
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anionic polymerization of phenyl-substituted isoprene derivatives: polymerization behaviour and cyclization-enabled fluorescence†
Moritz
Rauschenbach
a,
Laura
Stein
a,
Gregor M.
Linden
a,
Ramona
Barent
ab,
Katja
Heinze
 a and
Holger
Frey
*a
a and
Holger
Frey
*a
aDepartment of Chemistry. Johannes Gutenberg University, Duesbergweg 10-14, 55128 Mainz, Germany. E-mail: hfrey@uni-mainz.de
bMax Planck Graduate Center, Forum Universitatis 2, 55122 Mainz, Germany
First published on 10th July 2024
Abstract
1,3-Dienes are important monomers for living anionic polymerization. However, phenyl-substituted diene monomer structures have been rarely investigated. Based on DFT calculations and 13C NMR spectroscopy, a discrepancy in the reactivity of the two monomers 1-phenyl isoprene (1PhI) and 4-phenyl isoprene (4PhI) in anionic polymerization is expected. Starting from a Wittig reaction including an optimized extraction procedure, disubstituted 1,3-dienes were obtained that resulted in polymers with different degrees of 1,3-incorporation. The polymers have been characterized by 1H NMR spectroscopy and using different SEC conditions. Molecular weights of up to 48.8 kg mol−1 with narrow dispersities (Đ ≤ 1.13) were achieved. The addition of the modifier THF led to an initial increase of vinyl units as well as a loss of control over the polymerization of 4PhI. Increasing the THF concentration further resulted in a rather unusual decrease of the vinyl units and ended with more than 80% 1,4-units in pure THF. Copolymerizations with styrene (S) and isoprene (I), respectively, were tracked via in situ1H NMR kinetics. The observed ideally random copolymerizations of I and 1PhI as well as the gradient copolymers with S were further investigated via the synthesis of copolymers with a targeted Mn of 40 kg mol−1. In a subsequent reaction step, the homopolymers were cyclized using trifluoromethyl sulfonic acid inducing fluorescence properties. The different microstructures and substitution patterns of the original polymers differ in both emission maxima and quantum yields.
Introduction
Living anionic polymerization introduced by Michael Szwarc in 1956 offers excellent control over molecular weights and the dispersity Đ of polymers.1,2 It has been widely used for the polymerization of 1,3-dienes, such as 1,3-butadiene and isoprene, to generate polydiene materials for synthetic rubber. The elastic properties of these polydienes after crosslinking notably depend on their microstructure. Therefore, the main target of early studies of anionic polymerization of 1,3-dienes was the optimization of reaction parameters (e.g., initiator, solvent, further additives, and temperature) to obtain a high cis-1,4-content.3The copolymerization of 1,3-dienes, such as 1,3-butadiene (B) and isoprene (I), with styrene (S) affords a variety of possible polymer architectures.4,5 ABA triblock copolymers have attracted attention as thermoplastic elastomers (TPEs). This architecture relies on a flexible, low Tg polydiene midblock B (e.g., polyisoprene) and two outer polystyrene A blocks that act as crosslinks after cooling and vitrification.6 In 1966, Holden and Milkovich reported that the statistical copolymerization of isoprene and styrene in apolar solvents like cyclohexane results in block-like gradient copolymers that were later designated as “tapered”.7 The addition of a small quantity of THF with respect to the lithium-ion concentration leads to a change of the reactivity ratios and the respective monomer gradient. By varying the THF concentration, the incorporation of both monomers can be tuned to achieve ideally random incorporation, and even complete reversal of the molar composition is possible, as shown by detailed online NIR kinetics recently.8 However, the addition of THF as a “modifier” at the same time also influences the incorporation mode of isoprene (increasing the extent of 3,4-addition) and therefore has an undesired impact on the elastic properties. As described in many studies, an increase of the number of 1,2- and 3,4-units in polydienes is observed when increasing the polarity of the system.8–11
Only a few studies have been reported for phenyl-substituted butadiene derivatives.12,13 As an example, the 1-phenyl-1,3-butadiene (PhB) monomer has been polymerized via either anionic or catalytic approaches.14–17 It can be considered as a β-substituted styrene or as a 1-phenyl-substituted 1,3-butadiene.18 This combination of two very established monomers provides an intriguing perspective. Suzuki et al. systematically investigated the anionic polymerization of PhB regarding its copolymerization with butadiene and styrene as well as the microstructure with respect to various parameters.12,19–221H NMR studies of the resulting microstructure of PhB in different solvents revealed distinct behaviour compared to established dienes. The addition of aliquots of THF enhanced the formation of the vinylic microstructure. However, a 1,4-dominated microstructure was obtained when polymerization was performed in THF, confirmed by NMR analysis based on the chemical shifts of the aromatic protons of oligomeric structures in deuterated THF. The authors concluded that the negative charge is localized in the α-position of the phenyl ring to form the most stable anion as the active chain end. This explains why poly(1-phenyl butadiene) (PPhB) polymerized in THF shows predominantly 1,4-units.21
The phenyl-substituted polybutadienes showed a glass transition temperature (Tg) of around 30 °C.17 It is worth mentioning that the Tg is in between those of the structurally similar polyisoprene (Tg ≤ −65 °C)23 and polystyrene (Tg = 100 °C).11 Han et al. reported catalytic polymerization with a 3,4-content of 94%, resulting in a Tg of 82 °C, indicating a high dependency on the respective microstructure.24 In a cationic polymerization approach, phenyl butadiene underwent cyclization as a side-reaction. Fundamental studies report on the targeted cationic cyclization post-polymerization of polydienes to obtain an unsaturated polycyclic species.25–28 More recently, the cyclisation reaction was adapted to P(PhB) to maximize the Tg. Depending on the microstructure, the Tg of the cyclized material can reach nearly 200 °C, which is among the highest Tgs reported for aliphatic hydrocarbon polymers.29 Additionally, cyclized poly(phenylbutadiene) (cycP(PhB)) showed fluorescence.
Herein, we report the copolymerization of “phenyl isoprenes” with styrene and isoprene. For isoprene, the attachment of the phenyl ring can take place either at the 1- or the 4-position, resulting in a 1,2- or a 1,3-disubstituted 1,3-diene structure. While the 4-phenyl isoprene (4PhI) has been briefly described with respect to its anionic polymerization,30 to the best of our knowledge, the 1-phenyl isoprene (1PhI) has not been employed for anionic polymerization to date. In this work, we compare 1PhI and 4PhI on a theoretical basis using density functional theory (DFT) as well as 13C NMR spectroscopy to predict their reactivity in anionic polymerization. We use styrene and isoprene, respectively, for comparison. Both phenyl isoprene monomers are investigated in depth regarding their behaviour in anionic polymerization and are also copolymerized with both isoprene and styrene. For the determination of reactivity ratios, online 1H NMR spectroscopy was employed. We monitored the anionic copolymerization with the structurally related monomers styrene and isoprene in cyclohexane, respectively. Finally, polymer cyclization is explored according to literature procedures, aiming at fluorescent, high Tg materials based on the reported monomers.17,24,29,31
Results and discussion
Synthesis of phenyl-substituted isoprene derivatives
1PhI and 4PhI were synthesized via a one-step Wittig reaction starting from commercially available aldehyde and ketone structures, respectively. For the synthesis of 1PhI, using the potassium tert-butoxide monomer, yields exceeding 75% were achieved. However, the stronger base sodium hexamethyldisilyl amine (NaHMDS) is required to achieve similar yields for the ketone benzylidene acetone. A straightforward method was established for the separation of triphenylphosphine oxide including precipitation, centrifugation and subsequential distillation. The details are given in the ESI.†Since 1PhI and 4PhI can be viewed as β-substituted styrene derivatives, we estimate the reactivity of both monomers in analogy to previously described methods. 13C NMR spectroscopy was utilized to estimate the reactivity of para-substituted styrene derivatives by Hirao et al.32 The β-carbon shift identifies the electron charge density of the reactive vinyl bond. Increasing the polarization of the reacting carbon–carbon bond leads to increased reactivity. Therefore, it is in good agreement with the monomer reactivity in anionic polymerization. If 1PhI and 4PhI are viewed as styrene derivatives, the chemical shift of C1 as assigned in Scheme 1 was identified. Thus, 4PhI (δ = 117.45 ppm, CDCl3) is expected to be much more reactive compared to styrene (δ = 113.36 ppm, CDCl3), while 1PhI (δ = 113.02 ppm, CDCl3) should be slightly less reactive in anionic copolymerization in apolar media.
 | ||
| Scheme 1 Synthesis route to the phenyl-substituted isoprene derivatives. | ||
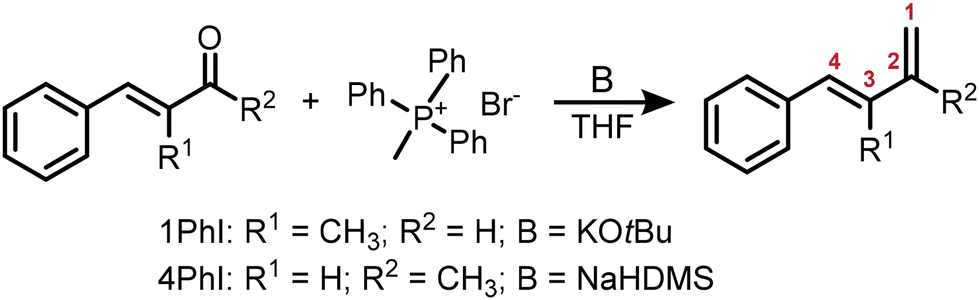
Furthermore, DFT calculations of both monomers were conducted to obtain further theoretical insights. Fig. 1 shows the relative electron densities of both 1PhI and 4PhI in comparison with the values calculated for isoprene (differences shown in brackets). Focusing on the free methylene groups, which are most likely to be attacked by nucleophilic reagents, 4PhI (−0.176e) exhibits a lower charge at this position compared to 1PhI (−0.193e). Hence, the nucleophilic attack as part of the propagation is prone to occur, supporting the expectation based on 13C NMR spectroscopy. Nevertheless, the electron densities of the reactive double bond show just a minor deviation from the 3,4-double bond in isoprene, which is assumed to be the one reacting in the anionic polymerization. This could be interpreted in terms of a fast crossover reaction in both directions.
 | ||
| Fig. 1 3D visualization of (a) 1PhI and (b) 4PhI, respectively, with the corresponding partial charges of the dienic carbon atoms calculated using DFT. In brackets, the deviations from the calculated electron densities of isoprene are given.59,60 | ||
Homopolymerization of PhIs with sec-BuLi in cyclohexane
Both monomers 1PhI and 4PhI can be interpreted as a structural combination of styrene and isoprene. To investigate whether their behaviour in the anionic polymerization resembles that of S or I, a series of homopolymers with increasing Mn has been synthesized. All polymerizations were carried out in cyclohexane, using sec-butyl lithium as an initiator at room temperature. Subsequently, the homopolymers were characterized via SEC, 1H NMR spectroscopy and DSC. Table 1 summarizes all characterization data of the obtained polymers, which possessed molecular weights in good agreement with the targeted values in a range of 4.6–48.8 kg mol−1 (SEC, eluent THF, and PS calibration) with low dispersities (Đ ≤ 1.13). The SEC-determined molecular weights are strongly dependent on the calibration standard employed.| Entry | M | M targn (kg mol−1) | M n (kg mol−1) | M n (kg mol−1) | Đ | 1,4-PhIc (%) | 1,2-PhIc (%) | T g (°C) |
|---|---|---|---|---|---|---|---|---|
| a Determined by SEC (THF, PI calibration, and RI detector). b Determined by SEC (THF, PS calibration, and RI detector). c Determined from the olefinic region of the 1H NMR spectra (400 MHz, CDCl3). | ||||||||
| 1 | 1PhI | 5 | 3.6 | 4.6 | 1.06 | 42 | 58 | 62 |
| 2 | 10 | 6.5 | 8.5 | 1.05 | 34 | 66 | 69 | |
| 3 | 20 | 13.6 | 18.4 | 1.04 | 35 | 65 | 65 | |
| 4 | 40 | 24.0 | 32.4 | 1.02 | 37 | 63 | 69 | |
| 5 | 4PhI | 5 | 3.8 | 4.7 | 1.13 | 85 | 15 | 47 |
| 6 | 10 | 10.7 | 13.6 | 1.07 | 94 | 6 | 47 | |
| 7 | 20 | 22.6 | 30.0 | 1.09 | 94 | 6 | 48 | |
| 8 | 40 | 48.8 | 64.8 | 1.11 | 94 | 6 | 48 | |
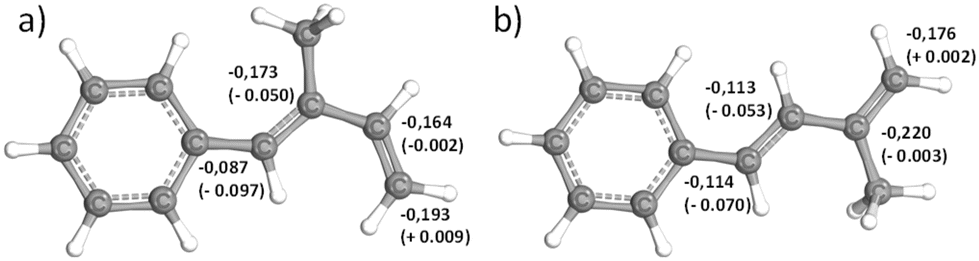
The microstructures of both P(1PhI) and P(4PhI) were investigated by 1H NMR spectroscopy in analogy to previously reported studies.12,17 The spectra (Fig. 2) show the olefinic signals used for the determination of the microstructures. The signals were assigned according to the reported microstructure of P(1-phenyl butadiene).12 The sharp signals of P(4PhI) indicate a highly defined composition, which is supported by the integration, showing 94% 1,4-units. This is consistent with the microstructure of polyisoprene obtained under these conditions.33 In comparison, the 1H NMR spectrum of P(1PhI) displays broad signals with a microstructure consisting of 66% 1,2-units. This result can be explained by a sterically hindered 1,4-addition reaction, as previously suggested for 1,1-disubstituted 1,3-dienes.34
 | ||
| Fig. 2 1H NMR spectra (CDCl3, 400 MHz) of (a) P(1PhI) and (b) P(P4PhI). | ||
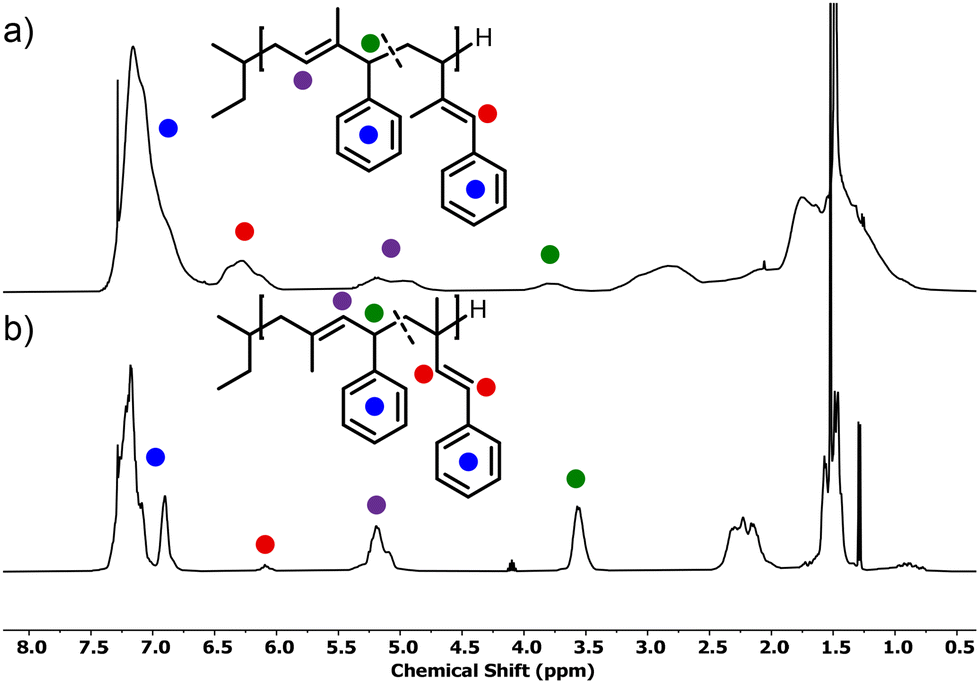
The SEC traces shown in Fig. 3 confirm the good control of the polymerization for both monomers, resulting in narrow, monomodal distributions. A high degree of agreement was observed between the targeted molecular weights and the results based on PI calibration, which can be taken as an indirect confirmation of the predominant 1,4-microstructure. In contrast, the prevalence of the 1,2-microstructure in P(1PhI) samples underlines why PS calibration yields values that exhibit closer proximity to theoretical predictions.
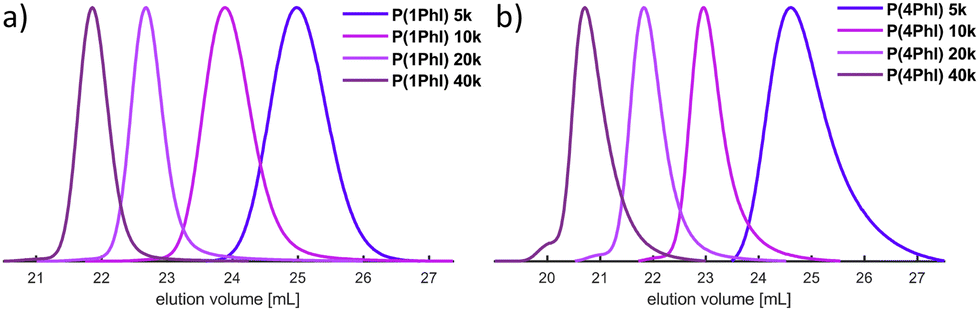 | ||
| Fig. 3 SEC traces of the synthesized homopolymers with increasing targeted Mn from (a) 1-phenyl isoprene and (b) 4-phenyl isoprene. | ||
Addition of polar additives
The polymerization of 1,3-dienes is known to depend on several parameters (e.g., chain end and monomer concentration, temperature, solvent, etc.) in terms of the regio-isomeric composition of the resulting polymers.8,35–38 The polarity of the solvent is a key parameter, since it determines the coordination of the chain end to the lithium ion.39 In numerous studies, it was shown that the addition of aliquots of THF in the polymerization of 1,3-dienes increases the content of vinylic units.8,9,40 Furthermore, the addition of THF accelerates propagation kinetics. This effect has been observed in both isoprene and styrene, where propagation reaches a maximum before further addition of THF decelerates propagation.41,42For 1-phenyl butadiene, it was reported that upon addition of a few aliquots of THF, the ratio of 1,2-units increases.12 Further increasing the content of THF and polymerization in pure THF led to a maximum of 90% 1,4-content. In a similar fashion, we conducted the polymerization of 1PhI and 4PhI in cyclohexane with 2 and 20 equivalents of THF with respect to the lithium-ion concentration. Furthermore, we carried out the polymerization reaction in pure THF at −78 °C. It should be noted that common dienes, such as myrcene and isoprene, are not capable of polymerizing under these conditions.43,44 As listed in Table 2, the addition of THF has an impact on the resulting microstructure as the ratio of vinyl units increases. In line with previous observations for PhB, in pure THF, more than 80% 1,4-units were formed. A tentative explanation might be given by the reactivity of the chain end and the resulting propagation rates. Strohmann et al. reported experiments with tert-butyl lithium and THF in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, revealing remarkable aggregation, leading to an increase in reactivity compared to the ratios 1:1 and 1:2.5.45 Consequently, the polymerization rate is enhanced by small amounts of THF, promoting the formation of vinylic units. The faster kinetics may also explain the broad distribution observed for entry 12. Carrying out the polymerization of 4PhI with 2 equivalents of THF with respect to the BuLi concentration did not result in dispersities lower than 1.4, as illustrated in Fig. S16.† Further increasing the polarity supports the formation of the most stable anion. For both 1PhI and 4PhI, the charge will be delocalized in the aromatic ring as demonstrated for PhB.21,22 Therefore, one observes the predominant formation of 1,4-units with increasing polarity of the system, as obvious from a comparison of the respective 1H NMR spectra (Fig. 4).
:2, revealing remarkable aggregation, leading to an increase in reactivity compared to the ratios 1:1 and 1:2.5.45 Consequently, the polymerization rate is enhanced by small amounts of THF, promoting the formation of vinylic units. The faster kinetics may also explain the broad distribution observed for entry 12. Carrying out the polymerization of 4PhI with 2 equivalents of THF with respect to the BuLi concentration did not result in dispersities lower than 1.4, as illustrated in Fig. S16.† Further increasing the polarity supports the formation of the most stable anion. For both 1PhI and 4PhI, the charge will be delocalized in the aromatic ring as demonstrated for PhB.21,22 Therefore, one observes the predominant formation of 1,4-units with increasing polarity of the system, as obvious from a comparison of the respective 1H NMR spectra (Fig. 4).
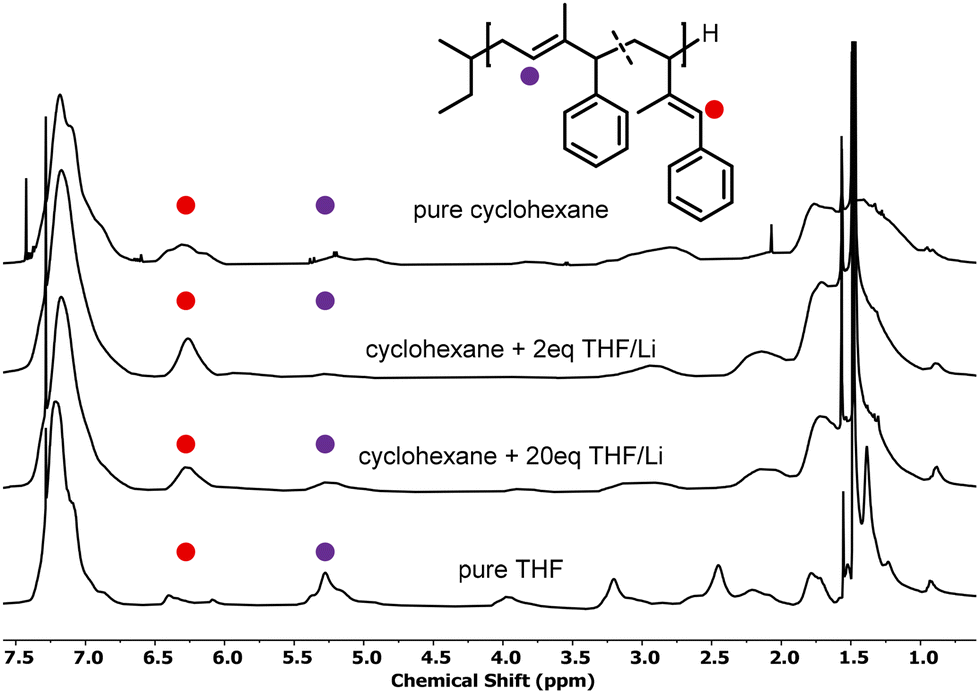 | ||
| Fig. 4 Stacked 1H NMR spectra of P(1PhI) polymerized with increasing concentrations of THF, resulting in microstructures with shifting ratios. | ||
| Entry | M | M targn (kg mol−1) | M n (kg mol−1) |
|
Đ | 1,4-PhIc (%) | 1,2-PhIc (%) | T g (°C) |
|---|---|---|---|---|---|---|---|---|
| a Polymerization at −78 °C. b Determined by SEC (THF, PS calibration, and RI detector). c Calculation using the respective olefinic signals of the 1H NMR spectra. | ||||||||
| 1 | 1PhI | 10 | 7.8 | 0 | 1.05 | 34 | 66 | 67 |
| 9 | 10 | 10.3 | 2 | 1.07 | 12 | 88 | 62 | |
| 10 | 10 | 11.1 | 20 | 1.08 | 33 | 67 | 53 | |
| 11a | 10 | 9.6 | Pure | 1.06 | 85 | 15 | 50 | |
| 5 | 4PhI | 10 | 13.9 | 0 | 1.09 | 94 | 6 | 56 |
| 12 | 10 | 7.1 | 2 | 1.44 | n.d. | n.d. | 73 | |
| 13 | 10 | 8.7 | 20 | 1.15 | 56 | 44 | 63 | |
| 14a | 10 | 9.9 | Pure | 1.08 | 82 | 18 | 50 | |

As expected, DSC measurements of the resulting polymers show a clear correlation between the glass transition and the respective microstructure. For example, 1PhI polymerized in THF with a high 1,4-content exhibited a lower Tg of 50 °C compared to 67 °C when polymerized in cyclohexane due to its more flexible backbone compared to P1PhI synthesized in cyclohexane, which is dominated by a 1,2-microstructure (Fig. S14†). As shown by the values in Table 2, the Tgs for the P1PhI samples prepared in different systems shift to higher temperature with increasing solvent polarity. P4PhI polymerized in either cyclohexane or THF always resulted in a high 1,4-content. Therefore, as shown in Fig. S15,† the Tgs of both samples are in the same range of 50–56 °C. A higher 1,2-content can raise the Tg to 73 °C.
In situ 1H NMR: copolymerisation with styrene as a comonomer
Living poly(1PhB) chain ends were not sufficiently reactive to initiate the polymerization of butadiene or styrene in THF. Reaching a maximum of 35% block efficiency, only bimodal distributions were reported. However, reverse monomer addition resulted in monomodal SEC traces, confirming that all living chain ends initiated 1PhB.20To investigate the statistical copolymerization behaviour of both monomers with styrene and isoprene, respectively, we conducted real-time 1H NMR spectroscopy to evaluate the reactivity ratios. First, copolymerization reactions of styrene with 1PhI and 4PhI, respectively, were conducted. During the statistical copolymerization, individual monomer peaks were traced to determine monomer conversion. Due to the absence of termination and transfer reactions in classical anionic polymerization, monitoring of the integrals can also be used to determine the relative position along the growing chains.46,47 The stacked 1H NMR spectra for the copolymerization of styrene and 1PhI are shown in Fig. 5 and in Fig. S18† for 4PhI. The monomer concentrations plotted as a function of time and total conversion, respectively, show preferential consumption of the phenyl isoprenes over styrene in both cases.
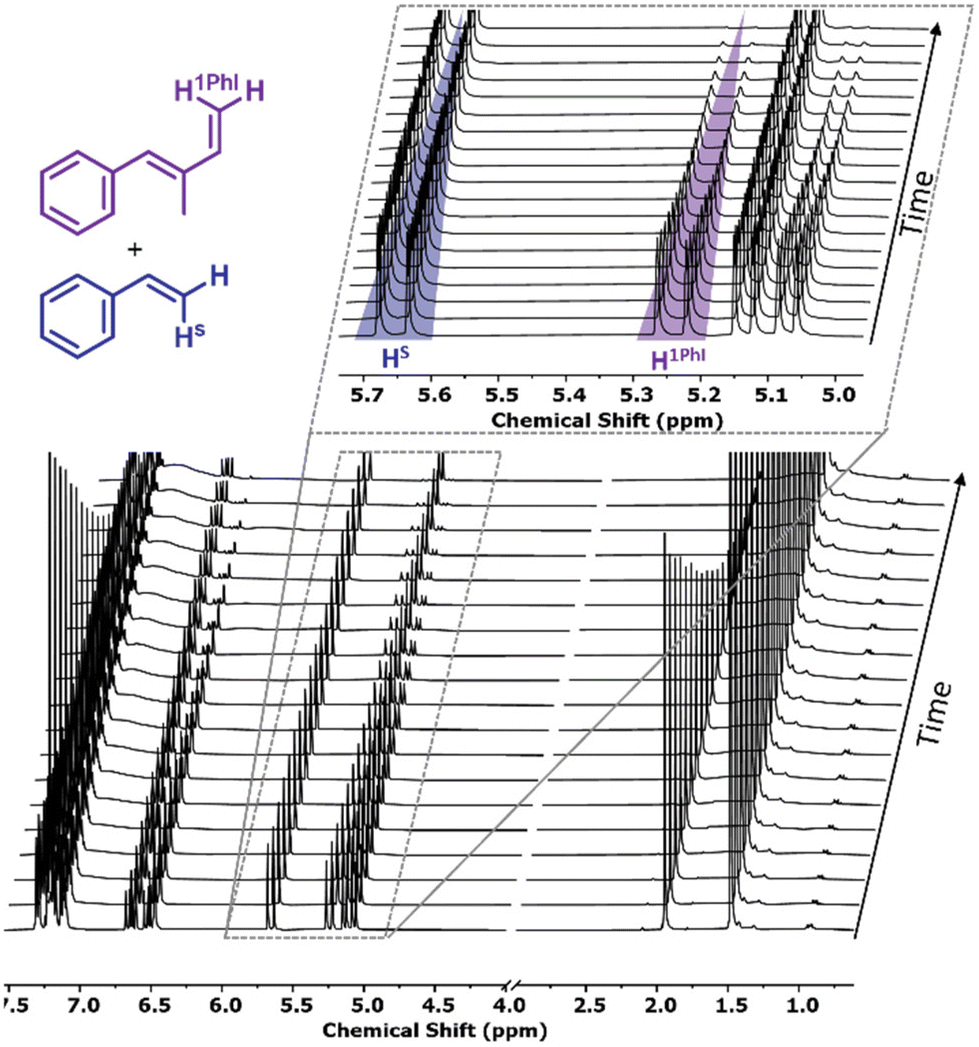 | ||
| Fig. 5 Stacked 1H NMR spectra (400 MHz, C6D12) of the copolymerization of styrene/1PhI as a function of time. The zoomed-in region shows the peaks tracked for the evaluation of the respective comonomer consumption. | ||
The collected data were used to calculate the reactivity ratios rPhI and rS, as listed in Table 3. They define the ratio between the rate of homopolymerization and the rate of the crossover reaction. Using the non-terminal model of Jaacks, the values were determined.48 Based on these evaluated parameters, plots of the relative comonomer position along the chain were generated. As illustrated in Fig. 6(b) and (c), for both systems, a pronounced gradient is observed. The steeper gradient for the 4PhI system confirms 4PhI to be more reactive, supporting the hypothesis derived from the β-C-shift. However, in contrast to the abovementioned assumption, 1PhI is also consumed faster, although the carbon shift indicated the opposite. This demonstrates that these simple considerations cannot be applied to diene systems. Reactivity ratios calculated using the terminal model of the Meyer–Lowry fit are given in the ESI.†
 | ||
| Fig. 6 (a) Monomer conversion for the copolymerization of S and 4PhI. Calculated molar composition profiles of S and (b) 1PhI and (c) 4PhI, respectively. | ||
In comparison with the reactivity ratios observed for the copolymerization of styrene with isoprene (substituted in the 2-position) and myrcene (Table 3), the di-substituted 1PhI and 4PhI display a less pronounced gradient. Nevertheless, it is worth noting that the 1,2-disubstituted 1PhI demonstrates higher reactivity ratios compared to previously reported 1,2-disubstituted dienes (i.e., trans- and cis-ocimene and 1-vinyl cyclohexene).52,53 To the best of our knowledge, 4PhI is the first 1,3-disubstituted 1,3-diene that has been investigated regarding its copolymerization kinetics with styrene.
In situ 1H NMR kinetics with isoprene as a comonomer
In analogy to the investigation of copolymerization with styrene, we also studied in situ1H NMR kinetics for copolymerization with isoprene (I). Due to the diene nature of both monomers, the respective peaks overlapped, as shown in the stacked spectra as a function of time in Fig. S21 and S22.† From the obtained data, the plots of the comonomer concentrations vs. the total conversion and against time are given in Fig. 7 and in the ESI.† To our surprise, the copolymerization of 4PhI with I did not result in plausible results, neither for the Jaacks nor for the Meyer–Lowry fit, when carried out in an equimolar ratio. Repeating the copolymerization under the same conditions or with a ratio of 70:30 did not improve the given results, which are illustrated in the ESI.† Using the Meyer–Lowry fit, in all cases the reactivity ratios were calculated to exceed 1, as summarized in Table S3.† In comparison, the obtained data do not reflect the theoretical Jaacks fit. Nevertheless, the stacked NMR spectra in Fig. S22† indicate a faster consumption of isoprene compared to 4PhI.
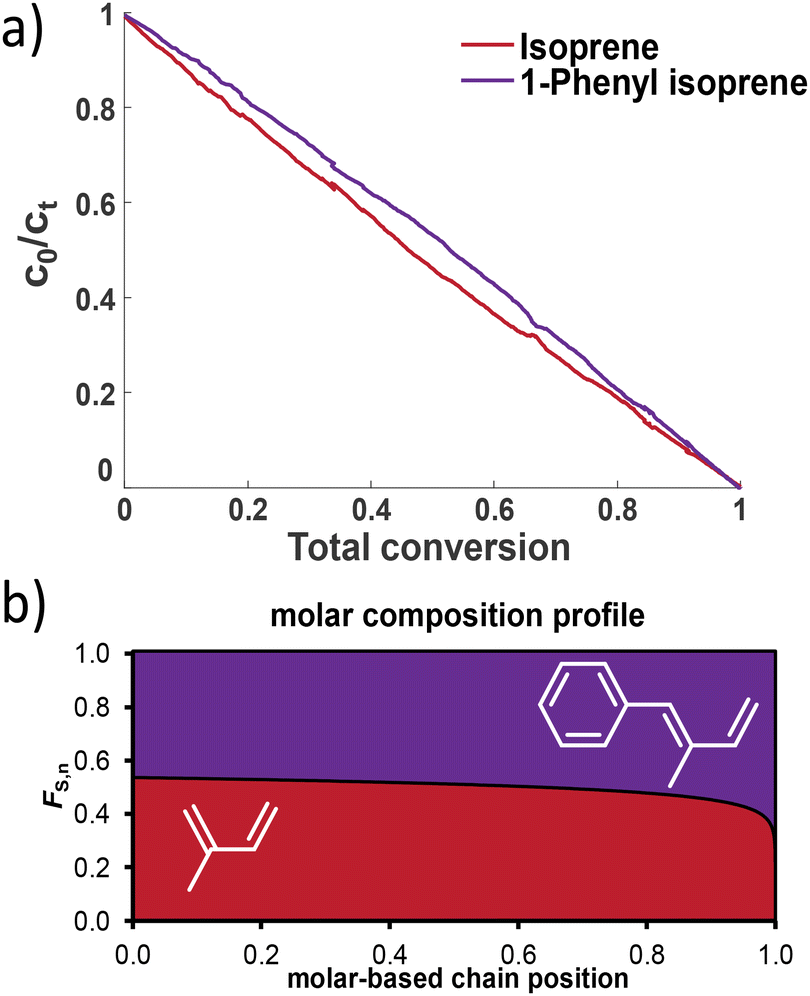 | ||
| Fig. 7 (a) Monomer conversion in the copolymerization of I and 1PhI and (b) the molar composition profile of the copolymer of I and 1PhI. | ||
The Jaacks fit was successfully used for 1PhI and isoprene, confirming the almost ideally random copolymerization with reactivity ratios of rI = 1.155 and r1PhI = 0.865. The cross-over reaction seems to be independent of the monomer. This might be explained by the similar electron densities calculated using DFT. A comparison with the known reactivity ratios using myrcene as another diene is given in Table 4.
| Monomer A | r A | r I |
|---|---|---|
| 1PhI | 0.865 | 1.155 |
| 4PhI | n.d. | n.d. |
| β-Myrcene50 | 4.4 | 0.23 |
Copolymerization with isoprene and styrene
The determined reactivity ratios may lead to unique material properties. Therefore, the copolymerizations were conducted on a larger scale (Fig. 8). A rather high molecular weight of 40 kg mol−1 was targeted to induce phase separation despite the gradient structure (Table 5). In all cases, monomodal distributions with a low dispersity (Đ < 1.11) were obtained. The stacked 1H NMR spectra in Fig. S28† show the characteristic signals of the monomers. The gradient structure in the anionic copolymerization of I and S can be visually tracked through the gradual colour change with monomer conversion. This results in a decreasing concentration of the colorless polydienyl chain ends and a gradually increasing concentration of orange polystyryl chain ends. For both copolymerization reactions with isoprene, an immediate coloration of the solution was observed after initiation. Throughout the polymerization, no colour change was observed. Therefore, as shown before, a significant number of polyphenylisoprenyl chain ends were present from the beginning. To lend further support to the completely random distribution of 1PhI throughout the chain when copolymerized with isoprene, we observed only one glass transition at 3 °C. The copolymer P(I-co-4PhI) exhibits a broad softening regime rather than a defined glass transition. Further drying in a high vacuum and the change of the heating rate did not clarify the inflection point. The soft gradients of the styrenic copolymers prevented phase segregation, resulting in merely one Tg >60 °C.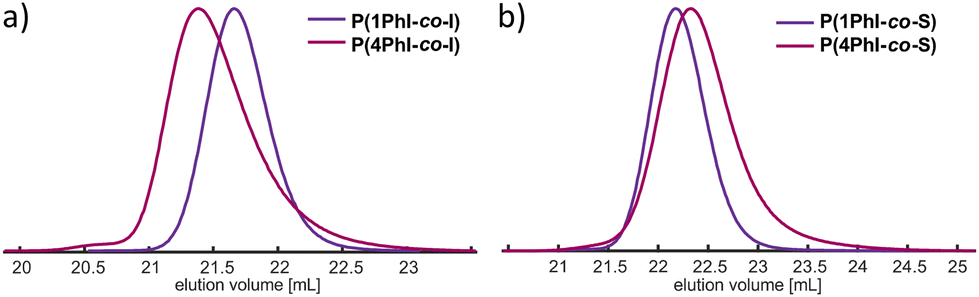 | ||
| Fig. 8 SEC traces of the copolymerization of 1PhI and 4PhI with (a) isoprene and (b) styrene. | ||
| Entry | Monomer (A) | Monomer (B) | M targn (kg mol−1) | M n,SEC (kg mol−1) | Đ | T g (°C) |
|---|---|---|---|---|---|---|
| a Determined by SEC (THF, PS calibration, and RI detector). | ||||||
| 15 | 1PhI | I | 40 | 33.8 | 1.04 | 3 |
| 16 | 1PhI | S | 40 | 35.6 | 1.05 | 67 |
| 17 | 4PhI | I | 40 | 37.9 | 1.09 | −10 |
| 18 | 4PhI | S | 40 | 29.7 | 1.11 | 61 |
Cyclization to enhance thermal properties and prepare fluorescent materials
The synthesized homopolymers show lower Tgs compared to polystyrene (Tg = 100 °C).11 However, since the phenyl rings are all adjacent to double bonds, cationic cyclization can be performed to alter the thermal properties. As already presented for butadiene-derived structures, the trifluoromethanesulfonic acid-initiated modification influences the thermal properties of these polymers drastically.16,17,24,29,31,54–56 Consequently, the cyclized P(PhI) structures are expected to exhibit strongly increased rigidity and thus high Tg values. The selected polymers (entries 4 and 8 of Table 1) with a targeted molecular weight of 40 kg mol−1 were cyclized using CF3SO3H in cyclohexane at room temperature (Scheme 2). After drying, amber-coloured powders were obtained. Since the samples remained soluble in a variety of solvents after the intramolecular cyclization, they were investigated by SEC and NMR analyses. The SEC traces shown in Fig. 9 and S33† present the expected shift to a higher elution volume. The SEC traces (Fig. S33†) show a signal for the flow rate marker toluene, indicating the presence of a polymer rather than degradation to small molecules. This discovery was described by Han et al. and attributed to the compression of the polymer chain and an increase in hydrodynamic radii.29 DSC measurements verified the expected increase of the Tg values (Table 6). The cyclization effect on the Tg values is stronger for cycP(1PhI), with Tg reaching 180 °C after cyclization. The Tg of cycP(4PhI) was determined to be 131 °C.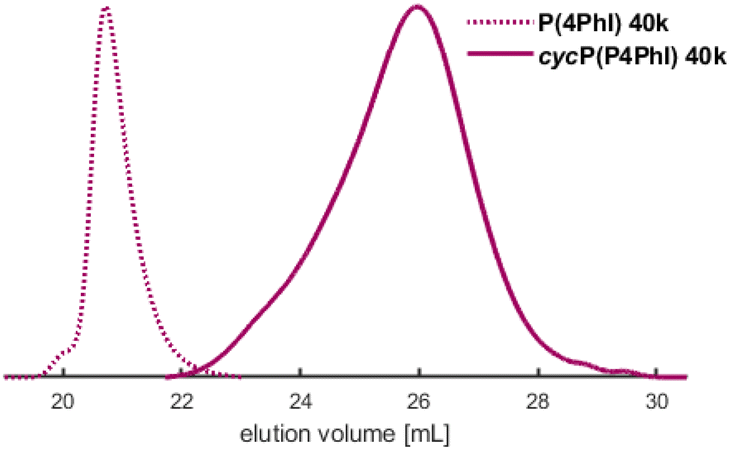 | ||
| Fig. 9 SEC traces of the cyclized P4PhI in comparison with the initial polymer (entry 8, Table 1). | ||
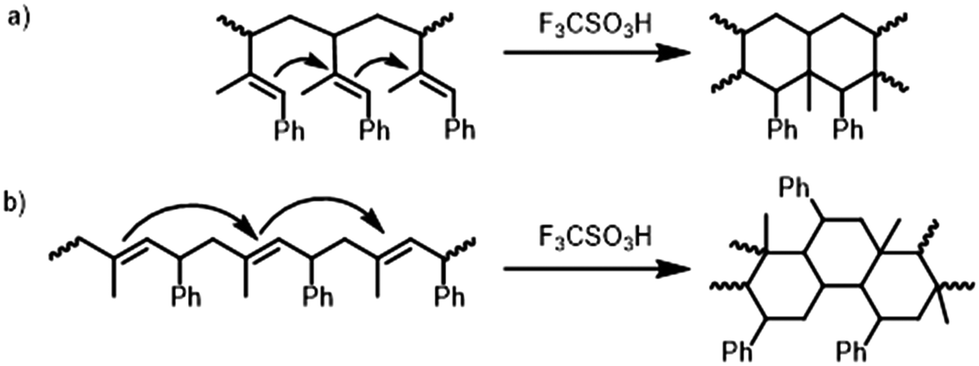 | ||
| Scheme 2 Proposed mechanism for the cyclization of the predominant microstructures of (a) P1PhI and (b) P4PhI in analogy to the literature.24 | ||
| Entry | Entry of the precursor | Polymer | M n (kg mol−1) | Đ | T g (°C) |
|---|---|---|---|---|---|
| a Determined by SEC (THF, PS calibration, and RI detector). | |||||
| 19 | 4 | P1PhI | 3.2 | 1.63 | 187 |
| 20 | 8 | P4PhI | 2.9 | 1.45 | 131 |
In agreement with the reported cycP(PhB) of Ma and coworkers, we observed that the cyclized P(PhI) samples show fluorescence when irradiated with UV light (Fig. 9).17 This observation is most likely explained by a clusterization-triggered emission.57 To quantify the photochemical properties dependent on presumed clusterization, absorption and emission properties were determined using four different concentrations (2, 0.2, 0.02, and 0.002 mg mL−1). The main questions raised were (i) whether the introduction of the methyl group leads to a shift in the emission maximum compared to the cycP(PhB) polymer due to different ordering and (ii) whether the differing initial microstructures of P1PhI and P4PhI might result in distinct emission properties. In Fig. 10a, the emission spectra of the solutions of cycP(P4PhI) with decreasing dilution are shown (λexc = 330 nm). Due to the high optical density and therefore self-absorption, the sample with the highest concentration shows a significantly reduced emission intensity compared to the other concentrations under the same conditions. At the same time, the emission maximum shifts from 375 nm to 395 nm (Table S4†). In contrast to previously reported interpretation clusterization-triggered emission,57 we propose that the emission maximum shifts apparently and its intensity decreases due to inner filter effects with increasing concentration. Going from cycP1PhI to cycP4PhI, a presumed impact of the initial microstructure is observable with the emission maximum shifting from 360 nm to 377 nm (Fig. 10b). The absolute quantum yields of both samples in the solid state were determined using an integrating sphere. In both cases, the observed self-absorption decreases the overall quantum yield. As it is higher for cycP4PhI (4.1%) than for cycP1PhI (2.9%), cycP4PhI is a more efficient candidate for utilization in organic light-emitting diodes (OLEDs).
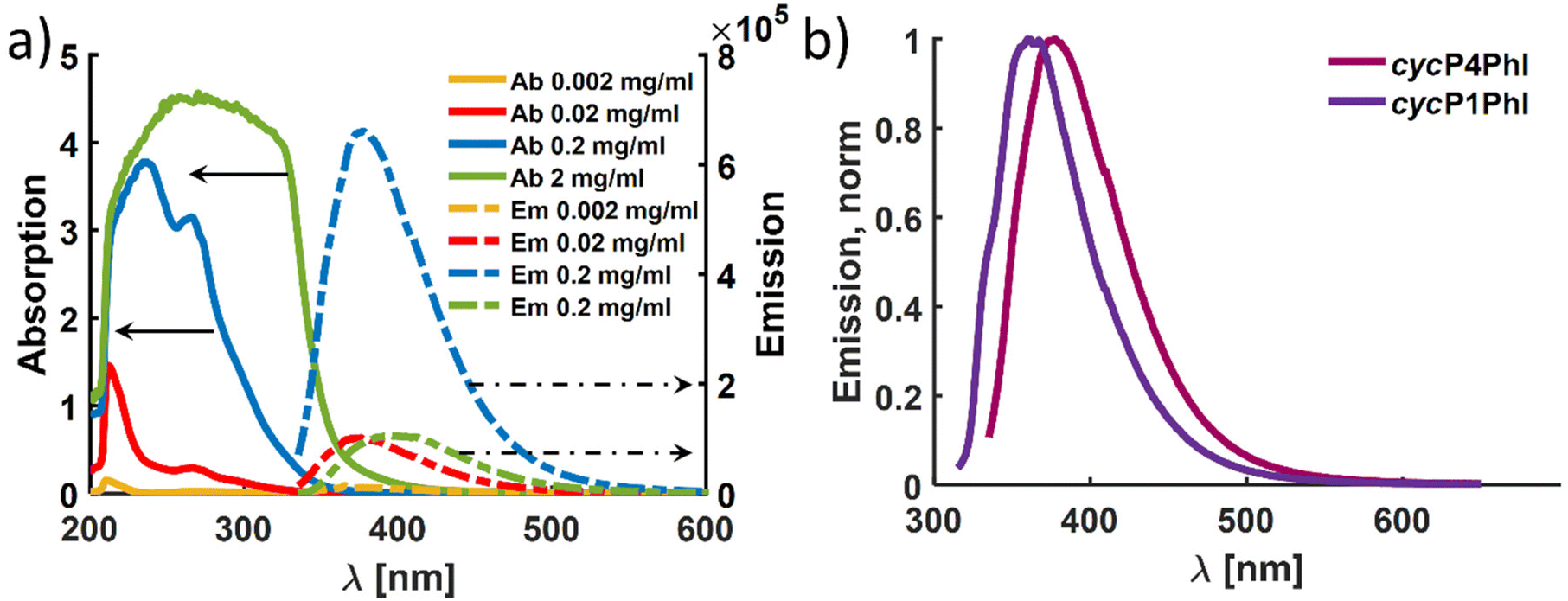 | ||
| Fig. 10 (a) Absorption (solid) and emission (dashed) spectra of cyclized P4PhI at different concentrations in toluene (λexc = 330 nm) and (b) normalized emission spectra of cycP1PhI and cycP4PhI in toluene (c = 0.2 mg mL−1, λexc,cycP1PhI = 310 nm and λexc,cycP4PhI = 330 nm). | ||
Experimental
Terminology
(E)-(2-Methylbuta-1,3-dien-1-yl)benzene and (E)-(3-methylbuta-1,3-dien-1-yl)benzene are abbreviated as 1PhI and 4PhI, respectively.Instrumentation
SEC measurements were performed using an Agilent 1100 Series system equipped with a SDV column set from PSS (SDV 103, SDV 105, and SDV 106). Tetrahydrofuran (THF) was used as the mobile phase (flow rate: 1 mL min−1) and as the solvent. The calibration standards polystyrene and polyisoprene were provided by PSS. Measurements were performed at 30 °C with both RI and UV (275 nm) detectors and toluene was used as a reference. The data analysis was carried out using PSS WinGPC UniChrom (V 8.31, Build 8417) software provided by PSS Polymer Standards Service GmbH. NMR spectra were recorded on a Bruker Avance 400 spectrometer at 400 MHz for 1H NMR and 103 MHz for 13C NMR. The determination of glass transition temperatures (Tg) was performed on a DSC 250 (TA Instruments) differential scanning calorimeter. Two heating cycles and one cooling cycle were conducted at a rate of 10 °C min−1. A detailed description of the DSC measurements and the description of the photochemical measurements are given in the ESI.†Monomer and polymer synthesis
Both novel monomers 1PhI and 4PhI were synthesized in a one-step Wittig reaction. Alterations to published procedures are given in the ESI† together with a detailed description of the polymerization conditions.Real-time 1H NMR
The measurements were conducted on a 400 MHz Bruker Avance spectrometer. All spectra are referenced internally to the residual proton signal of deuterated cyclohexane-d12. The polymerization mixtures were prepared in an argon-filled glove box. Monomers and solvents were purified over calcium hydride and trioctyl aluminum in the case of the monomers. The measurements were performed in a conventional NMR tube sealed with a rubber septum.Prior to initiation, a first spectrum was recorded and equilibrated to a temperature of 25 °C. Following the initiation using 30 μL of sec-butyl lithium (0.65 M in cyclohexane), the NMR experiment was started, in which every 30 s, a scan was performed over a period of 6 to 7 hours. By tracking the decrease of the respective monomer signals, determination of the reactivity ratios was achieved using NIREVAL software designed by our group.58
Cyclization
The cyclization reaction was performed according to the literature. Into a solution of 200 mg P(4PhI) dissolved in 25 mL cyclohexane, trifluorosulfonic acid (0.2 mL, 2 eq. per double bond in every repeating unit) was added. The dark solution was quenched after 1 hour using a 1 wt% aqueous sodium carbonate solution. After washing with water, the polymer was precipitated in methanol and dried under vacuum.Conclusion
In this work, the anionic polymerization of two isoprene derivatives, i.e., 1-phenyl isoprene and 4-phenyl isoprene, was introduced. 4PhI was investigated in more detail than in earlier reports, and 1PhI has not been described to date. The difference in the substitution pattern of 1,3-dienes was expected to result in different reactivities, as already indicated by their β-C-shifts and by DFT calculations. Butyl lithium-initiated polymerizations allowed for the synthesis of polymers with narrow molar mass distributions and good control over the molecular weights in a range of 4.6 to 48.8 kg mol−1. 1H NMR spectroscopy revealed the expected differences in the microstructures. This was further modified using THF, leading to unusual behaviour caused by the attached phenyl ring. Online NMR kinetics uncovered a preference for the phenyl-substituted isoprene over styrene, despite the initial theoretical results predicting reduced reactivity of 1PhI. Nevertheless, these predictions are in line with the data obtained from kinetics with isoprene, which showed random incorporation for 1PhI/I. DSC measurements of the respective compositions further revealed merely one glass transition. In a post-polymerization cyclization reaction, soluble materials with distinct photophysical properties were obtained. The characterization revealed an apparent dependency of the fluorescence maximum on the different microstructures of the starting materials due to self-absorption. As those materials also present a pronounced increase in rigidity, as indicated by the drastic increase of the glass transition temperature, they could be considered intriguing building blocks for block copolymers with an intramolecularly crosslinked high Tg block.Author contributions
Moritz Rauschenbach: conceptualization, data curation, investigation, methodology, validation, visualization, and writing. Gregor Linden, Ramona Barent, and Laura Stein: investigation, data curation, and methodology. Holger Frey: conceptualization, supervision, and writing – review and editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge A. H. E. Müller for valuable discussions and for critically reviewing the results of the online NMR kinetics. We thank the Deutsche Forschungsgemeinschaft DFG for funding via INST 247/1018-1 FUGG to Katja Heinze.References
- M. Szwarc, Nature, 1956, 178, 1168–1169 CrossRef CAS.
- A. Hirao, R. Goseki and T. Ishizone, Macromolecules, 2014, 47, 1883–1905 CrossRef CAS.
- N. Hadjichristidis and A. Hirao, Anionic Polymerization, Springer, Tokio, 2013 Search PubMed.
- F. S. Bates, M. A. Hillmyer, T. P. Lodge, C. M. Bates, K. T. Delaney and G. H. Fredrickson, Science, 2012, 336, 434–440 CrossRef CAS PubMed.
- K. Ntetsikas, V. Ladelta, S. Bhaumik and N. Hadjichristidis, ACS Polym. Au, 2022, 3, 158–181 CrossRef.
- W. Wang, W. Lu, A. Goodwin, H. Wang, P. Yin, N. G. Kang, K. Hong and J. W. Mays, Prog. Polym. Sci., 2019, 95, 1–31 CrossRef CAS.
- G. Holden and R. Milkovich, US Patent 3265765, 1966.
- M. Steube, T. Johann, H. Hübner, M. Koch, T. Dinh, M. Gallei, G. Floudas, H. Frey and A. H. E. Müller, Macromolecules, 2020, 53, 5512–5527 CrossRef CAS.
- D. A. H. Fuchs, H. Hübner, T. Kraus, B. J. Niebuur, M. Gallei, H. Frey and A. H. E. Müller, Polym. Chem., 2021, 12, 4632–4642 RSC.
- L. Shaw and L. R. Hutchings, Polym. Chem., 2020, 11, 7020–7025 RSC.
- A. H. E. Müller and K. Matyjaszewski, Controlled and Living Polymerizations: From Mechanisms to Applications, Wiley-VCH Verlag, Weinheim, 2010 Search PubMed.
- T. Suzuki, Y. Tsuji and Y. Takegami, Macromolecules, 1978, 11, 639 CrossRef CAS.
- T. Suzuki, Y. Tsuji, Y. Takegami and H. J. Harwood, Macromolecules, 1979, 12, 234 CrossRef CAS.
- S. Pragliola, M. Cipriano, A. C. Boccia and P. Longo, Macromol. Rapid Commun., 2002, 23, 356–361 CrossRef CAS.
- J. Lin, F. Wang, C. Zhang, H. Liu, D. Li and X. Zhang, RSC Adv., 2021, 11, 23184–23191 RSC.
- Y. Jiang, X. Kang, Z. Zhang, S. Li and D. Cui, ACS Catal., 2020, 10, 5223–5229 CrossRef CAS.
- H. Bai, L. Han, W. Li, C. Li, S. Zhang, X. Wang, Y. Yin, H. Yan and H. Ma, Macromolecules, 2021, 54, 1183–1191 CrossRef CAS.
- Y. Qi, Z. Liu, S. Liu, L. Cui, Q. Dai, J. He, W. Dong and C. Bai, Catalysts, 2019, 9, 97 CrossRef.
- T. Suzuki, Y. Tsuji, Y. Watanabe and Y. Takegami, Macromolecules, 1980, 13, 849–852 CrossRef CAS.
- Y. Tsuji, T. Suzuki, Y. Watanabe and Y. Takegami, Macromolecules, 1981, 14, 1194–1196 CrossRef CAS.
- T. Suzuki, Y. Tsuji, Y. Watanabe and Y. Takegami, Polym. J., 1979, 11, 651–660 CrossRef CAS.
- Y. Tsuji, T. Suzuki, Y. Watanabe and Y. Takegami, Polym. J., 1981, 13, 1099–1110 CrossRef CAS.
- J. M. Widmaier and G. C. Meyer, Macromolecules, 1981, 14, 450–452 CrossRef CAS.
- Y. Cai, J. Lu, D. Zuo, S. Li, D. Cui, B. Han and W. Yang, Macromol. Rapid Commun., 2018, 39, 1800298 CrossRef.
- R. K. Agnihotri, D. Falcon and E. C. Fredericks, J. Polym. Sci., Part A-1: Polym. Chem., 1972, 10, 1839–1850 CrossRef CAS.
- A. Priola, M. Bruzzone, F. Mistrali and S. Cesca, Angew. Makromol. Chem., 1980, 88, 1–19 CrossRef CAS.
- R. Y. Asami, K.-I. Hasegawa and T. Onoe, Polym. J, 1976, 8, 43–52 CrossRef CAS.
- J. Lal, Polymer, 1998, 39, 6183–6186 CrossRef CAS.
- Y. Cai, J. Lu, G. Jing, W. Yang and B. Han, Macromolecules, 2017, 50, 7498–7508 CrossRef CAS.
- J. Li and J. He, ACS Macro Lett., 2015, 4, 372–376 CrossRef CAS PubMed.
- Q. Lv, C. K. Yu, Q. Yin, J. Lu and B. Han, J. Macromol. Sci., Part A: Pure Appl.Chem, 2020, 57, 388–397 CrossRef CAS.
- T. Ishizone, A. Hirao and S. Nakahama, Macromolecules, 1993, 26, 6964–6975 CrossRef CAS.
- H. Hsieh and R. Quirk, Anionic polymerization: principles and practical applications, Dekker, New York, 1996 Search PubMed.
- S. Uchida, K. Togii, S. Miyai, R. Goseki and T. Ishizone, Macromolecules, 2020, 53, 10107–10116 CrossRef CAS.
- A. Forens, K. Roos, C. Dire, B. Gadenne and S. Carlotti, Polymer, 2018, 153, 103–122 CrossRef CAS.
- T. A. Antkowiak, A. E. Oberster, A. F. Halasa and D. P. Tate, J. Polym. Sci., Part A-1: Polym. Chem., 1972, 10, 1319–1334 CrossRef CAS.
- W. Gebert, J. Hinz and H. Sinn, Makromol. Chem., 1971, 144, 97–115 CrossRef CAS.
- D. J. Worsfold and S. Bywater, Macromolecules, 1978, 11, 582–586 CrossRef CAS.
- H. Hsieh, D. J. Kelley and A. V. Tobolsky, J. Polym. Sci., 1957, 26, 240–242 CrossRef CAS.
- C. A. Uraneck, J. Polym. Sci., Part A-1: Polym. Chem., 1971, 9, 2273–2281 CrossRef CAS.
- S. Bywater and D. J. Worsfold, Can. J. Chem., 1962, 40, 1564–1570 CrossRef CAS.
- M. Morton and L. J. Fetters, J. Polym. Sci., Part A: Gen. Pap., 1964, 2, 3311–3326 CrossRef.
- J. Bareuther, M. Plank, B. Kuttich, T. Kraus, H. Frey and M. Gallei, Macromol. Rapid Commun., 2021, 42, 2000513 CrossRef CAS PubMed.
- A. Garton, R. P. Chaplint and S. Bywater, Eur. Polym. J., 1976, 12, 697–700 CrossRef CAS.
- J. Kleinheider, T. Schrimpf, R. Scheel, T. Mairath, A. Hermann, K. Knepper and C. Strohmann, Chem. – Eur. J., 2024, 30, e202304226 CrossRef CAS PubMed.
- A. Natalello, M. Werre, A. Alkan and H. Frey, Macromolecules, 2013, 46, 8467–8471 CrossRef CAS.
- T. Johann, D. Leibig, E. Grune, A. H. E. Müller and H. Frey, Macromolecules, 2019, 52, 4545–4554 CrossRef CAS.
- V. Jaacks, Makromol. Chem., 1972, 161, 161–172 CrossRef CAS.
- S. P. Wadgaonkar, M. Wagner, L. A. Baptista, R. Cortes-Huerto, H. Frey and A. H. E. Müller, Macromolecules, 2023, 56, 664–677 CrossRef CAS.
- C. Hahn, M. Rauschenbach and H. Frey, Angew. Chem., Int. Ed., 2023, 62, e202302907 CrossRef CAS.
- E. Grune, T. Johann, M. Appold, C. Wahlen, J. Blankenburg, D. Leibig, A. H. E. Müller, M. Gallei and H. Frey, Macromolecules, 2018, 51, 3527–3537 CrossRef CAS.
- E. Grune, J. Bareuther, J. Blankenburg, M. Appold, L. Shaw, A. H. E. Müller, G. Floudas, L. R. Hutchings, M. Gallei and H. Frey, Polym. Chem., 2019, 10, 1213–1220 RSC.
- C. Wahlen, J. Blankenburg, P. Von Tiedemann, J. Ewald, P. Sajkiewicz, A. H. E. Müller, G. Floudas and H. Frey, Macromolecules, 2020, 53, 10397–10408 CrossRef CAS.
- K. Liu, F. Zhang, M. Sun, F. Xie, S. Ying, Z. Yang, C. Zhou, J. Xia, A. Li, K. Liu, F. Zhang, M. Sun, F. Xie, S. Ying, Z. Yang, C. Zhou, J. Xia and A. Li, Macromol. Chem. Phys., 2020, 221, 2000161 CrossRef CAS.
- A. Nakahara, K. Satoh and M. Kamigaito, Macromolecules, 2009, 42, 620–625 CrossRef CAS.
- A. Nakahara, K. Satoh, H. Saito and M. Kamigaito, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1298–1307 CrossRef CAS.
- H. Zhang, Z. Zhao, P. R. McGonigal, R. Ye, S. Liu, J. W. Y. Lam, R. T. K. Kwok, W. Z. Yuan, J. Xie, A. L. Rogach and B. Z. Tang, Mater. Today, 2020, 32, 275–292 CrossRef CAS.
- M. Steube, T. Johann, M. Plank, S. Tjaberings, A. H. Gröschel, M. Gallei, H. Frey and A. H. E. Müller, Macromolecules, 2019, 52, 9299–9310 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1998, 83, 735 CrossRef.
- T. Y. Nikolaienko, L. A. Bulavin and D. M. Hovorun, Comput. Theor. Chem., 2014, 1050, 15–22 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00601a |
| This journal is © The Royal Society of Chemistry 2024 |