
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the use of rigid 18-membered macrocycles with amide pendant arms for Pb(II)-based radiopharmaceuticals†
Charlene
Harriswangler
 a,
Brooke L.
McNeil
bc,
Isabel
Brandariz-Lendoiro
a,
Fátima
Lucio-Martínez
a,
Laura
Valencia
d,
David
Esteban-Gómez
a,
Caterina F.
Ramogida
*bc and
Carlos
Platas-Iglesias
*a
a,
Brooke L.
McNeil
bc,
Isabel
Brandariz-Lendoiro
a,
Fátima
Lucio-Martínez
a,
Laura
Valencia
d,
David
Esteban-Gómez
a,
Caterina F.
Ramogida
*bc and
Carlos
Platas-Iglesias
*a
aUniversidade da Coruña, Centro Interdisciplinar de Química e Bioloxía (CICA) and Departamento de Química, Facultade de Ciencias, 15071, A Coruña, Galicia, Spain. E-mail: carlos.platas.iglesias@udc.es
bDepartment of Chemistry, Simon Fraser University, Burnaby, BC, Canada. E-mail: caterina_ramogida@sfu.ca
cLife Sciences Division, TRIUMF, Vancouver, BC, Canada
dDepartamento de Química Inorgánica, Facultad de Ciencias, Universidade de Vigo, As Lagoas, Marcosende, 36310 Pontevedra, Spain
First published on 28th December 2023
Abstract
We report a detailed investigation on the use of chelators PYTAM, H4PYTAMGly, CHX-PYTAM and H4CHX-PYTAMGly for the complexation of Pb(II)-radioisotopes with potential use in the preparation of radiopharmaceuticals. The macrocyclic backbones from which the chelators are prepared, PYAN (3,6,10,13-tetraaza-1,8(2,6)-dipyridinacyclotetradecaphane) or CHX-PYAN ((41R,42R,101R,102R)-3,5,9,11-tetraaza-1,7(2,6)-dipyridina-4,10(1,2)-dicyclohexanacyclododecaphane), are readily synthesized in high yields using a Ba(II) template synthesis. The single difference between the two backbones is that while PYAN contains ethylene spacers, CHX-PYAN incorporates more rigid cyclohexyl spacers. The pendant arms incorporated into the backbone were strategically chosen, selecting amide pendants of different nature, compatible with the borderline Lewis acid character of Pb(II). To study the complexation of Pb(II), first, a detailed characterization of the non-radioactive complexes was carried out, including X-ray crystallography, NMR, and the determination of chelator protonation and stability constants of the complexes. Once the complexes were fully characterized and showed favourable properties towards Pb(II), a radiochemical study using the SPECT compatible radioisotope lead-203 was performed. Although all tested chelators sufficiently complexed lead-203 and presented excellent complex stability in human serum, it was found that novel chelator CHX-PYTAM was superior over the others due to its high kinetic inertness. This effect is due to increased rigidity of the complex with cyclohexyl spacers in the backbone and use of primary amides, over secondary amides, as pendant arms and thus this chelator is a promising candidate for future in vivo studies.
Introduction
There are a wide variety of radioactive metals that have potential use in radiopharmaceutical compounds, both for diagnostic and therapeutic applications.1 Within the family of diagnostic isotopes we can find positron (β+) emitters that can be used for positron emission tomography (PET) imaging, such as commonly used gallium-68 (68Ga, t1/2 = 67.7 min), or isotopes that directly emit gamma (γ-) rays for SPECT (single photon emission computed tomography) imaging, such as technetium-99m (99mTc, t1/2 = 6.00 h).2 Therapeutic isotopes, on the other hand, release cytotoxic radiation and can be divided into three categories depending on the type of radiation that is produced: alpha (α)-emitters, beta (β−)-emitters and Auger electron emitters.3Nowadays, the emerging field of theranostics combines both applications of radioisotopes in order to achieve a more personalized treatment.4 First, a diagnostic agent is administered to evaluate the uptake of the radiopharmaceutical and if the uptake is high, the therapeutic is administered subsequently. There are different combinations of isotopes that can be used in this procedure, “true theranostics”, same-element isotope pairs, and different-element pairs.5,6 True theranostic radionuclides emit both imaging compatible radiation and therapeutic particles, such as the β−-emitter lutetium-177 (177Lu, t1/2 = 6.64 days) which emits an accompanying γ-ray compatible with SPECT imaging. Although a true theranostic may be convenient, one of the drawbacks of the use of these isotopes, is the potential for off target effects if there is not significant uptake shown in the image.7 This is why, when evaluating the use of 177Lu for patient treatment, 68Ga is often used for pre-therapy imaging and uptake evaluation.8 However, when using a different-element pair, the pharmacokinetics of both radiopharmaceuticals may not be identical, which could be problematic if the update of the imaging tracer does not accurately reflect the biodistribution of the therapeutic pharmaceutical.5 This can be avoided by using a same element isotope pair, where two different isotopes of the same element are appropriate for either diagnosis or therapy, such as the lead-212 (212Pb, t1/2 = 10.6 h)/lead-203 (203Pb, t1/2 = 51.9 h) theranostic pair.9,10
The 212Pb/203Pb theranostic pair has emerged in recent years as a promising candidate for use in personalized theranostics, which has triggered efforts focused on finding chelators that are adequate to take advantage of the potential of this pair.10–15 The 203Pb-radionuclide emits a 279 keV (81%) γ-photon, and can be produced through proton bombardment of Tl on medical cyclotrons via either the 203Tl (p,n) 203Pb reaction or the 205Tl (p,3n) 203Pb reaction.10,16 The 212Pb-radionuclide emits β−-particles and, indirectly, an α-particle through the decay of its daughters bismuth-212 (212Bi, t1/2 = 60.6 min) or polonium-212 (212Po, t1/2 = 294.3 ns) and can be obtained via different generators based on isotopes from the thorium-228 (228Th, t1/2 = 1.9 years) decay series.17 The half-life of this isotope pairs nicely with the biological half-life of small biomolecules, like peptides, that can bind to targets such as prostate-specific membrane antigen (PSMA) and somatostatin receptors (SSTRs) which are overexpressed in some tumours.18–20 It should be noted that phase 1 clinical trial results using [212Pb]Pb-DOTAMTATE (bifunctional chelator [DOTAM] coupled to SSTR-targeting peptide [TATE]) have recently been published, in which SSTR-expressing neuroendocrine tumours are targeted, with very promising results, showing an objective radiologic response (ORR) of 80% compared to an ORR of 13% obtained with [177Lu]Lu-DOTATATE (bifunctional chelator [DOTA] coupled to SSTR-targeting peptide [TATE]).20
One of the most commonly used strategies to harness the decay properties of these metals in vivo is through the use of bifunctional chelators (BFCs), in which the radiometal of interest is coordinated by a chelator and attached, through a linker, to a biological targeting vector such as a peptide or an antibody.21,22 This allows for the radioactivity to be delivered selectively to the diseased tissue, while minimally damaging surrounding healthy tissue.23 Coordination chemistry plays a key role in the design of these BFCs, as it is necessary to tailor the properties of the chelator to the properties of each specific metal to obtain thermodynamically stable and kinetically inert complexes, to avoid the release of the metals in vivo and possible toxicity.24
When designing a new chelator, Pearson's hard–soft acid–base (HSAB) principle should be considered, so that donor atoms matching the metal ion are incorporated into the structure of the chelator.25 Stable complexation of lead(II), a borderline acid, is generally achieved using amide donor groups, borderline bases, with the CYCLEN (1,4,7,10-tetraazacyclododecane)-based macrocyclic derivative DOTAM being the leading chelator for this metal ion (Scheme 1).26,27 Macrocyclic chelators are often preferred as they are pre-organized in a way such that the entropic penalty associated with coordination is reduced, resulting in thermodynamically stable and kinetically inert complexes.28–30 In recent years, the effect of incorporating rigid spacers into the backbone of different acyclic ligands has been studied, in most occasions resulting in higher kinetic inertness.31–34 However, when studying the effect of the incorporation of a cyclohexyl spacer into the backbone of the 18-membered macrocycle H2MACROPA, the kinetic lability of the resulting lanthanide complexes actually increased.35
 | ||
| Scheme 1 Chelators discussed in this work and numbering scheme used for NMR spectral assignment. | ||
Concerning the use of macrocyclic chelators for biomedical applications, DOTA (2,2′,2′′,2′′′-(1,4,7,10-tetraazacyclododecane-1,4,7,10-tetrayl)tetraacetic acid) is the most commonly used system.36 This platform is based on the azamacrocycle CYCLEN, although macrocycles such as TACN (1,4,7-triazacyclononane) and CYCLAM (1,4,8,11-tetraazacyclotetradecane) have also been extensively investigated.37 The popularity of these systems is mainly due to the presence of amine nitrogen atoms that can be used to functionalise the macrocycle with additional pendant arms to increase the denticity of the systems. Apart from these systems, larger 18-membered macrocycles incorporating ether or pyridine donors, have been studied in recent years for the complexation of large radiometals.38–44 Although not yet reported for radiopharmaceutical uses, PYTAM (Scheme 1) is a decadentate macrocyclic chelator with four acetamide pendant arms, belonging to the PYAN family, first used for the complexation of lanthanides, resulting in very inert complexes.45,46
Herein, we present a study on the coordination ability towards [nat/203Pb]Pb(II) of PYTAM and another three new decadentate hexaazamacrocylic chelators. Additional rigidity was introduced into the PYTAM framework by incorporating two cyclohexyl units, giving rise to the new chelator CHX-PYTAM. To improve solubility of these chelators at more basic pH values, the acetamide groups were substituted with glycinate arms giving chelators H4PYTAMGly and H4CHX-PYTAMGly. The intermediate HSAB character of the primary and secondary amides, along with the pyridyl groups present in the structure of these chelators, is expected to pair nicely with coordination preference of Pb(II). The [natPb]Pb(II) complexes were prepared in situ and studied in solution through a combination of multinuclear (1H, 13C, 207Pb) NMR spectroscopy and DFT calculations. We also report the X-ray crystal structures of two of the complexes. Protonation constants of two of the chelators and the stability constants of the resulting Pb(II) complexes were determined through a combined spectrophotometric and potentiometric approach. Subsequently, radiolabelling and stability studies were performed with [203Pb]Pb(II) produced using a 13 MeV cyclotron. It should be noted that asymmetric functionalization of 3,6,10,13-tetraaza-1,8(2,6)-dipyridinacyclotetradecaphane, the backbone on which the chelators are based, has been recently described,42 paving the way for bifunctional systems based on this platform.
Results and discussion
Synthesis of the chelators
CHX-PYTAM was synthesized following a procedure similar to the one reported previously for PYTAM,45 through alkylation of macrocycle CHX-PYAN with 2-bromoacetamide (Scheme 2), yielding CHX-PYTAM as a white solid (40% yield). The CHX-PYTAM precursor was obtained by a Ba(II)-templated Schiff-base condensation reaction, following a slight modification of the method reported by Jackels.47 H4PYTAMGly and H4CHX-PYTAMGly were prepared through alkylation of the corresponding macrocyclic backbone with tert-butyl (2-chloroacetyl)glycinate followed by deprotection of the tert-butyl groups using TFA, affording the final chelators in 13% and 44% yields, respectively, over the two steps (see Experimental section for details).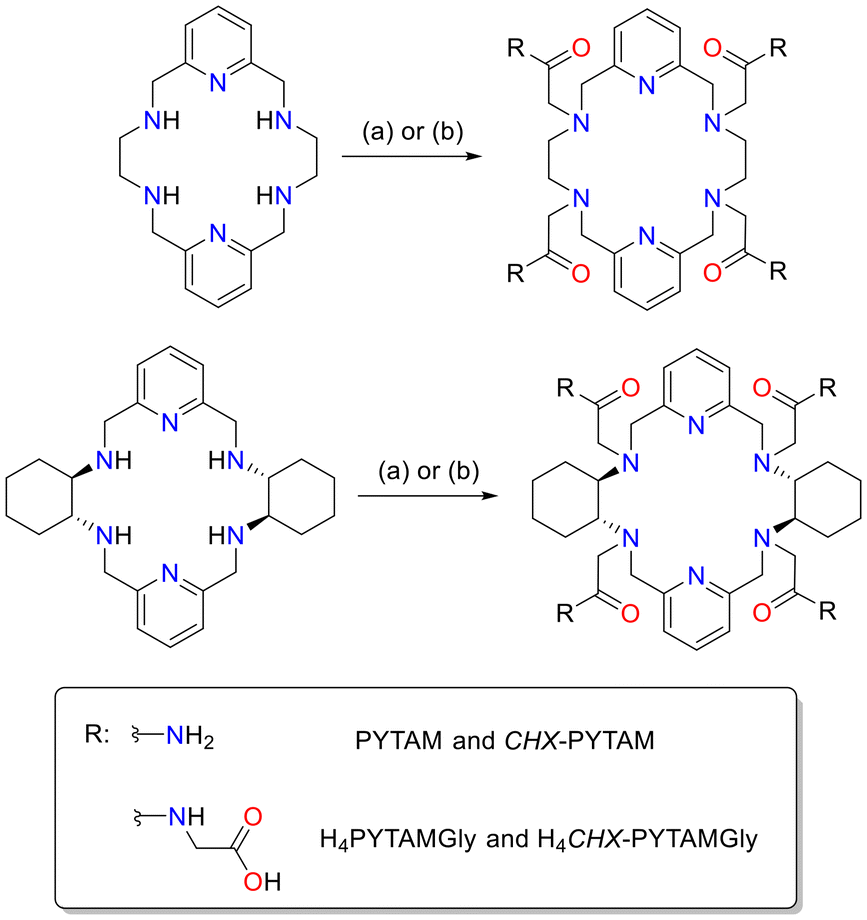 | ||
Scheme 2 General procedure for the synthesis of the chelators. (a) Bromoacetamide and Na2CO3 in acetonitrile. (b) tert-butyl (2-chloroacetyl)glycinate K2CO3 in acetonitrile, followed by 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 TFA:CH2Cl2. :1 TFA:CH2Cl2. | ||
X-ray crystal structures of [Pb(PYTAM)](PF6)2 and [Pb(H4PYTAMGly)](NO3)2·5H2O
Addition of excess KPF6 to an aqueous solution of the [Pb(PYTAM)]2+ complex and slow evaporation of an acidic solution of the [Pb(H4PYTAMGly)]2+ complex provided single crystals suitable for X-ray analysis (Fig. 1 and Fig. S1, S2, ESI†). Bond distances of the Pb(II) coordination environments are listed in Table 1. Compound [Pb(PYTAM)](PF6)2 crystallizes in the centrosymmetric orthorhombic Fddd space group, with the asymmetric unit showing only one quadrant of the molecule due to its crystallographically-imposed D2 symmetry. Crystals contain two non-coordinated PF6− anions involved in hydrogen-bonding interactions with the chelator amide N–H groups. On the other hand, [Pb(H4PYTAMGly)](NO3)2·5H2O crystallizes in the monoclinic P21/c space group and contains a network of water molecules interacting via hydrogen bonds with nitrate anions the protonated terminal carboxylate groups of the glycinate arms. | ||
| Fig. 1 ORTEP views (30%) of the cations present in the crystal structures of [Pb(PYTAM)](PF6)2 [(a)–(c)] and [Pb(H4PYTAMGly)](NO3)2·5H2O [(d)–(f)] with atom labelling. Hydrogen atoms bonded to C atoms, solvent molecules and anions are omitted for simplicity. | ||
| [Pb(PYTAM)](PF6)2 | [Pb(H4PYTAMGly)](NO3)2·5H2O | ||
|---|---|---|---|
| Pb(1)–N(1) | 2.646(6) | Pb(1)–N(1) | 2.650(2) |
| Pb(1)–N(4) | 2.623(2) | ||
| Pb(1)–N(2) | 2.732(5) | Pb(1)–N(2) | 2.733(2) |
| Pb(1)–N(3) | 2.7109(19) | ||
| Pb(1)–N(5) | 2.7663(19) | ||
| Pb(1)–N(6) | 2.753(2) | ||
| Pb(1)–O(1) | 2.791(4) | Pb(1)–O(1) | 2.8648(19) |
| Pb(1)–O(4) | 2.7828(17) | ||
| Pb(1)–O(7) | 2.6935(18) | ||
| Pb(1)–O(10) | 2.9217(17) | ||
The Pb(II) ion in [Pb(PYTAM)](PF6)2 is ten-coordinated by the chelator, which binds through the six N donor atoms of the macrocycle and the four amide O atoms. The Pb–N distances involving pyridyl N atoms (2.646(6) Å) are ∼0.09 Å shorter than those to amine N atoms (2.732(5) Å), a trend that is consistent with X-ray structures reported in the literature for eight-coordinate Pb(II) complexes.15,48 The distances between the metal ion and the O donor atoms (2.791(4) Å) are longer than those involving N donor atoms, a situation observed previously for complexes containing amide groups such as [Pb(DOTAM)]2+,49 or even negatively charged carboxylate groups (i.e. [Pb(DOTA)]2−).50
The Pb(II)-donor distances in [Pb(H4PYTAMGly)]2+ point to a slight distortion of the metal coordination environment compared to [Pb(PYTAM)](PF6)2 (Table 1). Indeed, one of the Pb–O distances is particularly short [Pb(1)–O(7) = 2.6935(18) Å], while the remaining three Pb–O distances are in the range of 2.78 to 2.92 Å. Similarly, the distances to amine N atoms differ by up to ∼0.06 Å. The deformation of the coordination sphere appears to be related to steric interactions involving methylene protons of neighbouring terminal acetic acid groups, which display H⋯H distances as short as 2.77 Å. The distortion of the metal coordination sphere is also reflected in the angle involving the two pyridyl N atoms and the metal ion, which is linear in [Pb(PYTAM)]2+ takes a value of 177.99(6)° in [Pb(H4PYTAMGly)]2+, with the pyridine rings being slightly bent towards the hemisphere of the metal coordination environment where O(7) and O(1) are situated. The slight alteration of the Pb(II) coordination sphere in [Pb(H4PYTAMGly)]2+ does not appear to be related to a significant activity of the 6s2 lone pair. In fact, previous works associated the stereochemical activity of the lone pair with a polarization of the 6s2 lone pair with a significant 6p contribution.51,52 However, the analysis of the natural bond orbitals using DFT in both [Pb(PYTAM)]2+ and [Pb(H4PYTAMGly)]2+ indicates that the Pb(II) lone pair has a 100% 6s character (Table S1, ESI†). This is in line with the general trend that holodirected structures are favoured by the presence of neutral donor atoms and high coordination numbers.51
The conformation of the macrocycles in these complexes is similar to those observed for lanthanide complexes based on the same macrocycle and functionalised with identical pendant arms.53–57 Two pendant arms attached to opposite N atoms on the macrocyclic structure are placed above the mean plane defined by the pyridine rings, while the remaining two pendant arms are placed below that plane. This conformation, together with the symmetrical coordination environment imposes a D2 symmetry for the [Pb(PYTAM)]2+ complex. Fig. 1 shows views of the structure of this complex along the three perpendicular C2 axes characterising the D2 point group. The five-membered chelates generated upon coordination of the ethylenediamine spacers adopt the same conformations, which can be represented as (λλ) or (δδ) (Fig. S3, ESI†).58,59 Furthermore, the four amine N atoms adopt identical configurations. The two structures are centrosymmetric, with both (λλ)-RRRR and (δδ)-SSSS enantiomers being present in the crystal.
Solution structure of the Pb(II) complexes
The Pb(II) complexes of the four chelators were prepared in situ by adding Pb(NO3)2 to a solution of the chelator in D2O. Upon addition of the metal salt, the pD dropped to low values, between 1 and 2. While 1H-NMR of the complexes with ethylene spacers show one species in solution, the complexes with cyclohexyl spacers do not show one unique species, presenting more complex spectra, at this pD (Fig. S4, ESI†). The pD was then raised to ∼7 using NaOD to record the NMR spectra of the complexes (Fig. 2). | ||
| Fig. 2 1H NMR spectra of [Pb(PYTAM)]2+ (pD = 6.5), PYTAM (pD = 1.4) [Pb(CHX-PYTAM)]2+ (pD = 7.4), CHX-PYTAM (pD = 1.7), [Pb(PYTAMGly)]2− (pD = 6.8), H4PYTAMGly (pD = 2.0), [Pb(CHX-PYTAMGly)]2+ (pD = 6.3) and H4CHX-PYTAMGly (pD = 2.0), recorded in D2O solution (300 MHz, 298 K). | ||
The 1H NMR spectra of the four complexes are characteristic of highly rigid structures, as evidenced by the well-resolved signals that can be observed in the aliphatic region, where doublets corresponding to AB systems associated with the geminal coupling of the protons of the CH2 groups appear (Fig. 2). These characteristic doublets are easily identified through their large 2J values (∼15 Hz) and roof effect. This is due to the rigidity of the system, which locks the protons of these groups into specific positions, making them no longer chemically equivalent. The NMR data in solution indicate the presence of a single complex species in each case. The number of 1H and 13C NMR signals point to an effective D2 symmetry of the complexes in solution. A full assignment of the 1H and 13C NMR spectra, achieved with the aid of 2D COSY, HSQC and HMBC experiments, along with J coupling values, is provided in the ESI (Tables S2–S4†). Noteworthy, the 1H NMR spectra of the free PYTAM and H4PYTAMGly chelators are characteristic of flexible molecules, with the signals due to CH2 groups being observed as singlets. The 1H NMR spectra of the corresponding cyclohexyl derivatives display broad signals, which reflects a certain degree of rigidity of the free chelator in solution.
Additional information on the structures of the complexes in solution was obtained from 2D 1H–207Pb heteronuclear multiple-quantum correlation (HMQC) spectra (Fig. 3).60 These spectra provide two strong cross-peaks relating the 207Pb resonance and the equatorial protons of the methylenic groups of the molecule (H4 and H5, see Scheme 1). Additionally, another strong cross peak is observed for one of the signals of the CH2 protons of the ethylene spacer (H7) in [Pb(PYTAM)]2+ and [Pb(PYTAMGly)]2−. The set of three geminal (axial) CH2 signals provide however weak cross peaks. This indicates that axial and equatorial protons provide rather different 3JPb–H coupling constants. Furthermore, the axial C–H protons (H7) of the cyclohexyl group in [Pb(CHX-PYTAM)]2+ and [Pb(CHX-PYTAMGly)]2− do not provide cross peaks with the 207Pb nucleus. Thus, we conclude that equatorial protons give much stronger cross peaks in the HMQC spectra than the axial counterparts. Equatorial CH2 protons are characterised by H–C–N–Pb dihedral angles of ∼160–170°, while axial protons give dihedral angles of ∼70–80°. This suggests that the 3JPb–H coupling constants involving methylenic protons follow a Karplus-like relationship with the dihedral H–C–N–Pb angle,61 as observed previously for properties originated by through-bond effects (i.e. spin densities).62 The strong 3JPb–H coupling involving equatorial protons is also evident by the presence of satellites in the 1H NMR spectra (Fig. 3) due to coupling with 207Pb (I = ½), which has a 22.6% natural abundance. These satellites provide 3J(1H–207Pb) couplings of ∼8–9 Hz, which are in the low range reported for Pb(II) complexes containing H–C–N(sp3) bonds (ca. 8–20 Hz).60,63 Larger coupling constants of up to ∼43 Hz were reported for 3J(1H–207Pb) couplings involving sp2 N donor atoms.64–66 Nevertheless, the presence of the 3J(1H–207Pb) coupling constants confirms the coordination of the macrocyclic unit to the Pb(II) ion and reveals a certain degree of covalence of the Pb–Namine bonds. We also notice that the pyridyl protons H2 give a sizeable 4J(1H–207Pb) coupling of ∼3 Hz, pointing to a rather strong coordination of the pyridyl N atom to the metal ion.
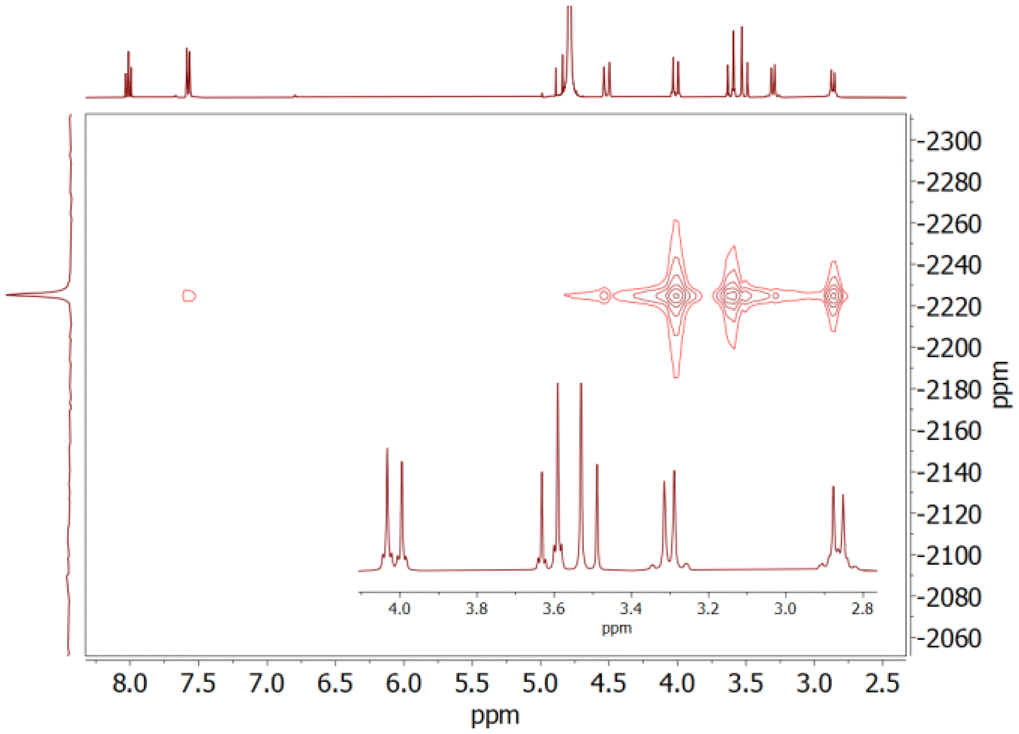 | ||
| Fig. 3 1H–207Pb HMQC spectrum of [Pb(PYTAM)]2+ (pD = 6.5) recorded in D2O solution (400 MHz, 298 K). The inset shows an expansion of the aliphatic region where satellites due to 3J(1H–207Pb) coupling are evident. | ||
The 207Pb NMR spectra of the complexes display signals in the range −2178 to −2255 ppm, with the cyclohexyl derivatives providing slightly more negative chemical shifts and the glycinate derivatives slightly more positive shifts (Fig. 4). The similar δ values observed for the four complexes point to comparable Pb(II) coordination environments. The 207Pb NMR signals observed here are shielded by ∼4000 ppm with respect to those reported for EDTA derivatives60,67 and [Pb(DOTAM)]2+.68 Positive chemical shift values were also reported for complexes with crown ether derivatives64 (the chemical shift of neat Pb(CH3)4 used as a reference corresponds to +2961 ppm versus the 1 M aqueous Pb(NO3)2 reference used here).67 The negative δ values found for the ten-coordinated Pb(II) ions reported here follow the general trend of an increasing shielding with higher coordination numbers.69 A similar situation was observed for Y(III) complexes, as ten-coordinated derivatives displayed unusually shielded 89Y resonances.70 In the latter case the experimental 89Y shifts could be approximated by the sum of the shielding contributions of the chelator donor atoms, and thus an increased coordination number results in a more important shielding effect. A similar behaviour may affect the 207Pb NMR shifts, though investigation of a wider range of complexes is required to find correlations between the number and type of donor atoms and the observed shifts.
 | ||
| Fig. 4 207Pb NMR spectra of [Pb(PYTAM)]2+ (pD = 6.5), [Pb(CHX-PYTAM)]2+ (pD = 7.4), [Pb(PYTAMGly)]2− (pD = 6.8), [Pb(CHX-PYTAMGly)]2+ (pD = 6.3) recorded in D2O solution (84 MHz, 298 K). | ||
Protonation constants and thermodynamic stability of the Pb(II) complexes of the glycinate derivatives
The protonation constants of the chelators and the stability constants of the lead(II) complexes were determined for the glycinate derivatives, as the chelators containing acetamide pendants presented solubility issues in basic media. Chelator protonation constants were measured using potentiometric and spectrophotometric titrations, following the absorption of the pyridyl groups at ca. 265 nm. Potentiometric titrations afforded three protonation constants for PYTAMGly4− above pH 5.0 and up to four protonation constants with logK values in the range ∼2.4 to 4.1. The assignment of the protonation constants was aided by spectrophotometric titrations (Fig. 5). Indeed, the absorption spectrum recorded at pH 1.30 displays a maximum at 263 nm and a shoulder at 267 nm. Increasing the pH above ∼4.0 provokes a slight shift of the absorption maximum to 265 nm as the band becomes more rounded and its intensity decreases. These spectral changes are compatible with the protonation of a pyridyl unit.34 The logK value obtained from spectrophotometric titrations (4.98) is in very good agreement with that determined by potentiometry (logK3 = 5.11(1)). The spectrum experiences further changes above pH ∼ 5.6, with the absorption maximum experiencing a slight shift to lower energies (267 nm) as its intensity increases. Spectrophotometric titrations afforded a single protonation constant of logK = 6.29(1), which coincides with the average of logK1 (6.70(1)) and logK2 (5.88(1)) obtained by potentiometry. We therefore assign these protonation constants to the amine groups of the macrocycle, though the significant spectral changes suggest that the pyridine units are also involved in these protonation processes. A comparison of the protonation constants logK1 and logK2 determined for PYTAMGly4− and DOTAMGly4− indicate that the amine groups of the 12-membered CYCLEN ring are considerably more basic than those of the 18-membered ring of PYTAMGly4−. The 18-membered macrocycle crown-4Py displays an intermediate behaviour.15 The protonation constants determined for PYAN using 0.1 M KNO3 and 0.1 M KCl as background electrolytes evidence that the incorporation of the amide pendants in PYTAMGly4− considerably reduces the basicity of the N atoms of the macrocycle (Table 2).71–73
 | ||
| Fig. 5 (a) UV-Vis absorption spectra recorded for a solution of PYTAMGly4− (10−4 M, I = 0.15 M NaClO4, 25 °C) at different pH values. The spectra highlighted in red, violet, and blue correspond to pH values of 1.30, 5.57 and 8.01, respectively. The inset shows the spectral changes at selected wavelengths and the fits of the data. (b) UV-Vis absorption spectra recorded to monitor the dissociation of the pre-formed Pb(II) complex with PYTAMGly4− (10−4 M, I = 0.15 M NaClO4, 25 °C) at different pH values. The spectra highlighted in red, and blue correspond to pH values of 0.82 and 3.58, respectively. The inset shows the spectral changes at 261 nm and the fit of the data. | ||
| PYTAMGly4− | CHX-PYTAMGly4− | MACROPA2−a |
Cyclen-4Pyb | Crown-4Pyb | DOTAMGly4−c |
PYANd | |
|---|---|---|---|---|---|---|---|
| a Data from ref. 78, 0.1 M KNO3, 25 °C. b Data from ref. 15, 0.1 M KCl, 25 °C. c Data from ref. 74, 0.1 M KCl, 25 °C. d Data from ref. 71, 0.1 M KNO3, 25 °C. | |||||||
| logK1 |
6.70(1) | 8.69(5) | 7.52 | 10.05 | 7.91 | 9.19 | 8.99 |
| logK2 |
5.88(1) | 7.80(7) | 7.03 | 7.53 | 7.33 | 6.25 | 8.22 |
| logK3 |
5.11(1) | 4.38(9) | 3.58 | 3.54 | 4.64 | 4.08 | 6.03 |
| logK4 |
4.08(1) | 3.66(9) | 2.47 | 2.23 | 4.00 | 3.45 | 5.34 |
| logK5 |
3.46(1) | 3.31(9) | 3.20 | 2.11 | |||
| logK6 |
3.13(1) | 2.62(9) | 1.40 | ||||
| logK7 |
2.46(1) | 2.05(9) | |||||
| logKPbL |
17.03(1) | 17.18(1) | 16.23 | 19.95 | 13.29 | ||
| logKPbHL |
4.58(2) | 4.691(2) | 2.86 | 2.06 | 5.22 | ||
| logKPbH2L |
3.57(4) | 3.66(7) | 3.50 | ||||
| logKPbH3L |
3.49(4) | 3.52(8) | |||||
| logKPbH4L |
2.87(3) | 3.09(6) | |||||
| pPb | 17.9 | 16.3 | 16.7 | 17.88 | 13.40 | ||
The protonation constants logK4−logK7 determined by potentiometry are not associated to noticeable spectral changes in the absorption spectra, and therefore can be safely assigned to the remote glycinate groups. Similar protonation constants were reported for the glycinate groups of DOTAMGly4−.74 The protonation of four non-interacting carboxylates is expected to have statistical separations of ΔlogK = 0.42, 0.36 and 0.42. The experimental protonation constants yield ΔlogK4,5 = 0.62, logK5,6 = 0.33 and logK6,7 = 0.67, in reasonable agreement with the values expected according to the statistical factor.75,76
The same combined methodology was used to determine the protonation constants of CHX-PYTAMGly4−. The constants involving the protonation of amine groups, logK1 and logK2, are about 2 logK units higher than those determined for PYTAMGly4− (Table 2). This is expected, as in general the substitution of ethyl for cyclohexyl groups significantly increases chelator basicity.31,77 The protonation constants logK3−logK6 are attributed to the protonation of the glycinate groups on the basis of the minor spectral changes observed in the absorption spectra (Fig. S6, ESI†). These protonation constants follow again reasonably well the trend expected according to the statistical factor. Interestingly, spectrophotometric experiments demonstrate that the protonation of the pyridine ring occurs at low pH, with a protonation constant of logK7 = 2.05(9). The rigidity of the macrocycle introduced by the presence of cyclohexyl rings appears to be responsible for the sharp decrease in the protonation constant of the pyridyl unit.
The stability constants of the Pb(II) complexes with PYTAMGly4− and CHX-PYTAMGly4− were determined using a combination of potentiometric and spectrophotometric titrations. Potentiometry was used to obtain the protonation constants of the complexes, while complex dissociation was investigated spectrophotometrically following the changes in the absorption band of the pyridyl groups. Of note, the amount of free Pb(II) at pH ∼2 is only 1.5 and 6.5% for PYTAMGly4− and CHX-PYTAMGly4−, respectively, preventing the determination of stability constants by direct potentiometry. The absorption band of the pyridyl rings of PYTAMGly4− at 263 nm undergoes a slight blue shift to 260 nm as its intensity increases as a result of complex formation (Fig. 5b). Similar spectral variations are observed for CHX-PYTAMGly4− (Fig. S6, ESI†). The fits of the data afforded the stability and protonation constants shown in Table 2. These studies showed that the incorporation of the cyclohexyl spacer did not have a large effect on the value of logKPbL affording values of 17.03 and 17.17 for [Pb(H4PYTAMGly)]2− and [Pb(H4CHX-PYTAMGly)]2−, respectively. These values are higher than those reported for the complexes with other potentially decadentate 18-membered macrocycles such as MACROPA2− and crown-4Py.15,78 Higher stability constants were reported for CYCLEN derivatives containing pyridyl rings such as CYCLEN-4Py.15 However, the low basicity of PYTAMGly4− and especially CHX-PYTAMGly4− renders rather high conditional stability at physiological pH. This can be conveniently assessed by calculating pPb values, defined as −log[Pb(II)]free for a metal ion concentration of 1 μM and a total chelator concentration of 10 μM.79 The pPb values obtained for PYTAMGly4− and CYCLEN-4py are virtually identical in spite of the three orders of magnitude difference in stability constants. The pPb value calculated for CHX-PYTAMGly4− is nearly two units lower than that of PYTAMGly4− as a result of the lower basicity of the latter, though comparable to that of MACROPA2−.
The speciation diagrams calculated from equilibrium constants (Fig. 6) indicate that complex protonation takes place below pH ∼ 7, with the non-protonated complex species being at nearly 100% abundance around neutral pH. Complex dissociation occurs below pH ∼ 2 and pH ∼ 3 for PYTAMGly4− and CHX-PYTAMGly4−, respectively. The two complexes show a very similar protonation behaviour, with four protonation constants associated to the glycinate groups. The ΔlogKPbHL,PbH2L values of 1.01 and 1.03 for the complexes with PYTAMGly4− and CHX-PYTAMGly4−, respectively, are higher than that expected according to the statistical effect (0.42), while the values of ΔlogKPbH2L,PbH3L < 0.15 are lower than the statistical value of 0.36.76 This suggests a certain negative cooperativity for the formation of the deprotonated complex species. We also note that the glycinate groups are characterized by higher protonation constants in the complexes compared with the free ligands. Complex formation forces the negatively charged glycinate groups to be in rather close proximity (Fig. 1), which likely favours their protonation.
 | ||
| Fig. 6 Speciation diagrams of the H4PYTAMGly:Pb(II) (a) and H4CHX-PYTAMGly4−:Pb(II) (b) systems calculated for [Pb(II)]tot = [L]tot = 1 mM (25 °C, I = 0.15 M NaClO4). | ||
Radiolabelling studies
Once the non-radioactive studies showed that these chelators presented appropriate properties for Pb(II) complexation, lead-203 radiolabelling studies were performed. [203Pb]Pb(II) was produced and purified according to the procedure reported by McNeil et al., without any modifications.80 Concentration dependent radiolabelling for all four chelators and DOTAM, the gold-standard for radiolabelling with lead(II) isotopes, was performed in NH4OAc buffer (0.1 M, pH 7) at five different chelator concentrations (10−4, 10−5, 10−6, 10−7 and 10−8 M). The reactions were followed at two time points (30 and 60 minutes) and two temperatures (room temperature: ∼25 °C, and 80 °C). The radiochemical yields (RCYs) were determined by spotting an aliquot of the reaction onto iTLC-SA plates and developed using 50 mM EDTA (pH 5) as the mobile phase.The RCYs observed for both temperatures at 60 min (Fig. 7 and Tables S4, S5, ESI†) improve slightly with respect to those recorded at 30 min (Fig. S8 and Tables S6, S7 ESI†). At room temperature (Fig. 7a and Table S5, ESI†), CHX-PYTAM is the only chelator that does not reach quantitative yields at 10−4 M, although the RCY is still high with a value of 97.2 ± 1.2% at this concentration. This effect is most likely due to the rigidity introduced by the cyclohexyl spacers, since at higher temperatures the yields are superior. At 80 °C (Fig. 7b and Table S6, ESI†), the RCYs of PYTAM and CHX-PYTAM are comparable to the values observed for DOTAM. The glycinate derivatives, however, behave differently, and their RCYs do not improve significantly as the labelling efficiency of these chelators is nearly identical at both room temperature and 80 °C.
 | ||
| Fig. 7 Radiolabelling studies using 203Pb (∼150 kBq, 0.1 M NH4OAc buffer, pH 7) (n = 3 for each data point). (a) Concentration dependent radiolabelling at room temperature after 60 min. (b) Concentration dependent radiolabelling at 80 °C after 60 min. | ||
Since 203Pb-radiolabelling of CHX-PYTAM at room temperature was not fully quantitative at the highest chelator concentration, labelling of this chelator was also performed at 37 °C (Table S9, ESI†), in addition to room temperature (∼25 °C) and 80 °C (Fig. 8). The radiochemical yields improve significantly at this temperature for all concentrations, with yields of nearly 100% for both 10−4 and 10−5 M (98.4 ± 0.4 and 97.3 ± 0.5%, respectively) after 30 minutes and even higher yields after 60 min (100% and 99.6 ± 0.6%, respectively). This result indicates that although labelling of 100% was not achieved at room temperature, the system is still quite promising for the design of bifunctional systems, as heating to 37 °C is still compatible with most biological targeting vectors.
 | ||
| Fig. 8 Temperature dependent 203Pb-radiolabelling (150 kBq, 0.1 M NH4OAc buffer, pH 7, 30 min) of CHX-PYTAM at different concentrations (n = 3 for each data point). | ||
Once the 203Pb-labelled complexes were prepared, challenge experiments were performed to assess the kinetic inertness of the 203Pb complexes. The complexes were formed at either room temperature and/or 80 °C to be representative of the tested radiolabelling conditions (Fig. 9 and Fig. S9, Tables S10–S14, ESI†). The purpose of this experiment was to evaluate the influence of complexation temperature on stability. For the stable Pb(II) and EDTA challenges, 20 equivalents of the respective chemical was added to the radiolabelling solution. DOTAM forms incredibly stable complexes in the studied competitive media, remaining 100% intact in all the conditions investigated. The complexes formed with PYTAM, H4PYTAMGly, and CHX-PYTAM presented excellent stability in presence of 20 equiv. of EDTA (Fig. 9b) over the course of over one half-life, showing no indications of transchelation. However, H4CHX-PYTAMGly did not remain intact, neither when prepared at room temperature or 80 °C. It may be possible that the steric hindrance introduced by the simultaneous presence of cyclohexyl spacer and acetylglycine pendant arms facilitates transchelation.
 | ||
| Fig. 9 Challenging studies of the 203Pb-labelled complexes (150 kBq, 0.1 M NH4OAc buffer, pH 7) (n = 3 for each data point). (a) Human serum stability challenge. (b) Excess EDTA challenge (20 equiv.). (c) Excess stable Pb(II) challenge (20 equiv.). | ||
When incubated with excess (20 equiv.) of stable Pb(II) (Fig. 9c), CHX-PYTAM is the only chelator that forms a complex that is stable under these conditions. The complex is 100% intact after 24 hours and after 72 hours the majority (93.2 ± 0.4%) of the radiocomplex remains intact. H4PYTAMGly is the least stable as within 2 hours, only 13.4 ± 2.0% of the complex remained intact at this time point. PYTAM and H4CHX-PYTAMGly, while not as stable as CHX-PYTAM, performed better than H4PYTAMGly, with similar results, as 37.0 ± 2.0% and 54.3 ± 1.5% of the complex is stable after 72 hours, respectively. It is surprising, after seeing the results of the competition with H4EDTA, that H4CHX-PYTAMGly performed better than H4PYTAMGly under these conditions, though transchelation and transmetallation occur through different mechanisms. The presence of glycinate groups likely favours the formation of the dinuclear intermediate responsible for the Pb(II)-assisted dissociation mechanism. The high stability of CHX-PYTAM is most likely due to the increase in rigidity introduced by the two cyclohexyl groups in the backbone and the use of acetamide pendant arms, which appears to be the ideal combination for complex stability. It should be noted though that the complex formed with DOTAM does, in fact, remain 100% stable. However, the 93.2 ± 0.4% of intact complex, observed for CHX-PYTAM at 72 hours, is the highest of all of the chelators presented in this work, and it should not present any issues in vivo as high concentrations of Pb(II) are not expected to be found in the body.
Finally, the kinetic inertness of the complexes was evaluated when incubated in human serum at 37 °C (Fig. 9a). CHX-PYTAM showed no dissociation over 72 hours while the other three chelators did drop slightly but remained above 95% in all cases. These results point towards all four complexes possibly being stable under in vivo conditions, although further studies would be necessary to confirm this hypothesis.
Conclusions
We have shown that macrocyclic chelators based on the 18-membered platform PYAN show promising properties for the development of Pb-based radiopharmaceuticals. Stable Pb(II) complexation was achieved by incorporating four amide pendant arms, leading to the formation of stable and rigid complexes with ten-coordinated Pb(II) ions. The inertness of the complexes is improved by incorporating rigid cyclohexyl spacers into the macrocyclic ring. The presence of bulky acetylglycine pendant arms appears to introduce some steric hindrance, while the chelator CHX-PYTAM displays excellent properties for the development of Pb-based radiopharmaceuticals. We are currently developing bifunctional derivatives of this platform to be able to further study the potential of these chelates.Experimental section
General considerations
Solvents and reagents were purchased from commercial sources and were used as supplied without further purification. Medium performance liquid chromatography (MPLC) was performed in a Puriflash XS 420 InterChim Chromatographer equipped with a UV-DAD detector and a 20 g BGB Aquarius C18AQ reversed-phase column (100 Å, spherical, 15 μm) using H2O and CH3CN with 0.1% TFA as the mobile phases. Semi-preparative high performance liquid chromatography (HPLC) was performed using a Jasco LC-4000 instrument equipped with a UV-4075 detector, in manual injection and collection mode, using a Fortis C18 column (5μm, 250 × 10 mm) and H2O and CH3CN with 0.1% TFA as the mobile phases, operating at a flow rate of 4.0 mL min−1. High-resolution electrospray-ionization time-of-flight ESI-TOF mass spectra were recorded in positive mode using a LTQ-Orbitrap Discovery Mass Spectrometer coupled to a Thermo Accela HPLC.1H and 13C NMR spectra of the chelators and their complexes were recorded on Bruker AVANCE III 300, Bruker AVANCE 400 or Bruker AVANCE 500 spectrometers. PYAN (3,6,10,13-tetraaza-1,8(2,6)-dipyridinacyclotetradecaphane), tert-butyl (2-chloroacetyl)glycinate and PYTAM (2,2′,2′′,2′′′-(3,6,10,13-tetraaza-1,8(2,6)-dipyridinacyclotetradecaphane-3,6,10,13-tetrayl)tetraacetamide) were prepared according to previously reported procedures.45,81,822,2′,2′′,2′′′-((2,2′,2′′,2′′′-(3,6,10,13-Tetraaza-1,8(2,6)-dipyridinacyclotetradecaphane-3,6,10,13-tetrayl)tetrakis(acetyl))tetrakis(azanediyl))tetraacetic acid (H4PYTAMGly)
PYAN (0.1869 g, 0.57 mmol), K2CO3 (0.4734 g, 3.43 mmol) and KI (0.0095 g, 0.057 mmol) were added to CH3CN (40 mL) while the mixture was heated to 50 °C. tert-Butyl (2-chloroacetyl)glycinate (0.5920 g, 2.85 mmol) was dissolved in CH3CN (15 mL) and added dropwise over the course of 2 hours after which the reaction was left stirring at 50 °C for 6 days after which the solvent was eliminated in the rotary evaporator. The product was isolated by suspending the oil in H2O (50 mL) and performing extractions with CHCl3 (3 × 50 mL). The organic phase was separated, dried using Na2SO4, and concentrated in the rotary evaporator. A yellow oil was obtained after drying in the vacuum line, which is dissolved in 20 mL of a 1:1 TFA:CH2Cl2 mixture and left stirring overnight. The solvent was then eliminated under a nitrogen flow and was washed with water (3 × 15 mL), obtaining a brown oil. This oil was dissolved in water (1.5 mL) and was purified by reverse phase using method A (Table S15†). The desired compound eluted at 36% CH3CN (retention time 8.6 min, 6.3 CV). The fraction of interest was lyophilized and an off-white solid was obtained (0.0887 g, 13% yield). 1H NMR (300 MHz, D2O, pD 2.0) δ (ppm) 8.31 (s, br, 2H), 7.80 (d, J = 7.4 Hz, 4H), 4.59 (s, 8H), 3.92 (s, 8 H), 3.85 (s, br, 8H), 3.48 (s, 8H).13C NMR (75 MHz, D2O, pD 2.0) δ (ppm) 172.96, 169.33, 150.57, 126.03, 55.12, 52.00, 41.15. Elemental analysis calcd (%) for C34H46N10 O12·4TFA: C 40.59, H 4.06, N 11.27; found: C 40.68, H 4.35, N 11.35. Experimental MS (ESI+, MeOH/H2O): m/z 413.1459, 424.1364, 825.2847; calculated for [C34H47N10 O12K]2+ 413.1501, calculated for [C34H47N10 O12NaK]2+ 424.1410, calculated for [C34H46N10 O12K]+ 825.2928.
(41R,42R,101R,102R)-3,5,9,11-Tetraaza-1,7(2,6)-dipyridina-4,10(1,2)-dicyclohexanacyclododecaphane (CHX-PYAN)
Synthesis adapted from a previously reported procedure.47 Pyridine-2,6-dicarbaldehyde (0.6250 g, 4.63 mmol) was dissolved in MeOH (150 mL) along with barium chloride dihydrate (0.4830 g, 2.32 mmol). (1R,2R)-Cyclohexane-1,2-diamine (0.5281 g, 4.62 mmol) was dissolved in MeOH (10 mL) and added dropwise to the refluxing reaction mixture over the course of 2 hours. The reaction was then left heating for an additional 3 hours after which it was cooled to 0 °C in an ice bath and NaBH4 (0.2122 g, 6.07 mmol) was added in small increments. The reaction mixture was allowed to reach room temperature, a second addition of NaBH4 (0.1062 g, 3.02 mmol) was made and the reaction was left stirring for 3 hours. The solvent was then eliminated in the rotary evaporator and water (50 mL, pH adjusted to 9 using NaOH) was added. Extractions with CHCl3 (3 × 50 mL) (50 mL) were performed and the combined organic phases were dried using NaSO4, filtered and concentrated in the rotary evaporator, obtaining a yellow solid (1.2429 g, yield 87%). 1H NMR (500 MHz, CDCl3) δ (ppm) 7.55 (s, 2H), 7.04 (d, 3J = 7.6 Hz, 4H), 4.10 (d, 2J = 15.4 Hz, 4H), 3.80 (d, 2J = 15.7 Hz, 5H), 2.17–1.95 (m, 8H), 1.63 (d, 2J = 7.3 Hz, 4H), 1.18–0.96 (m, 8H). 13C NMR (126 MHz, CDCl3) δ 137.19, 121.63, 59.56, 32.97, 24.99. Elemental analysis calcd (%) for C26H38N6·1.65CHCl3: C 52.64, H 6.23, N 12.97; found: C 52.58, H 6.33, N 13.31. Experimental MS (ESI+, MeOH/H2O): m/z 435.3206, 457.3022; calculated for [C26H39N6]+ 435.3231, calculated for [C26H39N6Na]+ 457.3050.2,2′,2′′,2′′′-((41R,42R,101R,102R)-3,5,9,11-Tetraaza-1,7(2,6)-dipyridina-4,10(1,2)-dicyclohexanacyclododecaphane-3,5,9,11-tetrayl)tetraacetamide (CHX-PYTAM)
CHX-PYAN·1.65CHCl3 (0.3008 g, 0.484 mmol) was dissolved in CH3CN (20 mL) and Na2CO3 (0.3204 g, 3.02 mmol) was added. 2-Bromoacetamide (0.2689 g, 1.95 mmol) was dissolved in CH3CN (10 mL) and added dropwise over the course of an hour. After stirring for three days, the solvent was evaporated, and the resulting yellow solid was then prepared for purification by MPLC. The solid was dissolved in water (1 mL) by lowering the pH to pH = 3 using HCl. This solution was purified by reverse phase using method B (Table S16†), and the desired compound eluted at 27% CH3CN (retention time 12.2 min, 8.9 CV). The fraction of interest was lyophilized and an off-white solid was obtained (180.0 mg, 40% yield). 1H NMR (400 MHz, D2O, pD = 1.74, 343.15 K) δ (ppm) 8.26 (s, 2H), 7.99 (d, 3J = 5.9 Hz, 2H), 7.60 (s, 2H), 5.19–4.85 (m, 3H), 4.69 (d, 2J = 15.3 Hz, 2H), 4.47 (d, 2J = 14.6 Hz, 2H), 4.17–3.75 (m, 5H), 3.60 (d, 2J = 13.6 Hz, 4H), 2.97 (d, 2J = 10.6 Hz, 2H), 2.83–2.56 (m, 2H), 2.53–2.29 (m, 4H), 2.09 (d, 2J = 10.1 Hz, 2H), 1.98–1.70 (m, 6H).13C NMR (101 MHz, D2O, pD = 1.74, 343.15 K) δ (ppm) 174.07, 169.61, 157.17, 151.74, 140.35, 126.54, 126.51, 123.26 (br), 62.93 (br), 61.57 (br), 58.33, 54.46 (br), 52.97, 52.28, 24.39, 24.14. Elemental analysis calcd (%) for C34H50N10O4·1.5TFA·1.5HBr: C 46.52, H 5.59, N 14.66; found: C 46.46, H 5.61, N 14.72. Experimental MS (ESI+, MeOH/H2O): m/z 332.2083, 663.4094, 685.3912; calculated for [C34H52N10O4]2+ 332.2081, calculated for [C34H51N10O4]+ 663.4089, calculated for [C34H50N10O4K]+ 685.3909.2,2′,2′′,2′′′-((2,2′,2′′,2′′′-((41R,42R,101R,102R)-3,5,9,11-Tetraaza-1,7(2,6)-dipyridina-4,10(1,2)-dicyclohexanacyclododecaphane-3,5,9,11-tetrayl)tetrakis(acetyl))tetrakis(azanediyl))tetraacetic acid (H4CHX-PYTAMGly)
CHX-PYAN·(CHCl3)1.65 (0.2000 g, 0.32 mmol), K2CO3 (0.3890 g, 2.81 mmol) and tert-butyl (2-chloroacetyl)glycinate (0.3920 g, 1.89 mmol) were added to CH3CN (30 mL) and left stirring at room temperature for 30 days, following the progress of the reaction using mass spectrometry. After this time, since the reaction was not evolving to form the tetraalkylated derivative, more tert-butyl (2-chloroacetyl)glycinate (0.0294 g, 0.14 mmol) was added, and the reaction was left stirring for another 30 days after which the solvent was concentrated in the rotary evaporator and the product was isolated by adding CHCl3 (30 mL) and H2O with the pH adjusted to 9 using NaOH (30 mL) and performing an additional two extractions with CHCl3 (30 mL). The organic phase was separated, dried using Na2SO4, and concentrated in the rotary evaporator. A yellow oil was obtained after drying in the vacuum line, which was dissolved in 10 mL of a 1:1 TFA:CH2Cl2 mixture and left stirring overnight. The solvent was then eliminated under a nitrogen flow and was washed with water (3 × 15 mL), obtaining a brown oil. This oil was dissolved in water (1.5 mL) and was purified by reverse phase using method C (Table S17†). The desired compound eluted at 41% CH3CN (retention time 12.4 min, 9.1 CV). The fraction of interest was lyophilized, and a white solid was obtained (185.6 mg, 44% yield). 1H NMR (400 MHz, D2O, pD = 10.2, 343.15 K) δ (ppm) 8.02 (t, 3J = 7.7 Hz, 2H), 7.64 (d, 3J = 7.5 Hz, 4H), 4.33–3.43 (m, 16H), 3.22 (s, 4H), 2.51 (d, 2J = 10.0 Hz, 4H), 2.28–1.99 (m, 5H), 1.76–1.37 (m, 6H).13C NMR (101 MHz, D2O, pD = 10.2, 343.15 K) δ (ppm) 176.34, 161.69, 158.74, 139.19, 123.26, 43.60, 25.67, 25.01. Elemental analysis calcd (%) for C42H58N10O12·3.5TFA·2H2O: C 44.25, H 4.96, N 10.53; found: C 44.02, H 4.71, N 10.76. Experimental MS (ESI+, MeOH/H2O): m/z 448.2192, 895.4324, 917.4142; calculated for [C42H60N10O12]2+ 448.2191, calculated for [C42H59N10O12]+ 895.4308, calculated for [C42H58N10O12K]+ 917.4128.
General procedure for the synthesis of the lead complexes
The Pb(II) complexes were prepared in situ by adding Pb(NO3)2 to a solution of each chelator in deuterated water (0.5 mL) in a molar ratio of 1:1.1 (H4L: Pb(NO3)2). At this point, the mixture was quite acidic so the pD is adjusted above pD = 6 using a diluted solution of NaOD while the formation of the complex was monitored using NMR.
Crystal structure determination
Compounds [Pb(PYTAM)](PF6)2 and [Pb(H4PYTAMGly)](NO3)2 were analysed by X-ray diffraction. Table S19† shows the crystallographic data and structure refinement parameters. Crystallographic data were collected on a Bruker D8 Venture diffractometer with a Photon 100 CMOS detector at 100 K with Mo-Kα radiation (λ = 0.71073 Å) generated by an Incoatec high brilliance microfocus source equipped with Incoatec Helios multilayer optics. The APEX483 software was used for collecting frames of data, indexing reflections, and the determination of lattice parameters, while SAINT84 was used for integration of intensity of reflections, and SADABS85 for scaling and empirical absorption correction. The structure was solved by dual-space methods using the program SHELXT.86 All non-hydrogen atoms were refined with anisotropic thermal parameters by full-matrix least-squares calculations on F2 using the program SHELXL-2014.87 Hydrogen atoms were inserted at calculated positions and constrained with isotropic thermal parameters. The crystal structure of [Pb(H4PYTAMGly)](NO3)2 shows a B level Alert in the checkcif due a short distance between two hydrogen atoms: one belonging to a water molecule (H5WA) and the other to a protonated glycinate group (H2A). The protonated glycinate group shows hydrogen bond interaction with the water molecule. Also, H5WA shows a hydrogen bond interaction with one nitrate group. These interactions determine the position of the hydrogens. CCDC 2305213 and 2305214 contain the supplementary crystallographic data.†Potentiometric and spectrophotometric measurements
Stability constants and chelator protonation constants were determined through potentiometric and spectrophotometric titrations using HYPERQUAD.88 All data were collected at 25 °C and NaClO4 was used as an inert electrolyte which is added to all solutions to maintain the ionic strength at a constant value of I = 0.15 M.Potentiometric titrations were carried out in dual-wall thermostated cell, using recirculating water. Nitrogen was bubbled on the surface to avoid CO2 absorption and the solutions were homogenised using magnetic stirring. The titrant was added using a Crison microBu 2030 automatic burette and the electromotive force (emf) was recorded using a Crison micropH 2000 pH meter connected to a Radiometer pHG211 glass electrode and a Radiometer REF201 reference electrode. A 1 mM solution of the chelator (10 mL) was added to the cell, and the pH of the solution was adjusted to 11 using NaOH. This solution was then titrated using a standard HClO4 solution to determine all pKa values in the same experiment. Once the solution reached an acidic pH, an equimolar amount of Pb(II) was added and the mixture was left stirring for one hour to ensure complex formation. The solution was titrated with a solution of NaOH to determine the protonation constants of the complex. At this point it became clear that the stability constant of the complex must be determined by spectrophotometry, as the dissociation occurs at pH values that are too low to be determined using the glass electrode.
To determine equilibrium constants, HYPERQUAD calculates the proton concentration at each point of the titration using emf values, E, and using the electrode calibration equation, E = E°′ + slog[H+], with E°′ representing the formal potential and s, the slope of the electrode. Both parameters are determined with a calibration of the electrode system, conducted by titrating HClO4 with NaOH, both with known concentrations and adjusted ionic strength, as [H+] is known for each calibration point and E is measured, with a lineal regression giving the values of E°′ and s.
Spectrophotometric titrations were performed using a Uvikon-XS (Bio-Tek Instruments) double-beam spectrophotometer and 1 cm path length quartz cuvettes, recording spectra between 220 and 300 nm. The spectra of each chelator at concentrations between 6–10 × 10−5 M at different pH values were used to obtain the pKa values of the chelators. The desired pH was obtained using different buffers (acetate, phosphate, borax) and for very low pH values, HClO4 was used directly (i.e. stability constant determination). Dissociation of the complexes was achieved at low pH values, and although equilibrium is reached quite quickly, the samples were equilibrated for a few hours before each measurement.
The ligand protonation constants reported in this work are defined as in eqn (1), where L represents a general ligand:
 | (1) |
The stability constants of the Pb(II) complexes and their protonation constants are defined as in eqn (2) and (3), respectively.
 | (2) |
 | (3) |
DFT calculations
DFT calculations were performed with the Gaussian 16 program package,89 with the TPSSh functional and the Def2-TZVPP basis set for chelator atoms. For Pb, we selected the small-core relativistic effective core potential of Dolg et al. (ECP60MDF),90 which incorporates 60 electrons in the core, together with the cc-pVTZ valence basis set, which has a (12s11p8d1f)/[5s4p3d1f] contraction scheme.91 Calculations were performed using crystallographic structures in which only the positions of H atoms were optimised. The integration grid was set with the integral = ultrafinegrid keyword. Natural bond orbital analysis was carried out with the NBO program (version 3.1)92 included in Gaussian.Radiolabelling
Caution: 203Pb emits ionizing radiation and should only be used in a facility in accordance with appropriate safety controls.All chelators were purified by HPLC before radiolabelling experiments to ensure high purity (Table S18, ESI†).
Stock solutions, with a concentration of 10−3 M of DOTAM, PYTAM, H4PYTAMGly, CHX-PYTAM and H4CHX-PYTAMGly were prepared in deionized water. Serial dilution was then used to prepare chelator solutions with concentrations between 10−4 M and 10−7 M. For the 203Pb labelling studies, a 10 μL aliquot of chelator (or deionized water for the negative control) was added to 78 μL of deionized water and 10 μL of 1 M NH4OAc (pH 7) and 2 μL of [203Pb]Pb(Cl)2 (150 kBq) were added and mixed to begin the reactions, which were performed in triplicate and left at either room temperature or 80 °C until measurement at 30 and 60 minutes. iTLC was used to determine radiochemical yields (RCY) of the reactions, using iTLC-SA (iTLC paper impregnated with silicic acid) plates (1.5 × 10 cm, baseline at 1.5 cm; Agilent Technologies), developed using 50 mM EDTA (pH 5). With this system, complexed 203Pb(II) remained at the baseline (Rf = 0) while uncomplexed 203Pb(II) migrated at the solvent front (Rf = 1) (representative iTLCs are included in ESI, Fig. S10–S16†). Aliquots of the reactions were taken at 30 and 60 min, spotted onto the plates, and developed. RCYs were then measured using a BioScan System 200 Scanner (Washington, DC) and quantified with WinScan software.
Human serum stability studies
The stability of the five radiocomplexes was determined by adding 90 μL of human serum to previously radiolabelled solutions (10−4 M chelator concentration, 90 μL) and the resulting mixture was incubated at 37 °C. Aliquots (10 μL) were taken at 24, 48 and 72 hours and spotted onto the iTLC-SA plates, developed, and then analysed using the previously reported procedure, to determine the amount of intact complex.EDTA and Pb(II) challenging studies
Stock solutions of EDTA (pH = 7) and Pb(OAc)2 were prepared with a concentration of 20 mM. Preformed 203Pb-complexes (10−4 M chelator concentration, 100 μL volume), prepared both at room temperature and 80 °C, were then challenged with a 20-fold excess of either EDTA or Pb(II) as 10 μL of the 20 mM solution was added to the radiolabelling reaction. Aliquots (10 μL) were taken at 0.083, 0.5, 1, 2, 24, 48 and 72 hours and spotted onto the iTLC-SA plates, which were then analysed using the previously reported procedure, to evaluate the inertness of the complexes.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. E.-G. and C. P.-I. thank Ministerio de Ciencia e Innovación (Grants PID2019-104626GB-I00 and PID2022-138335NB-I00) and Xunta de Galicia (ED431C 2023/33) for generous financial support. C. H. thanks Ministerio de Ciencia e Innovación (Grant PRE2020-092888) for funding her PhD contract and a short-term research visit to SFU and TRIUMF. C. P.-I. and C. H. thank Centro de Supercomputación de Galicia (CESGA) for providing supercomputer facilities. L. V. is indebted to CACTI (Universidade de Vigo) for X-ray measurements. C. F. R and B. L. M thank the Natural Sciences and Engineering Research Council (NSERC) of Canada for funding through the Discovery grants (RGPIN-2019-07207) and Canada Graduate Scholarships – Doctoral (CGS-D) programs, respectively. B. L. M thanks the TRIUMF cyclotron operators for their continued support with 203Pb production at TRIUMF. Funding for open access provided by Universidade da Coruña/CISUG.References
- T. I. Kostelnik and C. Orvig, Radioactive Main Group and Rare Earth Metals for Imaging and Therapy, Chem. Rev., 2019, 119, 902–956 CrossRef CAS PubMed.
- S. L. Pimlott and A. Sutherland, Molecular tracers for the PET and SPECT imaging of disease, Chem. Soc. Rev., 2010, 40, 149–162 RSC.
- D. E. Milenic and M. W. Brechbiel, Targeting of Radio-Isotopes for Cancer Therapy, Cancer Biol. Ther., 2004, 3, 361–370 CrossRef CAS PubMed.
- L. Filippi, A. Chiaravalloti, O. Schillaci, R. Cianni and O. Bagni, Theranostic approaches in nuclear medicine: current status and future prospects, Expert Rev. Med. Devices, 2020, 17, 331–343 CrossRef CAS PubMed.
- N. H. Álvarez, D. Bauer, J. Hernández-Gil and J. S. Lewis, Recent Advances in Radiometals for Combined Imaging and Therapy in Cancer, ChemMedChem, 2021, 16, 2909–2941 CrossRef PubMed.
- C. Miller, J. Rousseau, C. F. Ramogida, A. Celler, A. Rahmim and C. F. Uribe, Implications of physics, chemistry and biology for dosimetry calculations using theranostic pairs, Theranostics, 2022, 12, 232–259 CrossRef CAS PubMed.
- S. R. Banerjee, V. Kumar, A. Lisok, J. Chen, I. Minn, M. Brummet, S. Boinapally, M. Cole, E. Ngen, B. Wharram, C. Brayton, R. F. Hobbs and M. G. Pomper, 177Lu-labeled low-molecular-weight agents for PSMA-targeted radiopharmaceutical therapy, Eur. J. Nucl. Med. Mol. Imaging, 2019, 46, 2545–2557 CrossRef CAS PubMed.
- C. Kim, S. V. Liu, D. S. Subramaniam, T. Torres, M. Loda, G. Esposito and G. Giaccone, Phase I study of the 177 Lu-DOTA 0 -Tyr 3 -Octreotate (lutathera) in combination with nivolumab in patients with neuroendocrine tumors of the lung, J. Immunother. Cancer, 2020, 8, e000980 CrossRef PubMed.
- T. Rold, N. Okoye, E. Devanny, A. Berendzen, T. Dresser, T. Quinn and T. Hoffman, Pb-203/Pb-212 Evaluation as Theranostic Pair for Prostate Cancer Detection, Monitoring, and Treatment, J. Nucl. Med., 2020, 61, 229–229 Search PubMed.
- B. L. McNeil, A. K. H. Robertson, W. Fu, H. Yang, C. Hoehr, C. F. Ramogida and P. Schaffer, Production, purification, and radiolabeling of the 203Pb/212Pb theranostic pair, EJNMMI Radiopharm. Chem., 2021, 6, 6 CrossRef PubMed.
- C. G. Pippin, T. J. McMurry, M. W. Brechbiel, M. McDonald, R. Lambrecht, D. Milenic, M. Roselli, D. Colcher and O. A. Gansow, Lead(II) complexes of 1,4,7,10-tetraazacyclododecane-N,N′,N′′,N′′′-tetraacetate: solution chemistry and application to tumor localization with 203Pb labeled monoclonal antibodies, Inorg. Chim. Acta, 1995, 239, 43–51 CrossRef CAS.
- L. L. Chappell, E. Dadachova, D. E. Milenic, K. Garmestani, C. Wu and M. W. Brechbiel, Synthesis, characterization, and evaluation of a novel bifunctional chelating agent for the lead isotopes 203Pb and 212Pb, Nucl. Med. Biol., 2000, 27, 93–100 CrossRef CAS PubMed.
- A. Ingham, L. Wharton, T. El Sayed, L. Southcott, B. L. McNeil, M. B. Ezhova, B. O. Patrick, M. de G. Jaraquemada-Peláez and C. Orvig, H2ampa–Versatile Chelator for [203Pb]Pb2+, [213Bi]Bi3+, and [225Ac]Ac3+, Inorg. Chem., 2022, 61, 9119–9137 CrossRef CAS PubMed.
- A. Ingham, T. I. Kostelnik, B. L. McNeil, B. O. Patrick, N. Choudhary, M. de G. Jaraquemada-Peláez and C. Orvig, Getting a lead on Pb2+-amide chelators for 203/212Pb radiopharmaceuticals, Dalton Trans., 2021, 50, 11579–11595 RSC.
- B. L. McNeil, K. J. Kadassery, A. W. McDonagh, W. Zhou, P. Schaffer, J. J. Wilson and C. F. Ramogida, Evaluation of the Effect of Macrocyclic Ring Size on [203Pb]Pb(II) Complex Stability in Pyridyl-Containing Chelators, Inorg. Chem., 2022, 61, 9638–9649 CrossRef CAS PubMed.
- B. J. B. Nelson, J. Wilson, M. K. Schultz, J. D. Andersson and F. Wuest, High-yield cyclotron production of 203Pb using a sealed 205Tl solid target, Nucl. Med. Biol., 2023, 116–117, 108314 CrossRef CAS PubMed.
- R. G. Li, V. Y. Stenberg and R. H. Larsen, An Experimental Generator for Production of High-Purity 212Pb for Use in Radiopharmaceuticals, J. Nucl. Med., 2023, 64, 173–176 CrossRef CAS PubMed.
- S. R. Banerjee, I. Minn, V. Kumar, A. Josefsson, A. Lisok, M. Brummet, J. Chen, A. P. Kiess, K. Baidoo, C. Brayton, R. C. Mease, M. Brechbiel, G. Sgouros, R. F. Hobbs and M. G. Pomper, Preclinical Evaluation of 203/212 Pb-Labeled Low-Molecular-Weight Compounds for Targeted Radiopharmaceutical Therapy of Prostate Cancer, J. Nucl. Med., 2020, 61, 80–88 CrossRef CAS PubMed.
- J. C. dos Santos, M. Schäfer, U. Bauder-Wüst, W. Lehnert, K. Leotta, A. Morgenstern, K. Kopka, U. Haberkorn, W. Mier and C. Kratochwil, Development and dosimetry of 203Pb/212Pb-labelled PSMA ligands: bringing “the lead” into PSMA-targeted alpha therapy?, Eur. J. Nucl. Med. Mol. Imaging, 2019, 46, 1081–1091 CrossRef CAS PubMed.
- E. S. Delpassand, I. Tworowska, R. Esfandiari, J. Torgue, J. Hurt, A. Shafie and R. Núñez, Targeted α-Emitter Therapy with 212Pb-DOTAMTATE for the Treatment of Metastatic SSTR-Expressing Neuroendocrine Tumors: First-in-Humans Dose-Escalation Clinical Trial, J. Nucl. Med., 2022, 63, 1326–1333 CrossRef CAS PubMed.
- M. W. Brechbiel, Bifunctional Chelates for Metal Nuclides, Q. J. Nucl. Med. Mol. Imaging, 2008, 52, 166–173 CAS.
- E. W. Price and C. Orvig, Matching chelators to radiometals for radiopharmaceuticals, Chem. Soc. Rev., 2014, 43, 260–290 RSC.
- K. Zukotynski, H. Jadvar, J. Capala and F. Fahey, Targeted Radionuclide Therapy: Practical Applications and Future Prospects: Supplementary Issue: Biomarkers and their Essential Role in the Development of Personalised Therapies (A), Biomarkers Cancer, 2016, 8s2, BIC.S31804 CrossRef PubMed.
- E. Boros, B. V. Marquez, O. F. Ikotun, S. E. Lapi and C. L. Ferreira, in Ligand Design in Medicinal Inorganic Chemistry, John Wiley & Sons, Ltd, 2014, pp. 47–79 Search PubMed.
- R. G. Pearson, Hard and Soft Acids and Bases, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- H. Maumela, R. D. Hancock, L. Carlton, J. H. Reibenspies and K. P. Wainwright, The Amide Oxygen as a Donor Group. Metal Ion Complexing Properties of Tetra-N-acetamide Substituted Cyclen: A Crystallographic, NMR, Molecular Mechanics, and Thermodynamic Study, J. Am. Chem. Soc., 1995, 117, 6698–6707 CrossRef CAS.
- R. Hancock, Macrocyclic ligands with pendent amide and alcoholic oxygen donor groups, Coord. Chem. Rev., 1996, 148, 315–347 CrossRef CAS.
- R. Delgado, V. Félix, L. M. P. Lima and D. W. Price, Metal complexes of cyclen and cyclam derivatives useful for medical applications: a discussion based on thermodynamic stability constants and structural data, Dalton Trans., 2007, 2734–2745 RSC.
- T. J. Clough, L. Jiang, K.-L. Wong and N. J. Long, Ligand design strategies to increase stability of gadolinium-based magnetic resonance imaging contrast agents, Nat. Commun., 2019, 10, 1420 CrossRef PubMed.
- R. D. Hancock and A. E. Martell, Ligand design for selective complexation of metal ions in aqueous solution, Chem. Rev., 1989, 89, 1875–1914 CrossRef CAS.
- C. F. Ramogida, J. F. Cawthray, E. Boros, C. L. Ferreira, B. O. Patrick, M. J. Adam and C. Orvig, H2 CHX dedpa and H4 CHX octapa-Chiral acyclic chelating ligands for 67/68Ga and 111In radiopharmaceuticals, Inorg. Chem., 2015, 54, 2017–2031 CrossRef CAS PubMed.
- F. Lucio-Martínez, Z. Garda, B. Váradi, F. K. Kálmán, D. Esteban-Gómez, É. Tóth, G. Tircsó and C. Platas-Iglesias, Rigidified Derivative of the Non-macrocyclic Ligand H 4 OCTAPA for Stable Lanthanide(III) Complexation, Inorg. Chem., 2022, 61, 5157–5171 CrossRef PubMed.
- F. Lucio-Martínez, D. Esteban-Gómez, L. Valencia, D. Horváth, D. Szücs, A. Fekete, D. Szikra, G. Tircsó and C. Platas-Iglesias, Rigid H4OCTAPA derivatives as model chelators for the development of Bi(III)-based radiopharmaceuticals, Chem. Commun., 2023, 59, 3443–3446 RSC.
- R. Uzal-Varela, D. Lalli, I. Brandariz, A. Rodríguez-Rodríguez, C. Platas-Iglesias, M. Botta and D. Esteban-Gómez, Rigid versions of PDTA4− incorporating a 1,3-diaminocyclobutyl spacer for Mn2+ complexation: stability, water exchange dynamics and relaxivity, Dalton Trans., 2021, 50, 16290–16303 RSC.
- N. A. Thiele, J. J. Woods and J. J. Wilson, Implementing f-Block Metal Ions in Medicine: Tuning the Size Selectivity of Expanded Macrocycles, Inorg. Chem., 2019, 58, 10483–10500 CrossRef CAS PubMed.
- G. J. Stasiuk and N. J. Long, The ubiquitous DOTA and its derivatives: the impact of 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid on biomedical imaging, Chem. Commun., 2013, 49, 2732 RSC.
- R. E. Mewis and S. J. Archibald, Biomedical applications of macrocyclic ligand complexes, Coord. Chem. Rev., 2010, 254, 1686–1712 CrossRef CAS.
- A. Hu, E. Aluicio-Sarduy, V. Brown, S. N. MacMillan, K. V. Becker, T. E. Barnhart, V. Radchenko, C. F. Ramogida, J. W. Engle and J. J. Wilson, Py-Macrodipa: A Janus Chelator Capable of Binding Medicinally Relevant Rare-Earth Radiometals of Disparate Sizes, J. Am. Chem. Soc., 2021, 143, 10429–10440 CrossRef CAS PubMed.
- A. Hu, V. Brown, S. N. MacMillan, V. Radchenko, H. Yang, L. Wharton, C. F. Ramogida and J. J. Wilson, Chelating the Alpha Therapy Radionuclides 225Ac3+ and 213Bi3+ with 18-Membered Macrocyclic Ligands Macrodipa and Py-Macrodipa, Inorg. Chem., 2022, 61, 801–806 CrossRef CAS PubMed.
- N. A. Thiele, V. Brown, J. M. Kelly, A. Amor-Coarasa, U. Jermilova, S. N. MacMillan, A. Nikolopoulou, S. Ponnala, C. F. Ramogida, A. K. H. Robertson, C. Rodríguez-Rodríguez, P. Schaffer, C. Williams Jr., J. W. Babich, V. Radchenko and J. J. Wilson, An Eighteen-Membered Macrocyclic Ligand for Actinium-225 Targeted Alpha Therapy, Angew. Chem., Int. Ed., 2017, 56, 14712–14717 CrossRef CAS PubMed.
- K. J. Kadassery, A. P. King, S. Fayn, K. E. Baidoo, S. N. MacMillan, F. E. Escorcia and J. J. Wilson, H2BZmacropa-NCS: A Bifunctional Chelator for Actinium-225 Targeted Alpha Therapy, Bioconjugate Chem., 2022, 33, 1222–1231 CrossRef CAS PubMed.
- C. Harriswangler, L. Caneda-Martínez, O. Rousseaux, D. Esteban-Gómez, O. Fougère, R. Pujales-Paradela, L. Valencia, M. I. Fernández, N. Lepareur and C. Platas-Iglesias, Versatile Macrocyclic Platform for the Complexation of [natY/90Y]Yttrium and Lanthanide Ions, Inorg. Chem., 2022, 61, 6209–6222 CrossRef CAS PubMed.
- H. Yang, C. Zhang, Z. Yuan, C. Rodriguez-Rodriguez, A. Robertson, V. Radchenko, R. Perron, D. Gendron, P. Causey, F. Gao, F. Bénard and P. Schaffer, Synthesis and Evaluation of a Macrocyclic Actinium-225 Chelator, Quality Control and In Vivo Evaluation of 225Ac-crown-αMSH Peptide, Chem. – Eur. J., 2020, 26, 11435–11440 CrossRef CAS PubMed.
- L. Wharton, S. W. McNeil, H. Merkens, Z. Yuan, M. Van de Voorde, G. Engudar, A. Ingham, H. Koniar, C. Rodríguez-Rodríguez, V. Radchenko, M. Ooms, P. Kunz, F. Bénard, P. Schaffer and H. Yang, Preclinical Evaluation of [155/161Tb]Tb-Crown-TATE—A Novel SPECT Imaging Theranostic Agent Targeting Neuroendocrine Tumours, Molecules, 2023, 28, 3155 CrossRef CAS PubMed.
- T. Gambino, L. Valencia, P. Pérez-Lourido, D. Esteban-Gómez, M. Zaiss, C. Platas-Iglesias and G. Angelovski, Inert macrocyclic Eu 3+ complex with affirmative paraCEST features, Inorg. Chem. Front., 2020, 7, 2274–2286 RSC.
- G. Castro, G. Wang, T. Gambino, D. Esteban-Gómez, L. Valencia, G. Angelovski, C. Platas-Iglesias and P. Pérez-Lourido, Lanthanide(III) Complexes Based on an 18-Membered Macrocycle Containing Acetamide Pendants. Structural Characterization and paraCEST Properties, Inorg. Chem., 2021, 60, 1902–1914 CrossRef CAS PubMed.
- P. M. Michelle and S. C. Jackels, Helical hexaazamacrocyclic ligands containing pyridyl and (+ or −)-trans-diaminocyclohexyl groups: effect of ligand constraints upon metal ion binding, Inorg. Chim. Acta, 1996, 246, 301–310 CrossRef.
- L. M. P. Lima, M. Beyler, R. Delgado, C. Platas-Iglesias and R. Tripier, Investigating the Complexation of the Pb2+ /Bi3+ Pair with Dipicolinate Cyclen Ligands, Inorg. Chem., 2015, 54, 7045–7057 CrossRef CAS PubMed.
- R. D. Hancock, J. H. Reibenspies and H. Maumela, Structural Effects of the Lone Pair on Lead(II), and Parallels with the Coordination Geometry of Mercury(II). Does the Lone Pair on Lead(II) Form H-Bonds? Structures of the Lead(II) and Mercury(II) Complexes of the Pendant-Donor Macrocycle DOTAM (1,4,7,10-Tetrakis(carbamoylmethyl)-1,4,7,10-tetraazacyclododecane), Inorg. Chem., 2004, 43, 2981–2987 CrossRef CAS PubMed.
- J. W. Nugent, H.-S. Lee, J. H. Reibenspies and R. D. Hancock, Spectroscopic, structural, and thermodynamic aspects of the stereochemically active lone pair on lead(II): Structure of the lead(II) dota complex, Polyhedron, 2015, 91, 120–127 CrossRef CAS.
- L. Shimoni-Livny, J. P. Glusker and C. W. Bock, Lone Pair Functionality in Divalent Lead Compounds, Inorg. Chem., 1998, 37, 1853–1867 CrossRef CAS.
- D. Esteban-Gómez, C. Platas-Iglesias, T. Enríquez-Pérez, F. Avecilla, A. De Blas and T. Rodríguez-Blas, Lone-Pair Activity in Lead(II) Complexes with Unsymmetrical Lariat Ethers, Inorg. Chem., 2006, 45, 5407–5416 CrossRef PubMed.
- S. W. A. Bligh, N. Choi, C. F. G. C. Geraldes, S. Knoke, M. McPartlin, M. J. Sanganee and T. M. Woodroffe, A novel hexaaza macrocycle with methylenephosphonate pendant arms: a potential useful chelate for biomedical applications †, J. Chem. Soc., Dalton Trans., 1997, 4119–4126 RSC.
- L. Valencia, J. Martinez, A. Macías, R. Bastida, R. A. Carvalho and C. F. G. C. Geraldes, X-ray Diffraction and 1H NMR in Solution: Structural Determination of Lanthanide Complexes of a Py2N6Ac4 Ligand, Inorg. Chem., 2002, 41, 5300–5312 CrossRef CAS PubMed.
- G. Castro, G. Wang, T. Gambino, D. Esteban-Gómez, L. Valencia, G. Angelovski, C. Platas-Iglesias and P. Pérez-Lourido, Lanthanide(III) Complexes Based on an 18-Membered Macrocycle Containing Acetamide Pendants. Structural Characterization and paraCEST Properties, Inorg. Chem., 2021, 60, 1902–1914 CrossRef CAS PubMed.
- G. Castro, M. Regueiro-Figueroa, D. Esteban-Gómez, R. Bastida, A. Macías, P. Pérez-Lourido, C. Platas-Iglesias and L. Valencia, Exceptionally Inert Lanthanide(III) PARACEST MRI Contrast Agents Based on an 18-Membered Macrocyclic Platform, Chem. – Eur. J., 2015, 21, 18662–18670 CrossRef CAS PubMed.
- M. del C. Fernández-Fernández, R. Bastida, A. Macías, P. Pérez-Lourido, C. Platas-Iglesias and L. Valencia, Lanthanide(III) Complexes with a Tetrapyridine Pendant-Armed Macrocyclic Ligand: 1 H NMR Structural Determination in Solution, X-ray Diffraction, and Density-Functional Theory Calculations, Inorg. Chem., 2006, 45, 4484–4496 CrossRef PubMed.
- E. J. Corey and J. C. Bailar, The Stereochemistry of Complex Inorganic Compounds. XXII. Stereospecific Effects in Complex Ions 1, J. Am. Chem. Soc., 1959, 81, 2620–2629 CrossRef CAS.
- J. K. Beattie, Conformational analysis of tris(ethylenediamine) complexes, Acc. Chem. Res., 1971, 4, 253–259 CrossRef CAS.
- E. S. Claudio, M. A. ter Horst, C. E. Forde, C. L. Stern, M. K. Zart and H. A. Godwin, 207Pb− 1H Two-Dimensional NMR Spectroscopy: A Useful New Tool for Probing Lead(II) Coordination Chemistry, Inorg. Chem., 2000, 39, 1391–1397 CrossRef CAS PubMed.
- M. Karplus, Vicinal Proton Coupling in Nuclear Magnetic Resonance, J. Am. Chem. Soc., 1963, 85, 2870–2871 CrossRef CAS.
- A. Rodríguez-Rodríguez, D. Esteban-Gómez, A. de Blas, T. Rodríguez-Blas, M. Botta, R. Tripier and C. Platas-Iglesias, Solution Structure of Ln(III) Complexes with Macrocyclic Ligands Through Theoretical Evaluation of 1H NMR Contact Shifts, Inorg. Chem., 2012, 51, 13419–13429 CrossRef PubMed.
- M. Regueiro-Figueroa, D. Esteban-Gómez, C. Platas-Iglesias, A. De Blas and T. Rodríguez-Blas, Metal Ion Complementarity: Effect of Ring–Size Variation on the Conformation and Stability of Lead(II) and Cadmium(II) Complexes with Pendant–Armed Crowns, Eur. J. Inorg. Chem., 2007, 2007, 2198–2207 CrossRef.
- D. Esteban-Gómez, R. Ferreirós, S. Fernández-Martínez, F. Avecilla, C. Platas-Iglesias, A. De Blas and T. Rodríguez-Blas, Lateral Macrobicyclic Architectures: Toward New Lead(II) Sequestering Agents, Inorg. Chem., 2005, 44, 5428–5436 CrossRef PubMed.
- D. Esteban, F. Avecilla, C. Platas-Iglesias, J. Mahía, A. de Blas and T. Rodríguez-Blas, Lead(II) Complexes with Macrocyclic Receptors Derived from 4,13-Diaza-18-crown-6, Inorg. Chem., 2002, 41, 7170–7170 CrossRef CAS PubMed.
- D. Esteban, F. Avecilla, C. Platas-Iglesias, J. Mahía, A. de Blas and T. Rodríguez-Blas, Lead(II) Complexes with Macrocyclic Receptors Derived from 4,13-Diaza-18-crown-6, Inorg. Chem., 2002, 41, 4337–4347 CrossRef CAS PubMed.
- J. Sanchiz, P. Esparza, D. Villagra, S. Domínguez, A. Mederos, F. Brito, L. Araujo, A. Sánchez and J. M. Arrieta, Holo- and Hemidirected Lead(II) in the Polymeric [Pb4(μ-3,4-TDTA)2(H2O)2]·4H2O Complex. N,N,N‘,N‘-Tetraacetate Ligands Derived from o-Phenylenediamines as Sequestering Agents for Lead(II), Inorg. Chem., 2002, 41, 6048–6055 CrossRef CAS PubMed.
- F. Cuenot, M. Meyer, E. Espinosa, A. Bucaille, R. Burgat, R. Guilard and C. Marichal-Westrich, New Insights into the Complexation of Lead(II) by 1,4,7,10-Tetrakis(carbamoylmethyl)-1,4,7,10-tetraazacyclododecane (DOTAM): Structural, Thermodynamic, and Kinetic Studies, Eur. J. Inorg. Chem., 2008, 2008, 267–283 CrossRef.
- B. Wrackmeyer and K. Horchler, in Annual Reports on NMR Spectroscopy, Elsevier, 1990, vol. 22, pp. 249–306 Search PubMed.
- Y. Xing, A. K. Jindal, M. Regueiro-Figueroa, M. Le Fur, N. Kervarec, P. Zhao, Z. Kovacs, L. Valencia, P. Pérez-Lourido, R. Tripier, D. Esteban-Gómez, C. Platas-Iglesias and A. D. Sherry, The Relationship between NMR Chemical Shifts of Thermally Polarized and Hyperpolarized 89Y Complexes and Their Solution Structures, Chem. – Eur. J., 2016, 22, 16657–16667 CrossRef CAS PubMed.
- L. Branco, J. Costa, R. Delgado, M. G. B. Drew, V. Félix and B. J. Goodfellow, Metal complexes of dipyridine hexaaza macrocycles. Structural differences between 18- and 20-membered macrocycles on complexation, J. Chem. Soc., Dalton Trans., 2002, 3539–3550 RSC.
- K. I. Dhont, G. G. Herman, A. C. Fabretti, W. Lippens and A. M. Goeminne, Protonation and metal-ion complexation in aqueous solution by pyridine-containing hexaaza macrocycles, J. Chem. Soc., Dalton Trans., 1996, 1753 RSC.
- C. Cruz, S. Carvalho, R. Delgado, M. G. B. Drew, V. Félix and B. J. Goodfellow, Metal complexes of a dipyridine octaazamacrocycle: stability constants, structural and modelling studies, Dalton Trans., 2003, 3172–3183 RSC.
- Z. Baranyai, E. Brücher, T. Iványi, R. Király, I. Lázár and L. Zékány, Complexation Properties of N,N′,N′′,N′′′,-[1,4,7,10-Tetraazacyclododecane-1,4,7,10-tetrayltetrakis(1-oxoethane-2,1-diyl)]tetrakis[glycine] (H4dotagl). Equilibrium, Kinetic, and Relaxation Behavior of the Lanthanide(III) Complexes, Helv. Chim. Acta, 2005, 88, 604–617 CrossRef CAS.
- E. Graf, J. Harrowfield, J. Kintzinger, J. Lehn, J. Le Moigne and K. Rissanen, Protonation of a Spherical Macrotricyclic Tetramine: Water Inclusion, Allosteric Effect, and Cooperativity, ChemPlusChem, 2018, 83, 605–611 CrossRef CAS PubMed.
- J. Hamacek, M. Borkovec and C. Piguet, Simple thermodynamics for unravelling sophisticated self-assembly processes, Dalton Trans., 2006, 1473–1490 RSC.
- G. Tircsó, M. Regueiro-Figueroa, V. Nagy, Z. Garda, T. Garai, F. K. Kálmán, D. Esteban-Gómez, É. Tóth and C. Platas-Iglesias, Approaching the Kinetic Inertness of Macrocyclic Gadolinium(III)-Based MRI Contrast Agents with Highly Rigid Open-Chain Derivatives, Chem. – Eur. J., 2016, 22, 896–901 CrossRef PubMed.
- R. Ferreirós-Martínez, D. Esteban-Gómez, É. Tóth, A. de Blas, C. Platas-Iglesias and T. Rodríguez-Blas, Macrocyclic Receptor Showing Extremely High Sr(II)/Ca(II) and Pb(II)/Ca(II) Selectivities with Potential Application in Chelation Treatment of Metal Intoxication, Inorg. Chem., 2011, 50, 3772–3784 CrossRef PubMed.
- C. Paul-Roth and K. N. Raymond, Amide Functional Group Contribution to the Stability of Gadolinium(III) Complexes: DTPA Derivatives, Inorg. Chem., 1995, 34, 1408–1412 CrossRef CAS.
- B. L. McNeil, S. A. Mastroianni, S. W. McNeil, S. Zeisler, J. Kumlin, S. Borjian, A. W. McDonagh, M. Cross, P. Schaffer and C. F. Ramogida, Optimized production, purification, and radiolabeling of the 203Pb/212Pb theranostic pair for nuclear medicine, Sci. Rep., 2023, 13, 10623 CrossRef CAS PubMed.
- G. L. Rothermel, L. Miao, A. L. Hill and S. C. Jackels, Macrocyclic ligands with 18-membered rings containing pyridine or furan groups: preparation, protonation, and complexation by metal ions, Inorg. Chem., 1992, 31, 4854–4859 CrossRef CAS.
- M. M. Ali, M. Woods, E. H. Suh, Z. Kovacs, G. Tircsó, P. Zhao, V. D. Kodibagkar and A. D. Sherry, Albumin-binding PARACEST agents, JBIC, J. Biol. Inorg. Chem., 2007, 12, 855–865 CrossRef CAS PubMed.
- APEX4, Bruker AXS Inc., Madison, Wisconsin, USA, 2021 Search PubMed.
- SAINT Version 8.37A, Bruker AXS Inc., 2015 Search PubMed.
- G. M. Sheldrick, SADABS Version 2014/5, Bruker AXS Inc Search PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- P. Gans, A. Sabatini and A. Vacca, Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs, Talanta, 1996, 43, 1739–1753 CrossRef CAS PubMed.
- 16 Gaussian, C.01 Revision, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian Inc., Wallingford CT, 2016.
- B. Metz, H. Stoll and M. Dolg, Small-core multiconfiguration-Dirac–Hartree–Fock-adjusted pseudopotentials for post- d main group elements: Application to PbH and PbO, J. Chem. Phys., 2000, 113, 2563–2569 CrossRef CAS.
- K. A. Peterson, Systematically convergent basis sets with relativistic pseudopotentials. I. Correlation consistent basis sets for the post- d group 13–15 elements, J. Chem. Phys., 2003, 119, 11099–11112 CrossRef CAS.
- NBO Version 3.1, E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, 2003.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C, high-resolution crystal data and structure refinement details, thermodynamic constant determination and radiolabeling experiments. CCDC 2305213 and 2305214. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qi02354k |
| This journal is © the Partner Organisations 2024 |