
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual aggregation-induced emission enhancement (AIEE) and crosslink-enhanced emission (CEE) driven via halogen-bond-assisted cocrystallization and radical solid-phase polymerization†
Hong Tho
Le
 ,
Chelsea Violita
Stanley
and
Atsushi
Goto
*
,
Chelsea Violita
Stanley
and
Atsushi
Goto
*
School of Chemistry, Chemical Engineering and Biotechnology, Nanyang Technological University, 62 Nanyang Drive, 637459 Singapore, Singapore. E-mail: agoto@ntu.edu.sg
First published on 2nd July 2024
Abstract
Halogen bonding (XB) was used to drive aggregation-induced emission enhancement (AIEE) and crosslink-enhanced emission (CEE) in a concurrent manner. Weak luminophores and vinyl monomers were cocrystallized via XB to drive AIEE, the obtained monomer cocrystal solids were subsequently polymerized via free-radical solid-phase polymerization (SPP) to drive CEE. Weak luminophores containing bromine (Br) and vinyl monomers containing nitrogen (N) or oxygen (O) were combined to form XB-based monomer cocrystals (Br⋯N and Br⋯O bonds), which exhibited AIEE, and the subsequent polymerization of the obtained cocrystals enabled the weak luminophores to be incorporated into the polymer matrix. The resultant restriction of the vibrational and rotational motions of the luminophores led to CEE. The obtained luminophore-embedded emissive sheets exhibited stimuli-responsiveness to temperatures, pH, and solvents, and served as stimuli-responsive emissive polymers. The sheets also served as host–guest interactive materials.
Introduction
Aggregation-induced emission enhancement (AIEE) is an emission enhancement of non-luminous or weakly luminous molecules in their aggregation state.1–6 The aggregation can be driven by covalent bonds or noncovalent bonds such as π–π stacking, coordination, and hydrogen bonding.6–12 Among a number of factors, a main driving force of AIEE is the restriction of intramolecular motion in the aggregated state, preventing thermal decay and enhancing emission. Crosslink-enhanced emission (CEE) is a sub-category of AIEE and a recently proposed emission-enhancement mechanism related to polymers.13–15 Polymers are crosslinked using crosslinkers via covalent or noncovalent bonds, and the resultant restriction in the vibration and rotation of the polymer segment prevents thermal decay and enables polymer segments and crosslinkers to be more luminous.Halogen bonding (XB) is a noncovalent bond between electron-accepting halogens such as bromine (Br) and iodine (I) and electron-donating species such as nitrogen (N)- and oxygen (O)-containing molecules. XB has widely been used in supramolecular engineering and crystal engineering owing to its unique linear bonding angle (almost 180°).15–23 The use of XB in both AIEE and CEE has not yet extensively been studied, but some interesting studies have been reported. For example, Kim et al.24 studied the crystallization of a weakly luminous small molecule, i.e., 2,5-dihexyloxy-4-bromobenzaldehyde (Br6B) via XB, clearly demonstrating AIEE via XB in 2011. Separately, Kim et al. also reported the first CEE of luminophores embedded in polymers via XB in 2013.25 Br6B was embedded in amorphous glassy poly(methyl methacrylate) (PMMA) and other O-containing polymer matrices, resulting in enhanced emission (CEE) of Br6B via the XB (Br⋯O bond) of Br6B and the polymers.24,25
Herein, we describe the first dual use of XB to drive both AIEE and CEE. XB was used to suppress the motion of luminophores in monomer and polymer in solid phase. We used XB to cocrystallize weak luminophores and vinyl monomers to drive AIEE (Fig. 1A) and subsequently polymerize the obtained cocrystals via free-radical solid-phase polymerization (SPP) to drive CEE (Fig. 1B). We used Br-containing weak luminophores (1–5) as XB linkers and N- and O-containing vinyl monomers (6 and 7) (Fig. 1C). The weak luminophores and vinyl monomers were cocrystallized via XB (Br⋯N and Br⋯O bonds), restricting vibrational and rotational motions of the weak luminophores and hence enhancing the emission (AIEE) (Fig. 1A (step 1)). The obtained cocrystals were subsequently polymerized to form crosslinked structures, where polymers were noncovalently crosslinked by the luminophores (linkers) via XB (Fig. 1B (step 2)). As a result, the luminophores are tightly embedded in the polymer matrix, further restricting vibrational and rotational motions of the luminophores to enhance the emission (noncovalent CEE). We also used divinyl monomers to covalently crosslink the polymer matrix (Fig. 1B (step 3)), leading to even larger emission enhancement (covalent CEE). As applications, we modulated the emission intensity in response to temperature, pH, and solvents, demonstrating stimuli-responsive emissions. Stimuli-responsive emissive materials are useful for, e.g., anti-counterfeiting materials and luminescent sensors.26,27 We further studied sequential removal of the luminophores from the polymer matrix and embedment of other luminophores into the polymer matrix, demonstrating reversible host–guest interactions (using polymer matrixes as hosts and luminophores as guests).
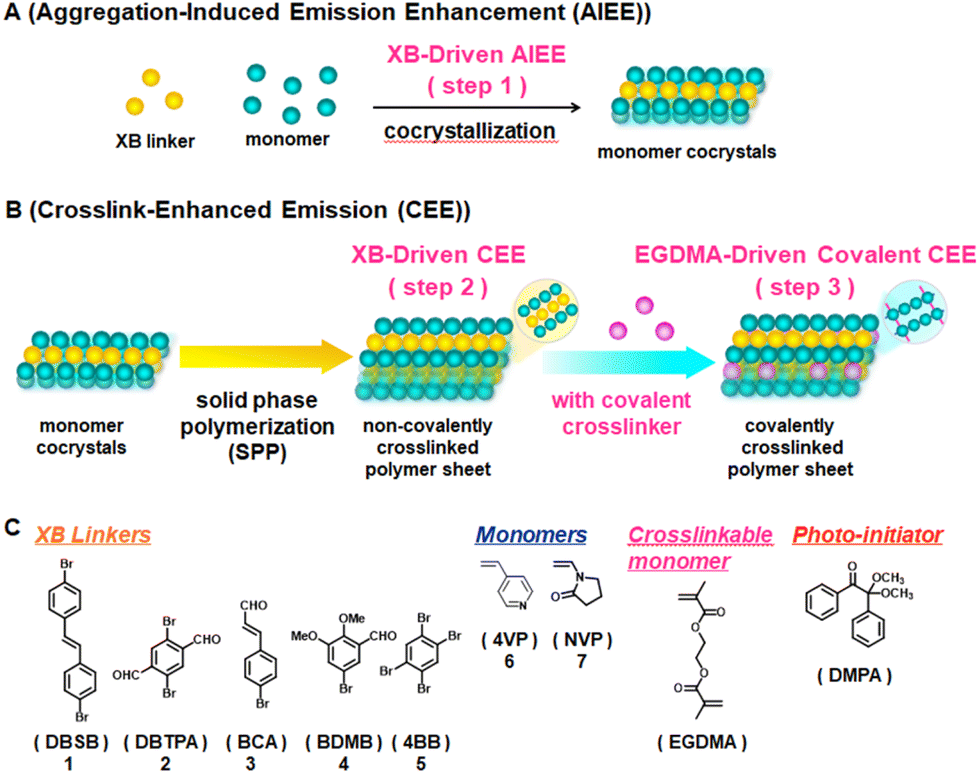 | ||
| Fig. 1 Schematic illustrations of (A) XB-induced AIEE, (B) XB-driven CEE and EGDMA-driven covalent CEE, and (C) structures of XB linkers, monomers, crosslinkable monomer, and photo-initiator used in this work. | ||
We previously synthesized a porous polymer material via SPP28–30 and studied one example of a host–guest interaction between the porous polymer material (host) and an external luminescent molecule (guest).30 However, we did not study AIEE or CEE in the previous work,30 since the external molecule was originally luminescent, and the luminescent mechanism was a single molecular luminescence (not AIEE or CEE). In the present work, we for the first time study AIEE and CEE via dual use of XB.
Results and discussion
Preparations of XB-based monomer cocrystal solids and their free-radical SPPs
We studied five XB linkers (weak luminophores), i.e., trans-4,4′-dibromostilbene (DBSB, 1), 2,5-dibromoterephthalaldehyde (DBTPA, 2), trans-4-bromocinnamaldehyde (BCA, 3), 5-bromo-2,3-dimethoxybenzaldehyde (BDMB, 4), and 1,2,4,5-tetrabromobenzene (4BB, 5), and two vinyl monomers (non-luminophores), i.e., 4-vinylpyridine (4VP, 6) and N-vinylpyrrolidone (NVP, 7) (Fig. 1C). The Br atom in the linker and the N or O atom in the monomer can form XB. We dissolved a linker (1 eq.), a monomer (1–4 eq.), a crosslinkable monomer (ethylene glycol dimethacrylate (EGDMA, Fig. 1C)) (0.4–1.6 eq. with a constant EGDMA/monomer ratio of 0.4), and a photo-initiator (2,2-dimethoxy-2-phenyl-acetophenone (DMPA, Fig. 1C)) (0.03 eq.) in dichloromethane (solvent). We subsequently evaporated dichloromethane and obtained four-component cocrystal solids in gram scales. The Br/monomer molar ratio was set to 1/1 for the stoichiometric XB formation, and hence the linker/monomer molar ratio was 1/1 to 1/4 depending on the number of Br in the linker. EGDMA can covalently crosslink the polymer (or the linker/polymer grain) and retain the resultant polymer shapes. DMPA can initiate the polymerization upon photoirradiation. The Raman spectra of the pure linker 5 and the linker/monomer cocrystal (5·6) of 5 and 6 showed a slight peak shift for the first C–Br stretch (from 221.2 cm−1 to 220.5 cm−1 (Raman)) after the cocrystallization (Fig. S5 in ESI†).21,31 Most of the other monomer cocrystals also showed slight peak shifts compared to the pure linkers 1–5 (except for 2·6 and 2·7) (Fig. S1–S5†), suggesting the formation of XB, although the peak shifts were slight and are not able to unequivocally prove the formation of XB.The obtained four-component solids were polycrystalline powders. Single cocrystals were not obtained probably because the linkers formed XB with not only monomer 6 or 7 (Br⋯N for 6 or Br⋯O for 7) but also EGDMA (Br⋯O) to some extents, which would disturb the long-range linker-monomer alignments. Thus, we were not able to analyze the molecular lattices using single X-ray crystallography. We instead analyzed the solids using powder X-ray diffraction (PXRD) (Fig. 2, S6, S7 and Table S2†). Compared to the PXRD peaks of the pure linkers, the four-component solids showed several new and shifted PXRD peaks, which would indicate the formation of linker/monomer cocrystallized grains via XB.
 | ||
| Fig. 2 Experimental PXRD patterns of pure linker 5 (orange), four-component monomer cocrystal solid (5·6) (blue), and polymer P(5·6) (green) obtained from free-radical SPP of the four-component monomer cocrystal solid 5·6, and their calculated PXRD patterns (grey) (overlapped with experimental PXRD patterns). | ||
We pressed the obtained solids using a hydraulic press to form sheets and conducted photo-initiated free-radical SPP of the sheets under UV-irradiation (λ = 365 nm) for 24 h in an argon atmosphere. After the SPP, we obtained the polymer sheets of poly(4-vinyl pyridine) (P4VP) from 6 and poly(N-vinylpyrrolidone) (PNVP) from 7, which were rigidified by the XB linker networks. Because of the use of the crosslinkable EGDMA, the obtained polymers were covalently crosslinked, retaining the sheet shapes even upon immersion in solvents. Because of the covalent crosslinking, we were not able to characterize the molecular weights of the obtained polymers. To obtain some information on the molecular weights of the primary chains, we conducted free-radical SPPs of three-component solids containing only monomers, linkers, and DMPA but without containing the crosslinkable EGDMA for the ten combinations (1·6, 2·6, 3·6, 4·6, 5·6, 1·7, 2·7, 3·7, 4·7, and 5·7), giving non-covalently crosslinked polymers. After 24 h of the SPP, the monomer conversions were 88–100%, and the peak-top molecular weights (Mp) of the non-crosslinked polymers determined by gel permeation chromatography (GPC) were 2100–93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (Table S1†). The monomer cocrystals (5·6 and 5·7) using linker 5 led to the high monomer conversions (99% and 97%, respectively) and the largest Mp values (93000 and 42000, respectively) of all ten combinations (Table S1†). Although the exact molecular weights of the primary polymer chains might be different between the four-component and three-component systems, the result suggests that the molecular weights of the primary polymer chains would be the largest for 5·6 and 5·7 even in the crosslinked four-component system.
000 (Table S1†). The monomer cocrystals (5·6 and 5·7) using linker 5 led to the high monomer conversions (99% and 97%, respectively) and the largest Mp values (93000 and 42000, respectively) of all ten combinations (Table S1†). Although the exact molecular weights of the primary polymer chains might be different between the four-component and three-component systems, the result suggests that the molecular weights of the primary polymer chains would be the largest for 5·6 and 5·7 even in the crosslinked four-component system.
We also studied the covalently crosslinked four-component polymer sheets obtained from 5·6 and 2·7 using PXRD (Fig. 2, S8A and Table S2†). 55% and 58% of the diffraction peaks remained even after the SPP for 5·6 and 2·7, respectively, demonstrating that the linker/monomer cocrystal (grain) structures were relatively largely retained even after the polymerizations. For comparative experiments, we conducted free-radical polymerizations of monomers 6 and 7 with EGDMA and DMPA in the solution phase (in dichloromethane (solvent)) but without using XB linkers, yielding crosslinked polymers. The obtained crosslinked polymers were analyzed with PXRD. Unlike the polymers obtained in the SPP, those obtained in the solution polymerizations gave only broad curves with no particular diffraction peaks (Fig. S8B and S8C†), suggesting amorphous (not polycrystalline) structures. The results suggest that the polymers obtained in the SPP were more aligned via XB.
XB-driven AIEE and CEE
We studied the emission of the pure linkers, four-component monomer solids, three-component non-covalently crosslinked polymer sheets, and four-component covalently crosslinked polymer sheets using photoluminescence (PL) spectroscopy to obtain emission spectra (Fig. 3 and S9–S17†) and confocal microscope (CM) to obtain emission images (Fig. S18†) in the solid state. The emission would consist of not only fluorescence but also phosphorescence. We used excitation wavelengths (λex) of 405 nm and 488 nm for both PL and CM studies. We determined emission intensities over the entire wavelength (λem = 420–585 nm and 503–635 nm) using PL and at specific wavelength (λem = 498 nm and 564 nm) using CM. In the present paper, emission intensities at λex (excitation) = 405 nm and 488 nm are termed I405 (arbitrary unit (a.u.)) and I488 (a.u.), respectively, and listed in Table 1 (PL) and Table S3† (CM). | ||
| Fig. 3 PL spectra with (A) λex = 405 nm and (B) λex = 488 nm for linker 5 (black solid lines), 4-component monomer cocrystal solid (5·6) (red solid lines), 3-component non-covalently crosslinked polymer P(5·6) (blue solid lines), 4-component covalently crosslinked polymer P(5·6) (green solid lines), linker 5 embedded in commercial P4VP (dotted purple lines), and linker 5 embedded in commercial PMMA (dotted orange lines). All the sample mass was 0.10 g with a thickness of 1 mm. | ||
| Entry | Modea | Sampleb | I 405 (a.u.)c | I 488 (a.u.)c | I 405 increasing ratio (times, ×) | I 488 increasing ratio (times, ×) | Entry | Modea | Sampleb | I 405 (a.u.)c | I 488 (a.u.)c | I 405 increasing ratio (times, ×) | I 488 increasing ratio (times, ×) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a 4-Comp monomer solid = four-component monomer solid (step 1), 3-comp polymer sheet = three-component polymer sheet (non-covalently crosslinked system) (step 2), and 4-comp polymer sheet = four-component polymer sheet (covalently crosslinked system) (step 3). b P denotes polymer. c I 405 and I488 values were determined from PL spectra area integrated at 420–585 nm and 503–635 nm, respectively. d Entry 21 = entry 1, entry 25 = entry 5, entry 29 = entry 9, entry 33 = entry 13, and entry 37 = entry 17. | |||||||||||||
| 1 | Pure linker | 1 | 1010 | 157 | 21d | Pure linker | 1 | 1010 | 157 | ||||
| 2 | 4-Comp monomer solid | 1·6 | 26800 |
2020 | ×27 from 1 | ×13 from 1 | 22 | 4-Comp monomer solid | 1·7 | 102000 |
1370 | ×101 from 1 | ×8.7 from 1 |
| 3 | 3-Comp polymer sheet | P(1·6) | 49400 |
3470 | ×1.8 from 1·6 | ×1.7 from 1·6 | 23 | 3-Comp polymer sheet | P(1·7) | 155000 |
5490 | ×1.5 from 1·7 | ×4.0 from 1·7 |
| 4 | 4-Comp polymer sheet | P(1·6) | 310000 |
29800 |
×12 from 1·6 | ×15 from 1·6 | 24 | 4-Comp polymer sheet | P(1·7) | 257000 |
26100 |
×2.5 from 1·7 | ×19 from 1·7 |
| 5 | Pure linker | 2 | 5050 | 368 | 25d | Pure linker | 2 | 5050 | 368 | ||||
| 6 | 4-Comp monomer solid | 2·6 | 18800 |
5280 | ×3.7 from 2 | ×14 from 2 | 26 | 4-Comp monomer solid | 2·7 | 33000 |
5510 | ×6.5 from 2 | ×15 from 2 |
| 7 | 3-Comp polymer sheet | P(2·6) | 38400 |
7200 | ×2.0 from 2·6 | ×1.4 from 2·6 | 27 | 3-Comp polymer sheet | P(2·7) | 91900 |
33200 |
×2.8 from 2·7 | ×6.0 from 2·7 |
| 8 | 4-Comp polymer sheet | P(2·6) | 150000 |
36100 |
×8.0 from 2·6 | ×6.8 from 2·6 | 28 | 4-Comp polymer sheet | P(2·7) | 134000 |
54000 |
×4.1 from 2·7 | ×9.8 from 2·7 |
| 9 | Pure linker | 3 | 1720 | 1870 | 29d | Pure linker | 3 | 1720 | 1870 | ||||
| 10 | 4-Comp monomer solid | 3·6 | 26100 |
11600 |
×15 from 3 | ×6.2 from 3 | 30 | 4-Comp monomer solid | 3·7 | 61500 |
11100 |
×36 from 3 | ×5.9 from 3 |
| 11 | 3-Comp polymer sheet | P(3·6) | 76500 |
11800 |
×2.9 from 3·6 | ×1.02 from 3·6 | 31 | 3-Comp polymer sheet | P(3·7) | 135000 |
71800 |
×2.2 from 3·7 | ×6.5 from 3·7 |
| 12 | 4-Comp polymer sheet | P(3·6) | 164000 |
31000 |
×6.3 from 3·6 | ×2.7 from 3·6 | 32 | 4-Comp polymer sheet | P(3·7) | 165000 |
99500 |
×2.7 from 3·7 | ×9.0 from 3·7 |
| 13 | Pure linker | 4 | 427 | 4.70 | 33d | Pure linker | 4 | 427 | 4.70 | ||||
| 14 | 4-Comp monomer solid | 4·6 | 1270 | 465 | ×3.0 from 4 | ×99 from 4 | 34 | 4-Comp monomer solid | 4·7 | 10900 |
2020 | ×26 from 4 | ×430 from 4 |
| 15 | 3-Comp polymer sheet | P(4·6) | 5800 | 663 | ×4.6 from 4·6 | ×1.4 from 4·6 | 35 | 3-Comp polymer sheet | P(4·7) | 30700 |
8950 | ×2.8 from 4·7 | ×4.4 from 4·7 |
| 16 | 4-Comp polymer sheet | P(4·6) | 16100 |
5360 | ×13 from 4·6 | ×12 from 4·6 | 36 | 4-Comp polymer sheet | P(4·7) | 88500 |
25000 |
×8.1 from 4·7 | ×12 from 4·7 |
| 17 | Pure linker | 5 | 358 | 0.152 | 37d | Pure linker | 5 | 358 | 0.152 | ||||
| 18 | 4-Comp monomer solid | 5·6 | 797 | 14.4 | ×2.2 from 5 | ×95 from 5 | 38 | 4-Comp monomer solid | 5·7 | 1190 | 31.7 | ×3.3 from 5 | ×210 from 5 |
| 19 | 3-Comp polymer sheet | P(5·6) | 3220 | 90.9 | ×4.0 from 5·6 | ×6.3 from 5·6 | 39 | 3-Comp polymer sheet | P(5·7) | 2020 | 356 | ×1.7 from 5·7 | ×11 from 5·7 |
| 20 | 4-Comp polymer sheet | P(5·6) | 3430 | 194 | ×4.3 from 5·6 | ×13 from 5·6 | 40 | 4-Comp polymer sheet | P(5·7) | 4030 | 1640 | ×3.4 from 5·7 | ×52 from 5·7 |
The PL study showed that, among the five pure linkers, pure linker 5 was the weakest luminophore with I405 = 358 and I488 = 0.152 (Table 1, entries 17 and 37 and Fig. 3 (black solid lines)). After the cocrystallization, the four-component monomer solid 5·6 became more emissive with I405 = 797 and I488 = 14.4 (Table 1, entry 18 and Fig. 3 (red solid lines)). The emission increased 2.2 and 95 times, respectively, because of the rigid alignment of linker 5 with monomer 6 in the cocrystal lattice, demonstrating XB-driven AIEE (Fig. 1A (step 1)). After the polymerization, the three-component non-covalently crosslinked polymer sheet gave stronger emission with I405 = 3220 and I488 = 90.9 (4.0 and 6.3 times increase from the monomer cocrystal), and the four-component covalently crosslinked polymer sheet gave even stronger emission I405 = 3430 and I488 = 194 and (4.3 and 13 times increase from the monomer cocrystal) (Table 1, entries 19 and 20, and Fig. 3 (blue and green solid lines)), because of the further restricted vibration and rotation of linker 5 through the polymerization. The results clearly demonstrate XB-driven CEE (noncovalent CEE) (Fig. 1B (step 2)) and further EGDMA-driven CEE (covalent CEE) (Fig. 1B (step 3)) both via SPP.
Such three-step emission increase was also observed for the other nine combinations (1·6, 2·6, 3·6, 4·6, 1·7, 2·7, 3·7, 4·7, and 5·7) by factors of 3.0–430 at step 1 (AIEE), 1.02–11 at step 2 (non-covalent CEE), and 2.5–52 at step 3 (covalent CEE) in the PL study (Table 1), suggesting a wide scope of linkers and monomers for the AIEE and CEE driven by XB and SPP. The emission enhancement would depend on various factors such as the number of XB per linker and the strength of XB. Of the studied ten combinations (1·6, 2·6, 3·6, 4·6, 5·6, 1·7, 2·7, 3·7, 4·7, and 5·7), 5·7 (Table 1, entries 38–40) showed the second largest emission enhancement (210 times) at step 1 (AIEE), the largest emission enhancement (11 times) at step 2 (non-covalent CEE), and the largest emission enhancement (52 times) at step 3 (covalent CEE), despite the originally weakest emission of pure linker 5. 5 bears more Br atoms (four Br atoms) than 1–4 (one or two Br atoms). The motion of 5 is restricted by four XB interactions, which would partly explain the observed particularly large emission enhancement.
We also confirmed the three-step increase in the CM study for all ten combinations by factors of 1.4–150 at step 1 (AIEE), 1.4–140 at step 2 (non-covalent CEE), and 1.8–170 at step 3 (covalent CEE) (Table S3†), although emission comparison is slightly more accurate by the PL study because the CM emission intensities are somewhat sensitive to sample roughness. To confirm that the emission enhancement was driven by XB and SPP, we carried out comparison experiments. A dichloromethane (solvent) solution of linker 5 and commercially available poly(methyl methacrylate) (PMMA) (molecular weight = ca. 50000) or P4VP (molecular weight = ca. 60000) was casted and dried to obtain 5 embedded in the polymer matrix (Fig. 3 (orange and purple dotted lines) and Table S4†). Compared to pure linker 5, the I405 and I488 values (Table S4†) increased by factors of 1.2 and 120 for 5 embedded in the PMMA matrix by casting (via CEE without XB), 5 and 570 for 5 embedded in the P4VP matrix by casting (via CEE with XB), and 9 and 600 for 5 embedded in the P4VP matrix by SPP (via CCE with XB via SPP) (non-covalently crosslinked system), respectively. The results mean that both XB and SPP drove emission enhancement because of more molecular alignment and more restriction of the motion of 5.
The emission intensity of a linker would depend on (1) the absorption efficiency (how the HOMO–LUMO gap of the linker is close to the excitation energy (3.06 eV for 405 nm excitation and 2.54 eV for 488 nm excitation)) and (2) the emission efficiency from the excited linker. The latter factor (emission efficiency) should depend on the restriction of the vibrational and rotational motions of the linker via XB and SPP. To probe the former factor (excitation efficiency), we conducted the density functional theory (DFT) calculations using the B3LYP/6-31G(d,p) method. The calculated HOMO–LUMO gaps of pure linkers increased in the order of 1 (3.951 eV) < 2 (4.069 eV) < 3 (4.243 eV) < 4 (4.502 eV) < 5 (5.440 eV) (Fig. S20†). The experimentally observed absolute emission intensities (Table 1, entries 1, 5, 9, 13 and 17) tended to be higher for more conjugated molecules (1–3) than less conjugated molecules (4 and 5), which is consistent with the DFT calculation that the excitation energies (2.54 or 3.06 eV) are closer to the HOMO–LUMO gaps of 1–3 (3.951–4.243 eV) than those of 4 and 5 (4.502–5.440 eV). The 5·6 and 5·7 linker-monomer pairs particularly narrowed the HOMO–LUMO gaps (by 1.093 eV and 0.651 eV, respectively (Fig. S29 and S30†)) because of the four Br atoms (the four XB interactions) in 5. For the 5·6 combination, we studied varied linker/monomer ratios (Fig. 4A–D and S31†). Compared to the pure linker 5, linker-monomer 5·6 pairs at 1/1, 1/2, and 1/4 (linker/monomer) ratios narrowed the HOMO–LUMO gap by 0.483 eV, 0.633 eV, and 1.093 eV, respectively, suggesting that a larger number of XB leads to a narrower HOMO–LUMO gap. We further studied a model polymer of 5·6 at a 1/2 linker/monomer ratio (Fig. 4E and S32†), where three linker molecules are linked to two short polymer chains with six monomer units. Compared to the model monomer cocrystal of 5·6 (at a 1/2 ratio) (Fig. 4C), the model polymer of 5·6 (at a 1/2 ratio) (Fig. 4E) further narrowed the HOMO–LUMO gap by 0.191 eV. Thus, the DFT calculation suggests that the experimentally observed emission enhancement (Table 1) was brought by increased absorption efficiencies by narrowed HOMO–LUMO gaps via XB and SPP as well as the anticipated increased emission efficiency by the restricted motions of the linkers via XB and SPP. The present DFT calculation considers interactions of pair molecules only. Actual interactions should be interactions of multiple molecules in the crystal or crystal grains. The actual HOMO–LUMO gaps would be narrower than those calculated in the present DFT study and would be even closer to the exploited excitation energies.
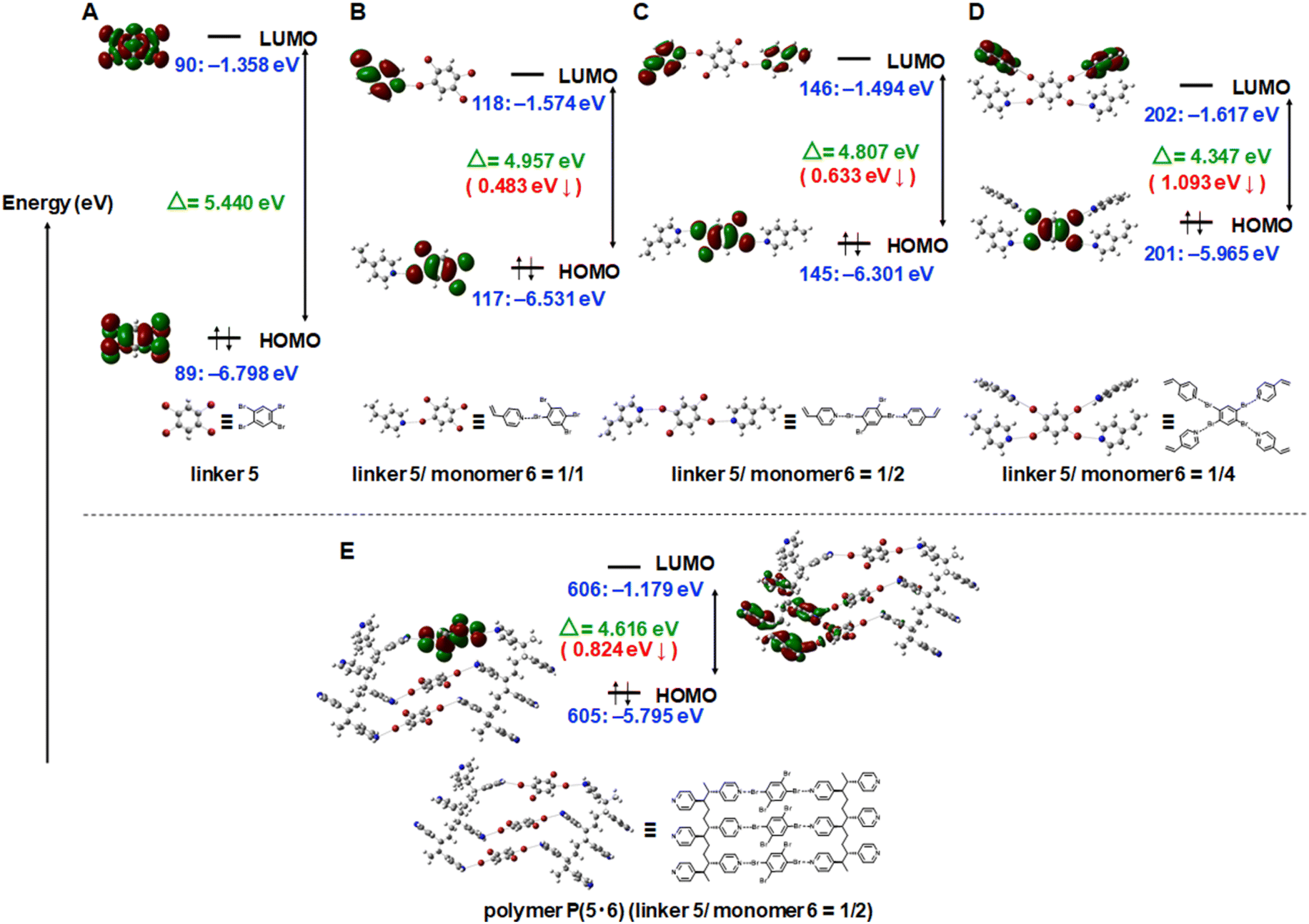 | ||
| Fig. 4 DFT calculations of HOMO and LUMO for (A) pure linker 5, monomer cocrystals 5·6 at the linker/monomer ratios of (B) 1/1, (C) 1/2, and (D) 1/4, and (E) model polymer P(5·6) at the linker/monomer ratio of 1/2, using B3LYP/6-31G(d,p) level of theory. The HOMO–LUMO gap is given in green. The decrease in the HOMO–LUMO gap from (A) to (B–E) is given in red. | ||
Emission-patterned polymer sheets
We focus on the 4-component polymer sheets below. We used photomasks to obtain emission-patterned polymer sheets by modulating the polymer formation (CEE) (Fig. 5 and S33†). We used two different photomasks, i.e., star 40 (Fig. 5A) and circle 50 (Fig. S33A†), where the numbers are the sizes (μm) of the masked star and circle patterns. The photomasks were inserted between the 4-component monomer cocrystal 5·6 sheet (Fig. 5B, C and S33B, S33C†) and the UV light source, allowing only the unmasked areas to be polymerized. After 24 h of polymerization, the polymerized sheets showed emission patterns in the CM images. Using the star 40 photomask (Fig. 5D and E†), we observed weaker emission in the non-polymerized masked star-shaped areas (I405 = 157 and I488 = 8.92) and stronger emission in the polymerized unmasked outer-star areas (I405 > 255 and I488 = 16.7) (Table 2, entry 1). The optical microscope (OM) image (Fig. 5F) showed no clear pattern, meaning that the observed emission patterns in the CM images were ascribed to CEE (formation or no formation of polymers) but not the topological change. Similarly, the circle 50 photomask gave emission patterns (Fig. S33D and S33E†), exhibiting weaker emission in the non-polymerized areas (I405 = 153 and I488 = 8.50) and stronger emission in the polymerized areas (I405 > 255 and I488 = 16.7) (Table 2, entry 2) and showing the versatility in photomasks. The results confirm the increase in the emission by CEE and also illustrate the fabrication of CEE in a patterning manner. Such PL-patterned materials with no visual change in the topology (conditional visibility) and no purification required might be useful for anti-counterfeiting applications, for example.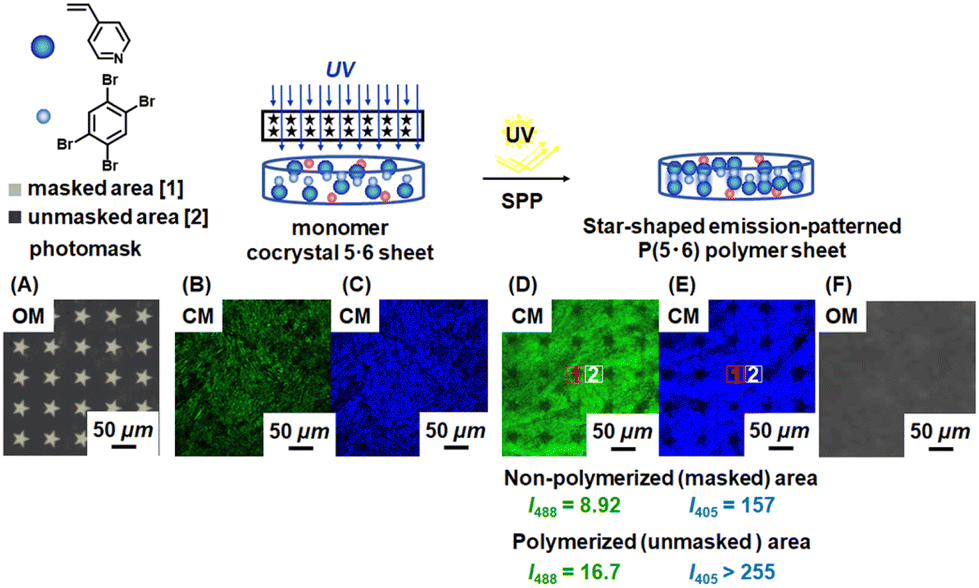 | ||
| Fig. 5 Schematic illustrations of the preparation of emission-patterned polymer sheet, (A) OM image of photomask star 40, CM images at (B) λex = 488 nm and (C) λex = 405 nm of monomer cocrystal (5·6) (before SPP), CM images at (D) λex = 488 nm and (E) λex = 405 nm and (F) OM image of polymer sheet P(5·6) (after SPP). | ||
| Entry | Study | Photomask/treatmenta | Sampleb | I 405 (a.u.)c | I 488 (a.u.)c |
|---|---|---|---|---|---|
| a Photomask for entries 1 and 2 and treatment for other entries. b Polymer was covalently crosslinked for all entries. c I 405 and I488 values were determined from CM emission intensities at 498 nm and 564 nm, respectively at a constant area (10 μm × 10 μm) for all entries. d Not analyzable (>255 a.u.) due to the detection limit of instrument (λex = 405 nm). e Table S3,† entry 20. f After cooling back from 150 °C to 22 °C. g Solution of I–C6F4–I (0.1 M) in dichloromethane. | |||||
| 1 | Patterning | Star 40 | P(5·6) | NAd (unmasked) | 16.7 (unmasked) |
| 157 (masked) | 8.92 (masked) | ||||
| 2 | Circle 50 | NAd (unmasked) | 16.7 (unmasked) | ||
| 153 (masked) | 8.50 (masked) | ||||
| 3 | Temperature-responsiveness | 22 °C (originale) | P(5·6) | NAd | 16.3 |
| 4 | 30 °C | 255 | 12.9 | ||
| 5 | 50 °C | 251 | 11.6 | ||
| 6 | 70 °C | 240 | 11.1 | ||
| 7 | 100 °C | 232 | 10.7 | ||
| 8 | 120 °C | 215 | 9.93 | ||
| 9 | 150 °C | 42.9 | 0.993 | ||
| 10 | 22 °Cf | NAd | 16.3 | ||
| 11 | pH-responsiveness | Originale | P(5·6) | NAd | 16.3 |
| 12 | 2.00 | 252 | 11.4 | ||
| 13 | 7.00 | NAd | 12.8 | ||
| 14 | 11.0 | NAd | 17.2 | ||
| 15 | Solvent-responsiveness | Originale | P(5·6) | NAd | 16.3 |
| 16 | Toluene | 222 | 4.19 | ||
| 17 | DMF | NAd | 4.26 | ||
| 18 | EtOH | NAd | 13.2 | ||
| 19 | I–C6F4–Ig | 232 | 9.11 | ||
| 20 | PhCF3 | NAd | 12.3 | ||
| 21 | CH2Cl2 | NAd | 17.1 | ||
| 22 | Host-guess interaction | Originale | P(5·6) | NAd | 16.3 |
| 23 | DMF (wash) | P4VP | NAd | 4.42 | |
| 24 | Embedment of 5 | P(5·6) | NAd | 16.5 | |
| 25 | DMF (wash) | P4VP | NAd | 4.43 | |
| 26 | Embedment of 2 | P(2·6) | NAd | 40.5 | |
| 27 | DMF (wash) | P4VP | NAd | 4.20 | |
| 28 | Embedment of 4 | P(4·6) | NAd | 37.3 | |
| 29 | DMF (wash) | P4VP | NAd | 4.53 | |
| 30 | Embedment of 3 | P(3·6) | NAd | 55.6 | |
| 31 | DMF (wash) | P4VP | NAd | 4.56 | |
| 32 | Embedment of 1 | P(1·6) | NAd | 48.8 | |
| 33 | DMF (wash) | P4VP | NAd | 4.44 | |
| 34 | Embedment using a mixture of 4 and 5 | NAd | 16.4 | ||
| 35 | Embedment using a mixture of 1–5 | NAd | 16.5 | ||
In this (patterning) and following studies, we used CM (not PL) for imaging and visualization purposes (Fig. 5–7) and thereby discuss I405 and I488 values determined using CM (Table 2). The CM emission intensities are slightly less accurate than the PL emission intensities but would still be valid enough for relative comparison.
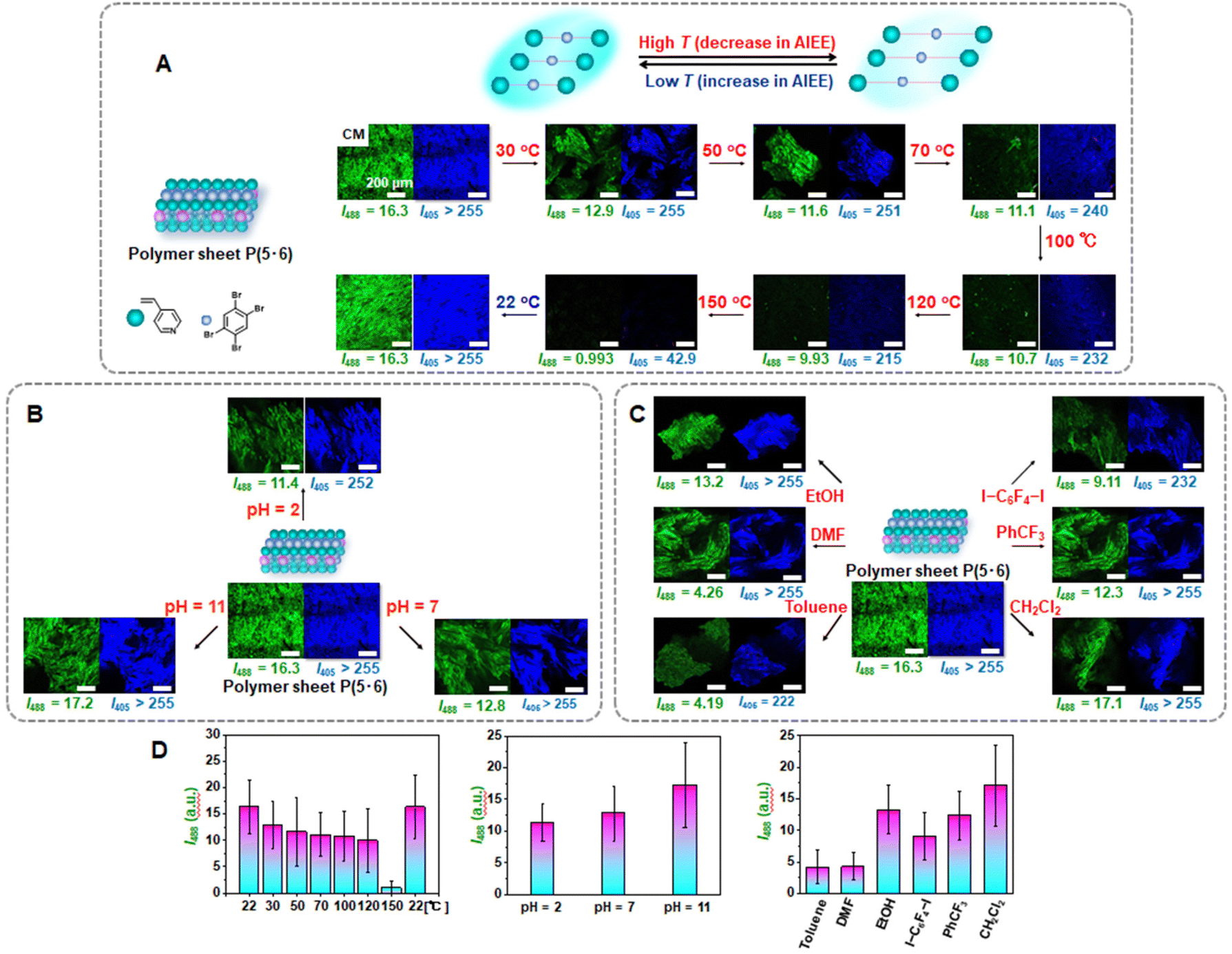 | ||
| Fig. 6 CM images at λex = 488 nm and 405 nm (scale bar: 200 μm) and I405 and I488 values of covalently crosslinked polymer P(5·6) (A) at various temperatures, (B) at various pH, and (C) in various solvents. (D) Bar graphs of I488 values in (A), (B) and (C). | ||
 | ||
| Fig. 7 CM images at λex = 488 nm (scale bar: 200 μm) and I488 values of covalently crosslinked polymer P(5·6) with 14 steps of unloading-loading of different linkers and bar graphs of I488 values in 14 steps. | ||
Stimuli-responsive emissive polymer sheets
We studied stimuli-responsive emission of the 4-component covalently crosslinked polymer sheet P(5·6) obtained from the 4-component monomer cocrystal 5·6. We used the noncovalent (reversible) character of XB. First, we studied temperature responsiveness based on different degrees of XB at different temperatures. In the dry solid state, the polymer sheet P(5·6) was gradually heated from 22 °C (room temperature) to 150 °C with intervals of 20–30 °C and subsequently cooled from 150 °C to 22 °C, as monitored with CM (Fig. 6A, D and Table 2, entries 3–10). Because the I405 value was saturated (>255) in most of the studied conditions, we discuss the I488 value below. The I488 value gradually decayed from 16.3 to 0.993 upon heating from 22 °C to 150 °C and recovered to 16.3 upon cooling to 22 °C.The decay in the emission intensity upon heating would be ascribed to the weaker XB upon heating and thereby caused less restricted motion of 5. The emission recovered upon cooling because of the recovered strong XB and restricted motion of 5. We examined thermal stability of the polymer sheet P(5·6) using thermogravimetric analysis (TGA). The polymer sheet had Td(10%) and Td(50%) values of 163 and 187 °C, respectively, where Td(10%) and Td(50%) are the decomposition temperatures at 10% and 50% weight losses (Fig. S34†). The result indicates that XB broken significantly at approximately 163 °C and that XB was still operative at the studied temperatures ≤150 °C. Thus, we were able to modulate the emission intensity by modulating the degree of XB by temperature.
Second, we studied pH-responsiveness (Fig. 6B and D and Table 2, entries 11–14). The polymer sheets P(5·6) were immersed in different pH-standard aqueous solutions ranging from pH = 2 to pH = 7 to pH = 11. The sheets were then taken out of the solutions, dried with a nitrogen flow, and analyzed with CM in the dry solid state. The polymer (P4VP) contained basic pyridyl groups. At low pH values, the pyridyl groups were protonated (quaternized) and had reduced binding abilities with linker 5, dissociating XB. Therefore, the I488 value dropped from 16.3 before the immersion to 11.4 after the immersion in the acidic (pH = 2) solution. After the immersion in the neutral (pH = 7) solution, the emission intensity (I488 = 12.8) increased compared to that at pH = 2 (I488 = 11.4), because more pyridyl groups were unprotonated and XB recovered. After the immersion in the basic (pH = 11) solution, the I488 value nearly fully recovered to 17.2, because the pyridyl groups are fully unprotonated and accessible to linker 5. Thus, the emission intensity was modulated by modulating the degree of XB by pH.
Third, we studied solvent-responsiveness (Fig. 6C and D and Table 2, entries 15–21). The polymer sheets P(5·6) were immersed in six different solvents containing aromatic ring and N, O, I, F, and Cl atoms, i.e., toluene, N,N-dimethyl formamide (DMF), ethanol (EtOH), 1,4-diiodotetrafluorobenzene (I–C6F4–I, 0.1 M) dissolved in CH2Cl2, trifluoromethylbenzene (PhCF3), and dichloromethane (CH2Cl2). Upon immersion, linker 5 could leak out of the polymer sheets. The polymer sheets were subsequently rinsed with CH2Cl2 to remove the residual linker 5 adsorbed on the surface of the sheets and dried. The emission intensity of the polymer sheet decreased in the order of EtOH > DMF ≥ toluene, meaning that linker 5 leaked more in the order of EtOH < DMF ≤ toluene, possibly because aromatic interaction of toluene with linker 5 and XB of DMF with linker 5 (N⋯Br–C) are stronger than XB of ethanol with linker 5 (O⋯Br–C).32 The I488 values after the immersion in toluene and DMF were 4.19 and 4.26, respectively, which were close to the background intensity, i.e., that (3.60) of the polymer (P4VP) synthesized in a solution phase using photo-initiator DMPA but not using linker 5 (Fig. S35†). Based on the nonluminous character of the polymer (P4VP), this background signal would originate from the photo-initiator fragment covalently attached to the polymer (P4VP) chain end. Therefore, linker 5 nearly fully leaked out of the polymer sheets in DMF and toluene. The emission intensity of the polymer sheet also decreased in the order of CH2Cl2 > PhCF3 > I–C6F4–I, meaning that linker 5 leaked more in the order of CH2Cl2 < PhCF3 < I–C6F4–I, possibly because the aromatic interaction of PhCF3 with linker 5 is stronger than the interaction of CH2Cl2 with linker 5. In the case of I–C6F4–I, linker 5 would be replaced with I–C6F4–I in the polymer sheet and hence leak out of the polymer sheet. The replacement is favorable, because the C–N⋯I bond (P4VP⋯I–C6F4–I) is stronger than the C–N⋯Br bond (P4VP⋯linker 5). The embedment of the non-luminescent I–C6F4–I would result in a large drop in the emission intensity of the polymer sheet. Thus, the polymer sheet showed solvent-dependent emission change.
We further explored host–guest interactions. We studied multiple cycles of loading and unloading of different XB linkers in the polymer sheet P(5·6) (Fig. 7 and Table 2, entries 22–35). The polymer sheet P(5·6) (I488 = 16.3) was first immersed in DMF to completely remove linker 5. The purified polymer sheet (I488 = 4.42) was re-embedded with linker 5 by immersing in a solution of 5, followed by rinsing with CH2Cl2 and drying. The I488 value recovered to 16.5, demonstrating the re-embedding of linker 5. We then studied the unloading-loading cycles using linkers 2, 4, 3, and 1 in a consecutive manner. After the loading, the I488 values changed to 40.5 for 2, 37.3 for 4, 55.6 for 3, and 48.8 for 1, demonstrating the cycled host–guest interactions using different guest molecules (linkers). However, the I488 values after the loading were somewhat lower than those (87.3, 61.3, 221, and 129) of the freshly embedded polymer sheets prepared from monomer/linker cocrystals via SPP (P(2·6), P(4·6), P(3·6), and P(1·6)) (Table S3,† entries 4, 8, 12 and 16). The lower emission intensities of the re-embedded polymer sheets suggest that not all of the voids were refilled owing to steric hindrance and possibly mismatched pore shapes. On the other hand, the I488 value after the re-embedding of linker 5 (16.5) (Table 2, entry 24) was totally recovered as compared with the freshly embeded polymer sheet P(5·6) (16.3) (Table 2, entry 22). We also studied the guest selectivity using mixtures of linkers. After we purified the polymer sheet (unloaded 1 after step 12 (Fig. 7)) (I488 = 4.44 (Table 2, entry 33)), we tested re-embedment of linkers using two mixtures. Mixture 1 consisted of linkers 4 and 5 (step 13) and Mixture 2 consisted of all linkers 1–5 (step 14) (Fig. 7 and Table 2, entries 34 and 35). In the presence of linker 5 in both mixtures, the polymer sheet showed I488 values at 16.4–16.5, which are similar to the original I488 value of the polymer sheet P(5·6) (16.3) but not to the values from the re-embedment of linkers 1–4 (37.3–55.6) (Fig. 7 and Table 2, entries 26, 28, 30 and 32). This indicates that the pore size was precisedly tuned by the linker 5, serving as a selective hosting material. The re-embedded I488 values increased in the order of 5 < 4 < 2 < 1 < 3. This order was consistent with the order (5 < 4 < 2 < 1 < 3) observed for the freshly embedded polymer sheets. The result demonstrates reversible host–guest interactions in a cycled manner owing to the noncovalent nature of XB.
Conclusions
The weak luminophores (linkers) became stronger luminophores via XB-driven cocrystallization with vinyl monomers (AIEE) and became even stronger luminophores via SPP of the obtained cocrystals to form XB-driven crosslinked polymers (noncovalent CEE) and EGDMA-driven crosslinked polymers (covalent CEE). The emission of the linkers was enhanced up to 430 times after the cocrystallization and further enhanced 11 and 52 times after the SPP to form noncovalently and covalently crosslinked polymers, respectively. By modulating CEE (formation of polymers) using photomasks, patterned emissive sheets were obtained. By exploiting changes in the degree (strength) of XB in response to temperature, pH, and solvents, stimuli-responsive emissive polymer sheets were obtained. The polymer sheet was also utilized as a host–guest interactive sheet. Current emissive materials contain metals or require extended conjugated systems obtainable via multi-step synthesis and purification in many cases. Our emissive materials are purely organic, are obtained from readily available linkers and monomers, are synthesized in a facile manner (via cocrystallization followed by polymerization without any purification), and therefore can serve as metal-free and readily accessible emissive materials possibly on large scales.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
H. T. L. performed all experiments; C. V. S. assisted in conceptual investigation and preliminary experiments; H. T. L. and A. G. contributed to the conceptualization, methodology, validation, and writing; A. G. contributed to funding acquisition, project administration, and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Academic Research Fund (AcRF) Tier 2 from Ministry of Education in Singapore (MOE-MOET2EP10121-0005). Authors thank Dr Xinjiang Zhang on the DFT theoretical calculations.References
- R. Hu, A. Qin and B. Z. Tang, Prog. Polym. Sci., 2020, 100, 101176 CrossRef CAS
.
- F. Würthner, Angew. Chem., Int. Ed., 2020, 59, 14192–14196 CrossRef PubMed
- X. Cai and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 9868–9886 CrossRef CAS PubMed
- X. Li, M. Li, M. Yang, H. Xiao, L. Wang, Z. Chen, S. Liu, J. Li, S. Li and T. D. James, Coord. Chem. Rev., 2020, 418, 213358 CrossRef CAS
- S. K. Behera, S. Y. Park and J. Gierschner, Angew. Chem., Int. Ed., 2021, 60, 22624–22638 CrossRef CAS PubMed
- Q. Peng and Z. Shuai, Aggregate, 2021, 2, e91 CrossRef CAS
- J. Ochi, K. Tanaka and Y. Chujo, Angew. Chem., Int. Ed., 2020, 59, 9841–9855 CrossRef CAS PubMed
- N. Yanai and N. Kimizuka, Angew. Chem., Int. Ed., 2020, 59, 10252–10264 CrossRef CAS PubMed
- W. L. Guan, J. F. Chen, J. Liu, B. Shi, H. Yao, Y. M. Zhang, T. B. Wei and Q. Lin, Coord. Chem. Rev., 2024, 507, 215717 CrossRef CAS
- M. Fang, J. Yang and Z. Li, Prog. Mater. Sci., 2022, 125, 100914 CrossRef CAS
- K. S. S. Kumar, Y. R. Girish, M. Ashrafizadeh, S. Mirzaei, K. P. Rakesh, M. H. Gholami, A. Zabolian, K. Hushmandi, G. Orive, F. B. Kadumudi and A. Dolatshahi-Pirouz, Coord. Chem. Rev., 2021, 447, 214135 CrossRef
- S. Xu, Y. Duan and B. Liu, Adv. Mater., 2020, 32, 1903530 CrossRef CAS PubMed
- S. Tao, S. Zhu, T. Feng, C. Zheng and B. Yang, Angew. Chem., Int. Ed., 2020, 59, 9826–9840 CrossRef CAS PubMed
- D. A. Tomalia, B. Klajnert-Maculewicz, K. A. M. Johnson, H. F. Brinkman, A. Janaszewska and D. M. Hedstrand, Prog. Polym. Sci., 2019, 90, 35–117 CrossRef CAS
- N. Gan, H. Shi, Z. An and W. Huang, Adv. Funct. Mater., 2018, 28, 1802657 CrossRef
- C. G. Wang, A. M. L. Chong, H. M. Pan, J. Sarkar, X. T. Tay and A. Goto, Polym. Chem., 2020, 11, 5559–5571 RSC
- D. Schilter, Nat. Rev. Chem., 2021, 5, 598–598 CrossRef CAS PubMed
- L. Turunen and M. Erdélyi, Chem. Soc. Rev., 2020, 49, 2688–2700 RSC
- A. M. S. Riel, R. K. Rowe, E. N. Ho, A. C. C. Carlsson, A. K. Rappé, O. B. Berryman and P. S. Ho, Acc. Chem. Res., 2019, 52, 2870–2880 CrossRef CAS PubMed
- R. Tepper and U. S. Schubert, Angew. Chem., Int. Ed., 2018, 57, 6004–6016 CrossRef CAS PubMed
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed
- A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686–2695 CrossRef CAS PubMed
- R. Hein and P. D. Beer, Chem. Sci., 2022, 13, 7098–7125 RSC
- O. Bolton, K. Lee, H. J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef CAS PubMed
- D. Lee, O. Bolton, B. C. Kim, J. H. Youk, S. Takayama and J. Kim, J. Am. Chem. Soc., 2013, 135, 6325–6329 CrossRef CAS PubMed
- Q. Kuang, X. Hou, C. Du, X. Wang and D. Gao, Phys. Chem. Chem. Phys., 2023, 25, 17759–17768 RSC
- M. Bayat, H. Mardani, H. Roghani-Mamaqani and R. Hoogenboom, Chem. Soc. Rev., 2024, 53, 4045–4085 RSC
- H. T. Le, C. G. Wang and A. Goto, Angew. Chem., Int. Ed., 2020, 59, 9360–9364 CrossRef CAS PubMed
- H. T. Le, C. G. Wang and A. Goto, Nat. Commun., 2023, 14, 171 CrossRef CAS PubMed
- H. T. Le and A. Goto, Cell Rep. Phys. Sci., 2021, 2, 100469 CrossRef CAS
- M. Erdelyi, Chem. Soc. Rev., 2012, 41, 3547–3557 RSC
- S. V. Rosokha, C. L. Stern, A. Swartz and R. Stewart, Phys. Chem. Chem. Phys., 2014, 16, 12968–12979 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00533c |
| This journal is © The Royal Society of Chemistry 2024 |