
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
“Diluting branches” put to work: from synthesis to properties control of multifunctional polymers derived from triphenylamine, fluorene and thiophene†
Ioana-Alexandra
Trofin
 ,
Catalin-Paul
Constantin
,
Mariana-Dana
Damaceanu
and
Radu-Dan
Rusu
*
,
Catalin-Paul
Constantin
,
Mariana-Dana
Damaceanu
and
Radu-Dan
Rusu
*
Petru Poni Institute of Macromolecular Chemistry, Romanian Academy, Electroactive Polymers and Plasmochemistry Department, Grigore Ghica Voda Alley, 41A, Iasi, 700487, Romania. E-mail: radu.rusu@icmpp.ro
First published on 2nd September 2024
Abstract
A series of highly branched polymers was synthesized by Suzuki polycondensation of thiophene-, fluorene-, and triphenylamine-based monomers, following the “A2 + B2 + C3” pathway. A “dilution of branches” approach allows branching density management and generates conjugated frameworks with linear, alkyl-decorated segments of various lengths. This strategy affects solubility, morphology, and thermal features and allows further control options. The building blocks are involved individually or in combinations in the main electronic transitions and charge and energy transfer processes, as confirmed by computational and experimental UV–vis and photoluminescence studies. The absorption and emission profiles are influenced by branching density, solvent, or the sample's physical state. The structural units’ particular arrangement in each branched construct regulates the redox patterns and electrochemical parameters. The polymers’ overall features and their variation with structure and branching density assemble the foundation for engaging conjugated materials for (opto)electronic applications.
Introduction
Over the past half-century, the field of conjugated polymers has put forward manifold gripping challenges, spanning several fields from fundamental and applied research alike. The core of these materials is an assembly of mobile electrons that drives macromolecules to take up, move, or accumulate electrical charges and further activates remarkable light absorption or emission and outstanding charge mobility or stabilization.1–4The overall synthetic provocation and the chromo- and electro-phore functions are the main incentives of a solid body of research that follows interconnected goals: to come close to structural perfection, to increase complexity and roles, to access processability by wet techniques, to boost the control degree over their chemical, optical, electronic and morphological features.4 The key principle is to master the chemical blueprint and structure–property correlations of such architectures as to tailor the functionalities and performance of optical and electronic materials and (light-emitting, photovoltaic, transistor, energy storage, sensor, electrochromic) devices based on them.3,5,6
Some of the most promising conjugated frameworks are based on thiophene, fluorene, and triphenylamine building blocks, each displaying optical- and electronic-related benefits. Thiophene-based polymers provide tailorable optical features, highly stable (un)doped states, reversible oxidation processes, accessible structural modification, and satisfying processability.6–8 Polyfluorenes and their derivatives are widely appreciated for excellent photo-optical conduct, superior luminescence quantum yields, photochemical stability, large p-type mobility with trap-free transport, and facile structural manipulation.9–11 Macromolecular constructs incorporating triphenylamine are equipped with attractive photo- and electro-activity, high electron mobility, stable redox behaviour, and good p-type transporting features.12–14 However, with such great potential come burdensome (optical, electronic, and processability-related) difficulties, and achieving the aforementioned goals is missing some key pieces, with conjugated polymers being far from large-scale employment.
Most of the work dedicated to solving the challenges of conjugated polymers is centred on one-dimensional, linear frameworks. Nevertheless, a divergent topological standpoint could furnish some solutions: hyper- and highly-branched polymers (HBPs; for a judicious disambiguation between the two terms, please see the references within this paragraph).15 HBPs bring a medley of compelling chemical and physical features derived from their particular, multi-dimensional structure of a rather globular nature. More importantly, this random distribution of dendritic, linear, and terminal units generates substantial advantages compared with the conjugated, linear analogues: dissimilar steric hindrance, distinct intermolecular interactions, abundant peripheral functional groups, prospects of good solubility, improved luminescent and charge-transport performances, alleviation of aggregation-induced quenching, new possibilities to modulate chemical, thermal, electrical and optical features.16–18 Additionally, the HBPs’ industrial relevance is increased by the well-established, convenient synthesis strategies and the facile (usually one-pot) preparation.19 Of course, the interconnected objectives detailed above for conjugated linear polymers also apply to HBPs and the detailed relationship between their architecture and the resulting features is the bottom line of research on the topic.
Building from these starting points, the present work introduces new, highly branched, completely conjugated frameworks simultaneously incorporating the three-starred players of conjugated linear macromolecules: thiophene, triphenylamine, and fluorene. Our goal was to broaden the structural access to this topological type of polymers since the literature on the topic only provides a few examples of HBPs, exclusively containing only one or pairs of the aforementioned building blocks. A controlled branching density approach was used in this regard, complemented by conjugated model compounds and theoretical calculations. The main research direction derived from the innovative focus on the diluting branches effect and consisted in evaluating the concurrence between structural elements or their distinct combinations within the branched architecture and some of the most important application-related characteristics. The resulting in-depth connections between design and solubility, thermal, optical, and electrochemical conduct generate the guidelines for developing particular conjugated HBPs as active materials in (opto)electronic technologies.
Experimental section
Starting materials and synthesis of thiophene-based monomer are detailed in the ESI.†Model compounds
Three model compounds (MC1, MC2, MC3) were synthesized starting from a mixture of 2-bromo-3-n-octyltiophene (0.058 g, 0.182 mmol), 4-bromotriphenylamine (0.05 g, 0.182 mmol), and 9,9-dihexyl-9H-fluorene-2,7-diboronic acid bis(pinacol) ester (0.091 g, 0.182 mmol). The starting compounds were charged carefully in a Schlenk flask to ensure complete transfer and solubilized in N,N-dimethylformamide (DMF, 8 mL). An aqueous NaHCO3 solution (0.152 g, 1.81 mmol in 3 mL of water), followed by the [1,1′-bis (diphenylphosphino) ferrocene] dichloropalladium(II), catalyst (Pd(dppf)Cl2, 6 mg, 5 mol%) were then added. The resulting light red suspension was submitted to three freeze–pump–thaw cycles. The mixture was refluxed for 12 h at 100 °C under nitrogen and monitored by TLC until completion. The reaction was quenched with water and extracted with chloroform (4 × 25 mL). The organic layer was washed with brine, dried over Na2SO4, and the solvent was evaporated under vacuum. The residue was purified by silica gel column chromatography eluting with hexane/dichloromethane (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture to give MC1, MC2, and MC3. The detailed 1H, 13C, COSY, HSQC, and HMBC NMR spectra of the three model compounds are presented in Fig. S1–S3.† Their UV spectra are displayed in Fig. S4.†
:1) mixture to give MC1, MC2, and MC3. The detailed 1H, 13C, COSY, HSQC, and HMBC NMR spectra of the three model compounds are presented in Fig. S1–S3.† Their UV spectra are displayed in Fig. S4.†
MC1: 1H NMR (CDCl3, 400.1 MHz, 25 °C, δ (ppm)): 7.78–7.74 (2H, d, J = 7.8 Hz, H8), 7.62–7.55 (8H, m, H5–H7), 7.35–7.27 (8H, m, H2), 7.23–7.14 (12H, m, H3, H4), 7.09–7.04 (4H, t, J = 7.3 Hz, H1), 2.1–1.95 (4H, m, H9), 1.35–1.2 (4H, m, H10), 1.2–1.01 (12H, m, H11–H13), 0.79–0.72 (6H, t, J = 6.7 Hz, H14).
MC2: 1H NMR (CDCl3, 400.1 MHz, 25 °C, δ (ppm)): 7.84–7.8 (1H, d, J = 7.87 Hz, H22), 7.79–7.75 (1H, d, J = 8.24 Hz, H12), 7.7–7.67 (1H, d, J = 7.85 Hz, H21), 7.65 (1H, s, H20), 7.47–7.41 (2H, m, H11, H13), 7.28–7.23 (7H, m, H10, H23, H26), 7.12–7.08 (6H, m, H24, H25), 7.05–6.98 (3H, m, H9, H27) 2.74–2.68 (2H, t, J = 7.69 Hz, H8), 2.11–1.99 (4H, m, H14), 1.68–1.60 (2H, m, H7), 1.38–1.25 (14H, m, H2–H6, H15), 1.2–1.02 (12H, m, H16–H18), 0.95–0.82 (3H, t, J = 6.19 Hz, H1), 0.85–0.71 (6H, t, J = 6.65 Hz, H19).
MC3: 1H NMR (CDCl3, 400.1 MHz, 25 °C, δ (ppm)): 7.75–7.72 (2H, d, J = 7.68 Hz, H12), 7.45–7.41 (4H, m, H11, H13), 7.24–7.23 (2H, d, J = 5.22 Hz, H10), 7.01–6.99 (2H, d, J = 5.2 Hz, H9), 2.71–2.67 (4H, t, J = 7.72 Hz, H8), 2.10–1.96 (4H, m, H14), 1.69–1.58 (4H, m, H7), 1.39–1.21 (24H, m, H2–H6, H15), 1.18–1.03 (12H, m, H16–H18), 0.95–0.87 (6H, t, J = 6.56 Hz, H1), 0.82–0.76 (6H, t, J = 6.82 Hz, H19).
Highly branched polymers
Three highly branched polymers (P1, P2, and P3) were synthesized by Suzuki polycondensation of tris(4-bromophenyl)amine, 9,9-dihexyl-9H-fluorene-2,7-diboronic acid bis(pinacol) ester and 2,5-dibromo-3-octylthiophene in different molar ratios. The reaction conditions (catalyst, time, temperature, solvent) were selected based on previous reports on this type of polymerization and monomers, showing them to provide high molar mass and yield values or acceptable solubility.11,20–22 A typical synthesis procedure is detailed below.P1: a 25 mL Schlenk flask (equipped with a magnetic stirring bar, nitrogen inlet and outlet, and a condenser) was charged with precisely weighed tris(4-bromophenyl)amine (0.06 g, 0.141 mmol), 2,5-dibromo-3-octylthiophene (0.05 g, 0.141 mmol) and 9,9-dihexyl-9H-fluorene-2,7-diboronic acid bis(pinacol) ester (0.177 g, 0.353 mmol). The monomers were dissolved in DMF (14 mL) and an aqueous NaHCO3 solution (0.296 g, 3.52 mmol in 5.6 mL of water) was added to form a white suspension. The Pd(dppf)Cl2 catalyst (0.0129 g, 5 mol%) was inserted, and the resulting light red reaction mixture was degassed by three freeze–pump–thaw cycles. The temperature was raised to 125 °C and the reaction proceeded by vigorous stirring for 72 h in inert conditions to yield a green-yellow suspension. After cooling to room temperature, the reaction mixture was filtered to isolate a green solid. This raw product was purified by washing it with hot MeOH (3 times) and dried to afford the P1 polymer as a green powder (91% yield, from which 34% was soluble in chloroform).
1H NMR (CDCl3, 400.1 MHz, 25 °C, δ (ppm)): 7.78–7.57 (m, Fl), 7.49–7.44 (m, TPA), 7.36–7.30 (m, Th), 2.78–2.72 (m, Th), 2.08–2.00 (m, Fl), 1.74–1.68 (m, Th), 1.39–1.25 (m, Fl, Th), 1.12–1.07 (m, Fl), 0.89–0.83 (m, Th), 0.78–0.75 (m, Fl).
P2: tris(4-bromophenyl)amine, (0.019 g, 0.4 mmol), 2,5-dibromo-3-octylthiophene (0.05 g, 0.141 mmol), 9,9-dihexyl-9H-fluorene-2,7-diboronic acid bis(pinacol) ester (0.101 g, 0.2 mmol), DMF (8 mL), NaHCO3 (0.169 g, 2.01 mmol), water (3.2 mL) and Pd(dppf)Cl2 (0.007 g, 5 mol%) were employed in the typical Suzuki polycondensation procedure detailed above to obtain P2 as a green polymer powder (91% yield, from which 55% soluble in chloroform).
1H NMR (CDCl3, 400.1 MHz, 25 °C, δ (ppm)): 7.82–7.57 (m, Fl), 7.51–7.48 (m, TPA), 7.35–7.30 (m, Th), 2.77–2.74 (m, Th), 2.09–2.00 (m, Fl), 1.74–1.66 (m, Th), 1.40–1.24 (m, Fl, Th), 1.14–1.06 (m, Fl), 0.89–0.86 (m, Th), 0.79–0.75 (m, Fl).
P3: tris(4-bromophenyl)amine, (0.009 g, 0.017 mmol), 2,5-dibromo-3-octylthiophene (0.05 g, 0.141 mmol), 9,9-dihexyl-9H-fluorene-2,7-diboronic acid bis(pinacol) ester (0.083 g, 0.16 mmol), DMF (6.6 mL), NaHCO3 (0.139 g, 1.65 mmol), water (2.7 mL) and Pd(dppf)Cl2 (0.007 g, 5% molar) were employed in the typical Suzuki polycondensation procedure detailed above to attain P3 as a green polymer powder (93% yield, from which 76% soluble in chloroform).
1H NMR (CDCl3, 400.1 MHz, 25 °C, δ (ppm)): 7.80–7.59 (m, Fl), 7.51–7.49 (m, TPA), 7.35–7.30 (m, Th), 2.78–2.72 (m, Th), 2.09–2.00 (m, Fl), 1.76–1.68 (m, Th), 1.39–1.25 (m, Fl, Th), 1.14–1.04 (m, Fl), 0.89–0.83 (m, Th), 0.80–0.73 (m, Fl).
Two notes must be mentioned: (1) no gelation was observed during polymerization; (2) the yield was determined ignoring the end functional groups whose nature is unclear.
Before any measurement, all polymers were extracted in chloroform using a Soxhlet installation. Two fractions were isolated: (1) an insoluble one, which was used for all measurements involving powdered samples, except solubility tests; (2) a highly soluble fraction, which was further used for all investigations involving solutions and coatings. Judging by the FTIR (for the insoluble fraction) and NMR (for the soluble one; its gluey nature was incompatible with FTIR analysis) investigations detailed later on, the two fractions have a similar composition, incorporating all three building blocks. Such behaviour is common for highly conjugated products of Suzuki polycondensation and, to an even larger extent, for hyperbranched, conjugated polymeric systems.11,16–19
The instruments and characterization techniques used in this study are detailed in the ESI.†
Results and discussion
Design and synthesis
We synthesized three new highly branched polymers (HBPs, P1, P2, and P3) based on thiophene (Th), fluorene (Fl), and triphenylamine (TPA) units by means of Suzuki polycondensation. An “A2 + B2 + C3” synthesis pathway was used based on design- and processability-related reasoning. First, the branching density is regulated and the distance between branching points is controlled by varying the molar ratio of the monomers via the so-called “dilution of branches” concept (Fig. 1).23 This concept is centred on the orderly reduction of the relative number of branching points (in this case, TPA units) from one highly branched polymeric framework to the other. A transition from a rather compact HBP to a more segmented one is consequently achieved by this route: P1 incorporates a high branching density and short linear fragments between the branching centres, whereas P3 has a (statistical) low degree of branching and long linear segments between branches. In this scenario, the building blocks’ intrinsic electronic and optical features are anticipated to be availed in different proportions. Second, this approach affords a gradual increase in the number of structural entities used to mitigate the solubility problem of such rigid, mostly planar constructs: the pivotal N atom from the propeller-shaped TPA and the short alkyl moieties anchored on Th (one octyl unit) and Fl (two hexyl units).4,14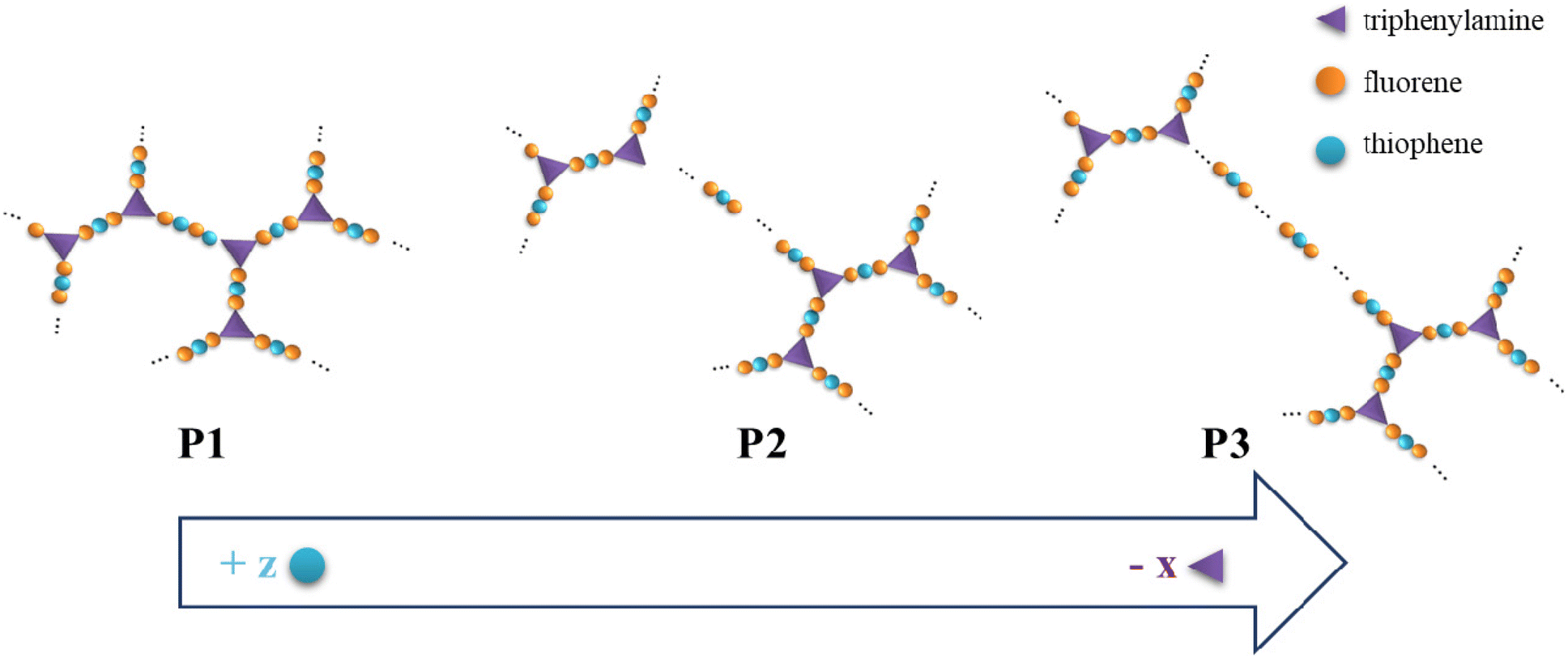 | ||
| Fig. 1 Graphical representation of the dilution of branches concept used in the design of P1–P3 HBPs. | ||
From another standpoint, we selected these three monomers also based on their commercial availability, which makes it possible for the HBPs to be produced at a larger scale and increases their applicative relevance.
The “A2 + B2 + C3” pathway used in the preparation of P1–P3 HBPs implies the one-pot Suzuki polycondensation of trifunctional TPA (C3) with bifunctional Fl (B2) and Th (A2) monomers in which the bis-pinacolato Fl derivative acts at each reaction step as a π-bridge between the other two heteroaromatic blocks. A homologous series with decreasing TPA:Fl and Fl:Th proportions was obtained by adjusting the ratios of the three monomers (Scheme 1).
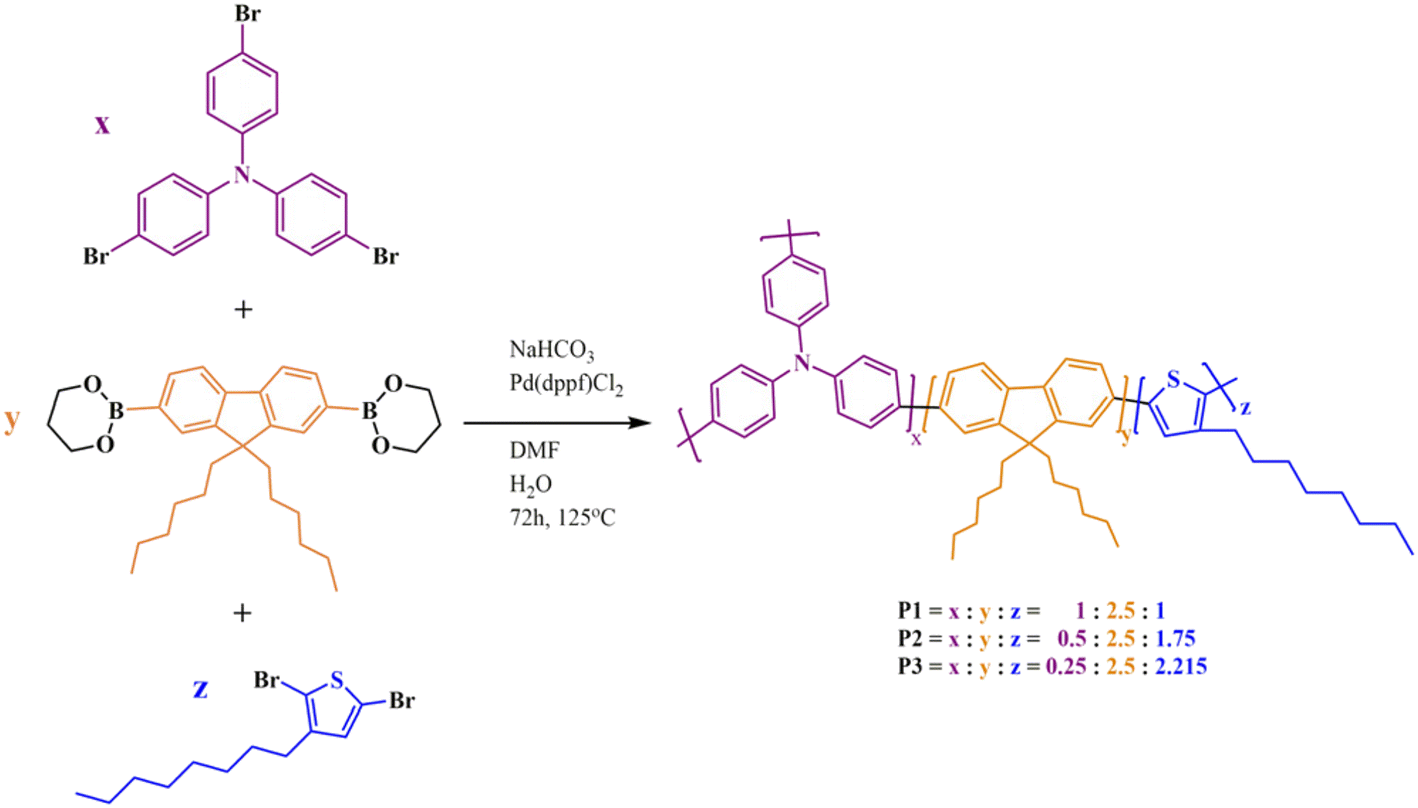 | ||
| Scheme 1 Synthesis of P1–P3 HBPs. Each building block is represented with a different color, while the complementary reacting groups are depicted in black. | ||
The complexity of such highly branched, largely aromatic architectures makes their investigation very challenging. Therefore, we designed model compounds as simplified versions of polymer fragments to ease the comprehension of some particular aspects of these HBPs, as it will be further detailed. The three model compounds (MC1, MC2, MC3) mimic terminal and linear units from the HBP framework and provide insights into the connectivity patterns within the polymer, thus guiding the elucidation of the actual highly branched structure. They were synthesized by the one-pot Suzuki–Miyaura cross-coupling of monofunctional TPA and Th with bifunctional Fl (Scheme 2).
 | ||
| Scheme 2 Synthesis of MC1–MC3 model compounds. | ||
Structural analysis
The structural confirmation of the P1–P3 HBPs was based on FT-IR and 1H NMR spectroscopies. The FT-IR and 1H NMR spectra of each HBP and the detailed assignment of every IR absorption band are displayed in Fig. S5 and S6 and in Table S1,† respectively.The overlapped FT-IR spectra of the three HBPs show the same envelope since the variations in the polymers’ structure are primarily determined by the different molar ratios of the same comonomers (Fig. 2). In the group frequency region, the FT-IR spectra exhibit absorption bands associated with the aromatic (stretching vibrations in the range of 3063–3023 cm−1) and aliphatic (asymmetric and symmetric stretching vibrations between 2931 and 2869 cm−1) C–H bonds within the polymer structure. These bonds present additional characteristic signals in the fingerprint region (in-plane deformation and rocking vibration at 1030 cm−1, in-plane bending vibrations at 1106 cm−1, and out-of-plane bending vibrations at 738 cm−1). The TPA unit (C–N stretching vibrations at 1314 cm−1) and Th ring (C–S–C in-plane stretching vibrations at 1376–1371 cm−1; Th-specific, in-plane and out-of-plane deformation vibrations at 637 and 622 cm−1) also display distinctive absorption bands in the same region.22,24
 | ||
| Fig. 2 Comparative FT-IR spectra of HBPs P1–P3. | ||
The FT-IR spectra show an increase in the relative intensities of the aliphatic C–H peaks (2841–2031, 1466, 1030, and 812 cm−1) with the Th content used as a branching diluent. A similar trend is observed for two other absorption peaks corresponding to Th-specific C–S bending (in the 800–600 cm−1 region). These changes confirm the variation of the HBPs’ branching density in the order P1 > P2 > P3, in accordance with the theoretical sequence proposed by design and implemented by using distinct molar ratios.
The NMR evaluation of the HBPs was performed in connection with the monomers and model compounds. The 1H NMR spectra of the latter (Fig. S1–S3†) were interpolated with the ones obtained for the soluble fraction of each polymer (Fig. 3 and Fig. S6†). They confirm the polymer structure and validate the successful incorporation of the starting TPA, Fl, and Th building blocks.
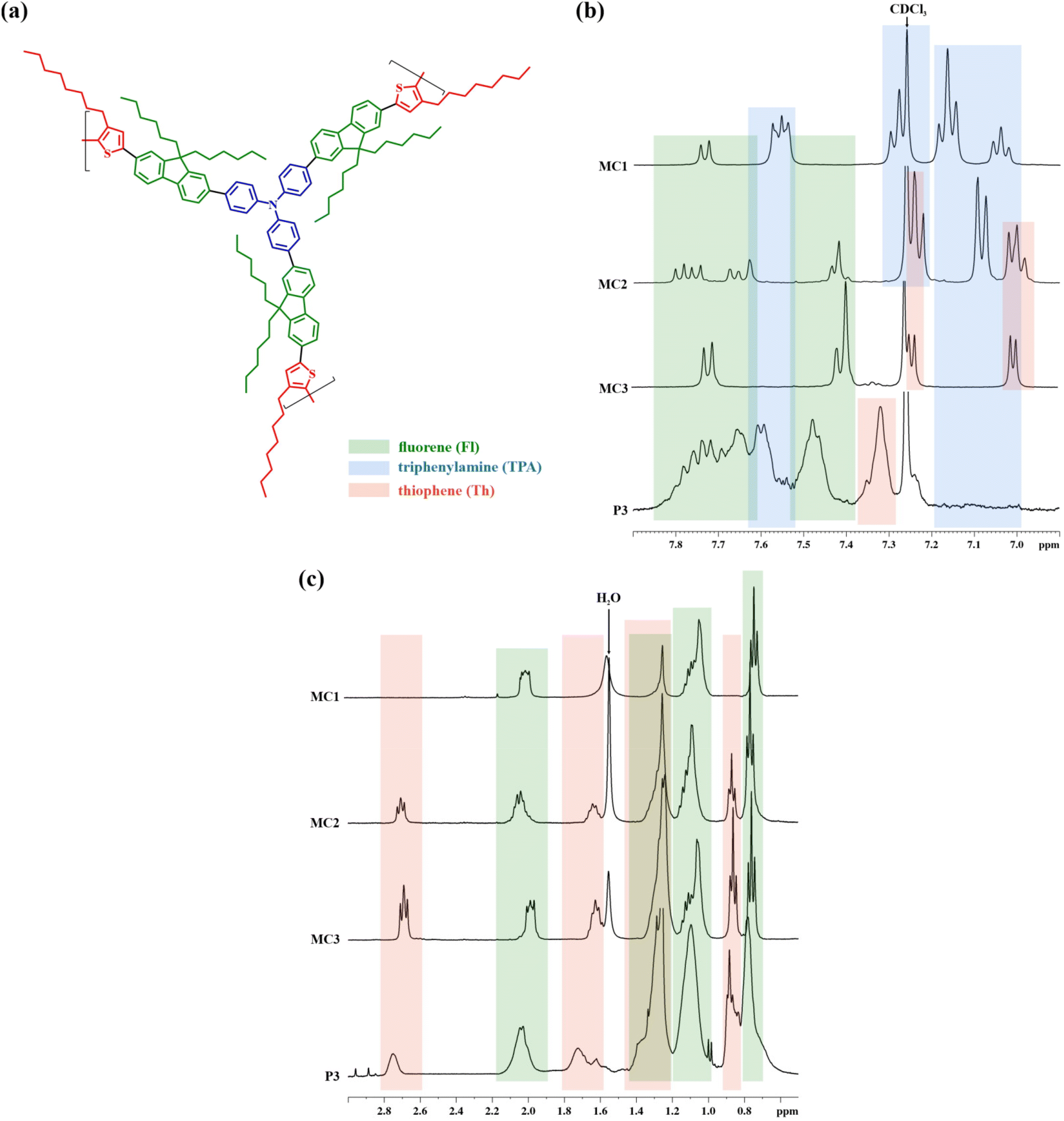 | ||
| Fig. 3 Polymer P3: a repetitive unit (the monomer ratio was not considered for clarity) (a); the aromatic (b) and aliphatic (c) regions of the 1H NMR spectrum as compared to those of MC1–MC3 model compounds. | ||
The symmetrical MC3 model compound comprises one Fl block interposed between two Th rings, readily identifiable by 1H NMR. Their specific signals are present at distinct resonance fields in the aromatic region: 7.75–7.35 ppm for Fl (green boxes in Fig. 3) and 7.25–6.95 ppm for the more electron-donating Th (pink boxes). Furthermore, in the aliphatic region, the signals of the methylene groups vicinal to Th (2.71–2.67 ppm) and Fl (2.00–1.96 ppm) show the expected 1:1 ratio, denoting the incorporation of these two monomers in a 2:1 ratio.
By replacing a Th ring with a TPA unit in the MC2 model compound, the symmetry disruption renders a splitting of the Fl signals from the highest resonance region and the appearance of new signals from the TPA unit (blue boxes in Fig. 3). Simultaneously, the ratio of the Th- and Fl-connected methylene signals changes to 1:2, evidencing the incorporation of the three monomers in a 1:1:1 ratio.
Finally, the complete replacement of Th with TPA in MC1 and the re-establishment of molecular symmetry is evidenced by the Fl-originating doublet signal (7.74–7.72 ppm) and the overlapping of its specific multiplet (MC3: 7.41–7.38 ppm) with the signals (7.57–7.53 ppm) of the aromatic rings from the connecting TPA. At the same time, the Th-adjoining methylene signal (2.71–2.67 ppm) disappears entirely from the spectra.
The extrapolation of the MC1–MC31H NMR data afforded a straightforward assignment of the signals from the polymers’ spectra. In the aromatic region, the multiplets placed at 7.83–7.63 ppm and 7.52–7.38 ppm (green boxes in Fig. 3) are attributed to the Fl block, while the ones in the range 7.64–7.51 ppm (blue box) are ascribed to the TPA branching core. The latter are associated with TPA-specific signals from the model compounds: H-5 from MC1 and H-23 from MC2 (Fig. S1 and S2†). Additionally, the absence of any peaks originating from unsubstituted aromatic rings belonging to the TPA unit, as the ones observed for MC1 (H-1, H-2 and H-3) and MC2 (H-25, H-26 and H-27) between 7.20 and 6.97 ppm (blue box), proves the complete substitution of TPA in all para positions. This pattern was observed for all three polymers and points towards TPA-based structural symmetry: TPA serves only as a branching unit, not as a linear or terminal one. More importantly, it certifies the branched nature of these polymers’ soluble fractions.
The ratio of integrated signals coming from methylene protons vicinal to Th and Fl was found to be 1:2 (P1), 1:2.5 (P2), and 1:3 (P3). This confirms the successful dilution of branches by adjusting the monomer ratio and the shift from a rather compact HBP (P1) to a more sparsely branched one with longer linear segments (P3).
Solubility and molar mass
As detailed in the Experimental section, the polymers were extracted in chloroform to separate a soluble fraction from an insoluble one, a widespread practice in the field of HBPs.22,24,25 As expected, the extraction yield was strongly dependent on the concentration of branching points: from 34% for P1 to 76% for P3. This conduct comes from the gradual increase in the number of structural entities used to mitigate the solubility issue and confirms the envisaged positive effect of the dilution of branches strategy.Qualitative solubility tests showed all HBPs’ soluble fractions readily soluble at room temperature in low boiling point solvents like chloroform, tetrahydrofuran, and dichloromethane (Table S2†). Therefore, these HBPs can be processed into thin films and coatings from convenient solvents, a noteworthy advantage for many practical applications requiring wet layer deposition techniques. Limited solubility was observed when switching to more (N-methylpyrrolidone, N,N-dimethylformamide, N,N-dimethylacetamide) or less polar (toluene, hexane) solvents, additional heating, time or both being necessary for the polymer samples’ dissolution. This behaviour is common for HBPs incorporating these three building blocks individually or in pairs.22,26,27
We did not observe any correlation between the branching density and the solubility determined by qualitative tests, which indicates smaller molecular mass values for the more compact polymers. Nevertheless, the least branched P3 showed superior solubility coming from the longer “hairy-rod” segments consisting of repeating Fl–Th substructures decorated with two hexyl and one octyl chain per segment. These flexible alkyl chains disrupt the coplanarity of the aromatic rings and lower the interaction and packing between the rigid rods. Furthermore, the smaller number of branching points imparts additional movement freedom to these linear segments and also contributes to superior solubility.
The macromolecular nature of P1–P3 was confirmed by GPC in CHCl3 against polystyrene standards. The three products show weight-average molar mass values (Mw), between 8.05 and 14.3 kg mol−1, and dispersity (Mw/Mn) values in the range 2.53–3.44 (Table S2†). While the Mw values might be considered rather low, it is known that TPA and Th alone do not render HBPs of higher Mw.26,27 Moreover, such values are common for (the whole material or soluble fraction of) HBPs incorporating paired combinations of the building blocks used herein.22,25–27 The evolution of the Mw values from P1 to P3 indicates that higher Mw values are accessible by further lowering the content of the trifunctional core and building more soluble, highly linear HBP structures, as also observed by other authors.28 On the other side, these values are not fully representative for the synthesized HBPs since they were obtained for the chloroform-soluble fraction of each polymer, and it is judiciously expected that the insoluble part incorporates a higher number of repeating units.22 More importantly, these molecular mass values should be treated as rough, minimum estimates and used only for cautious comparisons involving polymers of similar topologies. This is due to the large differences between the HBPs’ tree-like architecture and that of the linear standard used in terms of molecular density, hydrodynamic radius, chemistry, and even interactions with the column material.23
Morphology
Regardless of their linear or branched nature, the microscopic morphology of films and coatings developed from conjugated polymers is an important determinant of their macroscopic functions and critical properties as organic semiconductors.29 The few articles on HBPs incorporating some of the building blocks discussed herein show that, in the absence of crystallinity, the morphology of such materials varies between two extremes: from a highly porous surface to a very smooth one.30,31 The former usually comes with a large, variable specific surface area and is thus of interest for applications that require a high adsorption capacity, such as sensors. The latter is useful for electrochemical applications, given the high uniformity that promotes counterion diffusion, doping, and a larger contact area.The P1–P3 series is able to accommodate both morphological uttermost cases. Scanning electron microscopy (SEM) and atomic force microscopy (AFM) investigations show systematic variations in morphology and topography obtained in the same deposition conditions, depending on the branching dilution degree (Fig. 4).
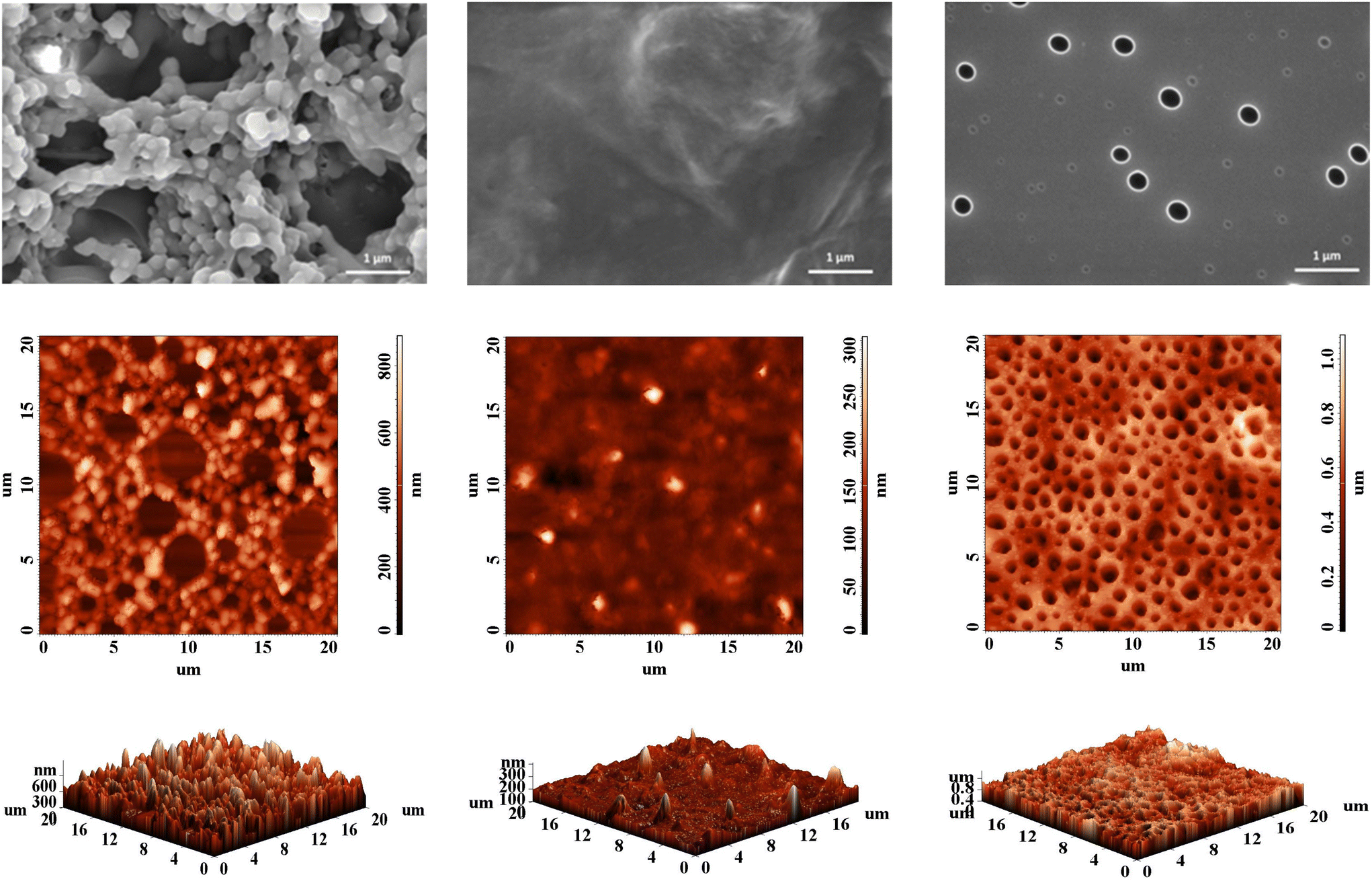 | ||
| Fig. 4 SEM micrographs (first row) and AFM images (second and third row, from left: P1, P2, P3) showing the morphology of HBPs thin coatings obtained by drop-casting from DCM solutions. | ||
P1 shows a highly porous, agglomerated, granular morphology, with large height variations, derived from the high content of branches based on TPA units lacking a planar backbone.32 The higher proportion of more rigid, planar Fl blocks and softening alkyl chains (Fl and Th) from P2 levels the surface morphology towards a smoother, irregular one with a waved appearance.33 Further drop in the TPA:Fl ratio, together with an increase in the less rigid Th content and a consistent softening from the high concentration of alkyl chains in P3 even the surface further to a very planar one, with a high density of holes having two types of size distribution. The origin of these holes is not completely clear, with the more mobile chains in P3 and the faster evaporation of the low boiling point solvent from the very smooth surface being a judicious explanation. The morphological trend is therefore consistent with the branching density and the length of the hairy rods connecting the branching points. At high branching density, the TPA influence is prevalent and imparts directionality to the π–π interactions between the heteroaromatic motifs. This effect abates once the branches are diluted, and the morphological arrangement is influenced more by alkyl chain softening and the composition of linear segments.
These large disparities in surface aspect and topography point towards the possibility of controlling the morphology and, consequently, other application-relevant features of the synthesized HBPs by fine-tuning the building blocks ratio.
Thermal behaviour
The thermal behaviour of the HBPs was investigated by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) experiments under a nitrogen atmosphere. Table S3† presents the most important thermal parameters.The HBPs did not reveal any thermal transition that could be correlated with a clear glass transition or melting phenomenon during DSC analysis up to 300 °C (Fig. S8†). This was an expected feature given the high Tg values (180–230 °C) of linear TPA–Fl polymers of similar Mw or no detectable Tg for HBPs based on paired combinations of the same building blocks reported by other studies.24,26,34,35 Such behaviour comes from the overall stiffness of the heteroaromatic polymer segments and their limited mobility within the highly branched framework. Moreover, the relatively opened, branched architecture and the extended rigid rods obstruct the participation of chain ends in any thermal relaxation.34 The TG and DTG curves (Fig. 5) show high thermal stability for all HBPs, with some peculiarities related to branching density, molecular packing, chain dimensions, and flexible units content.
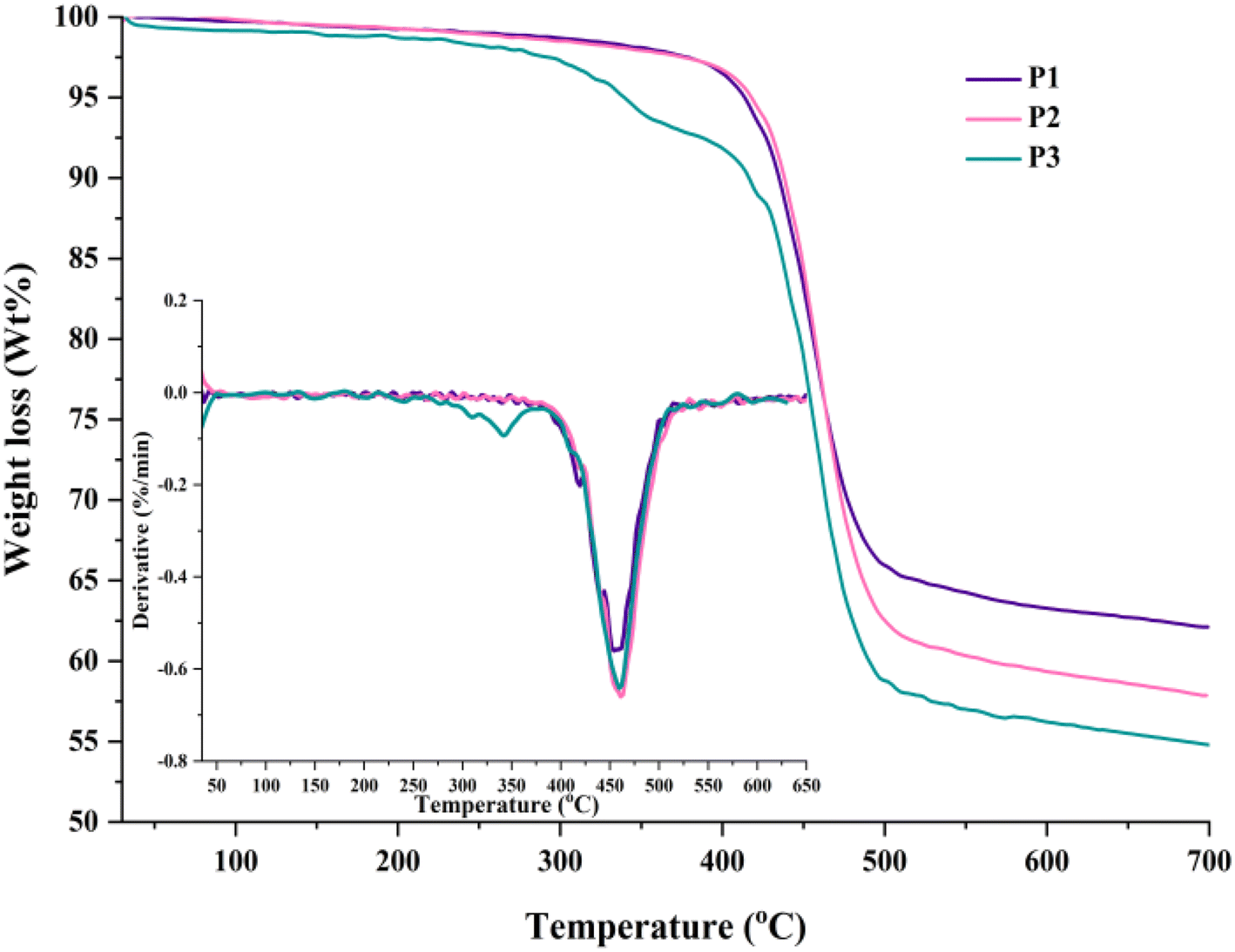 | ||
| Fig. 5 TGA and DTG (inset) curves for HBPs P1–P3. | ||
The polymers with higher branching densities, P2 and P3, started to decompose above 410 °C and lost 10% weight in the 434–438 °C range. The aromaticity-driven rigidity and branching-directed molecular packing are the elements responsible for these thermal parameters. The branching dilution effect expected when switching from P1 to P2 is compensated by the higher Mw values of the latter. However, this is not the case for the more segmented P3, which showed a two-step weight loss starting at much lower temperatures due to the reduced number of branching points, greater concentration of flexible alkyl chains, and subsequent disturbed molecular packing. Nevertheless, the longer hairy-rod segments still impart some stiffness, and P3 reaches 10% weight loss above 410 °C. The DTG curves mirror this conduct and evidence a multi-step degradation process along a maximum decomposition rate between 455 and 460 °C for all HBPs. Previous work has shown that the initial decomposition stages correspond to the degradation of the n-hexyl and n-octyl chains pendant to the Fl and Th units and the extrusion of terminal groups, respectively.11,34 The last decomposition step corresponds to the degradation of the main, heteroaromatic polymer chain. These results indicate additional pathways to finely tune the thermal conduct towards lower or higher decomposition onsets by further increasing the alkyl concentration via branching dilution/longer alkyl chains or by capping the bromide/boronic terminal groups with complementary monofunctional aromatics. Furthermore, a high char yield, between 54.6 and 62.11%, was attained at 700 °C due to the dominant aromatic nature of the HBPs (Table S3†). Its values agree with the branching density and the amount of aliphatic chains, P1 and P3 giving the highest and lowest values.
The high decomposition temperatures and lack of any thermal transitions serve as technological advantages for (opto)electronic devices that are produced and operate at high temperatures. The three polymers provide the thermal and consequent morphological and mechanical integrity for a stable performance and increased lifetime of such devices.
Electronic structure computation
The electron density distribution along the P1–P3 polymeric chains was evaluated by computational tools based on density functional theory (DFT). A simplified macromolecular segment consisting of a single, theoretical, repetitive polymeric unit incorporating all three building blocks was employed to expedite computation time and its ground-state geometries were fully optimized in vacuum. This optimized geometry was further used to evaluate their molecular orbitals (HOMO: highest occupied molecular orbital; LUMO: lowest unoccupied molecular orbital), which are presented in Fig. 6 together with the degenerated orbitals (HOMO−x; LUMO+y) involved in the primary transitions from the simulated UV–vis absorption spectra. | ||
| Fig. 6 The optimized geometry of a simplified HBP segment and a selection of its molecular orbitals, as computed with DFT using the B3LYP/6-31++G(d,p) basis set (isovalue of 0.3). | ||
The HOMO orbital exhibited uniform delocalization throughout the TPA unit, showcasing its electron-donor capability. Degenerated HOMO orbitals of lower energy, HOMO−1 and HOMO−2, were localized mostly on the Th–Fl substructure. The slightly higher density observed on the Th cycle denotes its role as a secondary electron-donating entity. Conversely, the LUMO orbital was localized on various regions of the macromolecular segment and particularly centred on the Th–Fl substructure acting as an electron-deficient assembly. An extended profile of the LUMO orbital was also distributed over the TPA-neighbouring benzene rings. Two other LUMO degenerations, LUMO+1 and LUMO+2, were confined on each arm of the branched structure. All these distributions induce significant oscillator strength to the charge transfer transitions involving HOMO and LUMO orbitals and their degenerations, as elaborated in the next sections. At the same time, the identified degenerated LUMO+5 orbital, with TPA-specific shape and distribution, is involved in the n–π* transition of this building block.
Experimental and theoretical UV–vis analysis
The optical behaviour of the HBPs was examined by UV–vis experiments carried out in various polarity-indexed, dilute solutions and on thin coatings obtained from different solvents. A summary of the most important data collected in this investigation can be found in Tables S4 and S5.†P1–P3 displayed almost identical, broad, and slightly unsymmetrical absorption profiles generated by specific electronic transitions involving each building block or combinations therefrom.The three HBPs showed one major absorption maxima peaking between 386 and 405 nm as a function of solvent, branching density, or sample's physical state. A small-scale (up to 7 nm variation) absorption dependence on solvent polarity points toward positive solvatochromic tendencies (Fig. S9†). This is most visible in the case of P1, with a gradual change in absorption maxima to longer wavelengths from toluene to NMP solutions. The magnitude of the bathochromic shift is shortened in the case of the more segmented HBPs, P2 and P3. Branching density also has some influence on the absorption maxima, a modest redshift (Δλabs = 1–6 nm) being discernible with branching dilution, both in solution (especially in less polar solvents like toluene, THF, and DCM) and in film state (Fig. 7 and Fig. S10†). Finally, the change from solution to condensed phase further alters the absorption maxima due to some reduced aggregation, as evidenced by the weak bathochromic shifts (up to 7 nm for films obtained from THF and 9 nm for films obtained from DCM) in absorption (Table S4†). Concurrently, the sole absorption maxima of P1–P3 are red-shifted with 30 and 50 nm compared to the one of HBPs based only on TPA–Fl and TPA–Th pairs.22,33 This is ascribed to a more extended π-conjugated architecture derived from longer rigid rod segments and the introduction of Fl units, respectively.
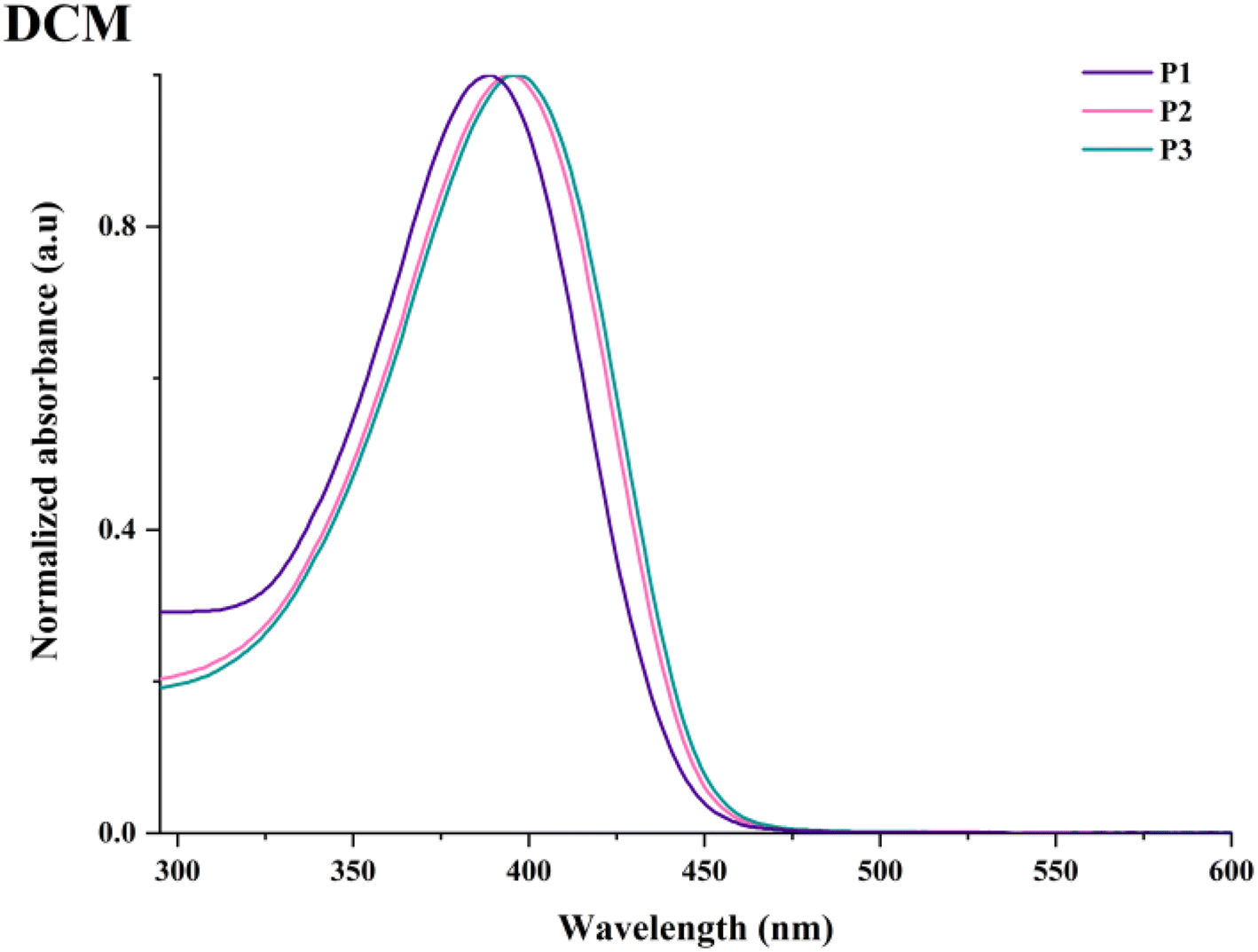 | ||
| Fig. 7 Experimental absorption spectra of P1–P3 in DCM solutions and coatings obtained therefrom. | ||
The experimental evaluation was complemented by a theoretical assessment to gain additional insights into the photo-optical behaviour and the nature of the main electronic transitions (Fig. 8). These are further useful to unfold energy transfer implications concerning photoluminescence, as detailed in the following section. The computed transitions show bathochromic shifts compared with experimental ones. This minor mismatch comes from using a simplified theoretical structure (trimeric entity) and calculations performed in vacuum. Therefore, any interaction with the environment is negligible, and the molecules adopt a more planar structure, favouring transitions at higher wavelengths.
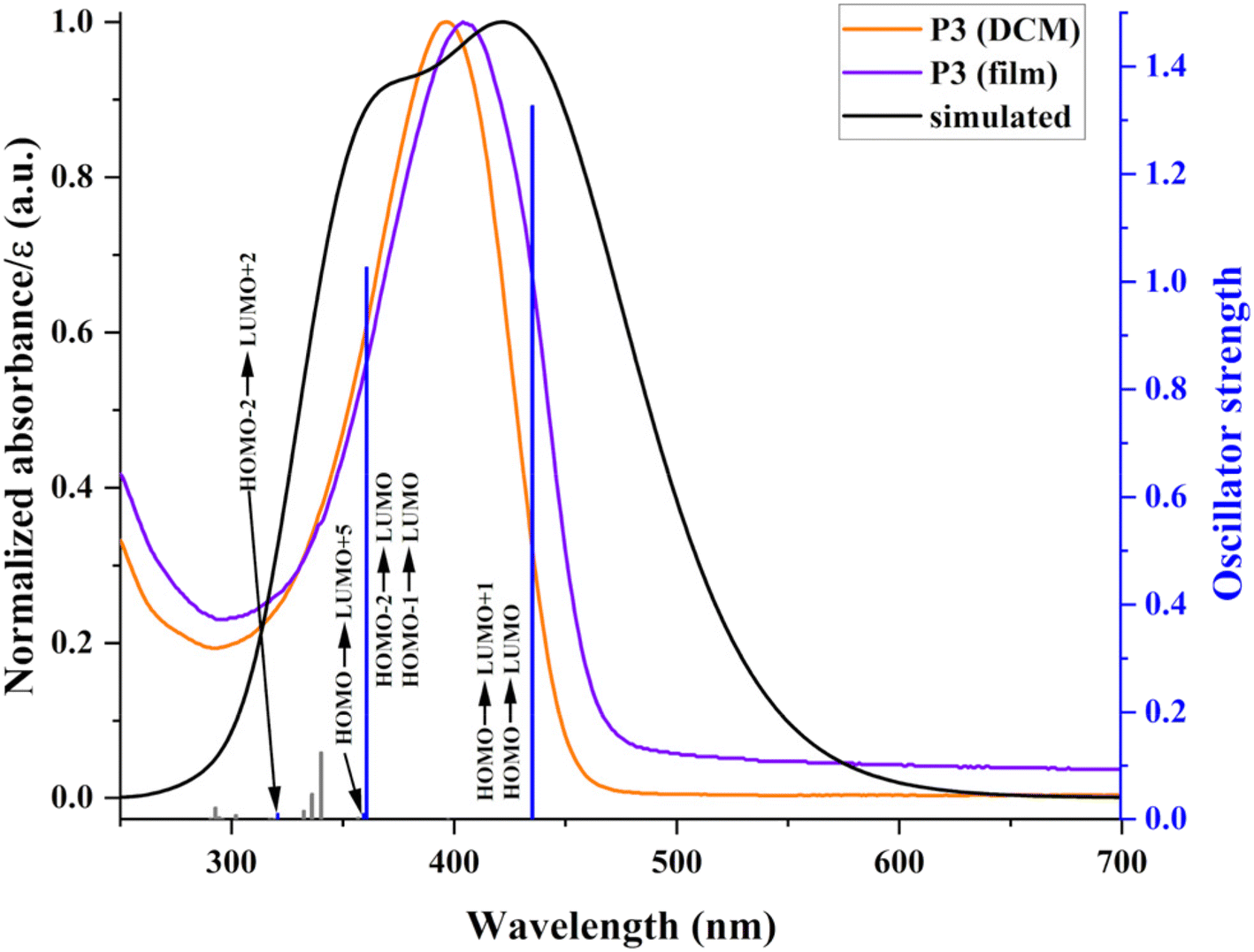 | ||
| Fig. 8 Experimental and simulated (at TD-DFT/B3LYP/6-31++G(d,p) level) absorption spectra and associated oscillator strengths of P3. | ||
The simulated spectra begin with unresolved absorptions close to 300 nm corresponding to localized π–π* transitions of individual aromatic rings and some conjugated structural elements of the polymers. These are followed by an absorption attributed to an n–π* transition involving the nonbonding electrons of TPA. The shape and localization of the associated orbitals (the HOMO and LUMO+5 degenerated orbitals are distributed on the TPA structure (Fig. 6)) confirm the nature of this transition. The main absorption band represents a combination of several transitions with a charge transfer character as based on the associated orbitals. The first two adjacent transitions at 435.4 and 435.2 nm take place between the TPA-located HOMO and the LUMO and LUMO+1 orbitals placed on the weak electron-accepting Th–Fl substructures. This correspondence is strengthened by the very small blue shift of the main experimental absorption bands. Under the main (simulated and experimental) absorption band, additional transitions are produced by excitations from HOMO−1 and HOMO−2 to LUMO, involving the Th rings as secondary electron donor units. These two main theoretical transitions have high oscillator strengths due to the favourable overlap between contributing orbitals. Ultimately, the present polymers exhibit good transparency in the visible domain, with low charge transfer implications, which is a solid advantage for (opto)electronic applications.
Photoluminescence investigations
The photoluminescent (PL) behaviour of the HBPs was assessed in the same conditions as the UV–vis experiments, with excitation being done at various wavelengths corresponding to the absorption maxima (Tables S4 and S5†).All polymers exhibited the same emission profile in the solution comprising one major maximum and two shoulder-like bands, with some small variations in band position or intensity as a function of solvent polarity, excitation wavelength, or branching density (Fig. 9, Fig. S11 and S12†).
 | ||
| Fig. 9 PL spectra of P1 in various solvents at the same excitation wavelength (left) and of P1–P3 in DCM solutions at different excitation wavelengths (right). Insets: associated chromaticity diagrams and polymer solutions’ images under UV light (365 nm). | ||
A narrow redshift (up to 6 nm) in emission maxima with solvent polarity was observed in the case of the highly branched P1, the other two polymers being less sensitive to environmental change. A similar spectral pattern appeared when switching to longer excitation wavelengths. The most significant difference was noticed in emission intensity as a function of both solvent polarity and excitation wavelength. A more intense emission was observed in less polar solvents (e.g., toluene) when excited at 295 nm due to a decline in polarity-induced emission quenching caused by an intramolecular charge transfer (ICT) involving a twisted conformation.36 However, this conduct was disrupted and no clear dependence appeared when the excitation was done at longer wavelengths (Fig. 9, Fig. S11 and S12†). These diverse interactions with the local environment during the excited state lifetime at various excitation energies originate from several elements that competitively contribute to the PL response: distinct fluorophoric units, structural arrangement variation, and divergent planarities in solutions of various polarities.
The branching density slightly affects the emission behaviour in solution, with some slight shifts (up to 7 nm variation) of the emission maxima appearing with branching dilution, especially in less polar solvents.
An energy transfer was noticed when excited at 295 nm, the wavelength characteristic of the π–π* transitions of individual aromatic rings. Their specific PL band overlaps with the ICT one, and the polymers experience only ICT emission. This assumption is validated by the polymers rendering the same emission band, with slightly higher intensities, upon excitation at longer wavelengths corresponding to the ICT absorption maxima (385–395 nm).
The reference-free, absolute fluorescence quantum yields of the polymers in NMP solutions decreased from 44.5% for P1 to 28.8% for P3, as detected by excitation at the ICT absorption maxima (Table S4†). The direct dependence of these moderate quantum yield values on the branching density illustrates the precise tuning achievable by controlling the building blocks’ ratio within HBPs of the same chemical blueprint. It also shows that a high branching dilution degree pushes the HBPs’ features closer to the ones of their linear analogues.
Similar spectral patterns were observed when the solvents were removed to obtain various coatings (Fig. 10, and Fig. S13†). The solid-state emission was red-shifted (Δλem = 32–48 nm) as compared with the one in solutions, and its intensity was reduced by one order of magnitude. The thin polymer coatings obtained from toluene displayed the most intense emission (up to 6× vs. films drop-cast from other media), attributable to a more relaxed macromolecular arrangement. Conversely, the polymer chains adopt a more planar conformation in films drop-cast from solvents of higher polarity, which results in more red-shifted emission bands and a higher propensity for aggregation-induced fluorescence quenching. This shows that the macromolecules adopt different degrees of planarity during solvent evaporation contingent on the polarity of the environment. Such behaviour enables further tuning of their photo-optical features by material processing. The solid-state experiments did not provide any straightforward correlation between the PL response (emission maxima or intensity) and branching density (Fig. 10 and Fig. S14†).
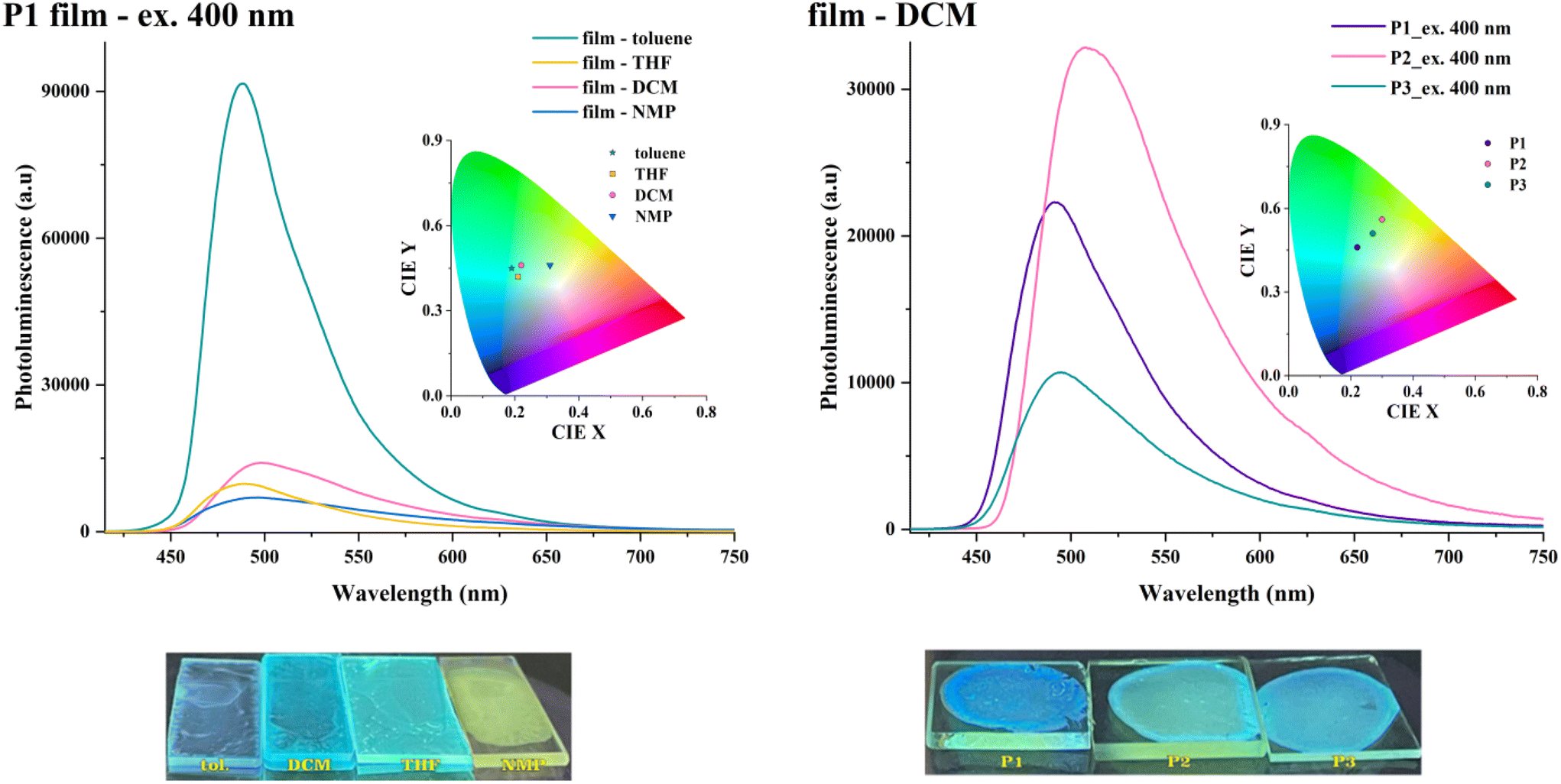 | ||
| Fig. 10 PL spectra of P1 films obtained from various solvents (left) and of P1–P3 films drop-cast from DCM (right). Insets: associated chromaticity diagrams. Images: films images under UV light (365 nm). | ||
The absolute fluorescence quantum yields of the HBPs polymers showed a severe contraction from solution to solid state, with values below 2% being obtained upon excitation at the ICT maxima (Table S5†). This fluorescence decrease is attributed to the formation of excimers and subsequent nonradiative decay via intermolecular energy transfer, particularly under molecular H-aggregation conditions.37 The lowest value corresponds to P3 and shows that an extended use of the diluting branches approach works against mitigating the fluorescence quenching limitation of linear conjugated polymers in solid-state.
At the same time, the emission maxima of P1–P3 show a significant bathochromic shift both in solution (20–30 nm) and in solid state (up to 75 nm) as compared with branched analogues based only on TPA–Fl and TPA–Th pairs.22 This further increases the colour domain achievable through HBPs based on these three building blocks and contributes to their applicative potential. The HBPs’ PL response covers broad areas of the blue and green colour domains of the chromaticity diagrams in solution and solid-state, respectively (Table S6†).
Electrochemical properties
The electrochemical characteristics of P1–P3 were assessed by cyclic voltammetry (CV) and differential pulse voltammetry (DPV) measurements to survey their potential as electroactive materials. Table S7† presents the most important parameters coming from these experiments (HOMO and LUMO energy values, bandgap energies, Eg). For correlative and interpretive purposes, the CV of each polymer is presented in comparison to its associated model compound (Fig. 11, Fig. S15†). | ||
| Fig. 11 Cyclic voltammetry (a) and differential pulse voltammetry (b) curves of P1–P3 thin films, and the comparative CV curves of polymer P1 and model compound MC1 (c). | ||
In the anodic region, the polymers revealed one or two reversible redox couples with particular distributions, patterns, and intensities associated with the generation of the TPA- and Th-derived oxidized species. Additionally, the polymers’ redox behaviour is aligned with that of associated model compounds. P1 showed two oxidation peaks at 0.73 and 0.95 V, while P2 and P3 registered only one peak at 1.02 and 0.89 V, respectively. This rather ambiguous nature of the polymers’ CV conduct required DPV experiments at a very low scan rate. Thus, it was possible to highlight linearity in the curves’ evolution as a function of the TPA amount and the resulting branching density. Polymer P3, which contains the lowest amount of TPA, presents an oxidation maximum at 1.07 V and two shoulders of the same nature at 0.83 and 1.21 V. Considering the large number of Th units within this HBP, the maximum and the shoulder at higher potential are associated with the oxidation of a Th–Fl–Th substructure. Hence, the shoulder at 0.83 V is linked to TPA oxidation, as further corroborated by the evolution of the DPV profiles of P1 and P2. Accordingly, the addition of TPA units from P3 to P1 increases the intensity of the oxidation at 0.83 V and generates another oxidation peak at a lower potential. Simultaneously, the magnitude of the oxidations from 1.07 V and 1.21 V starts to decline. As a result, P1 ends up showing up to 4 oxidation processes, at 0.66, 0.87, 1.02, and 1.26 V, respectively, the first two processes coming from two TPA units interconnected by one Fl block. Given its composition, the behaviour of P2 is expressed between the boundaries provided by the other two HBPs.
Correlations with the electrochemical response of the synthesized model compounds (also evaluated by CV and DPV experiments, Fig. S16†) further validated this assertion. For example, P1 and its associated model compound MC1 (having a TPA–Fl–TPA structure) show similar CV profiles, with small variations in oxidation potentials and CV patterns (Fig. 11c). The same trend is approximately observed for the P2/MC2 and P3/MC3 pairs (Fig. S15†). MC3 only shows oxidation processes of its two Th units (interconnected by a Fl block), comparable to those of P3. Accordingly, MC2's conduct is placed between the two extremes, similar to P2. The minor differences between the polymers and model compounds (in terms of maximum oxidation potentials, current intensities, and patterns) are generated by structural (Schemes 1 and 2) and experimental differences (polymer samples are deposited on ITO substrate vs. model compound are evaluated in DCM and TBAP solution).
The HBPs showed experimental HOMO energy values between −5.01 and −5.23 eV, and Eg values (estimated based on λonset values from optical measurements, see ESI†) between 2.67 and 2.73 eV. Based on these, the experimental LUMO energy values varied between −2.27 and −2.52 eV. All these values are typical for p-type conjugated polymers. The pattern of the HOMO and LUMO energy distribution is driven by the variation of the TPA (and, to a less extent, Th) content of each polymer and the corresponding branching density. In this regard, P1 showed the highest HOMO and LUMO energy values, while P3 revealed the lowest ones, even though Eg remained almost the same. This feature underlines a stronger p-type character for P1 as result of the higher electron-donating ability provided by the more consistent TPA amount.
Additionally, the particular structural arrangements of these electroactive HBPs provide superior oxidation potentials and energy level values compared with branched or linear analogues based only on TPA–Th or TPA–Fl pairs, respectively.34,35,38
Conclusions
We developed a series of HBPs based on thiophene, fluorene, and triphenylamine units by following the “A2 + B2 + C3” pathway. These templated conjugated frameworks ensure various benefits for the design and synthesis of organic semiconductor materials.The “dilution of branches” concept regulates branching density and subsequent transition from a rather compact HBP to a more segmented one, comprising longer “hairy-rod” sections. The approach mitigates the solubility issue of such highly aromatic constructs and affords the gradual exploitation of the intrinsic electronic and optical features of the three building blocks. Furthermore, the possibility to control the polymer coatings’ surface aspect and topography by finely tuning the monomers ratio allows to tailor their morphology depending on the envisaged application. The high decomposition temperatures and lack of any thermal transitions contribute to achieving (opto)electronic devices with stable performance and increased lifetime.
In-depth, computational and experimental photo-optical and electrochemical investigations (performed in comparison to model compounds or analogue frameworks based on pairs of the same building blocks) tackle a crucial issue in the field of HBPs as organic semiconductor materials: the comprehension of structure–property correlations. The building blocks or their combinations are precisely associated with the main electronic transitions and luminescent behaviour, together with charge and energy transfer implications. Moreover, the absorption and emission profiles display several particularities as a function of solvent, branching density, or the sample's physical state. The HBPs exhibit good transparency in the visible domain, and their emission covers a broad portion of the blue and green colour areas of the chromaticity diagrams in solution and solid-state, respectively. The fluorescence quantum yield values point towards some limitations of the branching dilution strategy. Cyclic voltammetry experiments aided by differential pulse voltammetry analysis reveal distinct oxidation patterns directly connected to the triphenylamine amount and the resulting branching density. They also show electrochemical parameters with values typical for p-type conjugated macromolecules.
In summary, the present work delivers the foundation for the predictable development and judicious control of a broad range of conjugated HBPs as active materials in (opto)electronic applications. Further structural engineering and processing endeavours are considered to address new queries derived from this work. These questions refer to solvent-induced morphology manipulation, solvato(fluoro)chromic features, fluorescence quenching control, redox stability, and optical- and energy-related capabilities.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant of the Romanian Ministry of Research, Innovation and Digitization, CNCS/CCCDI-UEFISCDI, project PN-III-P4-PCE-2021-1728 (contract no. 46/2022) within PNCDI III.References
- W. Rehwald and H. G. Kiess, Charge Transport in Polymers, Conjugated Conducting Polymers, 1992, pp. 135–173 Search PubMed.
- K. Okamoto and C. K. Luscombe, Polym. Chem., 2011, 2, 2424–2434 RSC.
- A. C. Grimsdale, K. Leok Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed.
- Z. Qiu, B. A. G. Hammer and K. Müllen, Prog. Polym. Sci., 2020, 100, 101179 CrossRef CAS.
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- X. Zhan and D. Zhu, Polym. Chem., 2010, 1, 409–419 RSC.
- T. P. Kaloni, P. K. Giesbrecht, G. Schreckenbach and M. S. Freund, Chem. Mater., 2017, 29, 10248–10283 CrossRef CAS.
- M.-D. Damaceanu, H.-D. Gilsing, B. Schulz, A. Arvinte and M. Bruma, RSC Adv., 2014, 4, 52467–52475 RSC.
- M. Leclerc, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2867–2873 CrossRef CAS.
- Q. Zhao, S. Liu and W. Huang, Macromol. Chem. Phys., 2009, 210, 1580–1590 CrossRef CAS.
- R.-D. Rusu and A. D. Schlüter, RSC Adv., 2014, 4, 57026–57034 RSC.
- A. Iwan and D. Sek, Prog. Polym. Sci., 2011, 36, 1277–1325 CrossRef CAS.
- H. J. Yen and G. S. Liou, Polym. Chem., 2018, 9, 3001–3018 RSC.
- R.-D. Rusu, M.-D. Damaceanu, S. Ursache and C.-P. Constantin, J. Photochem. Photobiol., A, 2023, 435, 114272 CrossRef CAS.
- Z. Zhou, N. Luo, X. Shao, H.-L. Zhang and Z. Liu, ChemPlusChem, 2023, 88, 202300261 CrossRef PubMed.
- C. Gao and D. Yan, Prog. Polym. Sci., 2004, 29, 183–275 CrossRef CAS.
- W. Wu, R. Tang, Q. Li and Z. Li, Chem. Soc. Rev., 2015, 44, 3997–4022 RSC.
- Y. Zheng, S. Li, Z. Weng and C. Gao, Chem. Soc. Rev., 2015, 44, 4091–4130 RSC.
- A. Ghosh, S. Banerjee and B. Voit, Aromatic Hyperbranched Polymers: Synthesis and Application, Advances in Polymer Science, 2014, pp. 27–124 Search PubMed.
- B. Amna, H. M. Siddiqi, A. Hassan and T. Ozturk, RSC Adv., 2020, 10, 4322–4396 RSC.
- M. M. Samy, M. Gamal Mohamed, S. U. Sharma, S. V. Chaganti, T. Hassan Mansoure, J.-T. Lee, T. Chen and S.-W. Kuo, Polymer, 2023, 264, 125541 CrossRef CAS.
- H. T. Nguyen, L.-T. T. Nguyen, T. T. Nguyen, A. T. Luu and T. Van Le, J. Polym. Res., 2014, 21, 552 CrossRef.
- B. I. Voit and A. Lederer, Chem. Rev., 2009, 109, 5924–5973 CrossRef CAS PubMed.
- M. Grigoras and L. Stafie, High Perform. Polym., 2009, 21, 304–314 CrossRef CAS.
- G. Wu, Y. Yang, C. He, X. Chen and Y. Li, Eur. Polym. J., 2008, 44, 4047–4053 CrossRef CAS.
- T. V. Richter, S. Link, R. Hanselmann and S. Ludwigs, Macromol. Rapid Commun., 2009, 30, 1323–1327 CrossRef CAS PubMed.
- M. Sun, J. Li, B. Li, Y. Fu and Z. Bo, Macromolecules, 2005, 38, 2651–2658 CrossRef CAS.
- T. Guo, L. Yu, B. Zhao, L. Ying, H. Wu, W. Yang and Y. Cao, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1043–1051 CrossRef CAS.
- Z.-F. Yao, J.-Y. Wang and J. Pei, Prog. Polym. Sci., 2023, 136, 101626 CrossRef CAS.
- J. Niu, L. Rao, P. Liu, W. Zhang, W. Zhang, Y. Zuo, K. Chai, S. Chen, J. Xu and X. Duan, Synth. Met., 2018, 246, 282–288 CrossRef CAS.
- X. Lv, S. Yan, Y. Dai, M. Ouyang, Y. Yang, P. Yu and C. Zhang, Electrochim. Acta, 2015, 186, 85–94 CrossRef CAS.
- K. Yamamoto, D. Suemasa, K. Masuda, K. Aita and T. Endo, ACS Appl. Mater. Interfaces, 2018, 10, 6346–6353 CrossRef CAS PubMed.
- L. Zhang, W. Zhan, Y. Dong, T. Yang, C. Zhang, M. Ouyang and W. Li, ACS Appl. Mater. Interfaces, 2021, 13, 20810–20820 CrossRef CAS PubMed.
- F. Wang, M. S. Wilson, R. D. Rauh, P. Schottland and J. R. Reynolds, Macromolecules, 1999, 32, 4272–4278 CrossRef CAS.
- C.-N. Chuang, C.-Y. Chang, C.-L. Chang, Y.-X. Wang, Y.-S. Lin and M. Leung, Eur. Polym. J., 2014, 56, 33–44 CrossRef CAS.
- H. Wu, L. Du, J. Luo, Z. Wang, D. L. Phillips, A. Qin and B. Z. Tang, J. Mater. Chem. C, 2022, 10, 8174–8180 RSC.
- M.-D. Damaceanu, C.-P. Constantin and L. Marin, Dyes Pigm., 2016, 134, 382–396 CrossRef CAS.
- C. Bathula, A. Appiagyei, H. Yadav, A. K. S. Ramesh, N. Shrestha, S. Shinde, H.-S. Kim, H. Kim, L. Reddy and A. Mohammed, Nanomaterials, 2019, 9, 1787 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00720d |
| This journal is © The Royal Society of Chemistry 2024 |