy2) (2.131(3) Å).15
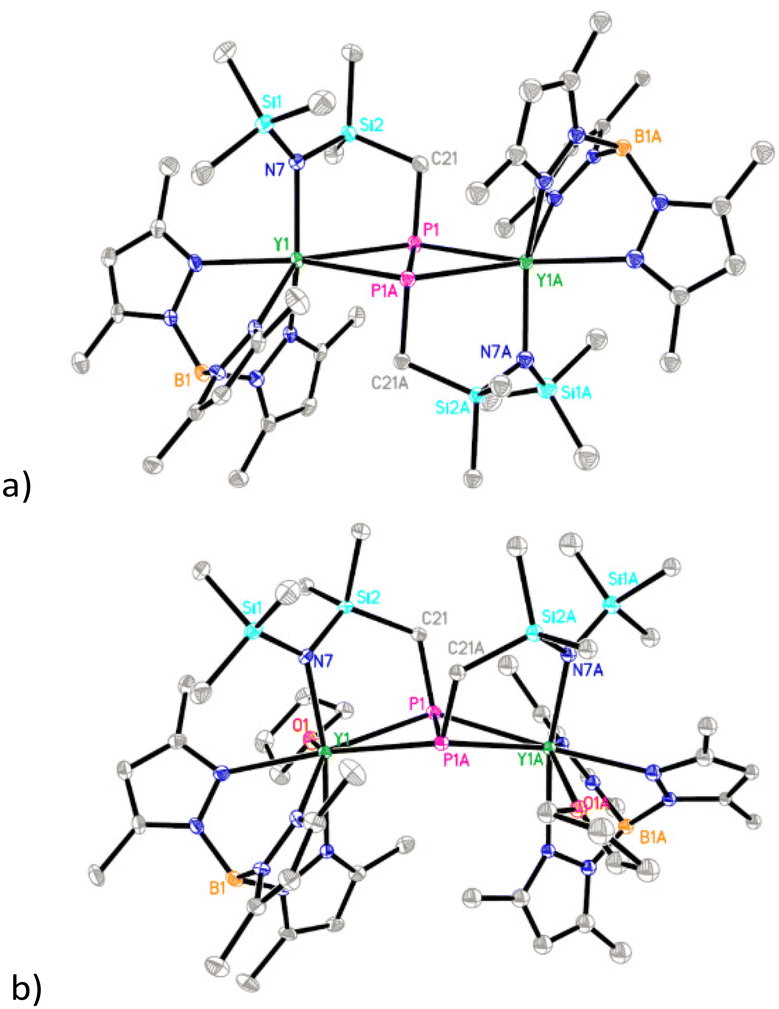 | ||
| Fig. 1 Molecular structures of 2 (a) and 2·THF (b) with 30% thermal ellipsoids. All hydrogen atoms are omitted for clarity. | ||
It should be noted that the chelating coordination of the pendant amido moiety might play a key role in determining the formation of 2. In our previous study,26 we found that the rare-earth dialkyl complex TpMe2LnBn2(THF) reacts with P4 to yield novel rare-earth norbornane-BnP74− and/or chain Bn4P64− complexes, accompanied by the formation of PBn3,26b and the rare-earth monoalkyl complex LYR(THF) (L = N,N′-2,6-diisopropylphenyl-1,4-diazabutadiene, R = CH2C6H4NMe2-2) reacts with P4 to give a rare-earth organic cyclo-P4 complex (LY·DMAP)2[1,2-R2-cyclo-P4], which further transforms into other polyphosphorus species through alkyl migration.26c These results demonstrate that appending a strongly coordinative amino substituent to the alkyl group, together with a coordination to Y3+ ions, surely offers a good stabilizing environment for 1,2-dialkyldiphosphanato dianion species.
CS2 is a cheap and manageable feedstock for the synthesis of sulfur-containing molecules, and the development of new reactivity patterns of CS2 and new methods for its activation under mild conditions is attracting scientific and industrial interest, including its use as a model for CO2 and its implication in catalytic degradation of CS2 pollutants.27,28 Despite tremendous advances in fragmented coupling reactions involving CS2, there is no report on the combined reorganization/cyclization of CS2 with other organic functional groups.28,29 Contrary to the metal complexes bearing other phosphorus-based ligands (e.g. phosphinidene and phosphinido),13b,30 treatment of 2 with two equiv. of CS2 in THF at room temperature led to the formation of the highly reorganized 2-sulfido-2,5-dihydro-1,2,5-thiadiphosphole-3,4-dithiolate dianion ligand that bridges the two yttrium atoms in a non-symmetric fashion (Scheme 3). Noticeably, the reaction works almost exclusively for the formation of 3. Our attempts to use 1 equiv. of CS2 for the isolation of the reaction intermediates afforded 3 with the recovery of significant quantities of the starting material 2, which was also detected from 31P NMR spectra at the end of the reaction. The X-ray structure of 3 (Fig. 2) reveals that the resulting thiadiphosphole ring was coplanar. The C41–C42 distance (1.332(6) Å) is a normal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond length. The P1–S3 and P2–S3 distances (2.157(2) and 2.100(2) Å, respectively) exhibit typical P–S single bond characteristics, whereas the P2–S4 distance (1.999(2) Å) falls in the range of the PS double bond lengths (1.949–2.09 Å).31 The Y2–S4 distance (2.805(2) Å) is longer than the Y2–S2 one (2.727(2) Å). The 31P NMR spectrum of 3 shows two characteristic signals at δ = 92.43 ppm (q, P1, 1JYP = 17.6 Hz) and 47.88 ppm (s, P2). The 13C {1H} NMR spectrum of 3 displays two sets of peaks at δ = 152.68 ppm (dd, 1JPC = 3.21 Hz, 2JYC = 1.56 Hz) and 152.21 ppm (d, 1JPC = 1.99 Hz), attributable to the resonances of the carbon atoms of the thiadiphosphole ring.
C double bond length. The P1–S3 and P2–S3 distances (2.157(2) and 2.100(2) Å, respectively) exhibit typical P–S single bond characteristics, whereas the P2–S4 distance (1.999(2) Å) falls in the range of the PS double bond lengths (1.949–2.09 Å).31 The Y2–S4 distance (2.805(2) Å) is longer than the Y2–S2 one (2.727(2) Å). The 31P NMR spectrum of 3 shows two characteristic signals at δ = 92.43 ppm (q, P1, 1JYP = 17.6 Hz) and 47.88 ppm (s, P2). The 13C {1H} NMR spectrum of 3 displays two sets of peaks at δ = 152.68 ppm (dd, 1JPC = 3.21 Hz, 2JYC = 1.56 Hz) and 152.21 ppm (d, 1JPC = 1.99 Hz), attributable to the resonances of the carbon atoms of the thiadiphosphole ring.
 | ||
| Scheme 3 Reaction of 1,2-dialkyldiphosphanato yttrium complex 2 with CS2. | ||
 | ||
| Fig. 2 Molecular structure of 3 with 30% thermal ellipsoids. All hydrogen atoms are omitted for clarity. | ||
The great success of alkene metathesis has stimulated research activity aimed at mapping the analogous reactivity modes of unsaturated carbon–heteroatom functionalities.32,33 However, CS2 metathesis reactions remain little explored. In contrast to the classical metatheses, the formation of 3 is not accompanied by the loss of any fragments. This finding provides a new probe for designing convergent metatheses that allow the simultaneous cleavage of multiple different chemical bonds and sequential incorporation of all fragments into a single organic architecture, without any fragment dispersion. To obtain an insight into the driving force for the reaction, the reaction was further investigated by theoretical calculations at the DFT level. The reaction profile appears to be quite complex (Fig. S34, ESI†). The reaction begins by the sequential addition of two CS2 molecules to the diphosphanato ligand. These electrophilic additions of CS2 to the phosphorus are kinetically accessible and the height of the barrier is only associated with the binding of the incoming CS2. This yields complex D, which is thermodynamically favorable (22.4 kcal mol−1). From there, one CS2 moiety can undergo a [2 + 2] addition to the P–P bond that is activated by the two electrophilic additions. This reaction is not assisted by the metal center, explaining that this is the rate determining step. After this migratory insertion, the C–S bond becomes strongly activated and can be easily cleaved to form a P–S–P bridge (complex F). The lack of stability of complex F allows further reaction. A kinetically accessible intramolecular cyclization transition state has been located, yielding a stable five-membered ring complex G (−30.1 kcal mol−1). This complex undergoes a facile metal-assisted intramolecular C–S bond activation, affording the formation of the very stable complex 3 (−77.1 kcal mol−1).
Diphosphorus ligands connected by a single atom have drawn considerable interest due to their complementary properties to wide bite-angle diphosphine ligands in homogeneous catalysis.34 Interestingly, the reaction of 2 with excess CO (1 atm) at 50 °C afforded the P–P and Y–P bond insertion product 4 (Scheme 4). The resulting carbonyl-bridged diphosphorus anion ligand features an additional degree of electron-delocalization control over the 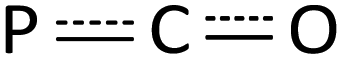 binding sites. In the 1H NMR spectra of 4, the two sets of signals for the methyl protons of Me3Si substituents (δ −0.13 and −0.09 ppm) are consistent with the observation that the Y3+ ions have two different coordination environments. Moreover, different chemical shifts assigned to the two TpMe2 ligands were observed in its 1H NMR spectrum. The 31P NMR spectrum shows two peaks at δ = 65.01 (d, 1JYP = 136.4 Hz) and −11.89 (d, 1JYP = 120.3 Hz) ppm. X-ray single crystal diffraction analyses of 4 (Fig. 3) clearly demonstrate the insertion of one CO molecule into the P–P bond to form a C–P single bond and a CP double bond. Complex 4 has a distinctive Y–O(phosphaenolate) distance (2.153(2) Å).
binding sites. In the 1H NMR spectra of 4, the two sets of signals for the methyl protons of Me3Si substituents (δ −0.13 and −0.09 ppm) are consistent with the observation that the Y3+ ions have two different coordination environments. Moreover, different chemical shifts assigned to the two TpMe2 ligands were observed in its 1H NMR spectrum. The 31P NMR spectrum shows two peaks at δ = 65.01 (d, 1JYP = 136.4 Hz) and −11.89 (d, 1JYP = 120.3 Hz) ppm. X-ray single crystal diffraction analyses of 4 (Fig. 3) clearly demonstrate the insertion of one CO molecule into the P–P bond to form a C–P single bond and a CP double bond. Complex 4 has a distinctive Y–O(phosphaenolate) distance (2.153(2) Å).
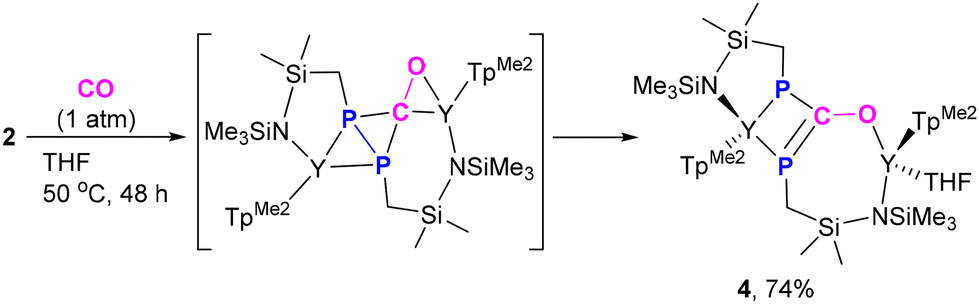 | ||
| Scheme 4 CO insertion into the P–P and Y–P bonds via an unusual Y–P addition and sequential phosphanyl migration pathway. | ||
 | ||
| Fig. 3 Molecular structure of 4 with 30% thermal ellipsoids. All hydrogen atoms are omitted for clarity. | ||
The catalytic carbonylation of organic substrates has become a powerful method in organic synthesis. Despite tremendous advances in this area, this process has mainly focused on late transition metals. Rare earth-mediated CO insertions have been largely unexplored. This is mainly due to the mismatch in the orbital energy between the hard rare earth ions and the soft carbon monoxide (or acyl) ligand, leading to an unstable acyl complex intermediate having a strong tendency toward dimerization or deoxygenative coupling.13b,35,36 Therefore, the formation of 4 is somewhat surprising. To explain the unusual reactivity of 2 toward CO, a DFT investigation of a possible reaction mechanism was carried out (see Fig. S35†). It is found that CO insertion into a Y–P bond followed by the migration of another P atom to the resulting acyl carbon is a knetically facile (highest barrier of 13.8 kcal mol−1) and thermodynamically favorable process (−33.9 kcal mol−1). This is quite different from the conventional oxidative-addition followed by CO insertion and sequential reductive-elimination pathways for transition-metal-mediated CO insertion into a single bond of organic substrates.
To validate our proposal of the preferential addition of the Y–P bond over the P–P bond, we carried out the reaction of 2 with PhNCS (Scheme 5). The addition of 2 equiv. of PhNCS to a THF solution of 2 resulted in an immediate color change, yielding a colorless solution, and storing the solution at room temperature for 48 h gave the Y–P bond insertion product 5 as colorless crystals in 93% isolated yield. In contrast to CS2 and CO, complex 5 did not undergo either isomerization or further reaction with excess PhNCS, even with prolonged heating at 70 °C. Regrettably, the low solubility of 5 precluded the acquisition of its NMR spectra. The same reactivity trend was observed when 2 reacted with 2 equiv. of PhNCO, giving the addition product 6. Recrystallization of 6 in THF/hexane led to the loss of one coordinated THF, giving compound 7, in which a different coordination situation of the diphosphine ligand was observed (Scheme 5). In contrast to 5, compounds 6 and 7 readily dissolve in THF. Notably, the transformation of 6 into 7 occurred slowly under vacuum conditions or exposure to nitrogen gas too. The 31P NMR spectrum of 7 shows two sets of signals at δ −39.64 (dd, JPP = 374, 12.8 Hz) and −26.71 (dm, 1JPP = 374 Hz) ppm, which may be attributed to the free and coordinated P atoms, respectively.20d The molecular structures of complexes 5–7 clearly indicate the formation of four-substituted diphosphine moieties (Fig. S28–S30†).
 | ||
| Scheme 5 Insertions of PhNCS, PhNCO and PhNNPh into the Y–P bond of 2. | ||
The 31P NMR monitoring data indicate that the reaction of 2 with azobenzene is much slower than those with CS2 and PhNCO, which likely resulted from the larger steric hindrance and the non-polarity effect of azobenzene.37,38 Compound 8 was sparingly soluble in THF-d8, enabling the acquisition of 1H NMR and 31P NMR data, but preventing characterization by 13C {1H} NMR spectra. The 31P NMR spectrum displays only a singlet at δ = 50.66 ppm, revealing that 8 is a symmetric structure as confirmed by X-ray crystallography (Fig. S31 in the ESI†).
The reaction of 2 with 0.5 equiv. of S8 in THF at room temperature gave the diphosphinodithioate complex 9 in 94% yield. Chalcogenylation is of great significance in adjusting the complexation ability and catalytic behaviour of diphosphines or in their potential utilization as synthetic intermediates.39a,40 Despite a large number of structural variants, such a substitution pattern for diphosphorus-containing species is still unknown due to the absence of suitable synthetic methods. Although the chemistry of phosphine sulfide and selenide ligands has been extensively investigated, the complexes of diphosphoranes containing P–P bonds are primarily limited to diphosphane mono- and di-sulfides/selenides.39b,40 The polychalcogenylations of either neutral or anionic P–P units with elemental sulfur (selenium) are typically accompanied by the P–P cleavage.21 To obtain a good insight into the formation process of 9, we firstly attempted the reaction of 2 with 0.25 equiv. of S8, but it generated a mixture of 9 and several unidentified products. Furthermore, the reaction of 2 with Se, which is known to have relatively modest reactivity compared with S8, was examined. 2 reacted with 3 equiv. of Se to yield the diphosphine triselenide complex 10 in 83% yield (Scheme 6). Despite several attempts, we were unable to gain the structural information of the product from a similar reaction of 2 with 4 equiv. of Se, owing to its low solubility even in polar organic solvents such as THF. The formation of 9 and 10 could be interpreted as a sulfur/selenium insertion into each Y–P bond of 2, followed by oxidative chalcogenylation of phosphorus. This further demonstrates that the reactivity of diphosphanato ligands toward small molecules may be adjusted by complexation of Y3+ ions. Complexes 9 and 10 were fully characterized; this information, including their single-crystal X-ray structures, can be found in the ESI (Fig. S32 and S33†).
 | ||
| Scheme 6 Oxidative chalcogenylation of 2 with S8 and Se. | ||
Experimental
General procedures
All manipulations involving air- and moisture-sensitive compounds were carried out with rigorous exclusion of air and water, using standard Schlenk techniques or a Vigor glovebox under an atmosphere of dinitrogen. THF, toluene and hexane were purified using an Mbraun SPS-800 solvent purification system, dried over fresh Na chips and stored in the glovebox. C6D6 and THF-d8 were obtained from Cambridge Isotope and dried over Na/K alloy prior to use. (TpMe2)YBn2(THF) was prepared according to the literature procedure.41 HN[Si(CH3)3]2, CS2, PhNCS and PhNCO were purchased from J&K and dried with 4 Å sieves. Other commercially available reagents were purchased and used without purification. P4 was prepared by sublimation of red phosphorus at 450 °C in a quartz tube under vacuum conditions and stored in the refrigerator of the glovebox. Organometallic samples for NMR spectroscopic measurements were prepared in the glovebox using J. Young valve NMR tubes. 1H NMR, 13C NMR and 31P NMR spectra were recorded using a Bruker Avance 400 MHz spectrometer (FT, 400 MHz for 1H; 100 MHz for 13C; and 161 MHz for 31P) at room temperature. Chemical shifts for 1H and 13C NMR were quoted in ppm referenced to the residual resonance of the deuterated solvents. H3PO4 (85%) sealed in a capillary was used as an external standard in 31P NMR analysis. Elemental analyses for C, H and N were carried out using a Vario EL III elemental analyzer. The resonances of the B–H bond in the TpMe2ligand are too broad to be observed in their1H NMR spectra.Caution: P4 is light-sensitive and highly flammable upon exposure to air. It should be handled carefully.Synthesis of 1
A THF solution of HN[Si(CH3)3]2 (3.22 g, 20.0 mmol) was added slowly to a stirred THF solution (50 mL) of TpMe2YBn2(THF) (6.40 g, 10.0 mmol) at room temperature. After stirring for 8 hours, all volatiles were removed under vacuum. The residue was washed twice with n-hexane (10 mL × 2) and dried under vacuum to give 1 as a white powder (5.67 g, 92% yield). Recrystallization of the white powder in a mixed solvent of THF and n-hexane afforded colorless crystals of 1 suitable for X-ray diffraction analysis. 1H NMR (400 MHz, C6D6, 25 °C): δ (ppm) 5.56 (s, 3H, 4-H-TpMe2), 3.56 (m, 4H, THF), 2.50 (s, 9H, 3-CH3-TpMe2), 2.10 (s, 9H, 5-CH3-TpMe2), 1.14 (m, 4H, THF), 0.79 (s, 6H, Si(CH3)2), 0.33 (s, 9H, Si(CH3)3), 0.13 (d, 2JYH = 1.6 Hz, 2H, SiCH2Y); 13C{1H} NMR (100 MHz, C6D6, 25 °C): δ (ppm) 150.13 (TpMe2), 145.49 (TpMe2), 105.91 (4-C-TpMe2), 71.27 (THF), 26.90 (d, 1JYC = 30.81 Hz, SiCH2Y), 25.29 (THF), 14.73 (CH3-TpMe2), 13.07 (CH3-TpMe2), 8.33 (s, Si(CH3)2), 8.32 (s, Si(CH3)2), 5.74 (Si(CH3)3). Elemental analysis: calcd (%) for C25H47N7BOSi2Y: C, 48.62; H, 7.67; N, 15.88. Found: C, 48.04; H, 7.74; N, 16.17.Synthesis and characterization of the diphosphanato yttrium complex 2 and 2·THF
P4 (31.0 mg, 0.25 mmol) was added to a stirred toluene solution (2 mL) of 1 (617 mg, 1.0 mmol) at ambient temperature. The color of the reaction mixture gradually changed from light yellow to reddish brown. After half an hour, a pale solid began to precipitate out. The mixture was continuously stirred overnight, and the precipitate was filtered under reduced pressure, washed with 2 mL of n-hexane and dried under vacuum to give 2 as a pale yellow powder (342 mg, 60% yield). Yellow crystals of 2 suitable for X-ray single crystal diffraction analysis were obtained by evaporation of the solvent from the solution of 2 in a 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of toluene and THF. 1H NMR (400 MHz, THF-d8, 25 °C): δ (ppm) 5.76 (s, 2H, 4-H-TpMe2), 5.74 (s, 2H, 4-H-TpMe2), 5.25 (s, 2H, 4-H-TpMe2), 2.67 (s, 6H, CH3-TpMe2), 2.59 (s, 6H, CH3-TpMe2), 2.38–2.33 (m, 24H, CH3-TpMe2), 1.29 (s, 4H, SiCH2P), 0.27 (s, 12H, Si(CH3)2), 0.15 (s, 18H, Si(CH3)3); 13C{1H} NMR (100 MHz, THF-d8, 25 °C): δ (ppm) 152.66 (TpMe2), 151.79 (TpMe2), 150.55 (TpMe2), 145.46 (TpMe2), 145.35 (TpMe2), 144.63 (TpMe2), 106.80 (4-C-TpMe2), 106.20 (4-C-TpMe2), 19.22 (CH3-TpMe2), 15.65 (CH3-TpMe2), 14.67 (m, SiCH2P), 13.31 (CH3-TpMe2), 13.18 (CH3-TpMe2), 13.06 (CH3-TpMe2), 8.43 (Si(CH3)2), 8.15 (Si(CH3)2), 6.15 (Si(CH3)3); 31P NMR (161 MHz, THF-d8, 25 °C): δ (ppm) −97.10 (t, 1JYP = 30 Hz). Elemental analysis: calcd (%) for C42H78N14B2Si4P2Y2: C, 43.76; H, 6.82; N, 17.00. Found: C, 42.86; H, 6.69; N, 17.33. It is noted that recrystallization of 2 in THF afforded 2·THF as yellow crystals. Elemental analysis: calcd (%) for C50H94N14B2O2Si4P2Y2: C, 46.30; H, 7.30; N, 15.12. Found: C, 45.66; H, 7.14; N, 15.37. However, the difficulty in redissolving the isolated crystals of 2·THF prevented its characterization by NMR spectroscopy.
:1 mixture of toluene and THF. 1H NMR (400 MHz, THF-d8, 25 °C): δ (ppm) 5.76 (s, 2H, 4-H-TpMe2), 5.74 (s, 2H, 4-H-TpMe2), 5.25 (s, 2H, 4-H-TpMe2), 2.67 (s, 6H, CH3-TpMe2), 2.59 (s, 6H, CH3-TpMe2), 2.38–2.33 (m, 24H, CH3-TpMe2), 1.29 (s, 4H, SiCH2P), 0.27 (s, 12H, Si(CH3)2), 0.15 (s, 18H, Si(CH3)3); 13C{1H} NMR (100 MHz, THF-d8, 25 °C): δ (ppm) 152.66 (TpMe2), 151.79 (TpMe2), 150.55 (TpMe2), 145.46 (TpMe2), 145.35 (TpMe2), 144.63 (TpMe2), 106.80 (4-C-TpMe2), 106.20 (4-C-TpMe2), 19.22 (CH3-TpMe2), 15.65 (CH3-TpMe2), 14.67 (m, SiCH2P), 13.31 (CH3-TpMe2), 13.18 (CH3-TpMe2), 13.06 (CH3-TpMe2), 8.43 (Si(CH3)2), 8.15 (Si(CH3)2), 6.15 (Si(CH3)3); 31P NMR (161 MHz, THF-d8, 25 °C): δ (ppm) −97.10 (t, 1JYP = 30 Hz). Elemental analysis: calcd (%) for C42H78N14B2Si4P2Y2: C, 43.76; H, 6.82; N, 17.00. Found: C, 42.86; H, 6.69; N, 17.33. It is noted that recrystallization of 2 in THF afforded 2·THF as yellow crystals. Elemental analysis: calcd (%) for C50H94N14B2O2Si4P2Y2: C, 46.30; H, 7.30; N, 15.12. Found: C, 45.66; H, 7.14; N, 15.37. However, the difficulty in redissolving the isolated crystals of 2·THF prevented its characterization by NMR spectroscopy.
Synthesis of 3
A cooled THF solution (5 mL) of CS2 (30.3 mg, 0.40 mmol) was added slowly to a stirred THF solution (10 mL) of 2 (230 mg, 0.20 mmol) at ambient temperature. The color of the mixture gradually changed from yellow to reddish brown. After stirring for 1 hour, the solvent was removed under vacuum and the residue was washed twice with n-hexane (10 mL × 2) and dried under vacuum to give 3 as a yellow powder (260 mg, 94% yield). Crystals suitable for X-ray analysis were obtained by recrystallization of 3 in toluene. 1H NMR (400 MHz, THF-d8, 25 °C): δ (ppm) 5.82 (s, 1H, 4-H-TpMe2), 5.81 (s, 1H, 4-H-TpMe2), 5.79 (s, 1H, 4-H-TpMe2), 5.76 (s, 1H, 4-H-TpMe2), 5.68 (s, 1H, 4-H-TpMe2), 5.57 (s, 1H, 4-H-TpMe2), 3.62 (m, THF), 2.65 (s, 3H, CH3-TpMe2), 2.51 (s, 3H, CH3-TpMe2), 2.47 (s, 3H, CH3-TpMe2), 2.41–2.40 (m, 18H, CH3-TpMe2), 2.37 (s, 3H, CH3-TpMe2), 2.28 (s, 3H, CH3-TpMe2), 2.22 (s, 3H, CH3-TpMe2), 1.94–1.81 (m, 4H, SiCH2P), 1.77 (m, THF), 0.48 (s, 3H, Si(CH3)2), 0.45 (s, 3H, Si(CH3)2), 0.30 (s, 3H, Si(CH3)2), 0.24 (s, 3H, Si(CH3)2), 0.00 (s, 9H, Si(CH3)3), −0.50 (s, 9H, Si(CH3)3); 13C{1H} NMR (100 MHz, THF-d8, 25 °C): δ (ppm) 152.67 (q, 1JPC = 3.21 Hz, 2JYC = 1.56 Hz, SCPS), 152.38 (TpMe2), 152.22 (d, 1JPC = 1.99 Hz, SCPS2), 151.70 (TpMe2), 151.28 (TpMe2), 150.69 (TpMe2), 150.66 (TpMe2), 150.08 (TpMe2), 147.16 (TpMe2), 146.76 (TpMe2), 146.16 (TpMe2), 145.42 (TpMe2), 144.32 (TpMe2), 106.82 (4-C-TpMe2), 106.68 (4-C-TpMe2), 106.58 (4-C-TpMe2), 106.45 (4-C-TpMe2), 106.37 (4-C-TpMe2), 106.27 (4-C-TpMe2), 68.02 (THF), 41.11 (d, 1JPC = 44.71 Hz, SiCH2P), 32.69 (d, 1JPC = 34.45 Hz, SiCH2P), 26.17 (THF), 16.52 (CH3-TpMe2), 16.15 (CH3-TpMe2), 15.58 (CH3-TpMe2), 15.50 (CH3-TpMe2), 14.49 (CH3-TpMe2), 14.36 (CH3-TpMe2), 13.18 (CH3-TpMe2), 13.09 (CH3-TpMe2), 13.04 (CH3-TpMe2), 12.93 (CH3-TpMe2), 12.85 (CH3-TpMe2), 12.77 (CH3-TpMe2), 9.27 (d, 3JPC = 7.28 Hz, Si(CH3)2), 8.66 (d, 3JPC = 11.6 Hz, Si(CH3)2), 6.41 (Si(CH3)2), 6.38 (Si(CH3)3), 6.25 (Si(CH3)2), 4.76 (Si(CH3)3); 31P NMR (161 MHz, THF-d8, 25 °C): δ (ppm) 92.43 (q, 1JYP = 17.6 Hz), 47.88 (s). Elemental analysis: calcd (%) for C48H86N14B2OSi4P2S4Y2: C, 41.86; H, 6.29; N, 14.24. Found: C, 41.14; H, 6.16; N, 14.45.Synthesis of 4
A THF solution (10 mL) of 2 (230 mg, 0.20 mmol) was degassed using freeze–pump–thaw cycles and then exposed to an excess of 1.0 atm of CO. The color of the reaction mixture gradually changed from yellow to orange. After stirring at 50 °C for two days, the solvent was removed under vacuum to give an orange solid. The residue was washed twice with hexane (5 mL × 2) and dried under vacuum, giving 4 as an orange powder (185 mg, 74% yield). The orange single crystals of 4 for X-ray diffraction analysis were obtained by solvent evaporation from a concentrated toluene solution (ca. 2 mL) of 4. 1H NMR (400 MHz, toluene-d8, 25 °C): δ (ppm) 5.61 (s, 2H, 4-H-TpMe2), 5.57 (s, 1H, 4-H-TpMe2), 5.50 (s, 2H, 4-H-TpMe2), 5.46 (s, 1H, 4-H-TpMe2), 3.67 (s, 4H, THF), 3.01 (s, 3H, CH3-TpMe2), 2.93 (s, 3H, CH3-TpMe2), 2.75 (s, 3H, CH3-TpMe2), 2.64 (s, 3H, CH3-TpMe2), 2.51 (s, 3H, CH3-TpMe2), 2.43 (s, 3H, CH3-TpMe2), 2.15–2.21 (m, 15H, CH3-TpMe2), 2.06 (s, 3H, CH3-TpMe2), 1.29–1.37 (m, 2H, SiCH2P), 1.33 (s, 4H, THF), 1.08–1.22 (m, 4H, SiCH2P), 0.66 (s, 3H, Si(CH3)2), 0.49 (s, 3H, Si(CH3)2), 0.41 (s, 6H, Si(CH3)2), −0.18 (s, 9H, Si(CH3)3), −0.22 (s, 9H, Si(CH3)3); 31P NMR (161 MHz, toluene-d8, 25 °C): δ (ppm) 65.01 (d, 1JYP = 136.4 Hz), −11.89 (d, 1JYP = 120.3 Hz). 1H NMR (400 MHz, C6D6, 25 °C): δ (ppm) 5.64 (s, 3H, 4-H-TpMe2), 5.53 (s, 2H, 4-H-TpMe2), 5.46 (s, 1H, 4-H-TpMe2), 3.78 (m, 4H, THF), 3.15 (s, 3H, CH3-TpMe2), 2.96 (s, 3H, CH3-TpMe2), 2.81 (s, 3H, CH3-TpMe2), 2.67 (s, 3H, CH3-TpMe2), 2.56 (s, 3H, CH3-TpMe2), 2.49 (s, 3H, CH3-TpMe2), 2.11–2.21 (m, 15H, CH3-TpMe2), 2.05 (s, 3H, CH3-TpMe2), 1.29–1.37 (m, 2H, SiCH2P), 1.17 (m, 2 + 4H, SiCH2P + THF), 0.74 (s, 3H, Si(CH3)2), 0.57 (s, 3H, Si(CH3)2), 0.45 (s, 6H, Si(CH3)2), −0.09 (s, 9H, Si(CH3)3), −0.13 (s, 9H, Si(CH3)3); 13C NMR (100 MHz, C6D6, 25 °C): δ (ppm) 151.55 (CO), 151.42 (TpMe2), 150.31 (TpMe2), 150.22 (TpMe2), 150.10 (TpMe2), 149.65 (TpMe2), 146.79 (TpMe2), 146.24 (TpMe2), 145.79 (TpMe2), 144.71 (TpMe2), 144.54 (TpMe2), 106.64 (4-C-TpMe2), 106.40 (4-C-TpMe2), 106.09 (4-C-TpMe2), 105.86 (4-C-TpMe2), 72.51 (THF), 25.14 (THF), 19.72 (d, 1JPC = 49.66 Hz, SiCH2P), 15.84 (d, 1JPC = 12.42 Hz, SiCH2P), 15.47 (CH3-TpMe2), 15.33 (CH3-TpMe2), 15.25 (CH3-TpMe2), 15.07 (CH3-TpMe2), 14.84 (CH3-TpMe2), 14.71 (CH3-TpMe2), 14.62 (CH3-TpMe2), 13.21 (CH3-TpMe2), 13.04 (CH3-TpMe2), 12.98 (CH3-TpMe2), 9.00 (Si(CH3)2), 8.44 (s, Si(CH3)2), 8.24 (s, Si(CH3)2), 5.81 (Si(CH3)3), 4.80 (Si(CH3)3), 1.43 (Si(CH3)2); 31P NMR (161 MHz, C6D6, 25 °C): δ (ppm) 66.56 (br s), −11.81 (br s). Elemental analysis: calcd (%) for C47H86N14B2O2Si4P2Y2: C, 45.05; H, 6.92; N, 15.65. Found: C, 44.33; H, 6.77; N, 15.75.Synthesis of 5
A cooled THF solution (3 mL) of PhNCS (27.5 mg, 0.20 mmol) was added slowly to a stirred THF solution (10 mL) of 2 (115.2 mg, 0.10 mmol) at ambient temperature. After stirring for 5 min, the reaction solution was left at room temperature for 2 days to give 5 as colorless crystals (145 mg, 93% yield). Regrettably, the low solubility of 5 precluded the acquisition of its NMR spectra. Elemental analysis: calcd (%) for C64H102N16B2O2Si4P2S2Y2: C, 49.10; H, 6.57; N, 14.32. Found: C, 49.74; H, 6.52; N, 14.59.Synthesis of 6 and 7
A cooled THF solution (5 mL) of PhNCO (47.6 mg, 0.40 mmol) was added slowly to a stirred THF solution (10 mL) of 2 (230 mg, 0.20 mmol) at ambient temperature. The color of the reaction solution gradually changed from yellow to colorless. After stirring at ambient temperature for 12 h, the solution was concentrated to ca. 3 mL under reduced pressure. After allowing to stand at room temperature for several days, colorless crystals of complex 6 were obtained (169 mg, 55% yield). However, if the reaction mixture was directly evaporated to dryness under vacuum, washing the residue with hexane three times (10 mL × 3) followed by crystallization in THF/hexane would lead to the loss of one coordinated THF, affording 7 as a crystalline solid (267 mg, 87% yield). Crystals suitable for X-ray diffraction analysis were obtained by gas phase diffusion of hexane into the THF solution of 7. Notably, retransformation of 7 into 6 is quite difficult to perform in THF at room temperature, whereas transformation of 6 into 7 occurred slowly under vacuum conditions or exposure to nitrogen gas too. For example, the 31P NMR monitoring data indicate the formation of a small amount of 7 after storing the solution of 6 in THF-d8 at room temperature for 1 week (Fig. S14†). Therefore, compound 6 is not characterized by EA and NMR spectra. For 7: 1H NMR (400 MHz, THF-d8, 25 °C): δ (ppm) 7.32 (d, 2H, 3JHH = 7.72 Hz, Ph), 7.03 (t, 2H, 3JHH = 7.32 Hz, Ph), 6.73 (t, 1H, 3JHH = 7.2 Hz, Ph), 6.27 (t, 1H, 3JHH = 7.08 Hz, Ph), 5.91 (m, 2H, Ph), 5.89 (s, 1H, 4-H-TpMe2), 5.82 (s, 1H, 4-H-TpMe2), 5.74 (s, 1H, 4-H-TpMe2), 5.59 (m, 2H, Ph), 5.54 (s, 1H, 4-H-TpMe2), 5.27 (s, 1H, 4-H-TpMe2), 5.27 (s, 1H, 4-H-TpMe2), 3.62 (m, 4H, THF), 2.57 (s, 6H, CH3-TpMe2), 2.49 (s, 3H, CH3-TpMe2), 2.45 (s, 3H, CH3-TpMe2), 2.35 (s, 12H, CH3-TpMe2), 2.31 (s, 3H, CH3-TpMe2), 2.24 (s, 3H, CH3-TpMe2), 2.04 (s, 3H, CH3-TpMe2), 1.89–1.86 (m, 2H, SiCH2P), 1.77 (m, 4H, THF), 1.61–1.54 (m, 2H, SiCH2P), 1.47 (s, 3H, CH3-TpMe2), 0.46 (s, 3H, Si(CH3)2), 0.44 (s, 3H, Si(CH3)2), 0.36 (s, 3H, Si(CH3)2), 0.21 (s, 3H, Si(CH3)2), −0.45 (s, 9H, Si(CH3)3), −0.67 (s, 9H, Si(CH3)3); 13C{1H} NMR (100 MHz, THF-d8, 25 °C): δ (ppm) 181.61 (t, 1JPC = 5.0 Hz, PCO), 165.83 (q, 1JPC = 3.0 Hz, PCN), 151.38 (TpMe2), 150.44 (TpMe2), 150.42 (TpMe2), 149.96 (TpMe2), 149.69 (TpMe2), 149.54 (TpMe2), 146.34 (TpMe2), 146.04 (TpMe2), 144.88 (TpMe2), 144.49 (TpMe2), 128.11 (Ph), 127.44 (Ph), 126.72 (Ph), 125.34 (Ph), 124.84 (Ph), 123.61 (Ph), 122.27 (Ph), 121.01 (Ph), 106.71 (4-C-TpMe2), 106.48 (4-C-TpMe2), 106.24 (4-C-TpMe2), 105.82 (4-C-TpMe2), 105.20 (4-C-TpMe2), 104.74 (4-C-TpMe2), 68.02 (THF), 26.16 (THF), 15.43 (CH3-TpMe2), 15.39 (CH3-TpMe2), 15.06 (CH3-TpMe2), 14.96 (CH3-TpMe2), 14.33 (CH3-TpMe2), 14.13 (CH3-TpMe2), 14.06 (d, 1JPC = 27.45 Hz, SiCH2P), 13.06 (d, 1JPC = 4.23 Hz, SiCH2P), 12.93 (CH3-TpMe2), 12.91 (CH3-TpMe2), 12.87 (CH3-TpMe2), 12.69 (CH3-TpMe2), 9.64 (d, 3JPC = 5.09 Hz, Si(CH3)2), 7.59 (d, 3JPC = 10.91 Hz, Si(CH3)2), 6.31 (Si(CH3)3), 6.17 (Si(CH3)2), 4.68 (Si(CH3)3), 3.81 (d, 3JPC = 4.28 Hz, Si(CH3)2); 31P NMR (161 MHz, THF-d8, 25 °C): δ (ppm) −26.71 (dm, 1JPP = 374 Hz, PY), −39.64 (dd, JPP = 374, 12.8 Hz, free P). Elemental analysis: calcd (%) for C60H94N16B2O3Si4P2Y2: C, 49.32; H, 6.48; N, 15.34. Found: C, 49.59; H, 6.34; N, 15.57.
Synthesis of 8
A THF solution (5 mL) of PhNNPh (39.6 mg, 0.20 mmol) was added slowly to a stirred THF solution (10 mL) of 2 (115 mg, 0.10 mmol) at ambient temperature. The colour of the reaction mixture gradually changed from yellow to greenish black. After stirring for 2 days, the solvent was removed under vacuum and the solid residue was washed twice with n-hexane (5 mL × 2) and dried under vacuum to give a yellow powder of 8 (141 mg, 85% yield). Yellow crystals suitable for X-ray diffraction analysis were obtained by solvent evaporation from a concentrated THF solution (ca. 2 mL) of 8. No satisfied 13C NMR data of 8 were obtained due to its poor solubility. 1H NMR (400 MHz, THF-d8, 25 °C): δ (ppm) 7.31 (d, 4H, 3JHH = 7.92 Hz, –C6H5), 6.97 (t, 3JHH = 7.88 Hz, 6H, –C6H5), 6.70 (br, 2H, –C6H5), 6.63 (t, 3JHH = 7.12 Hz, 2H, –C6H5), 6.30 (br, 2H, –C6H5), 6.13 (t, 3JHH = 6.4 Hz, 4H, –C6H5), 5.87 (s, 2H, 4-H-TpMe2), 5.82 (s, 2H, 4-H-TpMe2), 5.60 (s, 2H, 4-H-TpMe2), 3.60 (m, 4H, THF), 2.64 (s, 6H, CH3-TpMe2), 2.52 (s, 6H, CH3-TpMe2), 2.48 (s, 6H, CH3-TpMe2), 2.46 (s, 6H, CH3-TpMe2), 2.45 (s, 6H, CH3-TpMe2), 2.39–2.41 (m, 4H, SiCH2P), 2.11 (s, 6H, CH3-TpMe2), 1.77 (m, 4H, THF), 0.14 (s, 6H, Si(CH3)2), −0.38 (s, 18H, Si(CH3)3), −0.56 (s, 6H, Si(CH3)2); 31P NMR (161 MHz, THF-d8, 25 °C): δ (ppm) 50.66 (s). Elemental analysis: calcd (%) for C74H112N18B2O2Si4P2Y2: C, 53.56; H, 6.80; N, 15.19. Found: C, 53.27; H, 6.73; N, 15.27.Synthesis of 9
A THF solution (5 mL) of S8 (12.8 mg, 0.050 mmol) was added slowly to a stirred THF solution (10 mL) of 2 (115 mg, 0.10 mmol) at ambient temperature. After stirring for 5 min, the reaction solution was allowed to stand at room temperature for three days to give 9 as colorless crystals (120 mg, 94% yield). No satisfied NMR data of 9 were obtained due to its poor solubility even in heating THF-d8. Elemental analysis: calcd (%) for C42H78N14B2S4Si4P2Y2: C, 39.37; H, 6.14; N, 15.31. Found: C, 38.53; H, 6.03; N, 15.26.Synthesis of 10
Se powder (23.7 mg, 0.30 mmol) was added to a stirred THF solution (10 mL) of 2 (230 mg, 0.20 mmol) at ambient temperature. The color of the mixture gradually changed from greenish black to colorless. After stirring overnight and removing the solvent, a colorless solid was obtained, which was washed twice with hexane (10 mL × 2) and dried under vacuum to give 10 as a colorless powder (243 mg, 83% yield). Colorless crystals suitable for X-ray diffraction analysis were obtained by gas-phase diffusion of hexane into a concentrated THF solution (ca. 2 mL) of 10. 1H NMR (400 MHz, THF-d8, 25 °C): δ (ppm) 5.86 (s, 1H, 4-H-TpMe2), 5.83 (s, 3H, 4-H-TpMe2), 5.71 (s, 1H, 4-H-TpMe2), 5.60 (s, 1H, 4-H-TpMe2), 3.61 (m, 4H, THF), 2.75 (s, 3H, CH3-TpMe2), 2.63 (s, 6H, CH3-TpMe2), 2.60 (s, 3H, CH3-TpMe2), 2.50 (s, 3H, CH3-TpMe2), 2.44 (s, 12H, CH3-TpMe2), 2.42 (s, 3H, CH3-TpMe2), 2.34 (s, 3H, CH3-TpMe2), 2.30 (s, 3H, CH3-TpMe2), 2.12–2.27 (m, 4H, SiCH2P), 1.77 (m, 4H, THF), 1.47 (s, 3H, CH3-TpMe2), 0.42 (s, 3H, Si(CH3)2), 0.32 (s, 3H, Si(CH3)2), 0.30 (s, 6H, Si(CH3)2), −0.40 (s, 9H, Si(CH3)3), −0.50 (s, 9H, Si(CH3)3); 13C NMR (100 MHz, THF-d8, 25 °C): δ (ppm) 151.65 (TpMe2), 151.50 (TpMe2), 151.35 (TpMe2), 151.05 (TpMe2), 150.95 (TpMe2), 150.14 (TpMe2), 147.03 (TpMe2), 146.97 (TpMe2), 146.95 (TpMe2), 146.47 (TpMe2), 145.60 (TpMe2), 144.60 (TpMe2), 106.90 (4-C-TpMe2), 106.84 (4-C-TpMe2), 106.73 (4-C-TpMe2), 106.66 (4-C-TpMe2), 106.17 (4-C-TpMe2), 68.02 (THF), 34.68 (d, 1JPC = 5.34 Hz, SiCH2P), 34.49 (d, 1JPC = 5.87 Hz, SiCH2P), 26.16 (THF), 17.51 (CH3-TpMe2), 17.28 (CH3-TpMe2), 16.76 (CH3-TpMe2), 15.59 (CH3-TpMe2), 15.48 (CH3-TpMe2), 14.77 (CH3-TpMe2), 12.97 (CH3-TpMe2), 12.89 (CH3-TpMe2), 12.86 (CH3-TpMe2), 7.54 (d, 3JPC = 3.81 Hz, Si(CH3)2), 7.46 (overlap d, Si(CH3)2), 7.42 (d, 3JPC = 3.65 Hz, Si(CH3)2), 6.80 (d, 3JPC = 5.19 Hz, Si(CH3)2), 5.53 (Si(CH3)3), 4.98 (s, Si(CH3)3); 31P NMR (161 MHz, THF-d8, 25 °C): δ (ppm) 29.97 (d, 1JPP = 390 Hz), 23.68 (d, 1JPP = 390 Hz). Elemental analysis: calcd (%) for C46H86N14B2OSe3Si4P2Y2: C, 37.79; H, 5.93; N, 13.41. Found: C, 37.53; H, 5.81; N, 13.29.X-ray crystallographic analysis method
Suitable crystals were wrapped in mineral oil and then were frozen at 173 or 223 K. Data collections were performed on a Bruker SMART APEX (at 293 K) or Bruker SMART APEX (II) (at 173 or 293 K) diffractometer with a CCD area detector using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). Diffraction data were collected over the full sphere and corrected for absorption. Structure solutions were found with the SHELXS42 package using direct methods and were refined with the SHELXTL program43 against F2 using first isotropic and late anisotropic thermal parameters for all non-hydrogen atoms. Hydrogen atoms were placed at calculated positions and included in the structure calculation without further refinement of the parameters. The residual electron densities were of no chemical significance. Details of SQUEEZE are given in the cif files. Unfortunately, the precision of compounds 5 and 6 was limited by the poor quality of their crystals.Conclusions
In summary, we have developed a simple and effective method for the direct preparation of an amido-functionalized diphosphanato metal complex from P4 and the corresponding silyl-bridged amido/methylene yttrium complex. Moreover, it is found that the resulting diphosphanato yttrium complex displays some distinctive reactivities unprecedented in diphosphine chemistry, such as redox metathesis/cyclization of the P2 unit with CS2, and multichalcogenylations of the P2 unit with S8 and Se without the P–P bond cleavage, which provide access to previously unattainable but potentially useful classes of new polyfunctional diphosphine-based ligands in a convenient metal-coordinated form. The results presented here not only demonstrate that appending a strongly coordinative substituent to the alkyl ligand together with a coordination to rare earth ions is an efficient strategy for controlling the alkylated cleavage modes of P4, but also highlight the potential of the diphosphanato yttrium complex as a versatile P2 synthon in the development of new polyfunctional diphosphine ligands, achieved by the controllable Y–P and P–P bond insertions, P-based oxidation or their combination with other types of reactions. This valuable discovery opens up new pathways for the construction of cyclic and acyclic polyfunctional diphosphorus-containing organic ligands from white phosphorus and inexpensive, relatively simple and readily available small molecule substrates with perfect atom economy. This valuable discovery makes a previously unattainable class of dppm-like ligands, with varied substituents on the phosphorus donors, readily available, even those thought to be inaccessible due to the instability of the free diphosphinothioether. More research is currently underway to explore the chemistry of this underutilized class of polyfunctional diphosphine-based ligands.Author contributions
All authors have given approval to the final version of the manuscript. F. Z. and K. H. synthesized and analyzed all new compounds. J. Z. conceived the idea and supervised the work. J. Z. and X. Z. interpreted the results. I. R. and L. M. conducted DFT calculations. All authors contributed to the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 21871058, 22271051, and 22371049) and the 973 program (2015CB856600). LM is a member of the Institut Universitaire de France. Fudan University and the Chinese Academy of Science are acknowledged for financial support through visiting grants (LM). The authors also acknowledge the HPCs CALcul en Midi-Pyrénées (CALMIP-EOS grant 1415). We also greatly acknowledge Dr Yuejian Lin for single crystal X-ray diffraction analysis.References
- For reviews on the application of diphosphines in catalysis, see: (a) W. Tang and X. Zhang, New Chiral Phosphorus Ligands for Enantioselective Hydrogenation, Chem. Rev., 2003, 103, 3029 CrossRef CAS PubMed; (b) M. Berthod, G. Mignani, G. Woodward and M. Lemaire, Modified BINAP: The How and the Why, Chem. Rev., 2005, 105, 1801 CrossRef CAS PubMed; (c) Y.-M. Li, F.-Y. Kwong, W.-Y. Yu and A. S. C. Chan, Recent Advances in Developing New Axially Chiral Phosphine Ligands for Asymmetric Catalysis, Coord. Chem. Rev., 2007, 251, 2119 CrossRef CAS.
- For reviews on metal complexes bearing diphosphine ligands, see: (a) B. Chaudret, B. Delavaux and R. Poilblanc, Bisdiphenylphosphinomethane in Dinuclear Complexes, Coord. Chem. Rev., 1988, 86, 191 CrossRef CAS; (b) C. A. Besse, P. Aggarwal, A. C. Marschilok and K. J. Takeuchi, Transition-Metal Complexes Containing Trans-Spanning Diphosphine Ligands, Chem. Rev., 2001, 101, 1031 CrossRef PubMed; (c) M. Knorr and I. Jourdain, Activation of Alkynes by Diphosphine- and μ-Phosphido-Spanned Heterobimetallic Complexes, Coord. Chem. Rev., 2017, 350, 217 CrossRef CAS.
- (a) S. L. James, Phosphines as Building Blocks in Coordination-Based Self-Assembly, Chem. Soc. Rev., 2009, 38, 1744 RSC; (b) K. Konishi, M. Iwasaki and Y. Shichibu, Phosphine-Ligated Gold Clusters with Core+exo Geometries: Unique Properties and Interactions at the Ligand–Cluster Interface, Acc. Chem. Res., 2018, 51, 3125 CrossRef CAS PubMed.
- (a) M. Driess, A. D. Fantu, D. R. Powell and R. West, Synthesis, Characterization, and Complexation of an Unusual P2Si2 Bicyclobutane with Butterfly-Structure: 2,2,4,4-Tetramesityl-1,3-diphospha-2,4-disilabicyclo[1.1.0]butane, Angew. Chem., Int. Ed. Engl., 1989, 28, 1038 CrossRef; (b) J.-C. Hierso, R. Smaliy, R. Amardeil and P. Meunier, New Concepts in Multidentate Ligand Chemistry: Effects of multidentarity on Catalytic and Spectroscopic Properties of Ferrocenyl Polyphosphines, Chem. Soc. Rev., 2007, 36, 1754 RSC; (c) M.-N. Birkholz, Z. Freixab and P. W. N. M. van Leeuwen, Bite Angle Effects of Diphosphines in C–C and C–X Bond Forming Cross Coupling Reactions, Chem. Soc. Rev., 2009, 38, 1099 RSC; (d) M. Zablocka, A. Hameau, A.-M. Caminade and J.-P. Majoral, “Cage-Like” Phosphines: Design and Catalytic Properties, Adv. Synth. Catal., 2010, 352, 2341 CrossRef CAS; (e) S. S. Sen, S. Khan, H. W. Roesky, D. Kratzert, K. Meindl, J. Henn, D. Stalke, J.-P. Demers and A. Lange, Zwitterionic Si-C-Si-P and Si-P-Si-P Four-Membered Rings with Two-Coordinate Phosphorus Atoms, Angew. Chem., Int. Ed., 2011, 50, 2322 CrossRef CAS PubMed.
- (a) N. A. Piro, J. S. Figueroa, J. T. McKellar and C. C. Cummins, Triple-Bond Reactivity of Diphosphorus Molecules, Science, 2006, 313, 1276 CrossRef CAS PubMed; (b) A. Velian and C. C. Cummins, Synthesis and Characterization of P2N3−: An Aromatic Ion Composed of Phosphorus and Nitrogen, Science, 2015, 348, 1001 CrossRef CAS PubMed; (c) C. Hering-Junghans and E. Rivard, Accessing an Aromatic Diphosphatriazolate Anion by Formal Inorganic “Click” Chemistry, Angew. Chem., Int. Ed., 2015, 54, 10077 CrossRef CAS PubMed; (d) K. Helmdach, S. Ludwig, A. Villinger, D. Hollmann, J. Kösters and W. W. Seidel, Synthesis and Activation Potential of an Open Shell Diphosphine, Chem. Commun., 2017, 53, 5894 RSC; (e) A. J. Arggelles, S. Sun, B. G. Budaitis and P. Nagorny, Design, Synthesis, and Application of Chiral C2-Symmetric Spiroketal-Containing Ligands in Transition-Metal Catalysis, Angew. Chem., Int. Ed., 2018, 57, 5325 CrossRef PubMed.
- (a) O. Back, G. Kuchenbeiser, B. Donnadieu and G. Bertrand, Nonmetal-Mediated Fragmentation of P4: Isolation of P1 and P2 Bis(carbene) Adducts, Angew. Chem., Int. Ed., 2009, 48, 5530 CrossRef CAS PubMed; (b) D. Tofan and C. C. Cummins, Photochemical Incorporation of Diphosphorus Units into Organic Molecules, Angew. Chem., Int. Ed., 2010, 49, 7516 CrossRef CAS PubMed; (c) O. Back, B. Donnadieu, P. Parameswaran, G. Frenking and G. Bertrand, Isolation of Crystalline Carbene-Stabilized P2-Radical Cations and P2-Dications, Nat. Chem., 2010, 2, 369 CrossRef CAS PubMed; (d) I. Knopf, D. Tofan, D. Beetstra, A. Al-Nezari, K. Al-Bahily and C. C. Cummins, A Family of cis-Macrocyclic Diphosphines: Modular, Stereoselective Synthesis and Application in Catalytic CO2/Ethylene Coupling, Chem. Sci., 2017, 8, 1463 RSC.
- (a) H. Shimizu, I. Nagasaki, K. Matsumura, N. Sayo and T. Saito, Developments in Asymmetric Hydrogenation from an Industrial Perspective, Acc. Chem. Res., 2007, 40, 1385 CrossRef CAS PubMed; (b) J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Recent Advances in Transition Metal-Catalyzed Enantioselective Hydrogenation of Unprotected Enamines, Chem. Soc. Rev., 2012, 41, 4126 RSC; (c) J. Barwick-Silk, S. Hardy, M. C. Willis and A. S. Weller, Rh(DPEPhos)-Catalyzed Alkyne Hydroacylation Using β-Carbonyl-Substituted Aldehydes: Mechanistic Insight Leads to Low Catalyst Loadings that Enables Selective Catalysis on Gram-Scale, J. Am. Chem. Soc., 2018, 140, 7347 CrossRef CAS PubMed.
- J. R. Dilworth and N. Wheatley, The Preparation and Coordination Chemistry of Phosphorus Sulfur Donor Ligands, Coord. Chem. Rev., 2000, 199, 89 CrossRef CAS.
-
(a) L.-C. Liang, Metal Complexes of Chelating Diarylamido Phosphine Ligands, Coord. Chem. Rev., 2006, 250, 1152 CrossRef CAS;
(b) M. T. Whited and R. H. Grubbs, Late Metal Carbene Complexes Generated by Multiple C−H Activations: Examining the Continuum of MC Bond Reactivity, Acc. Chem. Res., 2009, 42, 1607 CrossRef CAS PubMed;
(c) M. J. Sgro and D. W. Stephan, Frustrated Lewis Pair Inspired Carbon Dioxide Reduction by a Ruthenium Tris(aminophosphine) Complex, Angew. Chem., Int. Ed., 2012, 51, 11343 CrossRef CAS PubMed;
(d) R. M. Bullock and M. L. Helm, Molecular Electrocatalysts for Oxidation of Hydrogen Using Earth-Abundant Metals: Shoving Protons Around with Proton Relays, Acc. Chem. Res., 2015, 48, 2017 CrossRef CAS PubMed;
(e) P. Bhattacharya, D. E. Prokopchuk and M. T. Mock, Exploring the Role of Pendant amines in Transition Metal Complexes for the Reduction of N2 to Hydrazine and Ammonia, Coord. Chem. Rev., 2017, 334, 67 CrossRef CAS.
- J.-P. Genet, T. Ayad and V. Ratovelomanana-Vidal, Electron-Deficient Diphosphines: The Impact of DIFLUORPHOS in Asymmetric Catalysis, Chem. Rev., 2014, 114, 2824 CrossRef CAS PubMed.
- M. S. Balakrishna, P. Chandrasekaran and P. P. George, Silicon Based Phosphines with P-Si-P, P-C-Si-C-P and P-O-Si-O-P Linkages and Their Coordination Chemistry and Catalytic Applications, Coord. Chem. Rev., 2003, 241, 87 CrossRef CAS.
- Y. Zhang, M. Schulz, M. Wächtler, M. Karnahl and B. Dietzek, Heteroleptic Diimine–Diphosphine Cu(I) Complexes as an Alternative towards Noble-Metal Based Photosensitizers: Design Strategies, Photophysical Properties and Perspective Applications, Coord. Chem. Rev., 2018, 356, 127 CrossRef CAS.
- (a) A. H. Cowley, D. M. Giolando, C. M. Nunn, M. Pakulski, D. Westmoreland and N. C. Norman, Synthesis and Reactivity of Mononuclear Molybdenum Phosphido Complexes, [Mo(CO)2{P(Cl)R}(η-C5H5)][R = CH(SiMe3)2 or NCMe2CH2CH2CH2CMe2]. X-Ray Crystal Structures of [Mo(CO)2{P(X)(NCMe2CH2CH2CH2CMe2)}(η-C5H5)](X = Cl or NMe2) and [Mo2(CO)4{μ-P2[CH(SiMe3)2]2}(η-C5H5)2], J. Chem. Soc., Dalton Trans., 1988, 2127 RSC; (b) Y. Lv, C. E. Kefalidis, J. Zhou, L. Maron, X. Leng and Y. Chen, Versatile Reactivity of a Four-Coordinate Scandium Phosphinidene Complex: Reduction, Addition, and CO Activation Reactions, J. Am. Chem. Soc., 2013, 135, 14784 CrossRef CAS PubMed.
- (a) M. Scheer, G. Balázs and A. Seitz, P4 Activation by Main Group Elements and Compounds, Chem. Rev., 2010, 110, 4236 CrossRef CAS PubMed; (b) B. M. Cossairt, N. A. Piro and C. C. Cummins, Early-Transition-Metal-Mediated Activation and Transformation of White Phosphorus, Chem. Rev., 2010, 110, 4164 CrossRef CAS PubMed; (c) M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, P4 Activation by Late-Transition Metal Complexes, Chem. Rev., 2010, 110, 4178 CrossRef CAS PubMed; (d) N. A. Giffin and J. D. Masuda, Reactivity of White Phosphorus with Compounds of the p-Block, Coord. Chem. Rev., 2011, 255, 1342 CrossRef CAS.
- N. Etkin, M. T. Benson, S. Courtenay, M. J. McGlinchey, A. D. Bain and D. W. Stephan, Niobium and Tantalum Diphosphanato Complexes: Synthesis, Structure, and NMR Studies of Cp2MH[(PR)2] (R = Ph, Cy, H), Organometallics, 1997, 16, 3504 CrossRef CAS.
- (a) R. Riedel, H.-D. Hausen and E. Fluck, Bis(2,4,6-tri-tert-butylphenyl)bicyclotetraphosphane, Angew. Chem., Int. Ed. Engl., 1985, 24, 1056 CrossRef; (b) A. Hübner, T. Bernert, I. Sänger, E. Alig, M. Bolte, L. Fink, M. Wagner and H.-W. Lerner, Solvent-Free Mesityllithium: Solid-State Structure and Its Reactivity towards White Phosphorus, Dalton Trans., 2010, 39, 7528 RSC.
- T. Arnold, H. Braunschweig, J. O. C. Jimenez-Halla, K. Radacki and S. S. Sen, Simultaneous Fragmentation and Activation of White Phosphorus, Chem. – Eur. J., 2013, 19, 9114 CrossRef CAS PubMed.
- (a) L. Xu, Y. Chi, S. Du, W.-X. Zhang and Z.-F. Xi, Direct Synthesis of Phospholyl Lithium from White Phosphorus, Angew. Chem., Int. Ed., 2016, 55, 9187 CrossRef CAS PubMed; (b) S. K. Ghosh, C. C. Cummins and J. A. Gladysz, A Direct Route from White Phosphorus and Fluorous Alkyl and Aryl Iodides to the Corresponding Trialkyl- and Triarylphosphines, Org. Chem. Front., 2018, 5, 3421 RSC.
- (a) A. R. Fox, R. J. Wright, E. Rivard and P. P. Power, Tl2[Aryl2P4]: A Thallium Complexed Diaryltetraphosphabutadienediide and its Two-Electron Oxidation to a Diaryltetraphosphabicyclobutane, Aryl2P4, Angew. Chem., Int. Ed., 2005, 44, 7729 CrossRef CAS PubMed; (b) S. Heinl, S. Reisinger, C. Schwarzmaier, M. Bodensteiner and M. Scheer, Selective Functionalization of P4 by Metal-Mediated C-P Bond Formation, Angew. Chem., Int. Ed., 2014, 53, 7639 CrossRef CAS PubMed; (c) J. E. Borger, A. W. Ehlers, M. Lutz, J. C. Slootweg and K. Lammertsma, Functionalization of P4 Using a Lewis Acid Stabilized Bicyclo[1.1.0]tetraphosphabutane Anion, Angew. Chem., Int. Ed., 2014, 53, 12836 CrossRef CAS PubMed; (d) M. Arrowsmith, M. S. Hill, A. L. Johnson, G. Kociok-Köhn and M. F. Mahon, Attenuated Organomagnesium Activation of White Phosphorus, Angew. Chem., Int. Ed., 2015, 54, 7882 CrossRef CAS PubMed; (e) J. E. Borger, A. W. Ehlers, M. Lutz, J. C. Slootweg and K. Lammertsma, Stabilization and Transfer of the Transient [Mes*P4]− Butterfly Anion Using BPh3, Angew. Chem., Int. Ed., 2016, 55, 613 CrossRef CAS PubMed; (f) S. Du, J. Yin, Y. Chi, L. Xu and W.-X. Zhang, Dual Functionalization of White Phosphorus: Formation, Characterization, and Reactivity of Rare-Earth-Metal Cyclo-P3 Complexes, Angew. Chem., Int. Ed., 2017, 56, 15886 CrossRef CAS PubMed.
- (a) O. J. Scherer, M. Ehses and G. Wolmershäuser, Activation of P4 and P2 by Transition Metal Complexes at Room Temperature, Angew. Chem., Int. Ed., 1998, 37, 507 CrossRef CAS; (b) J. S. Figueroa and C. C. Cummins, The Niobaziridine−Hydride Functional Group: Synthesis and Divergent Reactivity, J. Am. Chem. Soc., 2003, 125, 4020 CrossRef CAS PubMed; (c) E. B. Hulley, P. T. Wolczanski and E. B. Lobkovsky, [(silox)3M]2(μ:η1,η1-P2) (M = Nb, Ta) and [(silox)3Nb]2{μ:η2,η2-(cP3−cP3)} from (silox)3M (M = NbPMe3, Ta) and P4 (silox = tBu3SiO), Chem. Commun., 2009, 6412 RSC; (d) M. Demange, X.-F. Goff, P. L. Floch and N. Mézailles, P4 Activation with Pt0 Metal Centers: Selective Formation of a Dinuclear {Pt2(μ,η2:2-P2)} Complex, Chem. – Eur. J., 2010, 16, 12064 CrossRef CAS PubMed.
- (a) L. Weber, The Chemistry of Diphosphenes and Their Heavy Congeners: Synthesis, Structure, and Reactivity, Chem. Rev., 1992, 92, 1839 CrossRef CAS; (b) J. Y. Hu, W. Liu and W. X. Zhang, Direct functionalization of white phosphorus by organolithium reagents to organophosphorus compounds, Phosphorus, Sulfur Silicon Relat. Elem., 2022, 197(5–6), 398–407 CrossRef CAS; (c) G. Luo, S. Du, P. Wang, F. Liu, W. X. Zhang and Y. Luo, Fragmentation Mechanism of White Phosphorus: A Theoretical Insight into Multiple Cleavage/Formation of P−P and P−C Bonds, Chem. – Eur. J., 2020, 26, 13282 CrossRef CAS PubMed; (d) Z. J. Lv, Z. Huang, J. H. Shen, W. X. Zhang and Z. F. Xi, Well-Defined Scandacyclopropenes: Synthesis, Structure, and Reactivity, J. Am. Chem. Soc., 2019, 141, 20547 CrossRef CAS PubMed.
- (a) P. Coburger, S. Demeshko, C. Rödl, E. Hey-Hawkins and R. Wolf, Oxidative P-P-Bindungsaddition an Cobalt(−I): Bildung eines Low-spin-Cobalt(III)-Phosphanidokomplexes, Angew. Chem., Int. Ed., 2017, 56, 15871 CrossRef CAS PubMed; (b) Y.-E. Kim and Y. Lee, A P−P Bond as a Redox Reservoir and an Active Reaction Site, Angew. Chem., Int. Ed., 2018, 57, 14159 CrossRef CAS PubMed.
- (a) H. Schäfer, D. Binder and D. Fenske, Chelate-Stabilized Diphosphene and Diphosphorus Complexes of Nickel, Angew. Chem., Int. Ed. Engl., 1985, 24, 522 CrossRef; (b) S. Xin, H. G. Woo, J. F. Harrod, E. Samuel and A.-M. Lebuis, Synthesis and Crystal Structure of Some Novel Titanocene Phosphido Compounds by P−H Activation in the Presence of Hydrosilanes, J. Am. Chem. Soc., 1997, 119, 5307 CrossRef CAS; (c) P. M. Scheetz, D. S. Glueck and A. L. Rheingold, Rhodium-Catalyzed Isomerization of a Bis(secondary phosphine) to an Unsymmetrical Diphosphine via P–C Cleavage and P–P and C–H Bond Formation, Organometallics, 2017, 36, 3387 CrossRef CAS.
- (a) J. Hong, H. Tian, L. Zhang, X. Zhou, I. Rosal, L. Weng and L. Maron, Reversing Conventional Reactivity of Mixed Oxo/Alkyl Rare-Earth Complexes: Non-Redox Oxygen Atom Transfer, Angew. Chem., Int. Ed., 2018, 57, 1062 CrossRef CAS PubMed; (b) J. Zhou, L. Xiang, J. Guo, X. Leng and Y. Chen, Formation and Reactivity of a C-P-N-Sc Four-Membered Ring: H2, O2, CO, Phenylsilane, and Pinacolborane Activation, Chem. – Eur. J., 2017, 23, 5424 CrossRef CAS PubMed; (c) V. Radkov, T. Roisnel, A. Trifonov, J.-F. Carpentier and E. Kirillov, Tandem C(sp2)–OMe Activation/C(sp2)–C(sp2) Coupling in Early Transition-Metal Complexes: Aromatic C–O Activation beyond Late Transition Metals, J. Am. Chem. Soc., 2016, 138, 4350 CrossRef CAS PubMed; (d) M. E. Fieser, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, Dinitrogen Reduction via Photochemical Activation of Heteroleptic Tris(cyclopentadienyl) Rare-Earth Complexes, J. Am. Chem. Soc., 2013, 135, 3804 CrossRef CAS PubMed.
- (a) J.-B. Zhu, E. M. Watson, J. Tang and E. Y.-X. Chen, A Synthetic Polymer System with Repeatable Chemical Recyclability, Science, 2018, 360, 398 CrossRef CAS PubMed; (b) Y. Shao, F. Zhang, J. Zhang and X. Zhou, Lanthanide-Catalyzed Reversible Alkynyl Exchange by Carbon–Carbon Single-Bond Cleavage Assisted by a Secondary Amino Group, Angew. Chem., Int. Ed., 2016, 55, 11485 CrossRef CAS PubMed; (c) T. L. Lohr, Z. Li and T. J. Marks, Thermodynamic Strategies for C–O Bond Formation and Cleavage via Tandem Catalysis, Acc. Chem. Res., 2016, 49, 824 CrossRef CAS PubMed; (d) M. Nishiura, F. Guo and Z. Hou, Half-Sandwich Rare-Earth-Catalyzed Olefin Polymerization, Carbometalation, and Hydroarylation, Acc. Chem. Res., 2015, 48, 2209 CrossRef CAS PubMed.
- (a) Q. You, J. Zhang, F. Zhang, J. Cai and X. Zhou, Cooperative Rare-Earth/Lithium-Mediated Conversion of White Phosphorus, Chem. – Eur. J., 2023, e202203679 CrossRef CAS PubMed; (b) F. Zhang, K. Han, J. Cai, Z. Ye, J. Zhang, X. Zhou and Z. Li, Rare-Earth-Mediated Conversion of White Phosphorus into PBn3 and Highly Functionalized Norbornane-P7, Inorg. Chem. Front., 2024, 11, 478–486 RSC; (c) F. Zhang, J. Zhang, Z. Chen, L. Weng and X. Zhou, An Yttrium Organic Cyclo-P4 Complex and Its Selective Conversions, Inorg. Chem., 2019, 58, 8451 CrossRef CAS PubMed.
- (a) J. Andrez, J. Pécaut, P.-A. Bayle and M. Mazzanti, Tuning Lanthanide Reactivity Towards Small Molecules with Electron-Rich Siloxide Ligands, Angew. Chem., Int. Ed., 2014, 53, 10448 CrossRef CAS PubMed; (b) X.-F. Jiang, H. Huang, Y.-F. Chai, T. L. Lohr, S.-Y. Yu, W.-Z. Lai, Y.-J. Pan, M. Delferro and T. J. Marks, Hydrolytic Cleavage of Both CS2 Carbon–Sulfur Bonds by Multinuclear Pd(II) Complexes at Room Temperature, Nat. Chem., 2017, 9, 188 CrossRef CAS PubMed.
- (a) W. Petz, 40 Years of Transition-Metal Thiocarbonyl Chemistry and the Related CSe and CTe Compounds, Coord. Chem. Rev., 2008, 252, 1689 CrossRef CAS; (b) L. Wang, W. He and Z. Yu, Transition-Metal Mediated Carbon–Sulfur Bond Activation and Transformations, Chem. Soc. Rev., 2013, 42, 599 RSC.
- (a) H. Zhu, J. Chai, Q. Ma, V. Jancik, H. W. Roesky, H. Fan and R. Herbst-Irmer, A Seven-Membered Aluminum Sulfur Allenyl Heterocycle Arising from the Conversion of an Aluminacyclopropene with CS2, J. Am. Chem. Soc., 2004, 126, 10194 CrossRef CAS PubMed; (b) J. Ballmann, A. Yeo, B. A. MacKay, S. V. Rijt, B. O. Patrick and M. D. Fryzuk, Complete Disassembly of Carbon Disulfide by a Ditantalum Complex, Chem. Commun., 2010, 46, 8794 RSC; (c) S. I. Kalläne, T. Braun, M. Teltewskoi, B. Braun, R. Herrmann and R. Laubenstein, Remarkable Reactivity of a Rhodium(I) Boryl Complex towards CO2 and CS2: Isolation of a Carbido Complex, Chem. Commun., 2015, 51, 14613 RSC; (d) M. Falcone, L. Chatelain and M. Mazzanti, Nucleophilic Reactivity of a Nitride-Bridged Diuranium(IV) Complex: CO2 and CS2 Functionalization, Angew. Chem., Int. Ed., 2016, 55, 4074 CrossRef CAS PubMed.
- (a) K. Wang, G. Luo, J. Hong, X. Zhou, L. Weng, Y. Luo and L. Zhang, Homometallic Rare-Earth Metal Phosphinidene Clusters: Synthesis and Reactivity, Angew. Chem., Int. Ed., 2014, 53, 1053 CrossRef CAS PubMed; (b) F. Dielmann and G. Bertrand, Reactivity of a Stable Phosphinonitrene towards Small Molecules, Chem. – Eur. J., 2015, 21, 191 CrossRef CAS PubMed; (c) H. Tian, J. Hong, K. Wang, I. Rosal, L. Maron, X. Zhou and L. Zhang, Unprecedented Reaction Mode of Phosphorus in Phosphinidene Rare-Earth Complexes: A Joint Experimental–Theoretical Study, J. Am. Chem. Soc., 2018, 140, 102 CrossRef CAS PubMed.
- H. Heuclin, X. F. Goff and N. Mézailles, Mixed (P=S/P=O)-Stabilized Geminal Dianion: Facile Diastereoselective Intramolecular C-H Activations by a Related Ruthenium–Carbene Complex, Chem. – Eur. J., 2012, 18, 16136 CrossRef CAS PubMed.
- (a) T. M. Trnka and R. H. Grubbs, The Development of L2X2Ru=CHR Olefin Metathesis Catalysts: An Organometallic Success Story, Acc. Chem. Res., 2001, 34, 18 CrossRef CAS PubMed; (b) I. Nakamura and Y. Yamamoto, Transition-Metal-Catalyzed Reactions in Heterocyclic Synthesis, Chem. Rev., 2004, 104, 2127 CrossRef CAS PubMed; (c) S. Fustero, A. Simón-Fuentes, P. Barrio and G. Haufe, Olefin Metathesis Reactions with Fluorinated Substrates, Catalysts, and Solvents, Chem. Rev., 2015, 115, 871 CrossRef CAS PubMed.
- (a) J. R. Ludwig, P. M. Zimmerman, J. B. Gianino and C. S. Schindler, Iron(III)-Catalysed Carbonyl–Olefin Metathesis, Nature, 2016, 533, 374 CrossRef CAS PubMed; (b) M. A. Aljuhani, S. Barman, E. Abou-Hamad, A. Gurinov, S. Ould-Chikh, E. Guan, A. Jedidi, L. Cavallo, B. C. Gates, J. D. A. Pelletier and J.-M. Basset, Imine Metathesis Catalyzed by a Silica-Supported Hafnium Imido Complex, ACS Catal., 2018, 8, 9445 Search PubMed.
- R. J. Newland, J. M. Lynam and S. M. Mansell, Small Bite-Angle 2-Phosphinophosphinine Ligands Enable Rhodium-Catalysed Hydroboration of Carbonyls, Chem. Commun., 2018, 54, 5482 RSC.
- (a) W. J. Evans, K. J. Forrestal and J. W. Ziller, Synthesis and Structure of a Thermally Stable, Nonclassical, 7-Norbornadienyl Carbocation Obtained from (C5Me5)3Sm and CO, J. Am. Chem. Soc., 1995, 117, 12635 CrossRef CAS; (b) B. Wang, G. Luo, M. Nishiura, Y. Luo and Z. Hou, Cooperative Trimerization of Carbon Monoxide by Lithium and Samarium Boryls, J. Am. Chem. Soc., 2017, 139, 16967 CrossRef CAS PubMed.
- (a) W. J. Evans, D. S. Lee, J. W. Ziller and N. Kaltsoyannis, Trivalent [(C5Me5)2(THF)Ln]2(μ-η2:η2-N2) Complexes as Reducing Agents Including the Reductive Homologation of CO to a Ketene Carboxylate, (μ-η4-O2C-C=C=O)2−, J. Am. Chem. Soc., 2006, 128, 14176 CrossRef CAS PubMed; (b) T. Shima and Z. Hou, Hydrogenation of Carbon Monoxide by Tetranuclear Rare Earth Metal Polyhydrido Complexes. Selective Formation of Ethylene and Isolation of Well-Defined Polyoxo Rare Earth Metal Clusters, J. Am. Chem. Soc., 2006, 128, 8124 CrossRef CAS PubMed; (c) J. Cheng, M. J. Ferguson and J. Takats, Synthesis and Reaction of [(TpiPr2)LnH2]3 (Ln = Y, Lu) with CO: Trinuclear Cluster-Bound Propenolate en Route to Selective Formation of Propene, J. Am. Chem. Soc., 2010, 132, 2 CrossRef CAS PubMed.
- (a) M. H. Holthausen and J. J. Weigand, Preparation of the [(DippNP)2(P4)2]2+-Dication by the Reaction of [DippNPCl]2 and a Lewis Acid with P4, J. Am. Chem. Soc., 2009, 131, 14210 CrossRef CAS PubMed; (b) J. Cui, Y. Li, R. Ganguly, A. Inthirarajah, H. Hirao and R. Kinjo, Metal-Free σ-Bond Metathesis in Ammonia Activation by a Diazadiphosphapentalene, J. Am. Chem. Soc., 2014, 136, 16764 CrossRef CAS PubMed.
- (a) L.-X. Zhang, T. Suzuki, Y. Luo, M. Nishiura and Z. Hou, Cationic Alkyl Rare-Earth Metal Complexes Bearing an Ancillary Bis(phosphinophenyl)amido Ligand: A Catalytic System for Living cis-1,4-Polymerization and Copolymerization of Isoprene and Butadiene, Angew. Chem., Int. Ed., 2007, 46, 1909 CrossRef CAS PubMed; (b) J. Bravo, C. Cativiela, J. E. Chaves, R. Navarro and E. P. Urriolabeitia, 31P NMR Spectroscopy as a Powerful Tool for the Determination of Enantiomeric Excess and Absolute Configurations of α-Amino Acids, Inorg. Chem., 2003, 42, 1006 CrossRef CAS PubMed.
- (a) D. Tofan, B. M. Cossairt and C. C. Cummins, White Phosphorus Activation at a Metal–Phosphorus Triple Bond: a New Route to cyclo-Triphosphorus or cyclo-Pentaphosphorus Complexes of Niobium, Inorg. Chem., 2011, 50, 12349 CrossRef CAS PubMed; (b) D. Tofan, M. Temprado, S. Majumdar, C. D. Hoff and C. C. Cummins, Functionalization Reactions Characteristic of a Robust Bicyclic Diphosphane Framework, Inorg. Chem., 2013, 52, 8851 CrossRef CAS PubMed.
- P. Brunel, J. Monot, C. E. Kefalidis, L. Maron, B. Martin-Vaca and D. Bourissou, Valorization of CO2: Preparation of 2-Oxazolidinones by Metal–Ligand Cooperative Catalysis with SCS Indenediide Pd Complexes, ACS Catal., 2017, 7, 2652 CrossRef CAS.
- W. Yi, J. Zhang, F. Zhang, Y. Zhang, Z. Chen and X. Zhou, Versatile Reactivity of Scorpionate-Anchored Yttrium Dialkyl Complexes towards Unsaturated Substrates, Chem. – Eur. J., 2013, 19, 11975 CrossRef CAS PubMed.
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXL-2014, Program for Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 2014 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1879573 (1), 1879574 (2), 1879577 (2·THF), 1879583 (3), 1879582 (4), 1879576 (5), 1879581 (6), 1879575 (7·0.5C6H14), 1879580 (8), 1879579 (9), and 1879578 (10). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qi02680a |
| This journal is © the Partner Organisations 2024 |