
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Patchy stereocomplex micelles as efficient compatibilizers for polymer blends†
Roman
Schaller
a,
Marius
Schmidt
a,
Kristian
Schweimer
 bc and
Holger
Schmalz
*ad
bc and
Holger
Schmalz
*ad
aMacromolecular Chemistry, University of Bayreuth, Universitätsstraße 30, 95447 Bayreuth, Germany. E-mail: holger.schmalz@uni-bayreuth.de
bBiochemistry IV, Biophysical Chemistry, University of Bayreuth, Universitätsstraße 30, 95447 Bayreuth, Germany
cNorthern Bavarian NMR Center, University of Bayreuth, Universitätsstraße 30, 95447 Bayreuth, Germany
dBavarian Polymer Institute, Universitätsstraße 30, 95447 Bayreuth, Germany
First published on 9th July 2024
Abstract
Surface-compartmentalized polymer micelles (Janus and patchy micelles) have gained increasing attention as their unique properties open the way for various applications. While Janus micelles have been extensively studied, e.g. as compatibilizers in polymer blends, there are hardly any reports on the use of patchy micelles. In this study, we show that spherical micelles with a polylactide stereocomplex (SC) core and a patch-like microphase separated polystyrene/poly(tert-butyl methacrylate) (PS/PtBMA) corona are efficient compatibilizers for highly immiscible PS/PtBMA blends. The patchy SC micelles, prepared by stereocomplex-driven self-assembly (SCDSA) of enantiomeric diblock copolymers, improved the homogeneity of the blends and led to a significant reduction of the PS droplet size. We further employed SCDSA to selectively incorporate a fluorescent dye inside the SC micelle core without changing the shape or chemistry of the patchy corona. This allows the use of confocal scanning fluorescence microscopy to localize the patchy SC micelles, being predominantly assembled at the PS/PtBMA blend interface. Interestingly, the reduction in PS droplet size was comparable for blends compatibilized with patchy SC micelles and Janus micelles, but only for patchy SC micelles a monomodal droplet size distribution could be achieved. The outstanding interfacial activity of the patchy SC micelles can be attributed to their adaptive corona structure, resulting in a selective swelling/collapse of the respective miscible/immiscible corona patches at the blend interface.
Introduction
The unique corona structure of surface-compartmentalized polymer micelles (or particles) opens a wide variety of potential applications and, hence, has attracted significant attention in recent years.1–10 In general, surface-compartmentalized particles can be divided in Janus or patch-like (patchy) particles. Janus particles and micelles show a stringent biphasic geometry (two separate faces) of distinct chemistry and/or polarity,11–17 and have been utilized for various applications, e.g. as efficient particulate surfactants for emulsion stabilization,18–20 as biosensors and optical nanoprobes,21,22 in interfacial catalysis,23,24 and for superhydrophobic and anti-ice coatings.25,26 Moreover, attributable to their outstanding interfacial activity, Janus micelles have been intensively studied as compatibilizers in polymer blends, even under technologically relevant conditions.27–31On the other hand, patchy micelles feature a patch-like microphase-separated corona consisting of several compartments with different chemistry and/or polarity.1,2,5,32,33 In comparison to Janus micelles, patchy micelles have been considerably less studied with respect to applications and with a strong focus on hierarchical self-assembly.34–40 The patchy corona can be harnessed as template for the regioselective incorporation of nanoparticles opening applications in heterogeneous catalysis,41–44 or for the construction of hierarchical superstructures when combined with supramolecular self-assembly.45,46 Notably, worm-like patchy micelles show a comparable interfacial activity with respect to that of cylindrical Janus micelles.47 This is attributed to the ability of the patchy corona to adjust to the interface by selective swelling/collapse of the respective soluble/insoluble surface patches. Despite, there are only a few reports on utilizing patchy micelles or hybrids in the stabilization of emulsions or as compatibilizer in polymer blends.48,49 A highly efficient method for the preparation of defined one-dimensional (cylindrical, or worm-like) patchy micelles is crystallization-driven self-assembly (CDSA) or living CDSA,50–52 which allows control over length, length distribution, and corona chemistries. This involves commonly the use of ABC triblock terpolymers, featuring amorphous incompatible end blocks and a crystallizable middle block based on polyethylene or polyferrocenyldimethylsilane (PFS).32,53–55 Besides, tailoring the self-assembly kinetics or employing synergistic self-seeding gives access to patchy cylindrical micelles employing mixtures of PFS based diblock copolymers.56–59 By the latter approaches the use of triblock terpolymers, being more difficult to synthesize, can be circumvented.
We have previously introduced stereocomplex-driven self-assembly (SCDSA) of diblock copolymers bearing enantiomeric poly(L-lactide) (PLLA) and poly(D-lactide) (PDLA) core-forming blocks and highly incompatible polystyrene (PS) and poly(tert-butyl methacrylate) (PtBMA) corona-forming blocks as alternative and synthetically less demanding route for the preparation of patchy spherical micelles. These patchy stereocomplex (SC) micelles show a high interfacial activity and were applied as particulate surfactants in the stabilization of emulsions.49 SCDSA is a versatile method for the preparation of SC micelles with varying shapes and property profiles (e.g. enhanced hydrolytic stability), however, mostly SC micelles with a homogeneous corona have been reported so far.60–62 Interestingly, SCDSA can give rise to unexpected morphological transitions as reported for mixtures of cylindrical micelles with enantiomeric PLLA and PDLA cores, whereby the solubility of the employed diblock copolymers was found to play a vital role.63,64 SCDSA of block or graft copolymers with chemically different coil (amorphous) blocks has only scarcely been investigated and was employed for example for the preparation of non-covalent block copolymers or micelles with a mixed, thermo-responsive corona, respectively.65–68
Herein, we report on the application of patchy spherical SC micelles as efficient compatibilizers in immiscible PS/PtBMA polymer blends (Scheme 1). The improvement and up-scaling of the preparation method for the patchy SC micelle dispersions as well as the influence of corona chemistry (patchy vs. homogeneous corona) of the employed SC micelles and the compatibilizer content on the blend morphology will be addressed. Moreover, fluorescence labelling is introduced as an elegant method to localize the patchy SC micelles at the PS/PtBMA blend interfaces. Finally, the compatibilization efficiency of the patchy SC micelles is compared to that of Janus micelles.
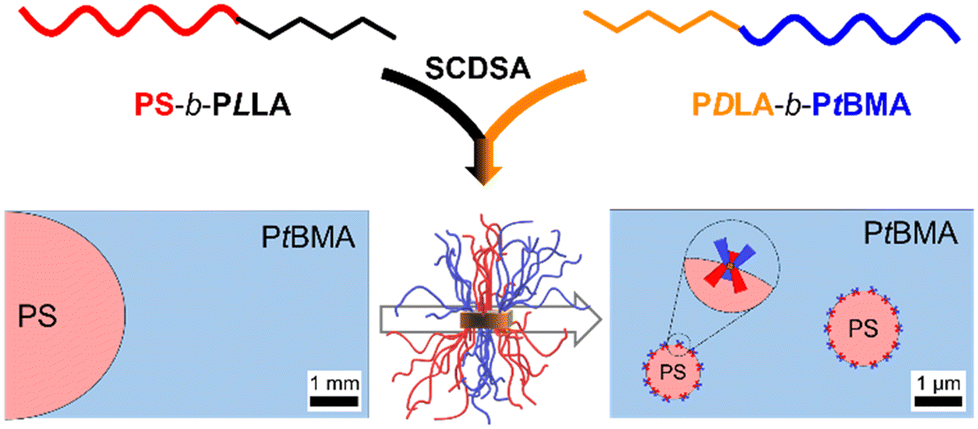 | ||
| Scheme 1 Schematic illustration of the preparation of patchy stereocomplex (SC) micelles via SCDSA of PS-b-PLLA and PDLA-b-PtBMA diblock copolymers and their application as compatibilizers in immiscible PS/PtBMA blends. | ||
Experimental
Materials
All chemicals were used as received unless otherwise noted. Ethylene oxide (Linde, 3.0) was stirred over calcium hydride (CaH2) at 0 °C for 3 h before being transferred into a storage ampoule. Prior to use ethylene oxide was additionally purified over n-butyllithium (n-BuLi) at 0 °C. Styrene (>99%, Sigma-Aldrich) was stirred over di-n-butylmagnesium (Bu2Mg) under nitrogen and condensed into a storage ampoule. Dichloromethane (DCM, ≥99.8% stabilized with amylene, analytical reagent grade, Thermo Scientific) used for lactide polymerization was dried by distillation over CaH2. Tetrahydrofuran (THF, 99.9%, Sigma-Aldrich) was dried by successive distillation over CaH2 and potassium and stored under nitrogen until use. 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU, >98%, TCI) was dried by distillation over CaH2. D- and L-lactide (≥99.8%, PURASORB® D and L, Corbion, Amsterdam, The Netherlands) were recrystallized from toluene and stored under nitrogen or argon until use. tert-Butyl methacrylate (tBMA, 98%, TCI) was passed through a column with basic aluminum oxide for ATRP or stirred over trioctylaluminum under nitrogen and condensed into a storage ampoule for anionic polymerization, respectively. N,N,N′,N″,N″-Pentamethyldiethylenetriamine (PMDETA, 99%, Sigma-Aldrich) was distilled prior to use. n-Pentane (technical) and isopropanol (technical) were purified by distillation. Copper(I) bromide (Cu(I)Br, 98%, Alfa Aesar) and copper(I) chloride (Cu(I)Cl, 99%, Sigma-Aldrich) were stirred in glacial acetic acid for 6 h, washed with glacial acetic acid and ethanol and dried in vacuo for 24 h. 1,1-Diphenylethylene (DPE, >98%, TCI) was stirred with sec-butyllithium (sec-BuLi) under argon atmosphere, distilled under vacuum and stored under argon until use. Pyridine (>99%, Carl Roth) was dried successively over KOH and CaH2 and distilled prior to use. Trioctylaluminum (25 wt% in hexane, Sigma-Aldrich), trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB, HPLC grade, Sigma-Aldrich), silver trifluoroacetate (AgTFA, >99.9%, Sigma-Aldrich), sodium trifluoroacetate (NaTFA, ≥99.5%, Fluka), sulforhodamine B acid chloride (pure, Thermo Scientific), Bu2Mg (1.0 M in heptane, Sigma-Aldrich), sec-BuLi (1.3 M in cyclohexane/hexane, Thermo Scientific), n-BuLi (1.6 M in hexanes, Thermo Scientific), α-bromoisobutyryl bromide (98%, Sigma-Aldrich), benzoic acid (p.a., AlppiChem), 4-dimethylaminopyridine (DMAP, 99%, Sigma-Aldrich), CaH2 (Merck), 2-hydroxyethyl 2-bromoisobutyrate (HEBiB, 95%, Sigma-Aldrich), deuterated chloroform (CDCl3, 99.8%, Deutero), deuterated cyclohexane (CH-d12, 99.5%, Deutero), deuterated dichloromethane (DCM-d2, 99.6%, Deutero), trimethylsilyl chloride (>98%, Sigma-Aldrich), ruthenium(III) chloride hydrate (ReagentPlus®, Sigma-Aldrich), sodium hypochlorite solution (NaOCl, 10–15 wt% in water, Sigma-Aldrich), chloroform (CHCl3, ≥99.8%, analytical reagent grade, Thermo Scientific), cyclohexane (CH, ≥99.8%, analytical reagent grade, Thermo Scientific), toluene (≥99.5%, AnalaR NORMAPUR ACS, Reag. Ph. Eur., VWR chemicals) and methanol (MeOH, ≥99.9%, analytical reagent grade, Thermo Scientific).Polymer syntheses
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v)), filtered and dried in vacuo (1 × 10−5 mbar).
:1 (v/v)), filtered and dried in vacuo (2 × 10−2 mbar) at 40 °C.
:1 (v/v)), filtered and dried in vacuo (1 × 10−5 mbar).
:1 (v/v)), filtered and dried in vacuo (2 × 10−2 mbar) at 40 °C.
SCDSA of diblock copolymer mixtures
Solutions of PS-b-PLLA and PtBMA-b-PDLA in DCM (c = 50 g L−1) were mixed together, employing a 1:1 weight ratio with respect to the enantiomeric PLA blocks. In a 50 mL round bottom flask, equipped with a magnetic stirring bar, the obtained mixture (2 mL) was added dropwise to CH (18 mL) over 2 min while stirring (approximately 350 rpm), resulting in a micelle dispersion with a concentration of c = 5.0 g L−1 and a final solvent ratio of CH/DCM = 9/1 (v/v). Subsequently, the dispersion was stirred for an additional 1 min and then aged at room temperature without stirring for at least one day. The dispersion was opened to air to evaporate the DCM over three days. To the remaining dispersion CH was added to restore the initial concentration before use. The same protocol was applied to the combination of PS-b-PLLA and PS-b-PDLA starting from solutions with concentrations of c = 50 g L−1. The dispersions of SC micelles in CH prepared by SCDSA are denoted as PS-sc-PLA-PtBMA micelles or PS-sc-PLA-PS micelles, respectively.
:1 weight ratio with respect to the enantiomeric PLA blocks. The dispersion of fluorescently labelled SC micelles was prepared the same way as described above and is denoted as PS-sc-PLA-PtBMA_RB micelles.
Preparation of polymer blends
In a hydrophobized snap lid vial (by reacting with trimethylsilyl chloride) PS and PtBMA were dissolved in CH or a SC micelle dispersion (c = 5 g L−1 in CH) with an overall polymer concentration of c = 100 g L−1. The amount of micelles was varied from 0, 1, 3, 5, 7 to 10 wt% while the ratio of PS/PtBMA was kept constant at 30/70 (w/w). The samples were shaken at 45 °C (200 rpm, thermo shaker HLC MKR-13, Ditabis) for 16 h to ensure complete dissolution of the polymers. Then, the solvent was allowed to evaporate slowly over 1 week while shaking gently (200 rpm) at 45 °C. The films were obtained by smashing the vial after freezing in liquid nitrogen, followed by drying in vacuo (2 × 10−2 mbar). For the blend compatibilized with 7 wt% Janus micelles, which were formed by self-assembly of the PS-b-PLLA-b-PtBMA triblock terpolymer in CH, the same procedure as described for blends with SC micelles as compatibilizers was employed.Methods
Proton nuclear magnetic resonance ( 1 H NMR) spectroscopy was conducted with a Bruker Ultrashield-300 spectrometer (300 MHz) at 20 °C. The chemical shift (δ) was determined relatively to the residual solvent signal of CDCl3 (δ (1H) = 7.26 ppm).2D 1 H NMR nuclear Overhauser effect spectroscopy (NOESY). 1H NMR spectra of the patchy SC micelle dispersion in CH-d12 (c = 5 g L−1) were recorded at 298 K on a Bruker Avance IIIHD 600 MHz spectrometer. The 2D 1H NOESY spectrum (mixing time 250 ms) was recorded with 96 scans and 2048 × 128 complex data points. The spectral width was 12 kHz and the recycle delay 2 s. The patchy SC micelle dispersion was prepared as described above using deuterated solvents for SCDSA.
Size exclusion chromatography (SEC) was carried out on SEC 1200/1260 Infinity systems (Agilent Technologies, Santa Clara, CA, USA) equipped with a SDV gel precolumn (particle size = 5 μm, PSS, Mainz, Germany). CHCl3-SEC was performed with a SDV linear XL gel column (particle size = 5 μm) with porosity range from 102 to 105 Å (PSS, Mainz, Germany) using CHCl3 (HPLC grade) as eluent. The samples were measured at a flow rate of 0.5 mL min−1 at 23 °C, employing a refractive index (RI) detector (Agilent Technologies, Santa Clara, CA, USA). THF-SEC was performed with four SDV gel columns (particle size = 5 μm) with porosity range from 102 to 105 Å (PSS, Mainz, Germany). THF (HPLC grade) was used as eluent and the samples were measured at a flow rate of 1.0 mL min−1 at 40 °C. For detection a RI detector (Agilent Technologies, Santa Clara, CA, USA) or a variable wave-length detector (VWD, Agilent Technologies, Santa Clara, CA, USA) were used. All samples were dissolved and filtered through a 0.2 μm PTFE filter before analyses. A calibration with narrowly distributed PS standards (PSS calibration kit) and toluene (HPLC grade) as internal standard were used in all cases.
A confocal WITec Alpha 300 RA + Raman imaging system equipped with a UHTS 300 spectrometer and a back-illuminated Andor Newton 970 EMCCD camera together with the WITec Suite SIX 6.1 software package were employed for Raman measurements. All spectra were acquired with an excitation wavelength of λ = 532 nm, using integration times of 0.3–0.35 s and a laser intensity of 20 mW for Raman imaging. For large area Raman imaging (typically 180 × 150 µm2) a 50× long working distance objective (Zeiss LD EC Epiplan-Neofluar Dic 50×, numerical aperture NA = 0.55) and a step size of 0.5 µm pixel−1 were used. Raman imaging with a 100× objective (Zeiss EC Epiplan-Neofluar Dic 100×, NA = 0.9, typically 50 × 50 µm2) was conducted with a step size of 0.2 µm pixel−1. All spectra were corrected for cosmic ray spikes and subjected to a background removal routine. The spatial distribution of PS and PtBMA was determined using the True Component Analysis tool of the WITec Project SIX 6.1 software. The sizes of the PS domains were determined with the software Fiji by evaluating at least 100 PS droplets from multiple positions.73 Samples of the micelle dispersions were prepared by dropping small amounts (3 × 10 µL) of the dispersion onto a glass slide followed by drying in vacuo (3 h, 20 mbar). Raman spectra of the dried SC micelle dispersions were acquired with the 100× objective employing a laser intensity of 20 mW and integration times of 0.5–1 s (100 accumulations).
Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-ToF MS) was carried out with a Bruker Daltonics autoflex maX (Bruker, Germany) equipped with a smartbeam-II solid state laser and operating in linear mode. The samples were prepared by the dried droplet method, employing DCTB as matrix and AgTFA in the case of PS and NaTFA in the case of PtBMA as ionization agents. All components were dissolved in THF (HPLC grade) at a concentration of c = 10 g L−1 and mixed together in a volumetric ratio of matrix/sample/ionization agent = 20/3/1.
Differential scanning calorimetry (DSC) was performed on a Netzsch DSC 204 F1 Phoenix instrument using aluminum crucibles (pierced lid) and nitrogen as protecting gas. The temperature range was selected from 0 °C to 220 °C employing a scanning rate of 10 K min−1.
Dynamic light scattering (DLS) was performed using a Zetasizer Nano S (Malvern Panalytical, UK) instrument equipped with a He–Ne laser (λ = 632.8 nm) and a detector placed at θ = 173°. Unless otherwise noted, the micelle dispersions were diluted to a concentration of c = 0.5 g L−1 and measured at 25 °C in quartz glass cuvettes (10.0 mm path length), which were sealed with Teflon caps to prevent solvent evaporation. The samples measured at 45 °C were equilibrated for 30 min inside the measurement chamber to assure constant temperature. The data were evaluated using the Malvern Zetasizer software (version 7.13) and the implemented distribution analysis fit option. The results are displayed as normalized intensity weighted size distributions, and the micelle sizes are reported as apparent hydrodynamic diameter (Dh,app) determined from the average of three replicate measurements.
Transmission electron microscopy (TEM) was carried out with a Zeiss/LEO EM922 Omega and a JEOL JEM-2200FS field emission TEM. All microscopes are energy filtering transmission electron microscopes (EFTEMs), operated at an acceleration voltage of 160 kV and 200 kV, respectively. Zero-loss filtered micrographs (ΔE ≈ 0 eV) were taken with a bottom mounted CCD camera (Ultrascan 1000, Gatan) in case of the Zeiss/LEO EM922 Omega and a bottom mounted CMOS camara system (OneView, Gatan) for the JEOL JEM-2200FS. The images were processed with a digital image processing software (Digital Micrograph DM 2.3 and 3.11, Gatan). For TEM analyses of the micelle dispersions, samples were diluted to a concentration of c = 0.1 g L−1 and stirred for 30 min at room temperature. Subsequently, 10 µL of the respective dispersion was applied to a carbon-coated copper grid and residual solvent was removed directly by blotting with a filter paper followed by drying of the coated copper grid in vacuo (24 h, 1 × 10−5 mbar). For TEM investigation of the polymer blend, the sample was cut into ultrathin sections (cut thickness: 50–60 nm) under cryogenic conditions (T = −140 °C) using a Leica UC7 ultramicrotome together with a EM FC7 cryochamber (Leica, Wetzlar, Germany). For RuO4 staining the samples were treated for 7 min with RuO4 vapor, which was formed in situ from RuCl3 hydrate and an aqueous NaOCl solution. After staining, the samples were stored for at least 1 h in a fume hood to ensure that any not reacted RuO4 was completely removed.
Scanning electron microscopy (SEM) measurements were done with a Zeiss Ultra Plus at an acceleration voltage of 1–3 kV and with SE2 and backscattered electron (BSE) detectors. Prior to analyses the samples were stained for 15 min with RuO4 vapor, following the same procedure as described for TEM. The manually fractured surfaces were fixed on an aluminum stub via electrically conductive tape and sputtered with a thin layer of platinum of around 2 nm with a Cressington sputter coater 208HR. The sizes of the PS domains were determined with the software Fiji.73 To this end, the area of at least 200 PS droplets from multiple positions were measured using the free hand selection tool and converted to the respective equivalent diameter assuming a perfect spherical cross-section.
A commercial optical microscope MicroTime 200 (PicoQuant) was used for the fluorescence experiments. The setup contained a laser diode operated in pulsed mode with a repetition rate of 20 MHz and an excitation wavelength of λ = 560 nm (LDH-D-TA-560B, PicoQuant). The excitation intensities were adjusted to approximately 1 µW. Radiation from the laser was directed through a single-mode optical fiber into the main optical unit, reflected by a dichroic mirror (ZT488/561rpc, AHF/Croma) into an inverted confocal optical microscope, and focused onto the sample using a super-apochromatic water immersion objective (UPLSAPO60XW, NA = 1.2, Olympus). Fluorescence images were acquired by scanning the focal spot over 250 μm × 250 μm on the sample using a galvo scanner (FLIMbee unit, PicoQuant). The signal from the sample was transmitted to the dichroic mirror and passed through a long pass filter (561 LP Edge Basic, Semrock). The signal was detected by a single-photon counting avalanche diode (SPCM-AQRH-14-TR, Excelitas) and a time-correlated single-photon counting unit (TCSPC TimeHarp 260 PICO Dual, PicoQuant, temporal resolution of 250 ps) was used to collect fluorescence data. The fluorescence images were evaluated using the commercial software SymPhoTime 64 (Picoquant). The PS/PtBMA (30/70 (w/w)) blend sample (3 wt% compatibilizer) was prepared as described above using a PS-sc-PLA-PtBMA_RB micelle dispersion.
Results and discussion
Up-scaling of patchy micelle preparation via SCDSA
Well-defined and long-term stable patchy micelles with a shamrock-like microphase-separated PS/PtBMA corona were prepared by SCDSA of diblock copolymers with enantiomeric PLLA and PDLA blocks and highly incompatible corona-forming PS and PtBMA blocks (Scheme 1). For the use as blend compatibilizers the preparation of patchy SC micelles had to be scaled up, as in our previous work the maximum concentration achieved was only c = 1 g L−1 in cyclohexane (CH), being impractical for the preparation of blends by solvent casting.49 The employed PS-b-PLLA (S169LLA106) and PS-b-PDLA (S156DLA106) diblock copolymers (subscripts denote number-average degrees of polymerization) were synthesized by organo-catalyzed ring-opening polymerization (ROP) of the respective enantiomeric lactide with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) starting from PS-OH macroinitiators, which were prepared by living anionic polymerization of styrene followed by end-capping with ethylene oxide (Scheme S1A and B†). For the synthesis of PDLA-b-PtBMA (DLA100tBMA132) a combination of ROP of D-lactide and atom transfer radical polymerization (ATRP) of tBMA was employed, utilizing 2-hydroxyethyl 2-bromoisobutyrate as bifunctional initiator (Scheme S1C†). The success of the diblock copolymer syntheses was followed by a combination of proton nuclear magnetic resonance (1H NMR) spectroscopy (Fig. S1†) and size exclusion chromatography (SEC), whereby monomodal distributions and low dispersities (Đ < 1.2) were obtained in all cases (Fig. 1). The syntheses are described in detail in the Experimental section and additional characterization is provided in the ESI (Fig. S2 and Table S1†).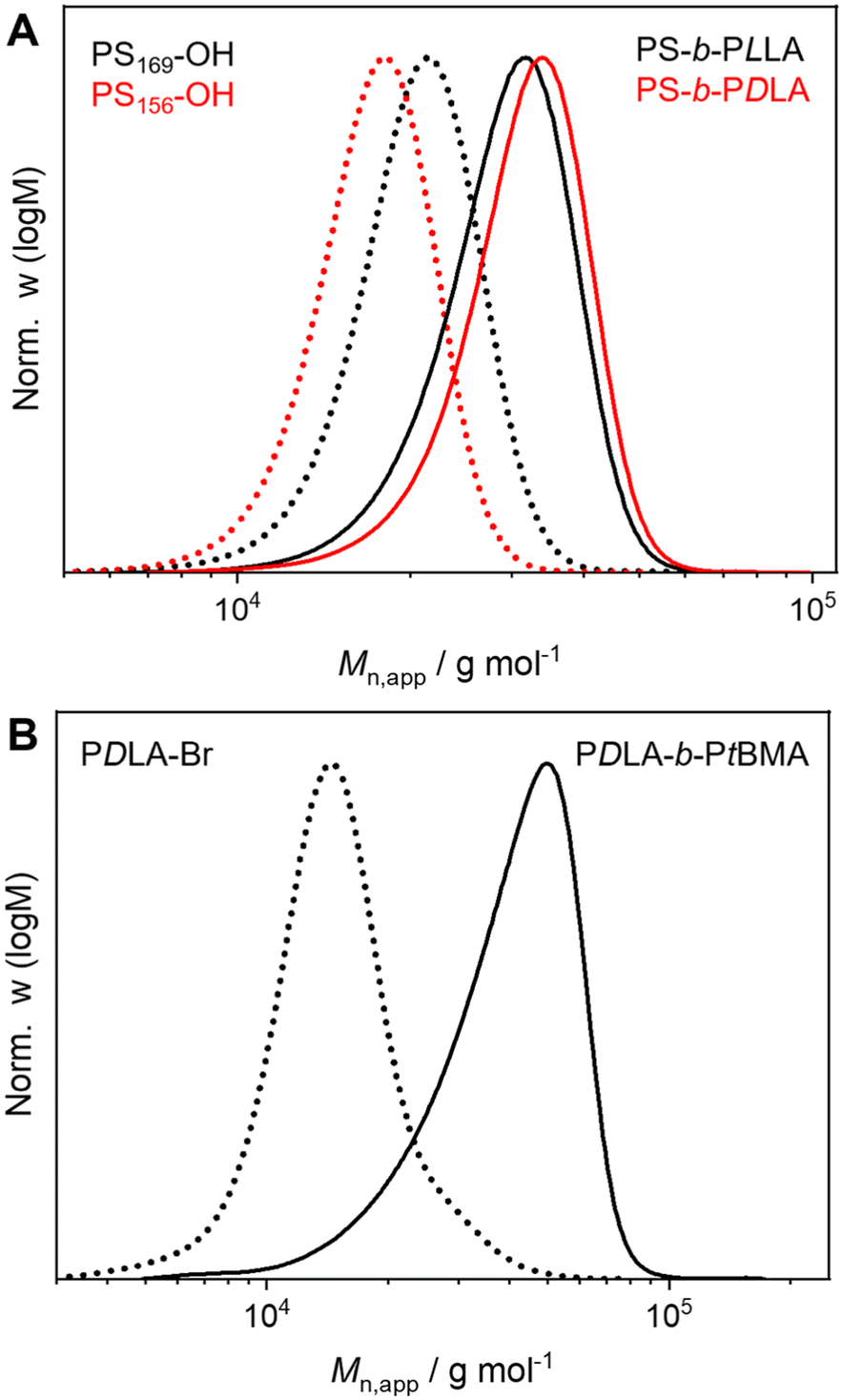 | ||
| Fig. 1 Apparent molecular weight distributions of (A) PS-b-PLLA (black, solid line) with PS169-OH (black, dotted line) precursor, PS-b-PDLA (red, solid line) with PS156-OH (red, dotted line) precursor and (B) PDLA-b-PtBMA (solid line) with PDLA-Br (dotted line) precursor (CHCl3-SEC, PS calibration). | ||
For SCDSA a mixture of the enantiomeric diblock copolymers (PS-b-PLLA and PDLA-b-PtBMA, 1/1 (w/w) with respect to the enantiomeric PLLA/PDLA blocks) in DCM was added drop-wise to an excess of CH, a selective solvent for the corona forming PS and PtBMA blocks. This resulted in a dispersion of spherical PS-sc-PLA-PtBMA micelles with an insoluble PLLA/PDLA SC core and a soluble, patchy PS/PtBMA corona. As shown before, the corona structure of the PS-sc-PLA-PtBMA micelles is influenced by the final CH/DCM ratio in the dispersion and, hence, the extend of dilution upon micelle formation.49 SC micelles with a well-defined patchy corona were obtained at a volume ratio of CH/DCM = 9/1 and a final concentration of c = 1 g L−1, whereby at higher dilution a mixture of Janus and patchy micelles was obtained. Accordingly, the ratio was kept at CH/DCM = 9/1 (v/v) in this study and the desired higher concentration of the SC micelle dispersion was achieved by increasing the concentration of the initial diblock copolymer mixture in DCM from c = 10 g L−1 to 50 g L−1 (V = 2 mL). After aging for one day under ambient conditions the remaining DCM was allowed to slowly evaporate over 3 days. Since DCM is a good solvent for all blocks its removal is essential for complete SC formation and also enhances corona segregation. After refilling with CH to compensate the simultaneous loss of CH during DCM evaporation 20 mL of the final PS-sc-PLA-PtBMA micelle dispersion with a concentration of c = 5 g L−1 were obtained (100 mg SC micelles per batch). Dynamic light scattering (DLS) and transmission electron microscopy (TEM) were used to prove that the increase in concentration does not compromise the formation of well-defined, spherical patchy SC micelles. DLS shows a monomodal size distribution of the SC micelles with an apparent hydrodynamic diameter of Dh,app = 103 ± 40 nm, and the dispersion was stable even for 8 months with no signs of agglomeration as would be indicated by a shift or substantial broadening of the size distribution (Fig. 2A, respective autocorrelation functions are shown in Fig. S3†). The size of the SC micelles is comparable to the size obtained in our previous study employing a lower concentration of c = 1 g L−1 (Dh = 104 ± 30 nm) and enantiomeric diblock copolymers with comparable block lengths.49 This supports the conclusion that an increase in concentration does not significantly alter the SCDSA behavior of the diblock copolymers. No significant shift of the size distribution to lower values was observed upon diluting the SC micelle dispersion from c = 5 g L−1 to c = 0.1 g L−1 (Fig. S4†), which is also consistent with the presence of single SC micelles. The dispersion was also stable after thermal annealing at T = 45 °C for 30 min, showing only a slight increase of the hydrodynamic diameter (Dh,app = 119 ± 41 nm). This is important, as the blends were cast at 45 °C to ensure proper solubility of PS and PtBMA during film formation. The increased micelle size can be attributed to an increased solubility and stretching of the PS chains, since CH is a theta-solvent for PS with a theta temperature of Tθ = 34 °C.75
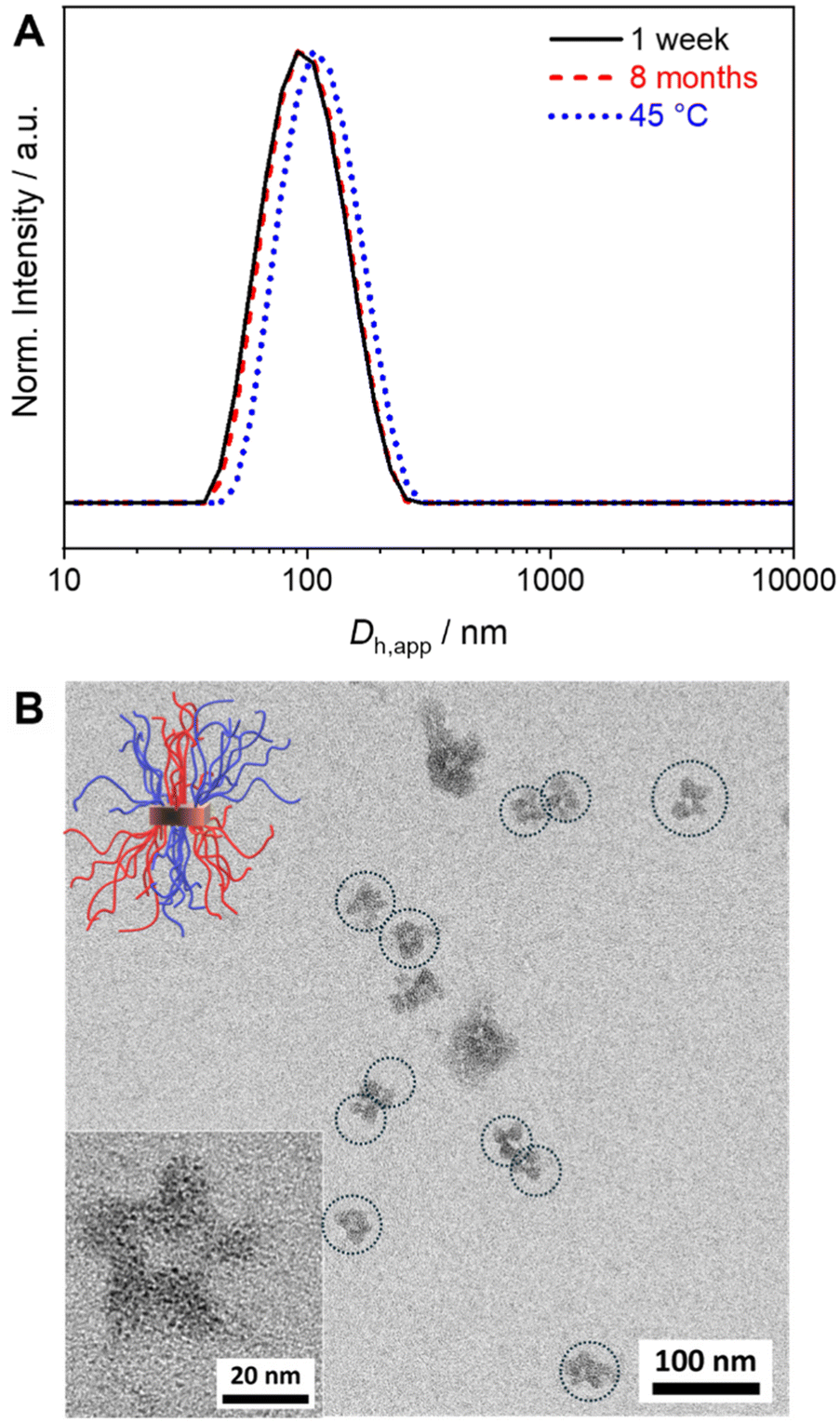 | ||
| Fig. 2 (A) Hydrodynamic diameter distributions from DLS for SC micelles prepared from PS-b-PLLA/PDLA-b-PtBMA mixtures (c = 5 g L−1, CH) after aging for 1 week, 8 months, and at 45 °C. (B) TEM micrographs of the micelles with a patchy PS/PtBMA corona (PS selectively stained with RuO4). The dimensions of the single micelles in the partially aggregated structures are indicated by dotted circles for means of clarity and the sketch illustrates the patchy structure of the corona. | ||
The TEM micrograph shown in Fig. 2B confirms the formation of spherical SC micelles with a patch-like microphase separated corona. The PS domains in the corona appear dark due to selective staining with RuO4 vapor, whereas the semicrystalline SC core as well as the PtBMA domains in the corona appear bright. The micelles tend to form small aggregates on the carbon-coated TEM grid. This is a drying artifact from sample preparation, as DLS shows a monomodal size distribution (Fig. 2A), and is typically observed for patchy micelles.75
To prove the microphase separation within the corona in solution a 2D 1H nuclear Overhauser effect spectroscopy (NOESY) experiment was conducted on the PS-sc-PLA-PtBMA micelles prepared in CH-d12. No cross-peaks were observed between 1H NMR signals of PS [δ = 7.5–6.5 ppm (aromatic protons)] and PtBMA [δ = 0.9 ppm (methyl group, polymer backbone)], confirming that the PS and PtBMA chains are demixed in the corona (Fig. S5†). In case of a mixed corona the close proximity of PS and PtBMA units would give rise to cross-polarization and the appearance of respective cross-peaks in the 2D 1H NOESY spectrum.
Dispersions of PS-sc-PLA-PS micelles with a homogeneous PS corona used as reference in the blend compatibilization studies were prepared analogously to the method described above for the patchy SC micelles. In comparison, the PS-sc-PLA-PS micelles also show a narrow size distribution with comparable Dh,app = 126 ± 44 nm, however, in TEM the PS corona now appears homogeneously dark (Fig. S6†). The observable agglomeration on the TEM grid can again be attributed to drying artifacts arising from sample preparation, as DLS shows a narrow size distribution.
To prove that self-assembly is indeed driven by SC formation between the enantiomeric PLLA/PDLA blocks Raman spectroscopy on the dried PS-sc-PLA-PtBMA and PS-sc-PLA-PS micelle dispersions was conducted. A characteristic shift of the carbonyl stretching vibration from ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) ≈ 1770–1773 cm−1 for the individual diblock copolymers to lower wavenumbers ( ≈ 1750–1752 cm−1) was observed, confirming SC formation (Fig. S7†).62,76 In addition, differential scanning calorimetry (DSC) on the freeze-dried PS-sc-PLA-PtBMA micelle dispersion shows a melting temperature of Tm = 203 °C (Fig. S8†), which is typical for polylactide stereocomplexes.61,65,66
≈ 1770–1773 cm−1 for the individual diblock copolymers to lower wavenumbers ( ≈ 1750–1752 cm−1) was observed, confirming SC formation (Fig. S7†).62,76 In addition, differential scanning calorimetry (DSC) on the freeze-dried PS-sc-PLA-PtBMA micelle dispersion shows a melting temperature of Tm = 203 °C (Fig. S8†), which is typical for polylactide stereocomplexes.61,65,66
Patchy SC micelles as compatibilizers in PS/PtBMA blends
After the successful scale-up of the preparation method, the efficiency of the patchy SC micelles as compatibilizers in immiscible PS/PtBMA blends was tested. This blend system was chosen not only because of the matching chemistry with the corona blocks of the patchy SC micelles, but rather because of the high incompatibility of the two polymers (Flory–Huggins interaction parameter for PS/PtBMA: χ = 0.08–0.10).77 Hence, PS/PtBMA blends are an ideal model system to study the performance of patchy SC micelles, as differences in interfacial activity (e.g. patchy vs. homogeneous corona) will result in distinctly altered blend morphologies. The used homopolymers were synthesized by living anionic polymerization and have molecular weights of Mn (PS) = 39400 g mol−1 and Mn (PtBMA) = 93700 g mol−1 as determined by matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-ToF MS). Details on the syntheses procedures and molecular weight characterization can be found in the ESI (Scheme S2, Fig. S1, S2, S9 and Table S1†). All blends were cast from CH at T = 45 °C under gentle shaking to ensure proper solubility of the polymers upon film formation. At lower temperatures a demixing of the phases during the evaporation process was observed due to the comparably high theta temperature of PS in CH of Tθ = 34 °C.74 The respective neat PS/PtBMA (30/70 (w/w)) blend showed macrophase separation into millimeter sized PS and PtBMA regions, as revealed by a combination of optical microscopy and Raman spectroscopy (Fig. 3), manifesting the high incompatibility of this blend system.
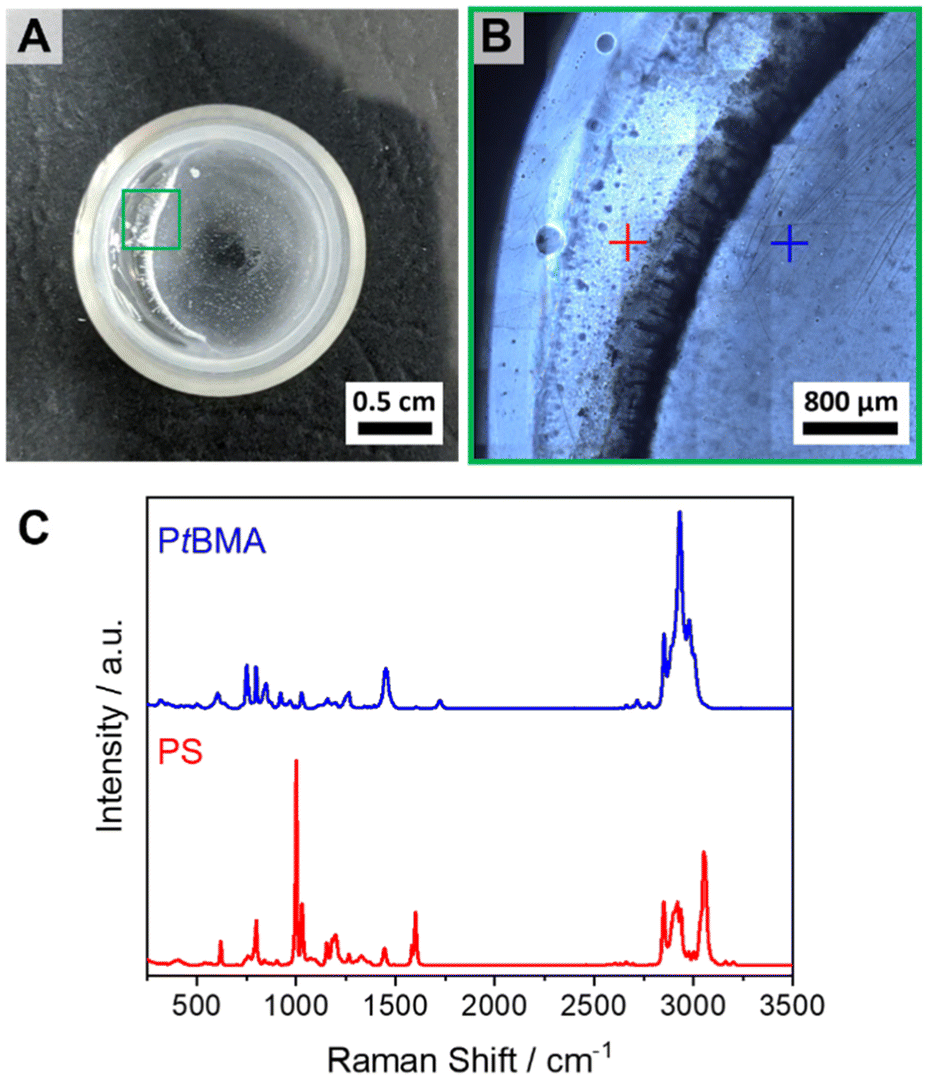 | ||
| Fig. 3 (A) Optical image of the neat PS/PtBMA (30/70 (w/w)) blend. (B) Optical microscopy image taken at the area depicted in green in (A). (C) Raman spectra of the PtBMA (blue) and PS (red) phases taken at the respective positions indicated in (B). | ||
First, the influence of the corona chemistry of the SC micelles on their compatibilization efficiency was studied. Fig. 4 shows the component distributions extracted from Raman imaging for a PS/PtBMA (30/70 (w/w)) blend compatibilized with 7 wt% patchy PS-sc-PLA-PtBMA micelles and of the respective blend with 7 wt% PS-sc-PLA-PS micelles having a homogeneous PS corona. As both SC micelles have comparable hydrodynamic diameters in CH (PS-sc-PLA-PtBMA: Dh,app = 103 ± 40 nm; PS-sc-PLA-PS: Dh,app = 126 ± 44 nm) an influence of micelle size on the blend morphology can be ruled out.
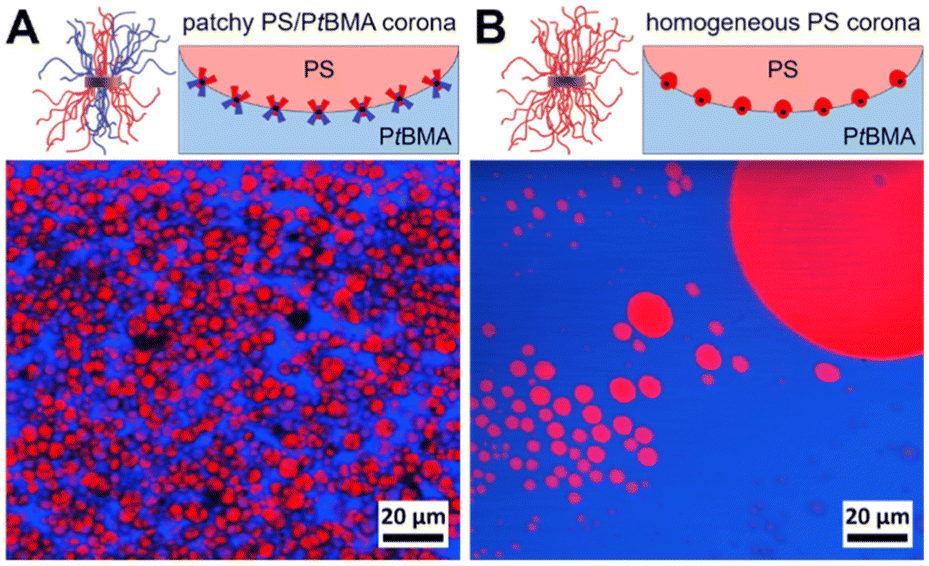 | ||
| Fig. 4 Spatial component distributions extracted from Raman imaging for PS/PtBMA (30/70 (w/w)) blends compatibilized with (A) 7 wt% patchy PS-sc-PLA-PtBMA micelles and (B) 7 wt% PS-sc-PLA-PS micelles with homogeneous PS corona. The domains colored in red represent PS droplets being dispersed in a continuous PtBMA matrix (depicted in blue). The sketches depict the adaption of the SC micelle corona according to their miscibility/immiscibility with the blend components. | ||
In both blends the PS phase (depicted in red in Fig. 4) is dispersed as droplets in a continuous PtBMA matrix, as might have been expected from the asymmetric blend composition with PS being the minority phase. However, the blend compatibilized with patchy SC micelles shows a more homogeneous dispersion of the PS droplets in the PtBMA matrix (Fig. 4). Notably, statistical evaluation of the PS droplet sizes (Fig. S10A and E†) revealed that the size of the PS droplets is significantly smaller and their size distribution narrower (DPS = 3.9 ± 1.3 µm) as compared to the blend containing SC micelles with a homogeneous PS corona (DPS = 46.8 ± 66.4 µm). This higher compatibilization efficiency can be attributed to the ability of the patchy PS/PtBMA corona to adapt to the PS/PtBMA blend interface by expansion/collapse of the respective miscible/immiscible corona blocks, as indicated in the insets of Fig. 4. Hence, the amphiphilicity of the patchy corona in combination with the Pickering effect of nanoparticles is decisive for the observed high interfacial activity, leading to a decrease in interfacial tension and accordingly PS droplet size. In contrast, for the SC micelles with a homogeneous PS corona only the Pickering effect contributes to the stabilization, which makes them considerably less effective.
Next, the content of patchy PS-sc-PLA-PtBMA micelles in the blend was systematically varied from 1–10 wt% to find the optimum content with respect to blend homogeneity and reduction in PS droplet size. At a filler content of 1 wt% already no macrophase separation of the two blend components was visible anymore in the component distribution extracted from Raman imaging. However, the blend still showed a bimodal PS droplet size distribution with rather large PS droplets having diameters up to DPS ≈ 100 µm (Fig. 5A and Fig. S10B†). With increasing amount of patchy micelles as compatibilizer the PS droplet size decreased but the size distributions were still comparably broad and bimodal up to a content of 5 wt% (Fig. 5B and C, histograms in Fig. S10C and D†). Starting with 7 wt% a monomodal size distribution of the dispersed PS domains within the PtBMA matrix was obtained, and the average PS droplet diameter decreased substantially to DPS = 3.9 ± 1.3 µm (Fig. 5D and Fig. S10E†). Increasing the amount of patchy micelles to 10 wt% did not result in a further decrease in PS droplet size (Fig. 5E and Fig. S10F†), indicating that 7 wt% is the optimum compatibilizer content for the investigated PS/PtBMA (30/70 (w/w)) blends. In the Raman images of the blends with 7 and 10 wt% patchy SC micelles acquired at smaller step sizes the homogeneous dispersion of the PS droplets is also clearly visible (Fig. S11†).
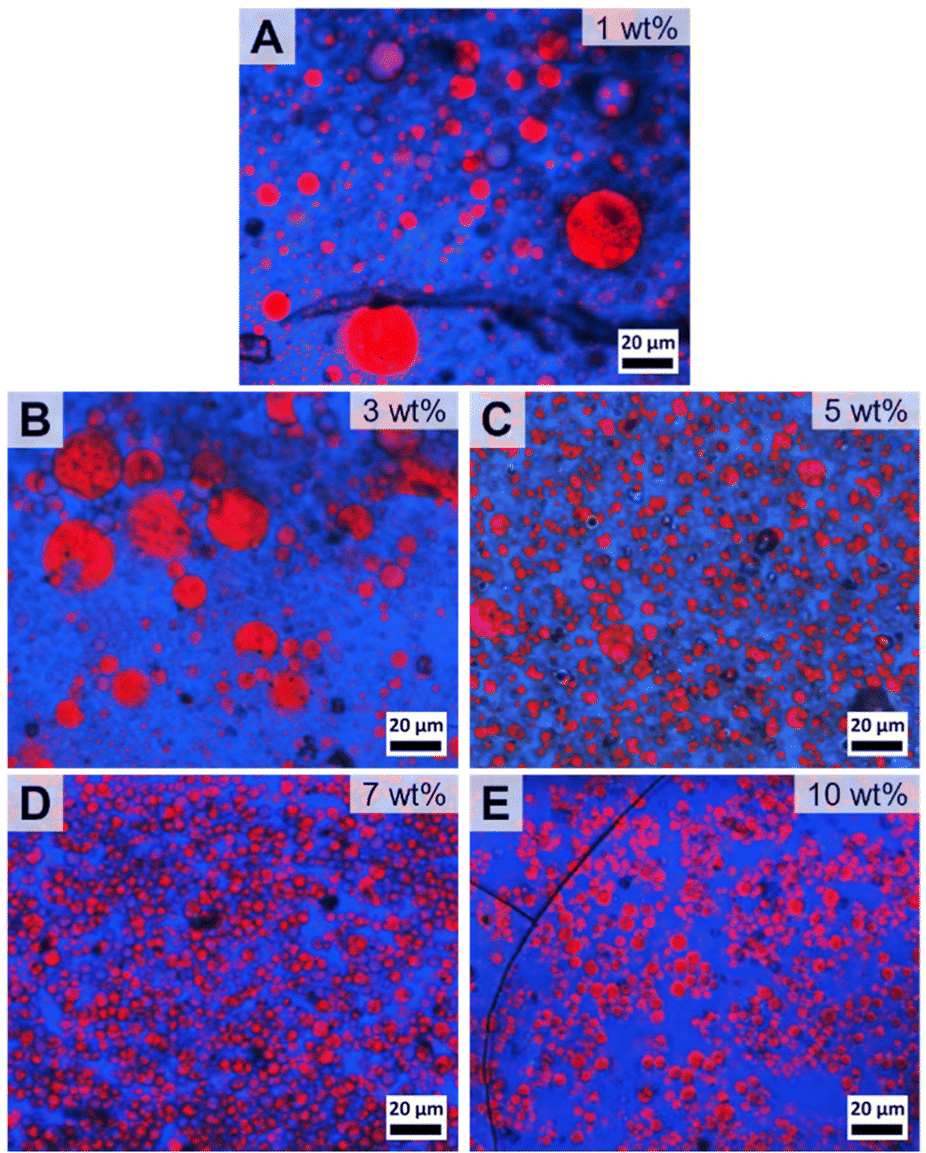 | ||
| Fig. 5 Spatial component distribution extracted from Raman imaging for PS/PtBMA (30/70 (w/w)) blends compatibilized with patchy PS-sc-PLA-PtBMA micelles with various concentrations: (A) 1, (B) 3, (C) 5, (D) 7 and (E) 10 wt%. The domains colored in red represent PS droplets being dispersed in a continuous PtBMA matrix (depicted in blue). | ||
The blends with 3–10 wt% patchy micelles were further characterized by scanning electron microscopy (SEM) to determine the exact size and size distribution of the dispersed PS droplets in the PS/PtBMA (30/70 (w/w)) blends (Fig. 6). Confocal Raman imaging gives direct laterally resolved chemical information, enabling a clear assignment of the blend phases without the need of contrast enhancement by staining as commonly necessary for electron microscopy. However, lateral resolution is restricted by the wavelength of the used laser (here λ = 532 nm) and the characteristics of the employed objective, resulting in a maximum resolution of ca. 0.3–0.4 µm for a 100× objective (NA = 0.9). Accordingly, especially for the blends with higher contents of patchy micelles the size of the PS droplets is in the µm-range and might probably be overestimated by Raman imaging. Moreover, with SEM it is more convenient to scan larger areas, which is necessary to evaluate the size and size distribution of the larger PS droplets in blends with a bimodal size distribution. For SEM measurements the samples were stained with RuO4 to enhance contrast between the dispersed PS droplets (appear bright) and the surrounding PtBMA matrix, and for size evaluation at least 200 PS droplets from different positions were counted. SEM confirms the conclusions drawn from Raman imaging that with increasing amount of patchy micelles the PS droplet size decreases and the size distribution becomes monomodal for micelle contents ≥7 wt%. Especially in the overview SEM images shown in Fig. S12† the bimodal PS droplet size distribution for the blend with 3 and 5 wt% patchy micelles becomes more obvious. The large PS droplets partially feature droplet-like PtBMA inclusions that were less obvious in the respective Raman images (Fig. 5B and C). For the blends with higher micelle contents (7 and 10 wt%) no inclusions within the considerably smaller PS droplets were observed. In good accordance with Raman imaging the strongest reduction in PS droplet size and the narrowest size distribution was observed for a micelle content of 7 wt%, yielding an average PS droplet diameter of DPS = 2.5 ± 0.9 µm (Fig. 6C and E). Again, no further change in size and size distribution was noticeable upon further increasing the micelle content to 10 wt% (DPS = 2.6 ± 0.9 µm, Fig. 6D and E). Notably, the sizes determined from SEM are systematically lower as compared to that determined by Raman imaging, which is attributed to the aforementioned limited lateral resolution for Raman imaging. Hence, Raman and SEM analyses reveal the same trend, that is a reduction in size and a narrowing in size distribution with increasing content of patchy SC micelles as compatibilizer. Nevertheless, a combination of both methods is recommended to ensure a proper phase assignment, as staining procedures employed for electron microscopy studies might give rise to artifacts in case staining is not sufficiently selective for one of the blend components and might vary from sample to sample and method used.
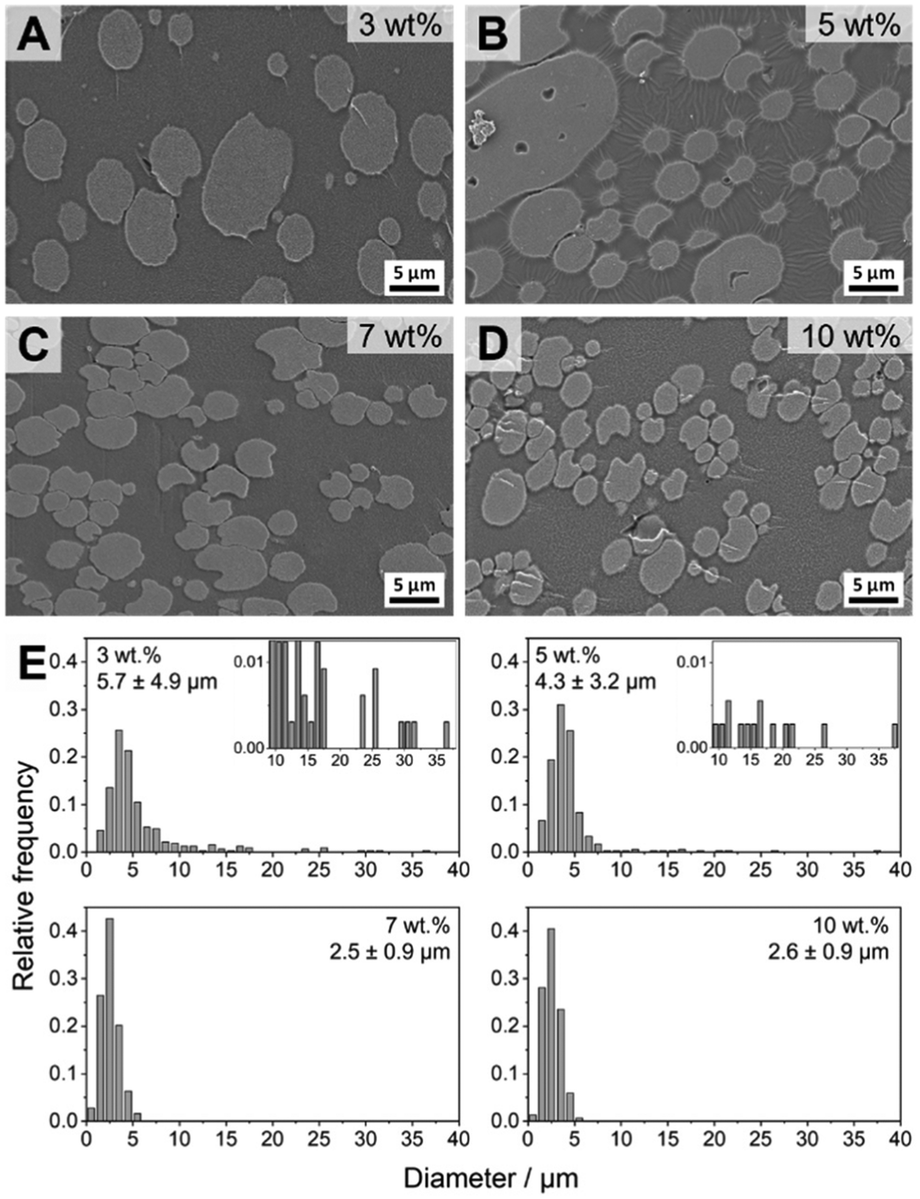 | ||
| Fig. 6 SEM images of the fracture surfaces of PS/PtBMA (30/70 (w/w)) blends compatibilized with different amounts of PS-sc-PLA-PtBMA micelles: (A) 3, (B) 5, (C) 7 and (D) 10 wt%. The PS domains were selectively stained with RuO4 vapor to enhance contrast and appear bright. (E) Corresponding histograms of PS droplet diameter distributions. | ||
Localizing patchy micelles at the blend interface
The excellent compatibilization efficiency of the patchy SC micelles is attributed to their high interfacial activity resulting in an accumulation at the PS/PtBMA blend interfaces and, hence, reducing the interfacial tension and accordingly the PS droplet size. In order to prove the presence of patchy SC micelles at the PS/PtBMA interface, confocal scanning fluorescence microscopy (FM) was performed employing fluorescently labelled patchy SC micelles. For fluorescence labelling the PS-sc-PLA-PtBMA micelles were prepared by co-assembly of the enantiomeric diblock copolymers with a small amount of a sulforhodamine B labelled PDLA62 homopolymer (PDLA62-RB, Fig. 7A). Details on synthesis and molecular characterization of PDLA62-RB are provided in the Experimental section and ESI (Scheme S3, Fig. S13 and Table S1†). The incorporation of the fluorescently labelled PDLA62-RB homopolymer does not affect the SCDSA to well-defined spherical micelles as DLS shows a monomodal size distribution for the PS-sc-PLA-PtBMA_RB micelles with an apparent hydrodynamic diameter of Dh,app = 88 ± 30 nm (Fig. S14A,† respective autocorrelation function is depicted in Fig. S14B†), being slightly lower compared to that of the non-labelled micelles (Dh,app = 103 ± 40 nm, Fig. 2A). In addition, the overall morphology and patch-like compartmentalized PS/PtBMA corona of the micelles are not altered as revealed by TEM (Fig. S14C†). This underpins the potential of SCDSA to selectively incorporate different guest molecules inside the SC core of the micelles, without changing the shape and chemistry of the micelle corona.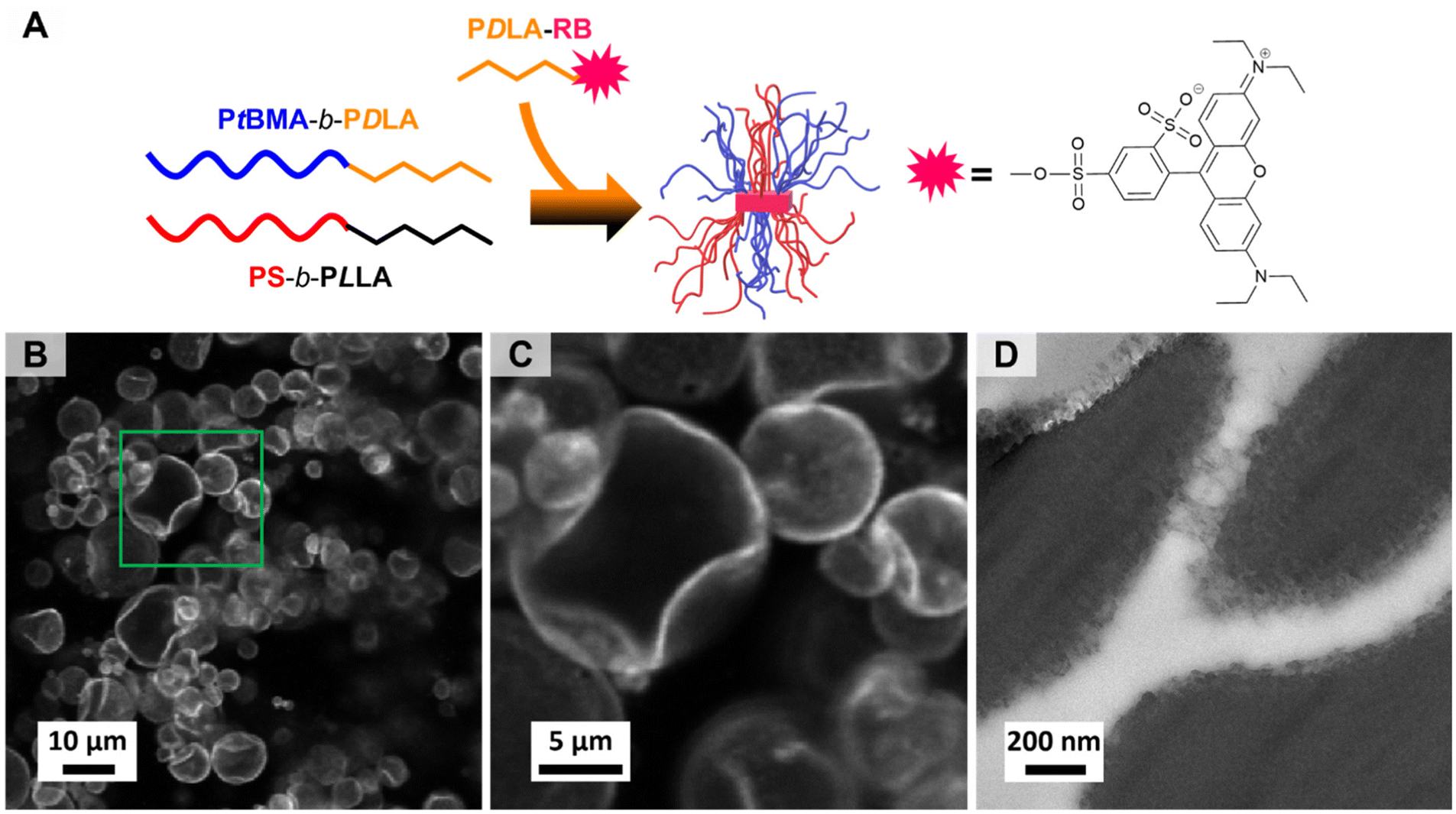 | ||
| Fig. 7 (A) Schematic illustration of the preparation of fluorescently labelled PS-sc-PLA-PtBMA_RB micelles via SCDSA. (B and C) Confocal scanning FM images of a PS/PtBMA (30/70 (w/w)) blend compatibilized with 3 wt% fluorescently labelled PS-sc-PLA-PtBMA_RB micelles. (D) TEM micrograph of a PS/PtBMA (30/70 (w/w)) blend compatibilized with 7 wt% patchy PS-sc-PLA-PtBMA micelles (PS selectively stained with RuO4). | ||
Fig. 7B and C show the confocal scanning FM images of a respective PS/PtBMA (30/70 (w/w)) blend compatibilized with 3 wt% of fluorescently labeled PS-sc-PLA-PtBMA_RB micelles, prepared by solvent casting. The low compatibilizer content of 3 wt% was deliberately chosen as larger PS droplets are expected to form, which makes it easier to visualize the interface. From the confocal FM image it can be clearly deduced that the patchy SC micelles are preferentially located at the PS/PtBMA interface, resulting in a bright appearing rim around the dispersed PS droplets.
In addition, TEM was used to visualize the patchy SC micelles at the PS/PtBMA interface. Accordingly, ultrathin slices were cryo-cut from a PS/PtBMA (30/70 (w/w)) blend compatibilized with 7 wt% patchy PS-sc-PLA-PtBMA micelles, followed by selective staining of the PS droplets/patches with RuO4 vapor. Hence, the PS droplets appear dark and are embedded in the bright appearing PtBMA matrix, as shown in Fig. 7D (additional TEM micrographs are presented in Fig. S15†). The interface between the two blend phases shows a fringe-like structure with clusters of smaller, dark appearing patches. In contrast, for a neat (without compatibilizer) PS/PtBMA blend sharp interfaces would be expected in order to reduce energetically unfavorable interactions between the highly incompatible blend components. Hence, the observed dark appearing clusters resemble the patchy SC micelles being assembled at the PS/PtBMA interface. Nonetheless, it is stressed that the conclusion drawn from TEM was only possible in combination with the results from confocal FM. From the TEM results alone it is not possible to unambiguously attribute the fringe-like structure at the interface to the presence of patchy SC micelles.
Comparison with Janus-type micelles
As pointed out in the introduction, mainly Janus-type micelles have been studied so far for the compatibilization of polymer blends. Hence, the question arises whether the compatibilization efficiency of the spherical patchy SC micelles is comparable or might even be better with respect to spherical Janus micelles. To address this point, we have synthesized a PS-b-PLLA-b-PtBMA triblock terpolymer that forms spherical Janus-type micelles upon self-assembly in CH. The triblock terpolymer was synthesized starting from a S169LLA96 diblock copolymer by end-functionalization with an ATRP initiator moiety (2-bromoisobutyryl group) followed by ATRP of tBMA in toluene (Scheme S4†), resulting in a narrowly distributed S169LLA96T200 triblock terpolymer (Đ = 1.18, Table S1†). The SEC traces of the PS-b-PLLA-Br diblock copolymer precursor and the PS-b-PLLA-b-PtBMA triblock terpolymer as well as the respective 1H NMR spectra can be found in the Supplemantary Information (Fig. S16†).The Janus-type PS-b-PLLA-b-PtBMA micelles were prepared by a simple heating and cooling protocol in CH (c = 5 g L−1). The dispersion was annealed for 2 h at 45 °C followed by cooling to 25 °C. DLS (Fig. 8A) shows that the obtained micelles are smaller (Dh,app = 50 ± 20 nm, respective autocorrelation functions are shown in Fig. S17†) with respect to the patchy PS-sc-PLA-PtBMA micelles prepared by SCDSA (Dh,app = 103 ± 40 nm). From the TEM micrograph in Fig. 8B it becomes obvious that the morphology of the micelle corona has changed, showing predominantly a Janus-type structure with a dark appearing PS hemisphere (selectively stained with RuO4 vapor). The PtBMA hemisphere is hardly recognizable due to insufficient contrast to the carbon layer on the TEM grid combined with partial electron beam induced degradation. The PLLA core of the micelles again appears bright. It is noted that the Janus structure of the micelles is only visible when the micelles have been deposited on the carbon-coated grid in the proper orientation. In case the PS (or PtBMA) hemisphere points to the surface of the grid the Janus structure will not be visible.
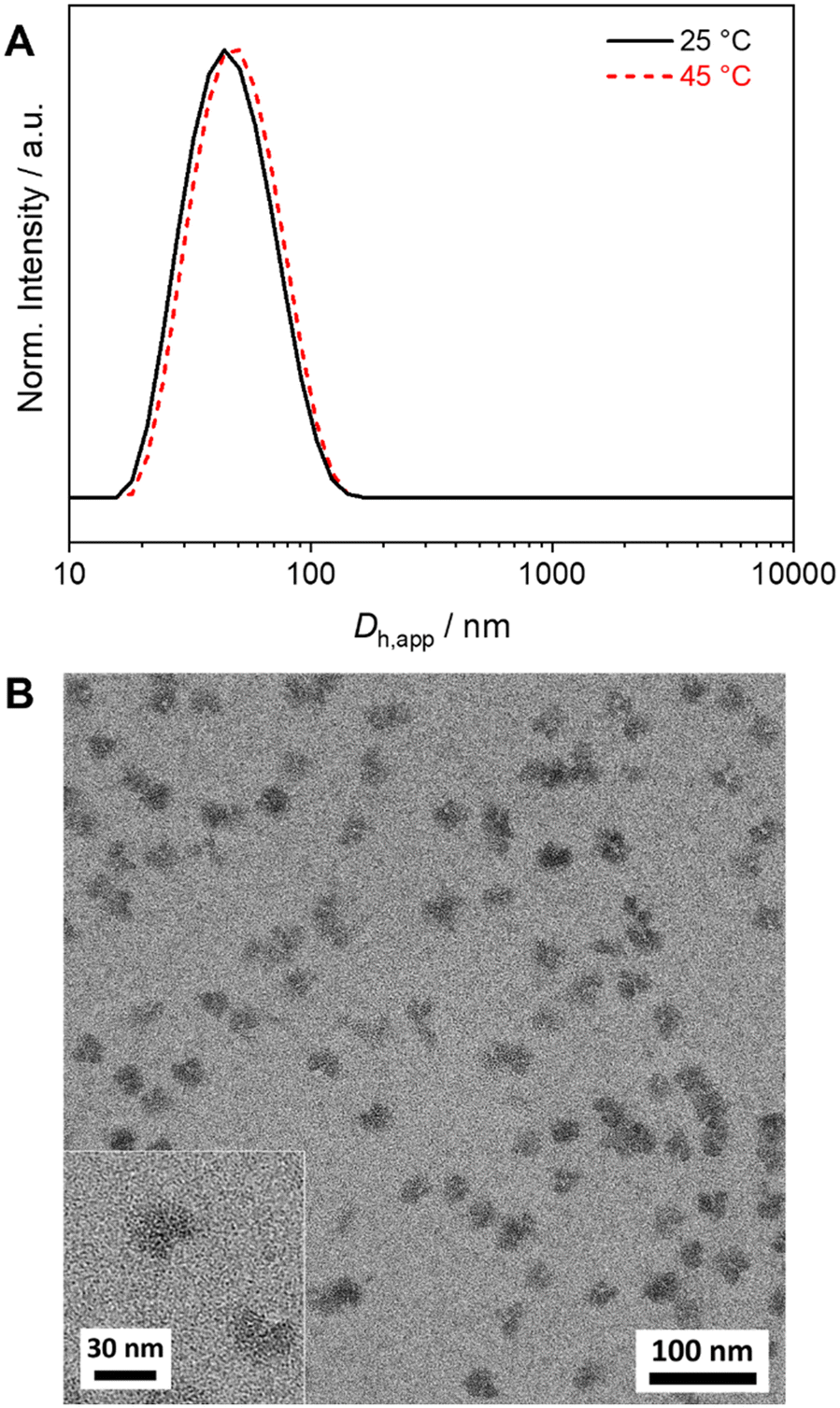 | ||
| Fig. 8 (A) Hydrodynamic diameter distributions from DLS for S169LLA96T200 triblock terpolymer micelles (c = 5 g L−1, CH). (B) TEM micrographs of the micelles with a Janus-type corona made of dark appearing PS and bright appearing PtBMA hemispheres (PS selectively stained with RuO4). | ||
Fig. 9A and B show the component distribution extracted from Raman imaging and the respective SEM micrograph for a PS/PtBMA (30/70 (w/w)) blend compatibilized with 7 wt% PS-b-PLLA-b-PtBMA Janus micelles. The average PS droplet size of DPS = 2.6 ± 1.8 μm is comparable to that obtained when employing the same amount of patchy PS-sc-PLA-PtBMA micelles (DPS = 2.5 ± 0.9 µm). However, the size distribution is significantly broader and bimodal for the Janus micelles compatibilized blend (Fig. 9C). The latter becomes obvious both in the Raman imaging data as well as the SEM micrograph by the presence of significantly larger PS droplets with diameters up to DPS ≈ 10–15 µm, which were not observed for the blend compatibilized with patchy SC micelles (Fig. 5D, 6C and E). Hence, patchy micelles seem to be superior with respect to achieving a homogeneous size distribution of the dispersed PS droplets, whereby the size reduction of the dispersed PS droplets is comparable.
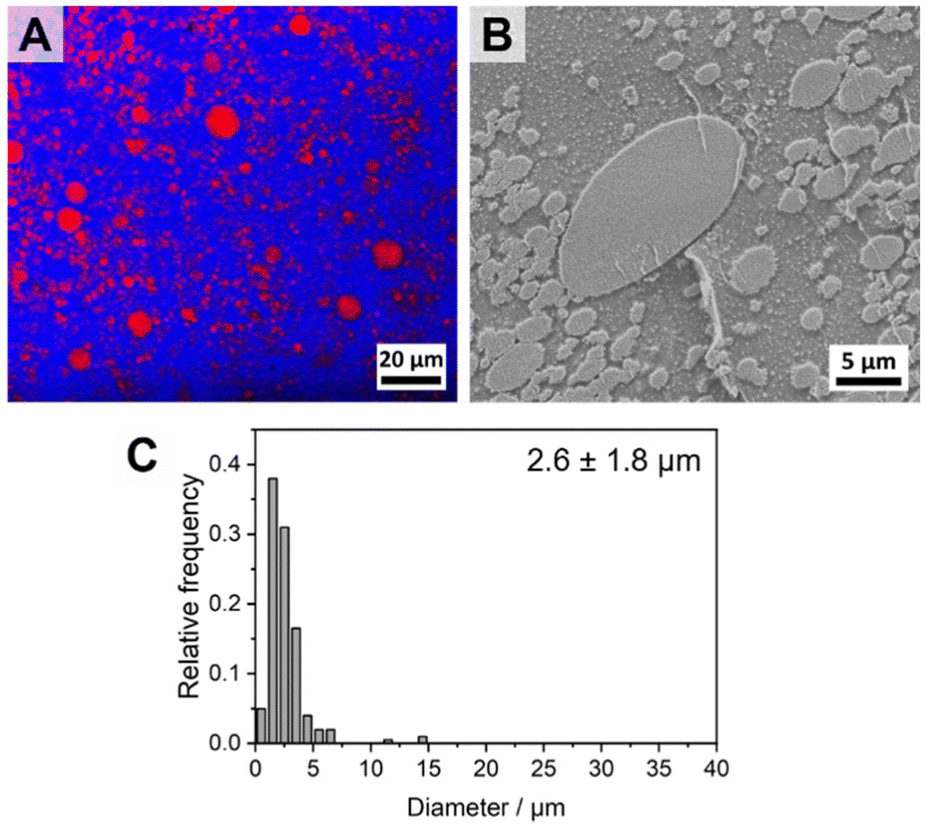 | ||
| Fig. 9 (A) Spatial component distribution extracted from Raman imaging (PS droplets are depicted in red and the continuous PtBMA matrix in blue) and (B) SEM micrograph for a PS/PtBMA (30/70 (w/w)) blend compatibilized with 7 wt% PS-b-PLLA-b-PtBMA Janus micelles. (C) Corresponding histogram of PS droplet diameter distribution. | ||
Conclusions
This work demonstrates that SCDSA of enantiomeric PLA-based diblock copolymers to well-defined and long-term stable patchy spherical micelles can be scaled-up without compromising their properties, allowing their application as compatibilizers in solvent-cast polymer blends. The patchy stereocomplex (SC) micelles feature a patch-like compartmentalized PS/PtBMA corona and have proven highly efficient in the compatibilization of strongly immiscible PS/PtBMA blends. With an increasing compatibilizer content the homogeneity of the blends increased and concomitantly the size of the dispersed PS droplets, being embedded in the PtBMA matrix, was significantly reduced. This is attributed to the unique corona structure of the patchy SC micelles, being capable to adapt to their surrounding by selective collapse/expansion of the respective miscible/immiscible corona patches. SCDSA also opens the way to incorporate different guest molecules inside the SC core of the micelles (exemplary shown for a fluorescence dye), without changing the shape and chemistry of the micelle corona. This makes SCDSA highly valuable for the construction of surface-compartmentalized micelles with tailored functionalities. The modularity of the SCDSA approach allows an easy variation of the corona-forming blocks, which might open the use of patchy SC micelles as compatibilizers in various polymer blend systems. To this end, alternative large-scale preparation methods need to be developed in future.Data availability statement
All data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Financial support by the German Research Foundation (project SCHM2428/3-1) is gratefully acknowledged. We thank Prof. Andreas Greiner for helpful discussions, Rika Schneider for SEC measurements, Dr Lisa Günther and Werner Reichstein for help with confocal fluorescence imaging measurements, and Annika Pfaffenberger for ultramicrotome cutting. R. S and M. S. acknowledge support by the Graduate School of University of Bayreuth. M. S. thanks the Elite Study Program Macromolecular Science within the Elite Network of Bavaria (ENB) for support. We acknowledge support from the keylabs “Electron and Optical Microscopy” and “Synthesis and Molecular Characterization” of the Bavarian Polymer Institute (BPI) at the University of Bayreuth, and Corbion for the donation of D- and L-lactide.References
- W. Li, H. Palis, R. Mérindol, J. Majimel, S. Ravaine and E. Duguet, Chem. Soc. Rev., 2020, 49, 1955–1976 RSC.
- A. K. Pearce, T. R. Wilks, M. C. Arno and R. K. O'Reilly, Nat. Rev. Chem., 2021, 5, 21–45 CrossRef CAS PubMed.
- K. Zhang, M. Jiang and D. Chen, Prog. Polym. Sci., 2012, 37, 445–486 CrossRef CAS.
- A. H. Gröschel and A. H. E. Müller, Nanoscale, 2015, 7, 11841–11876 RSC.
- J. Du and R. K. O'Reilly, Chem. Soc. Rev., 2011, 40, 2402 RSC.
- U. Tritschler, S. Pearce, J. Gwyther, G. R. Whittell and I. Manners, Macromolecules, 2017, 50, 3439–3463 CrossRef CAS.
- I. W. Wyman and G. Liu, Polymer, 2013, 54, 1950–1978 CrossRef CAS.
- M. Karayianni and S. Pispas, J. Polym. Sci., 2021, 59, 1874–1898 CrossRef CAS.
- I. Mirza and S. Saha, ACS Appl. Bio Mater., 2020, 3, 8241–8270 CrossRef CAS.
- A. O. Moughton, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2012, 45, 2–19 CrossRef CAS.
- Q. Yang and K. Loos, Polym. Chem., 2017, 8, 641–654 RSC.
- A. Walther and A. H. E. Müller, Chem. Rev., 2013, 113, 5194–5261 CrossRef CAS PubMed.
- Y. Shao, Y. Ye, D. Sun and Z. Yang, Macromolecules, 2022, 55, 6297–6310 CrossRef CAS.
- C. Marschelke, A. Fery and A. Synytska, Colloid Polym. Sci., 2020, 298, 841–865 CrossRef CAS.
- X. Fan, J. Yang, X. J. Loh and Z. Li, Macromol. Rapid Commun., 2019, 40, 1800203 CrossRef.
- R. Deng, F. Liang, J. Zhu and Z. Yang, Mater. Chem. Front., 2017, 1, 431–443 RSC.
- G. Agrawal and R. Agrawal, ACS Appl. Nano Mater., 2019, 2, 1738–1757 CrossRef CAS.
- A. Walther, M. Hoffmann and A. H. E. Müller, Angew. Chem., 2008, 120, 723–726 CrossRef.
- Y. Lan, J. Choi, H. Li, Y. Jia, R. Huang, K. J. Stebe and D. Lee, Ind. Eng. Chem. Res., 2019, 58, 20961–20968 CrossRef CAS.
- L. C. Bradley, W.-H. Chen, K. J. Stebe and D. Lee, Curr. Opin. Colloid Interface Sci., 2017, 30, 25–33 CrossRef CAS.
- S. Karadkar, A. Tiwari and A. C. Chaskar, Int. Nano Lett., 2023, 13, 93–115 CrossRef PubMed.
- Y. Yi, L. Sanchez, Y. Gao and Y. Yu, Analyst, 2016, 141, 3526–3539 RSC.
- C. Chen, L. Zhang, N. Wang, D. Sun and Z. Yang, Macromol. Rapid Commun., 2023, 44, 2300280 CrossRef CAS.
- J. Wu, C. Feng, X. Ma, S. Liu, C. Zhang, J. Han, L. Wang and Y. Wang, ACS Appl. Polym. Mater., 2024, 6, 1740–1750 CrossRef CAS.
- Y. Bao, J. Chang, Y. Zhang and L. Chen, Chem. Eng. J., 2022, 446, 136959 CrossRef CAS.
- A. Kirillova, L. Ionov, I. V. Roisman and A. Synytska, Chem. Mater., 2016, 28, 6995–7005 CrossRef CAS.
- A. Walther, K. Matussek and A. H. E. Müller, ACS Nano, 2008, 2, 1167–1178 CrossRef CAS PubMed.
- F. I. Seyni and B. P. Grady, Colloid Polym. Sci., 2021, 299, 585–593 CrossRef CAS.
- H. L. He and F. X. Liang, Chin. J. Polym. Sci., 2023, 41, 500–515 CrossRef CAS.
- R. Bahrami, T. I. Löbling, H. Schmalz, A. H. E. Müller and V. Altstädt, Polymer, 2017, 109, 229–237 CrossRef CAS.
- R. Bahrami, T. I. Löbling, A. H. Gröschel, H. Schmalz, A. H. E. Müller and V. Altstädt, ACS Nano, 2014, 8, 10048–10056 CrossRef CAS.
- C. Hils, I. Manners, J. Schöbel and H. Schmalz, Polymers, 2021, 13, 1481 CrossRef CAS PubMed.
- I.-S. Jo, S. Lee, J. Zhu, T. S. Shim and G.-R. Yi, Curr. Opin. Colloid Interface Sci., 2017, 30, 97–105 CrossRef CAS.
- A. H. Gröschel, A. Walther, T. I. Löbling, F. H. Schacher, H. Schmalz and A. H. E. Müller, Nature, 2013, 503, 247–251 CrossRef.
- T. I. Löbling, O. Borisov, J. S. Haataja, O. Ikkala, A. H. Gröschel and A. H. E. Müller, Nat. Commun., 2016, 7, 12097 CrossRef.
- A. Piloni, A. Walther and M. H. Stenzel, Polym. Chem., 2018, 9, 4132–4142 RSC.
- Y. Lu, J. Lin, L. Wang, L. Zhang and C. Cai, Chem. Rev., 2020, 120, 4111–4140 CrossRef CAS.
- B. Jin, Y. Chen, Y. Luo and X. Li, Chin. J. Chem., 2023, 41, 93–110 CrossRef CAS.
- Z. Zhang and S. C. Glotzer, Nano Lett., 2004, 4, 1407–1413 CrossRef CAS PubMed.
- D. J. Lunn, J. R. Finnegan and I. Manners, Chem. Sci., 2015, 6, 3663 RSC.
- J. Schöbel, C. Hils, A. Weckwerth, M. Schlenk, C. Bojer, M. C. A. Stuart, J. Breu, S. Förster, A. Greiner, M. Karg and H. Schmalz, Nanoscale, 2018, 10, 18257–18268 RSC.
- C. Hils, M. Dulle, G. Sitaru, S. Gekle, J. Schöbel, A. Frank, M. Drechsler, A. Greiner and H. Schmalz, Nanoscale Adv., 2020, 2, 438–452 RSC.
- J. Schöbel, M. Burgard, C. Hils, R. Dersch, M. Dulle, K. Volk, M. Karg, A. Greiner and H. Schmalz, Angew. Chem., Int. Ed., 2017, 56, 405–408 CrossRef.
- J. Schöbel, M. Karg, D. Rosenbach, G. Krauss, A. Greiner and H. Schmalz, Macromolecules, 2016, 49, 2761–2771 CrossRef.
- A. Frank, C. Hils, M. Weber, K. Kreger, H. Schmalz and H.-W. Schmidt, Angew. Chem., Int. Ed., 2021, 60, 21767–21771 CrossRef CAS PubMed.
- A. Frank, M. Weber, C. Hils, U. Mansfeld, K. Kreger, H. Schmalz and H. W. Schmidt, Macromol. Rapid Commun., 2022, 43, 2200052 CrossRef CAS.
- J. Schmelz, D. Pirner, M. Krekhova, T. M. Ruhland and H. Schmalz, Soft Matter, 2013, 9, 11173 RSC.
- T. Gegenhuber, M. Krekhova, J. Schöbel, A. H. Gröschel and H. Schmalz, ACS Macro Lett., 2016, 5, 306–310 CrossRef CAS.
- R. Schaller, C. Hils, M. Karg and H. Schmalz, Macromol. Rapid Commun., 2023, 44, 2200682 CrossRef CAS.
- L. MacFarlane, C. Zhao, J. Cai, H. Qiu and I. Manners, Chem. Sci., 2021, 12, 4661–4682 RSC.
- J. Ma, G. Lu, X. Huang and C. Feng, Chem. Commun., 2021, 57, 13259–13274 RSC.
- S. Ganda and M. H. Stenzel, Prog. Polym. Sci., 2020, 101, 101195 CrossRef CAS.
- J. Schmelz, M. Karg, T. Hellweg and H. Schmalz, ACS Nano, 2011, 5, 9523–9534 CrossRef CAS.
- A. M. Oliver, J. Gwyther, M. A. Winnik and I. Manners, Macromolecules, 2018, 51, 222–231 CrossRef CAS.
- A. M. Oliver, R. J. Spontak and I. Manners, Polym. Chem., 2019, 10, 2559–2569 RSC.
- J. Xu, H. Zhou, Q. Yu, I. Manners and M. A. Winnik, J. Am. Chem. Soc., 2018, 140, 2619–2628 CrossRef CAS.
- S. Song, X. Liu, E. Nikbin, J. Y. Howe, Q. Yu, I. Manners and M. A. Winnik, J. Am. Chem. Soc., 2021, 143, 6266–6280 CrossRef CAS.
- J. Xu, H. Zhou, Q. Yu, G. Guerin, I. Manners and M. A. Winnik, Chem. Sci., 2019, 10, 2280–2284 RSC.
- J. R. Finnegan, D. J. Lunn, O. E. C. Gould, Z. M. Hudson, G. R. Whittell, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2014, 136, 13835–13844 CrossRef CAS PubMed.
- Z. Li, B. H. Tan, T. Lin and C. He, Prog. Polym. Sci., 2016, 62, 22–72 CrossRef CAS.
- R. M. Michell, V. Ladelta, E. Da Silva, A. J. Müller and N. Hadjichristidis, Prog. Polym. Sci., 2023, 146, 101742 CrossRef CAS.
- H. Tsuji, Adv. Drug Delivery Rev., 2016, 107, 97–135 CrossRef CAS.
- Y. Xie, W. Yu, T. Xia, R. K. O'Reilly and A. P. Dove, Macromolecules, 2023, 56, 7689–7697 CrossRef CAS.
- L. Sun, A. Pitto-Barry, N. Kirby, T. L. Schiller, A. M. Sanchez, M. A. Dyson, J. Sloan, N. R. Wilson, R. K. O'Reilly and A. P. Dove, Nat. Commun., 2014, 5, 5746 CrossRef CAS.
- V. Ladelta, K. Ntetsikas, G. Zapsas and N. Hadjichristidis, Macromolecules, 2022, 55, 2832–2843 CrossRef CAS.
- A. Arkanji, V. Ladelta, K. Ntetsikas and N. Hadjichristidis, Polymers, 2022, 14, 2431 CrossRef CAS.
- J. Wu, Y. Chen, H. Wang and Y. Li, Giant, 2023, 14, 100150 CrossRef CAS.
- S. H. Kim, J. P. K. Tan, F. Nederberg, K. Fukushima, Y. Y. Yang, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2009, 42, 25–29 CrossRef CAS.
- W. Yu, M. Inam, J. R. Jones, A. P. Dove and R. K. O'Reilly, Polym. Chem., 2017, 8, 5504–5512 RSC.
- C. Cheng, E. Khoshdel and K. L. Wooley, Macromolecules, 2005, 38, 9455–9465 CrossRef CAS.
- S. Karanam, H. Goossens, B. Klumperman and P. Lemstra, Macromolecules, 2003, 36, 3051–3060 CrossRef CAS.
- F. A. Plamper, H. Becker, M. Lanzendörfer, M. Patel, A. Wittemann, M. Ballauff and A. H. E. Müller, Macromol. Chem. Phys., 2005, 206, 1813–1825 CrossRef CAS.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- Physical Properties of Polymers Handbook, ed. J. E. Mark, Springer New York, 2nd edn, 2007 Search PubMed.
- J. Schmelz, A. E. Schedl, C. Steinlein, I. Manners and H. Schmalz, J. Am. Chem. Soc., 2012, 134, 14217–14225 CrossRef CAS PubMed.
- X. Hou, Q. Li and A. Cao, Macromol. Chem. Phys., 2013, 214, 1569–1579 CrossRef CAS.
- G. G. Du Sart, R. Rachmawati, V. Voet, G. A. van Ekenstein, E. Polushkin, G. Ten Brinke and K. Loos, Macromolecules, 2008, 41, 6393–6399 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00449c |
| This journal is © The Royal Society of Chemistry 2024 |