
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the thermal properties of L-lactide/ε-caprolactone chain shuttled copolymers via catalyst selection†
Xavier
Mosca
a,
Lucas
Perchery
 a,
Marc
Bria
b,
Julien
De Winter
c,
Gregory
Stoclet
d,
Till
Bousquet
a,
Lydie
Pelinski
a,
Fanny
Bonnet
*d and
Philippe
Zinck
*a
a,
Marc
Bria
b,
Julien
De Winter
c,
Gregory
Stoclet
d,
Till
Bousquet
a,
Lydie
Pelinski
a,
Fanny
Bonnet
*d and
Philippe
Zinck
*a
aUniv. Lille, CNRS, Centrale Lille, Univ. Artois, UMR 8181 – UCCS – Unité Catalyse et Chimie du Solide, F-59000 Lille, France. E-mail: philipe.zinck@univ-lille.fr
bPlateforme RMN, Univ. Lille, CNRS, Centrale Lille, Univ. Artois, F-59650, Villeneuve d'Ascq, France
cInterdisciplinary Centre for Mass Spectrometry, Organic Synthesis and Mass Spectrometry Laboratory, University of Mons-UMONS, 23 Place du Parc, 7000 Mons, Belgium
dUniv. Lille, CNRS, INRAE, Centrale Lille, UMR 8207 – UMET – Unité Matériaux et Transformations, F-59000 Lille, France. E-mail: fanny.bonnet@univ-lille.fr
First published on 10th September 2024
Abstract
Chain shuttling copolymerisation (CSP) is a synthetic strategy allowing the one-pot, one-step formation of block copolymers. Initially developed in the frame of coordinative polymerisation of olefins and conjugated dienes, it was recently transferred to the ring-opening polymerisation of cyclic esters. In this contribution, we report six new catalytic systems able to perform the chain shuttling copolymerisation of L-lactide (L-LA) with ε-caprolactone (ε-CL) and to tune the thermal properties of the resulting copolymers. They are based on amino(bis)phenolate supported aluminium complexes bearing different pendant donor arms ((Al(O2NL)OBn), L = NEt2 (2a), NBn2 (2b), Py (2c), Mor (2d)). A Mannich reaction allowed the ligands synthesis. The two new alkoxide complexes 2a and 2b were obtained by reaction of the protonated ligands with trimethylaluminium followed by benzyl alcohol in reasonable yield, as well as two already described compounds 2c and 2d. Initially assessed as catalysts for L-LA and ε-CL homopolymerisations and statistical copolymerisation, the aluminium compound bearing pyridine as a donor arm (2c) resulted in a high selectivity toward lactide. 2c together with yttrium and aluminium alkoxides, also known for their selectivity for lactide, were successfully assessed for the chain shuttling copolymerisation of L-LA with ε-CL in combination with the three other amino(bis)phenolate supported aluminium complexes that showed a higher selectivity toward ε-CL. Chain shuttling copolymerization via transalkoxylation between two different metals, Y and Al, is achieved for the first time, therefore extending the range and scope of cyclic esters CSP. Such an alteration of the nature of the catalysts allowed fine tuning of the thermal properties of the chain shuttled copolymer, as shown by a variation of the glass transition temperature (Tg) of the soft block over ca. 25 °C without changing the catalysts ratio and feed of the reaction.
1. Introduction
Global warming has become a growing concern in recent years. Using biomass as a raw material rather than oil may seem appropriate in this context. In the field of plastic/polymer materials, the use of bio-sourced poly(lactic acid) also called polylactide (PLA) emerges as a promising solution. PLA is a biodegradable, compostable and biocompatible aliphatic polyester, with mechanical properties close to those of some oil-based plastics. It allows its use in a wide range of applications such as food packaging, biomedicine or 3D printing, among others.1–4 The presence of two stereocentres in the lactide monomer, commonly used for the production of PLA, allows the possibility to control the tacticity of the final material depending on the initiator and/or the stereoisomer selected. Poly(L-lactide) (PLLA), a semi-crystalline material, is of high interest as it shares similar properties with polyethylene terephthalate (PET) and polystyrene (PS). However, its inherent brittleness, with low elongation at break and impact resistance limits its applications.5 To overcome these limitations, PLLA is often used in the form of blend with other polymers and in particular poly(ε-caprolactone) (PCL)6–8 which displays a much higher elongation at break of up to 300%. Copolymerisation of L-lactide (L-LA) with other monomers can also be explored for this purpose, notably statistical. Random copolymer of L-LA and ε-caprolactone (ε-CL) can be synthesised via ring-opening copolymerisation using metal-based catalysts,9 organocatalysts10 or enzymes.11Chain-shuttling copolymerisation (CSP) has emerged as an interesting strategy to produce block copolymers in a one-pot, one-step process.12–18 CSP is based on the simultaneous use of two metal-based catalysts able to work in tandem via the use of a chain transfer agent allowing the shuttling of the polymer chain between the two active sites (Fig. 1). The two initiators have to be able to copolymerise the co-monomers in the same experimental conditions with a different reactivity ratio, resulting in blocks with different compositions arranged in an alternating fashion. We extended this concept, initially developed based on coordinative copolymerisation of ethylene with α-olefins,12 to the ring opening polymerisation of cyclic esters.15 This led to the formation of block copolymers composed of PLLA rich semi-crystalline hard blocks and amorphous poly(L-lactide-co-ε-caprolactone) soft blocks. The catalytic system involved commercial Al(OiPr)3 that barely inserts ε-CL19 combined to an amino(bis)phenolate-supported aluminium complex (see Scheme 1 for the general structure of amino(bis)phenolate ligands) able to insert significant amount of the lactone without significant occurrence of transesterification. The glass transition temperatures (Tg) of the soft block ranged from −8 to +16 °C. For some applications, it can be interesting to reach lower Tg, which can be achieved by producing soft blocks with a higher content of ε-CL. We therefore planned to design catalysts able to statistically copolymerise LA and ε-CL with a higher insertion of ε-CL.
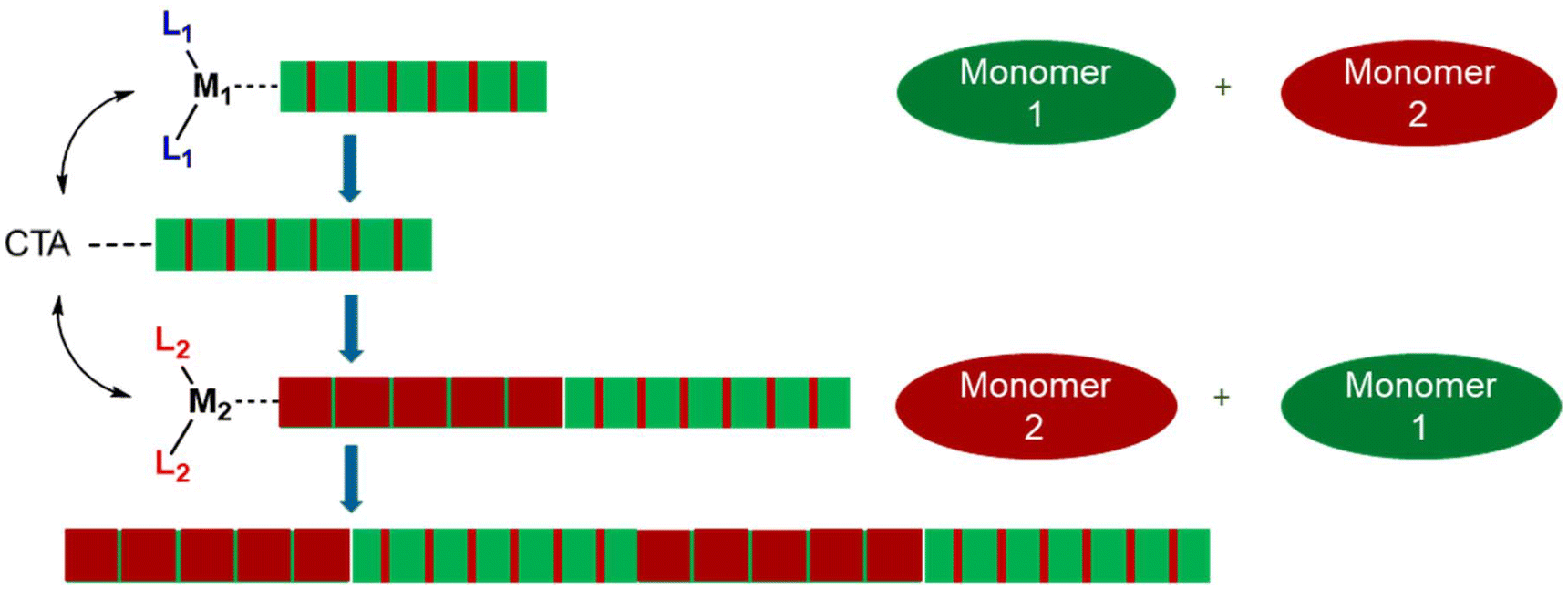 | ||
| Fig. 1 Chain Shuttling Copolymerisation (CSP). Where M is the catalyst metal, L the ligands and CTA the chain transfer agent. | ||
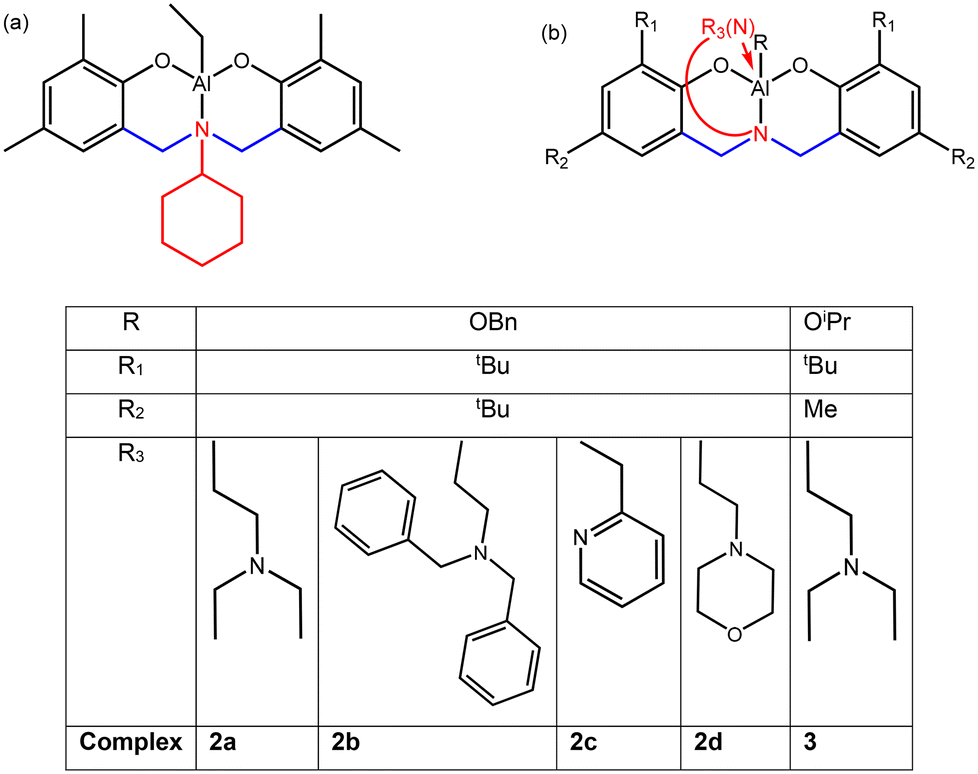 | ||
| Scheme 1 Aluminium supported amino(bis)phenolate complexes used in our preliminary work (a) and bearing a pendant donor arm considered in the present work for chain shuttling copolymerization (2a, 2b, 2c and 2d) and in the literature for homo- and statistical copolymerizations (3 after ref. 25 and 27) (b). | ||
ε-CL homopolymerisation is generally known to be more efficient than the L-LA one, which may notably be attributed to the increased steric hindrance of lactide due to the methyl groups. However, this trend reverses in copolymerisation, which is usually ascribed to the fact that lactide is more coordinating to oxophilic metals,20,21 leading to gradient or even block copolymers starting from an equimolar mixture of the monomers with certain systems.22 Possible strategies to modify the reactivity ratio consist in varying steric hindrance around the active centre of the complex and/or altering the ligand electron density. In general, increasing the hindrance around the initiator active site tends to promote a higher ε-CL insertion.20
Amino(bis)phenolate ligands are particularly interesting for this purpose, as their structure (see Scheme 1) and synthetic route allow a wide variability of the ligand framework. They have been used with aluminium metals to design active catalysts for the ROP of cyclic esters,23,24 and later, for LA/ε-CL statistical copolymerisation, with the potential to control the selectivity. In addition to the R1 and R2 substituents, the nature of the amino side arms plays a crucial role. For instance, it has been reported that using pyridine as a side arm results in very low ε-CL insertion in the course of a LA/ε-CL statistical copolymerization, whereas employing amino(bis)phenolate Al complexes bearing a tertiary amine leads to significantly higher lactone insertions, with R1 = tBu, R2 = Me and R = OiPr (complex 3).25 This was rationalized by DFT calculations, which revealed a difference of activation energy for the insertion of the co-monomers into Al-caprolactoyl/Al-lactyl bonds.26 In addition, regarding our purpose, amino(bis)phenolate supported aluminium complexes have been previously reported to achieve chain shuttling copolymerisation with an aluminium trisalkoxide.15
In the present study, we have assessed a variety of amino(bis)phenolate supported aluminium systems for the CSP of L-LA and ε-CL. The ligands were designed based on the following assumptions. We selected structures (shown in Scheme 1) bearing:
- –tBu as R1 and R2 substituents as a literature survey tends to show that steric hindrance might increase the insertion of ε-CL in the course of metal catalysed L-LA/ε-CL statistical copolymerisation.
- Amino side-arms with a group allowing a coordination to the aluminium centre. Indeed, such complexes with e.g. tertiary alkylamine allows an insertion of ε-CL higher25 than that afforded by the cyclohexyl substituted complex used in our preliminary work (Scheme 1a).15
This includes complexes already described in the literature (2c and 2d see ref. 24) but not assessed for the L-LA/ε-CL statistical copolymerisation to our knowledge, as well as new complexes (2a and 2b). They are first assessed in a comparative way for the homopolymerisation of both monomers, followed by statistical copolymerisation attempts. By combining those amino-(bis)phenolate-supported aluminium complexes, either among themselves or with aluminium and yttrium alkoxides, we report in this contribution six new catalytic systems able to perform the L-LA/ε-CL chain shuttling copolymerisation, leading to various Tg for the soft blocks. As such, the thermal properties of the copolymers can be tuned by selecting the proper CSP catalytic combination.
2. Results and discussion
2.1 L-Lactide and ε-CL polymerisation
Prior to statistical and chain shuttling copolymerisation, the 2a–d complexes were tested for the polymerisation of the monomers. Experiments representative of the ROP of L-LA in toluene at 100 °C in the presence of the different initiators are presented in Table 1. All initiators were found to be active with significant conversions. Complex 2c led to full conversion in less than 1 h (entry 3) while 53–66% conversions were obtained for 2a, 2b and 2d in 24 h. 2b shows the lowest conversion with 53%, while 2a and 2d have relatively similar conversions around 65%. The activity ranking was found as follow: 2c (Py) ≫ 2a (NEt2) ≅ 2d (Mor) > 2b (NBn2). The higher activity of complex 2c with a pyridine donor arm agrees with previous studies of the literature, and was attributed to the lower steric hindrance on the pyridine side arm.28 The lower activity of 2b observed in this study aligns with this trend. 2d was reported to be less active than 2c in bulk at 120 °C,24 although to a lesser extent. In addition, amino(bis)phenolate Al isopropoxyde bearing a dialkyl amine pendant arm with methyl and tert-butyl substituted phenol in the para and ortho positions respectively (shown as 3 in Scheme 1) were found to be inactive at 70 °C vs. the active pyridine substituted analogue.27| Entrya | Complex | Monomer | M/Ib | t | T (°C) | Conv.d (%) |
M
n calcd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) e (g mol−1) e (g mol−1) |
M
n expf (g mol−1) |
Đ |
|---|---|---|---|---|---|---|---|---|---|
| a Polymerisations conducted in toluene at 1 M (mol L−1). b Monomer/initiator molar ratio. c Time. d Conversion determined by 1H NMR in CDCl3. e M n calcd = (50 × 144 × conversion)/100 for PLA and ([ε-CL]/[Al] × 114 × conversion)/100. f Number-average molecular weight determined by size exclusion chromatography in THF with 0.58 as correction factor for PLA,32 0.56 as correction coefficient factor for PCL33 and dispersity. | |||||||||
| 1 | 2a | L-LA | 50 | 24 h | 100 | 66 | 4700 | 4800 | 1.07 |
| 2 | 2b | L-LA | 50 | 24 h | 100 | 53 | 3800 | 2400 | 1.12 |
| 3 | 2c | L-LA | 50 | 1 h | 100 | 100 | 7200 | 7100 | 1.06 |
| 4 | 2d | L-LA | 50 | 24 h | 100 | 62 | 4500 | 4200 | 1.08 |
| 5 | 2a | ε-CL | 500 | 10 min | 50 | 91 | 51900 |
48700 |
1.11 |
| 6 | 2b | ε-CL | 1000 | 5 min | 30 | 66 | 75300 |
50600 |
1.12 |
| 7 | 2c | ε-CL | 500 | 10 min | 50 | 1 | 600 | n.d | n.d |
| 8 | 2d | ε-CL | 1000 | 5 min | 30 | 85 | 97000 |
63200 |
1.29 |
The activity of the 2a, 2b and 2d amino(bis)phenolate aluminum complexes for the polymerization of lactide in solution is rather modest compared to conventional initiators. For example, the polymerization of lactide by salen aluminum complexes leads to 62% conversion in toluene at 70 °C after 22 h for M/I = 75, 0.6 M.29 Regarding another conventional catalyst, yet more used in the bulk, the polymerization of lactide mediated by Sn(Oct)2 leads to 95% yield in toluene at 115 °C after 24 h for M/I = 1000, 1 M.302c leads in turn to more significant conversion.
According to SEC data, Mn ranging from 2400 to 7100 g mol−1 were obtained. The experimental Mn were found to be close to the calculated ones. The polymerisations were found to be well-controlled with dispersities in the range 1.06 to 1.12. Based on the MALDI mass spectra, a good control over the mass parameters, in agreement with NMR and SEC results, and end-groups fidelity can be confirmed. Indeed, the main distribution is characterised by polylactide initiated by benzyl alcohol. Moreover, most of the mass spectra shows mass difference of 144 Da between signals of the main distribution, confirming a low amount of transesterification reaction (see Fig. SI1–4†). It is worth to note that mass spectra seem to present a huge amount of low mass ions but those distributions are significantly overestimated compare to the main one.31
The polymerisation of ε-CL conducted with 2a–d as the initiators was then investigated in toluene at 30 or 50 °C (Table 1). Entries 5 and 7 were performed at 50 °C in 10 min for monomer/initiator ratio of 500 while entries 6 and 8 were conducted at 30 °C in 5 min for a monomer/initiator ratio of 1000 due to the difference of reactivity of the complexes for the polymerisation of ε-CL. Narrow molar mass distributions with dispersities around 1.1 to 1.3 were obtained. The activity of the initiators could be rated as follow: 2d (Mor) > 2b (NBn2) ≫ 2a (NEt2) ≫ 2c (Pyr). The inactivity of complex 2c aligns with previous studies of the literature, while 2d was reported as significantly active in toluene at 25 °C for ε-caprolactone.24 Similarly, amino(bis)phenolate Al isopropoxyde bearing a dialkyl amine pendant arm with methyl and tert-butyl substituted phenol in the para and ortho positions respectively (shown as 3 in Scheme 1) was found much more active than the pyridine-substituted analogue at 70 °C in toluene.27 Generally, the results suggest that the polymerisation of ε-CL is more influenced by the nature of the initiator pendant donor arm than for L-LA.
2.2 L-LA/ε-CL statistical copolymerisation
Entries representative of the statistical ring-opening copolymerisation of ε-CL and L-LA in toluene at 100 °C mediated by the different initiators are given in Table 2. All complexes were found to be active. A very high selectivity for L-LA was observed for 2c, while 2a, 2b and 2d were found to insert more ε-CL than L-LA starting from an equimolar mixture of the co-monomers, with similar conversions. This is noteworthy regarding targeted chain shuttled materials. Indeed, the amino(bis)phenolate aluminium complex bearing a cyclohexyl moieties reported in our preliminary work did insert less ε-CL than L-LA, with ca. 80/60 conversions for L-LA and ε-CL respectively at 70 °C in toluene.15 The higher selectivity for L-LA obtained using 2cvs.2a is in line with the literature, as amino(bis)phenolate Al isopropoxyde bearing a dialkyl amine pendant arm with methyl and tert-butyl substituted phenol in the para and ortho position respectively (shown as 3 in Scheme 1) was found to convert much more ε-CL than the pyridine-substituted analogue at 100 °C in toluene.25Mn ranging from 7500 to 8800 g mol−1 were obtained along with narrow molar mass distributions (dispersities Đ in the range 1.05 to 1.17). It is difficult to rationalize the structure/activity/selectivity relationships. However, steric hindrance seems to be an influential factor. Indeed, 2c is less sterically hindered than 2a, 2b and 2d and leads to the highest activity for the polymerization of lactide, and to the lowest activity for the polymerization of ε-CL. In statistical copolymerization, it leads to the lowest insertion of ε-CL.| Entrya | Complexa | L-LA/ε-CL Conv.b (%) | t (h) |
M
n calcc (g mol−1) |
M
nd (g mol−1) |
Đ |
|---|---|---|---|---|---|---|
| a Copolymerisations conducted at 100 °C in toluene at 1 M (mol L−1) with ([L-LA] + [ε-CL])/[Al] = 100 with [L-LA] = [ε-CL]. b Determined by 1H NMR in CDCl3. c M n calc = (50 × 144 × L-LA conversion)/100 + (50 × 114 × ε-CL conversion)/100. d Number-average molecular weight determined by size exclusion chromatography in THF corrected as Mn = (Mn raw × 0.56 × ε-CL conversion)/100 + (Mn raw × 0.58 × L-LA conversion)/100.34 | ||||||
| 9 | 2a | 64/80 | 24 | 9200 | 7500 | 1.10 |
| 10 | 2b | 58/78 | 24 | 8800 | 8800 | 1.15 |
| 11 | 2c | 99/3 | 4 | 7400 | 7600 | 1.05 |
| 12 | 2d | 63/81 | 24 | 9200 | 7700 | 1.17 |
NMR is a powerful tool for the structural analysis of poly(L-lactide-co-ε-caprolactone) copolymers. In particular, 13C NMR provides some information regarding the sequences. L is defined as the lactyl unit and LL as the lactidyl unit. From the MALDI of the L-LA homopolymerisation, we have seen that transesterification does not occur significantly, with mainly multiple of 144 Da, i.e. mostly LL sequences. For the statistical copolymerisation, if transesterification inside a lactidyl LL sequence occurs, one may see CLC sequences at 170.8 ppm,35 which is not the case here as shown in Fig. SI5–7 provided in the ESI section.† Thus, we can reasonably assume that only LL lactidyl sequences are present in our copolymers.
In addition, 1H NMR allows to determine the composition and the percentage of lactidyl units neighbouring a caprolactoyl unit and vice versa. The distribution of the dyads provided in Table 3 is derived from Fig. 2. Signal representative of the caprolactoyl units at 4–4.2 ppm (2H, –CH2O) can be used to quantitatively determine the percentage of C–C vs. C–LL. Then, given that there are as many LL–C dyads in the 5.05–5.25 ppm zone corresponding to 1H PLA as C–LL dyads, one can deduce the amount of LL–LL dyads. From that, we can now determine the lactidyl and caprolactoyl number-average sequence length from the work of Herbert:36
| LLL = (2LL–LL + LL–C)/LL–C | (1) |
| LC = (2C–C + LL–C)/LL–C | (2) |
 | ||
| Fig. 2 1H NMR spectrum in CDCl3 of poly(L-LA-co-ε-CL) synthetized using 2a (entry 9 -crude product) in the 4–5.5 ppm region. | ||
| Entry | Initiators | L-LAa mol% | (LL–LL)b mol% | (LL–C)b mol% | (C–C)b mol% |
L
LLc |
L
Cd |
η |
|---|---|---|---|---|---|---|---|---|
| a Composition determined by 1H NMR in CDCl3. b Dyad distribution (see text). c Lactidyl number-average sequence length determined using eqn (1). d Caprolactoyl number-average sequence length determined using eqn (2). e Randomness coefficient determined using eqn (3). | ||||||||
| 9 | 2a | 47.5 | 25.2 | 22.2 | 30.4 | 3.3 | 3.7 | 0.44 |
| 10 | 2b | 35.5 | 8.3 | 27.2 | 37.3 | 1.6 | 3.7 | 0.59 |
| 12 | 2d | 37.1 | 10.5 | 26.5 | 36.4 | 1.8 | 3.7 | 0.57 |
The randomness coefficient η can further be calculated from eqn (3).36
| η = (LL–C)/(2 × L-LA × CL)/100 | (3) |
The values are provided in Table 3.
Complex 2a leads to a lactidyl number-average sequence length of 3.3, which is higher than the values of 1.6–1.8 found for 2b and 2d. The latter also have a randomness coefficient around 0.57–0.59 vs. 0.44 for the former. If the order of magnitude of 0.5 indicates a moderately blocky character,‡ a difference of microstructure can be observed between these two groups, with a higher random character for 2b and 2d. They also exhibit higher ε-CL insertions.
2.3 L-LA/ε-CL chain shuttling copolymerisation
Regarding chain shuttling copolymerisation, the combination of two catalysts exhibiting different selectivities towards the co-monomers were targeted. From the previous section, it can be seen that 2c has a very high selectivity for lactide, making it a suitable catalyst to produce the hard block. 2a, 2b and 2d insert much more ε-CL, which is desirable to produce soft blocks. Therefore, three experiments were conducted where 2c was combined to the three other complexes, under similar experimental conditions for comparison (entries 13–17 in Table 4). The higher activity of 2c prompted us to put the soft block catalysts in excess. A chain shuttling agent was not introduced at this step, as we highlighted in our previous study15 that the shuttling can operate without. Introducing it would result in a reduction in molar mass of the final materials. It should also be noted that average number-average molecular weights (from ca. 6000 to 13000 g mol−1) were targeted at this step to be able to perform DOSY (Diffusion Order NMR Spectroscopy) analyses, which is crucial to prove the occurrence of chain shuttling copolymerisation (see here after).
| Entrya | Hard block Initiator (HBI) | Soft block Initiator (SBI) | HBI/SBI/LA/ε-CL Mol. ratiob | L-LA/ε-CL Conv.c (%) | t (h) |
M
n calcd (g mol−1) |
M
ne (g mol−1) |
Đ | (LL–LL)f mol% | (LL–C)f mol% | (C–C)f mol% |
L
LLg |
L
Ch |
η |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Copolymerisations conducted at 100 °C in toluene at 1 M (mol L−1). b Corresponding to the metal ratio: [Y] or [Al] HBI/[Al] SBI/LA/CL. c Determined by 1H NMR in CDCl3. d For entries 13–17 Mn calc = ((500 × 144 × L-LA conversion)/100 + (500 × 114 × -CL conversion)/100)/6; for entry 18 Mn calc = ((250 × 144 × L-LA conversion)/100+ (250 × 114 × -CL conversion)/100)/8; for entries 19–21 Mn calc = ((HBI/SBI/LA/CL mol. ratio × 144 × L-LA conversion)/100 + (HBI/SBI/LA/CL Mol. ratio × 114 × -CL conversion)/100)/7.6 (the 7.6 denominator corresponds to the number of initiating alkoxide moieties: 5 for the 5 equiv. of soft initiator, plus 2.6 for the yttrium hard block initiator. Indeed, the latter is a cluster with 13 alkoxides moieties for 5 yttrium atoms, which makes an average of 2.6 per Y). e Number-average molecular weight determined by size exclusion chromatography in THF corrected as Mn = (Mn raw × 0.56 × ε-CL conversion) + (Mn raw × 0.58 × L-LA) and dispersity. f Dyad distribution (see text). g Lactidyl number-average sequence length determined using eqn (1). h Caprolactoyl number-average sequence length determined using eqn (2). i Randomness coefficient determined using eqn (3). | ||||||||||||||
| 13 | 2c | 2a | 1/5/500/500 | 79/48 | 16 | 14000 |
12200 |
1.08 | 69.0 | 9.8 | 11.4 | 15.1 | 3.3 | 0.29 |
| 14 | 2b | 1/5/500/500 | 33/35 | 16 | 7300 | 3900 | 1.12 | 47.2 | 16.3 | 20.3 | 6.8 | 3.5 | 0.35 | |
| 15 | 51/61 | 30 | 11800 |
7000 | 1.08 | 46.7 | 16.8 | 24.8 | 6.3 | 3.2 | 0.34 | |||
| 16 | 2d | 1/5/500/500 | 46/39 | 16 | 9200 | 6800 | 1.10 | 54.6 | 15.1 | 15.3 | 8.2 | 3.0 | 0.36 | |
| 17 | 64/45 | 24 | 12000 |
9600 | 1.11 | 58.2 | 14.9 | 12.8 | 7.9 | 2.9 | 0.32 | |||
| 18 | Al(OiPr)3 | 2a | 1/5/250/250 | 92/51 | 6 | 5900 | 6300 | 1.13 | 70.2 | 9.6 | 10.5 | 15.6 | 3.2 | 0.30 |
| 19 | OY5(OCH(CH3)2)13 | 1/5/250/250 | 94/59 | 6 | 6700 | 6500 | 1.17 | 70.1 | 6.4 | 17.1 | 22.8 | 6.3 | 0.18 | |
| 20 | 2d | 1/5/250/250 | 46/30 | 6 | 3300 | 2600 | 1.48 | 59.7 | 12.3 | 15.4 | 19.4 | 5.8 | 0.21 | |
| 21 | 1/5/500/500 | 93/67 | 16 | 13800 |
13700 |
1.23 | 65.4 | 6.8 | 20.9 | 20.2 | 7.1 | 0.17 | ||
All three combinations lead to significant conversion. Considering that 2c is highly selective for L-LA and that the soft block initiator polymerises both L-LA and ε-CL, it was anticipated that the conversion for L-LA would exceed that of ε-CL when both initiators are involved, which indeed was the case. Regarding the activity, the following ranking could be established from entries 13, 14 and 16: 2a (NEt2) > 2b (NBn2) > 2d (Mor). Narrow molar mass distributions were obtained in all cases, with dispersities close to 1.1. The molar mass distributions are monomodal (see Fig. SI8–10 in the ESI section†), which agrees with the occurrence of chain shuttling.
DOSY analyses of the copolymers were then performed. The measurement of the diffusion coefficient allows to confirm whether lactyl and caprolactoyl units are within the same macromolecule or if it is a mixture of two different (co-)polymers that would have been produced independently by the two different initiators. In the former case, lactyl and ε-caprolactoyl units will share the same diffusion coefficient, while in the latter case, two different coefficients will be detected. A typical DOSY analysis presented in Fig. 3 shows a single diffusion coefficient, consistent with the occurrence of transalcoxylation in the course of the polymerisation. This is the case for all three systems (see Fig. SI14 and 15 in the ESI section† for the two other DOSY analyses). Moreover, as observed before in the course of the statistical copolymerisation, 13C NMR analysis indicated the absence of LCL enchainments indicating the absence of significant transesterification reactions.
 | ||
| Fig. 3 DOSY analysis of a chain shuttled copolymer synthetized using 2a and 2c (entry 13). | ||
In order to potentially extend the scope of L-LA/ε-CL chain shuttling copolymerisation, we further assessed Al(OiPr)3 as hard block catalyst, as it successfully performed the CSP reaction with the amino(bis)phenolate-supported aluminium complex bearing the cyclohexyl pendant arm in our previous work (see Scheme 1 and ref. 15). Entry 18 and related DOSY experiment represented Fig. SI16† shows that the Al(OiPr)3/2a combination is also efficient for L-LA/ε-CL CSP. We finally assessed if amino(bis)phenolate aluminium complexes could be associated to other metal centres to conduct CSP. In that frame, yttrium alkoxides were selected due to their established efficacy as catalysts for the ROP of cyclic esters with a high selectivity towards lactide in the course of the L-LA/ε-CL statistical copolymerisation.37,38 Representative experiments are shown as entries 19–21 in Table 4 with 2a and 2d as soft block initiators. As observed using 2c as the hard block initiator, the activity in the presence of 2d was found to be lower. Higher molar mass can be obtained by increasing the monomers over initiators ratio, as shown in entry 21. To our delight, the DOSY experiments (Fig. SI17 and 18 in the ESI section†) show that the transalkoxylation is efficiently occurring between yttrium and amino(bis)phenolate aluminium alkoxides. By comparing entry 21 to entry 16, it can be seen that the activity is significantly higher using the yttrium precursor as the hard block catalyst than 2c. We also conducted a model 1H NMR study that shows that the amino(bis)phenolate moieties remains on the aluminum center even after heating an equimolar mixture of OY5(OCH(CH3)2)13 and 2a as a case study at 80 °C in deuterated benzene (found in the ESI section as part 7 and Fig. SI19 and SI20†).
Finally, the lactidyl and caprolactoyl number-average sequence lengths were also determined for the chain shuttled copolymers for the sake of comparison. It can be seen that, due to the high selectivity of the hard block catalysts for the lactide comonomer, the lactidyl number-average sequence lengths are significantly higher than those obtained for the statistical copolymer, in the 6.3–22.8 range vs. 1.6–3.3 respectively. The randomness coefficient, as a consequence, is lower for the chain shuttled copolymers (0.17–0.36 vs. 0.44–0.59 for the statistical copolymers).
2.4 Thermal properties of chain shuttled copolymers
The thermal properties of some samples were finally characterised, and are given in Table 5, while the DSC thermograms are provided Fig. 4. It should be noted that, due to the low molar mass requirements for DOSY analyses, not all samples from Table 4 could easily be precipitated. From Table 4, it can be seen that in the presence of 2c as the hard block catalyst, the highest molar masses were obtained in combination with 2a, which was thus selected to study the thermal properties. The samples obtained from entry 13 show two glass transition temperatures together with a melting point. The lower Tg is at −14 °C starting from an equimolar mixture of the co-monomers. In comparison, Al(OiPr)3 leads to a higher soft block Tg of 2 °C in similar conditions (entry 21), with higher melting point and melting enthalpy. The lower molar mass obtained for the sample synthesised with Al(OiPr)3 can be ascribed to the presence of three alkoxide initiating groups on the aluminium centre vs. one for 2c. The lowest glass transition temperatures were obtained for the copolymers synthesised with the yttrium alkoxide initiator combined to 2a or 2d, around −22 to −24 °C starting from an equimolar mixture of the co-monomers (entries 22 and 23). The glass transition temperature of the soft blocks follow the ε-CL content of the sample: the highest for the former, the lowest the Tg. Regarding the hard block, the Tg are all in the same range. The chain shuttling between aluminum amino(bis)phenolate and yttrium allows to reach microstructures with a lower Tg for the soft block and higher melting points. This can be further linked to longer lactidyl and caprolactoyl number-average sequence lengths, and to an overall lower randomness coefficient. It may also somehow be linked to higher activity and selectivity of the yttrium hard block initiator toward lactide (see Table SI3†). This might further lead to a wider range of mechanical properties. As a matter of fact, tuning of the thermal properties of the chain shuttled copolymer can be realised by changing the type of catalysts, with a variation of the Tg of the soft block over ca. 25 °C starting from a similar equimolar mixture of the co-monomers. Fine analyses of the phase segregation states and the mechanical properties of those materials will be reported in a forthcoming paper. | ||
| Fig. 4 Thermograms of the chain shuttled copolymers. | ||
| Entrya | Hard block initiators | Soft block initiators | Conv.b LA/ε-CL (%) | t (h) |
M
n calcc (g mol−1) |
M
nd (g mol−1) |
Đ |
T
g1e (°C) |
T
g2e (°C) |
T
me (°C) |
ΔHe (J g−1) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Copolymerisations conducted at 100 °C in toluene at 1 M (mol L−1) with metal ratio: [Y] or [Al] HBI/[Al] SBI/LA/CL = 1/5/500/500. b Determined by 1H NMR in CDCl3. c For entry 13 Mn calc = ((500 × 144 × L-LA conversion)/100 + (500 × 114 × -CL conversion)/100)/6; for entry 21 Mn calc = ((500 × 144 × L-LA conversion)/100 + (500 × 114 × -CL conversion)/100)/8; for entries 22–23 Mn calc = ((500 × 144 × L-LA conversion)/100 + (500 × 114 × CL conversion)/100)/7.6 (the 7.6 denominator corresponds to the number of initiating alkoxide moieties: 5 for the 5 equiv. of soft initiator, plus 2.6 for the yttrium hard block initiator. Indeed, the latter is a cluster with 13 alkoxides moieties for 5 yttrium atoms, which makes an average of 2.6 per Y). d Number-average molecular weight determined by size exclusion chromatography in THF corrected as Mn = (Mn raw × 0.56 × ε-CL conversion) + (Mn raw × 0.58 × L-LA) and dispersity. e Glass transition temperatures, melting temperature and melting enthalpy determined by Differential Scanning Calorimetry (DSC). | |||||||||||
| 13 | 2c | 2a | 79/48 | 16 | 14000 |
12200 |
1.10 | −14 | 46 | 74 | 5.4 |
| 21 | Al(OiPr)3 | 2a | 60/22 | 6 | 6500 | 7000 | 1.08 | 2 | 49 | 117 | 17 |
| 22 | OY5(OCH(CH3)2)13 | 2a | 92/69 | 16 | 13900 |
13600 |
1.27 | −22 | 44 | 120 | 10.5 |
| 23 | 2d | 93/67 | 16 | 13800 |
13700 |
1.23 | −24 | 44 | 129 | 15 | |
3. Conclusion
We have reported in this contribution six new catalytic systems for the L-LA/ε-CL chain shuttling copolymerisation. They are based on four different amino(bis)phenolate complexes bearing a pendant donor arm in the form of a tertiary alkylamine (2a), a tertiary dibenzylamine (2b), a pyridine (2c) and a morpholine (2d) functional group. The corresponding ligands were synthesised by a Mannich condensation procedure. Two new alkoxide complexes and two already reported ones were obtained in reasonable yield through a two-steps treatment with trimethylaluminium followed by benzyl alcohol. They were initially investigated for the polymerisation of L-LA and ε-CL. The polymerisation of the latter was found to be much faster than the former, and barely dependant on the nature of the pendant donor arm. Complex 2c led to the highest activity regarding the polymerisation of L-LA. Narrow molar mass distributions were obtained in most cases with dispersities around 1.1.Statistical copolymerisations of L-LA and ε-CL were successfully conducted in the presence of all complexes with dispersities below 1.2. Complex 2c showed a high selectivity toward lactide, while 2a, 2b and 2d led to ε-CL conversions higher than L-LA. This selectivity differences prompted us to assess the chain shuttling copolymerisation of L-LA and ε-CL using 2a, 2b and 2d in combination with several hard block initiators including 2c, Al(OiPr)3 and OY5(OCH(CH3)2)13. The high selectivity of the two latter complexes for lactide was known from the literature. All systems were found to able to perform the chain shuttling copolymerisation. To our knowledge, this is the first description of amino(bis)phenolate complexes bearing a pendant arm for CSP. Additionally, we report for the first time the L-LA/ε-CL chain shuttling copolymerisation between two different metals, i.e. Al and Y. This allows tuning of the thermal properties of the chain shuttled copolymers, as shown by a variation of the Tg of the soft block over 25 °C by changing the catalytic system without altering the reaction feed. The fine microstructure and properties of the so-formed materials will be discussed in a forthcoming paper.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The National Research Agency, ANR, is gratefully acknowledged for funding this project (ANR PLANAVETTE, ANR-21-CE06-0024). The Chevreul Institute (FR 2638), the Ministère de l'Enseignement Supérieur de la Recherche et de l'Innovation (MESRI), the CNRS, and the Région Hauts de France are acknowledged for supporting and partially funding this work. Aurélie Malfait and Maxence Epina are acknowledged for SEC measurements. The authors also acknowledge Dr Phoebe Lowy for careful proof-reading of the manuscript.References
- R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS.
- A. P. Gupta and V. Kumar, Eur. Polym. J., 2007, 43, 4053–4074 CrossRef CAS.
- X. Pang, X. Zhuang, Z. Tang and X. Chen, Biotechnol. J., 2010, 5, 1125–1136 CrossRef CAS.
- V. Nagarajan, A. K. Mohanty and M. Misra, ACS Sustainable Chem. Eng., 2016, 4, 2899–2916 CrossRef CAS.
- R. M. Rasal, A. V. Janorkar and D. E. Hirt, Prog. Polym. Sci., 2010, 35, 338–356 CrossRef CAS.
- G. Maglio, A. Migliozzi, R. Palumbo, B. Immirzi and M. G. Volpe, Macromol. Rapid Commun., 1999, 20, 236–238 CrossRef CAS.
- R. Dell'Erba, G. Groeninckx, G. Maglio, M. Malinconico and A. Migliozzi, Polymer, 2001, 42, 7831–7840 CrossRef.
- R. V. Castillo, A. J. Müller, J.-M. Raquez and P. Dubois, Macromolecules, 2010, 43, 4149–4160 CrossRef CAS.
- N. Nomura, A. Akita, R. Ishii and M. Mizuno, J. Am. Chem. Soc., 2010, 132, 1750–1751 CrossRef CAS PubMed.
- L. Mezzasalma, J. D. Winter, D. Taton and O. Coulembier, Green Chem., 2018, 20, 5385–5396 RSC.
- H. Ö. Düşkünkorur, A. Bégué, E. Pollet, V. Phalip, Y. Güvenilir and L. Avérous, J. Mol. Catal. B: Enzym., 2015, 115, 20–28 CrossRef.
- D. J. Arriola, E. Carnahan, P. D. Hustad, R. L. Kuhlman and T. T. Wenzel, Science, 2006, 312, 714–719 CrossRef.
- L. Pan, K. Zhang, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2011, 50, 12012–12015 CrossRef.
- A. Valente, G. Stoclet, F. Bonnet, A. Mortreux, M. Visseaux and P. Zinck, Angew. Chem., Int. Ed., 2014, 53, 4638–4641 CrossRef PubMed.
- J. Meimoun, C. Sutapin, G. Stoclet, A. Favrelle, P. Roussel, M. Bria, S. Chirachanchai, F. Bonnet and P. Zinck, J. Am. Chem. Soc., 2021, 143, 21206–21210 CrossRef PubMed.
- H. Gao, S. Chen, B. Du, Z. Dai, X. Lu, K. Zhang, L. Pan, Y. Li and Y. Li, Polym. Chem., 2022, 13, 245–257 RSC.
- A. Valente, A. Mortreux, M. Visseaux and P. Zinck, Chem. Rev., 2013, 113, 3836–3857 CrossRef PubMed.
- R. Mundil, C. Bravo, N. Merle and P. Zinck, Chem. Rev., 2024, 124, 210–244 CrossRef.
- C. Jacobs, P. Dubois, R. Jerome and P. Teyssie, Macromolecules, 1991, 24, 3027–3034 CrossRef.
- E. Stirling, Y. Champouret and M. Visseaux, Polym. Chem., 2018, 9, 2517–2531 RSC.
- A. Duda, T. Biela, J. Libiszowski, S. Penczek, D. Mecerreyes and R. Jérôme, Polym. Degrad. Stab., 1998, 59, 215–222 CrossRef.
- K. Matsubara, K. Eda, Y. Ikutake, M. Dan, N. Tanizaki, Y. Koga and M. Yasuniwa, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2536–2544 CrossRef.
- O. Wichmann, R. Sillanpää and A. Lehtonen, Coord. Chem. Rev., 2012, 256, 371–392 CrossRef.
- E. D. Cross, G. K. Tennekone, A. Decken and M. P. Shaver, Green Mater., 2013, 1, 79–86 CrossRef.
- P. Chumsaeng, S. Haesuwannakij, A. Virachotikul and K. Phomphrai, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1635–1644 CrossRef.
- Y. Wongnongwa, S. Haesuwannakij, K. Udomsasporn, P. Chumsaeng, A. Watcharapasorn, K. Phomphrai and S. Jungsuttiwong, Polymer, 2023, 281, 126065 CrossRef.
- K. Phomphrai, P. Chumsaeng, P. Sangtrirutnugul, P. Kongsaeree and M. Pohmakotr, Dalton Trans., 2010, 39, 1865–1871 RSC.
- Y. Wongnongwa, S. Haesuwannakij, K. Udomsasporn, P. Chumsaeng, A. Watcharapasorn, K. Phomphrai and S. Jungsuttiwong, Polymer, 2023, 281, 126065 CrossRef.
- N. Spassky, M. Wisniewski, C. Pluta and A. Le Borgne, Macromol. Chem. Phys., 1996, 197, 2627–2637 CrossRef.
- S. M. Weidner, A. Meyer and H. R. Kricheldorf, Polymer, 2022, 255, 125142 CrossRef.
- P. Lloyd, K. Duddaby, J. Varney, E. Scrivener, P. Derrick and D. Haddleton, Eur. J. Mass Spectrom., 1995, 1, 293 CrossRef.
- A. Kowalski, A. Duda and S. Penczek, Macromolecules, 1998, 31, 2114–2122 CrossRef.
- Ionic Polymerizations and Related Processes, ed. J. E. Puskas, A. Michel and S. Barghi, Springer, Netherlands, Dordrecht, 1999 Search PubMed.
- M. Save, M. Schappacher and A. Soum, Macromol. Chem. Phys., 2002, 203, 889–899 CrossRef.
- D. Dakshinamoorthy and F. Peruch, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2161–2171 CrossRef.
- I. R. Herbert, in NMR Spectroscopy of Polymers, ed. R. N. Ibbett, Springer, Netherlands, Dordrecht, 1993, pp. 50–79 Search PubMed.
- V. Simic, S. Pensec and N. Spassky, Macromol. Symp., 2000, 153, 109–121 CrossRef.
- W. M. Stevels, M. J. K. Ankoné, P. J. Dijkstra and J. Feijen, Macromol. Chem. Phys., 1995, 196, 1153–1161 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00671b |
| ‡ A value of 0 indicates a diblock character, while a value of 1 indicates a fully random copolymer. |
| This journal is © The Royal Society of Chemistry 2024 |