
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Well-defined poly(2-isopropenyl-2-oxazoline) brushes provide fouling resistance and versatility in surface functionalization†
Manisha
Singh
 ,
Lenka
Poláková
,
Andres
de los Santos Pereira
,
Ognen
Pop-Georgievski
,
Jan
Svoboda
,
Tomáš
Riedel
,
Sachin
Gupta
,
Zdeňka
Sedláková
,
Vladimír
Raus
and
Rafał
Poręba
*
,
Lenka
Poláková
,
Andres
de los Santos Pereira
,
Ognen
Pop-Georgievski
,
Jan
Svoboda
,
Tomáš
Riedel
,
Sachin
Gupta
,
Zdeňka
Sedláková
,
Vladimír
Raus
and
Rafał
Poręba
*
Institute of Macromolecular Chemistry, Czech Academy of Sciences, Heyrovského nám. 2, 162 00 Prague 6, Czech Republic. E-mail: poreba@imc.cas.cz
First published on 23rd July 2024
Abstract
Poly(2-isopropenyl-2-oxazoline) (PIPOx), a biocompatible polymer amenable to clean and orthogonal post-polymerization modifications, has recently emerged as a suitable candidate for the preparation of functional polymer brushes via surface-initiated reversible-deactivation radical polymerization (SI RDRP). However, the field currently lacks a universal SI RDRP method that would provide a straightforward control over the PIPOx brush thickness and be applicable to non-planar surfaces. Herein, we designed an aqueous, metallic copper-mediated RDRP (Cu(0)-RDRP) protocol for SI grafting of IPOx that manifests an excellent degree of temporal control over the PIPOx brush thickness up to more than 100 nm. The superior kinetic control was achieved through the use of an all-chlorine initiation/catalytic Cu(0)-RDRP system and careful ligand selection, demonstrating a clear advantage over previous approaches based on brominated initiators. Additionally, we found that using neat water as a reaction medium for the Cu(0) catalyst generation in the standard disproportionation step significantly accelerates the brush growth. Importantly, a surface plasmon resonance analysis demonstrated for the first time the high resistance of PIPOx brushes against non-specific protein fouling, as documented by a significant (96%) decrease in protein deposition from undiluted blood plasma and negligible adsorption from fetal bovine serum and other protein solutions. Finally, we showcased in model scenarios the versatility of the prepared well-defined PIPOx brushes by modifying them with suitable functional carboxylic acids under mild conditions in order to subsequently synthesize graft copolymer brushes or trigger a CuAAC click reaction. Our results highlight PIPOx brushes as an attractive platform for the fabrication of low-fouling, multifunctional surfaces.
Introduction
Grafting of polymer brushes onto surfaces represents an important technique for engineering the physicochemical characteristics of interfaces and introducing desired functionalities.1–3 Surfaces modified with polymer brushes have found applications in diverse fields, including medical diagnostics,4–6 environmental monitoring,7 or cell-harvesting technologies.8–10 2-Isopropenyl-2-oxazoline (IPOx) has recently emerged as a new monomer for polymer brush synthesis, attracting considerable attention due to its dual functionality that combines a polymerizable double bond with a reactive 2-oxazoline ring.11,12 Poly(2-isopropenyl-2-oxazoline) (PIPOx) can thus serve as a universal post-polymerization modification platform13,14 where the pendent 2-oxazoline groups are amenable to highly orthogonal and byproduct-free reactions with thiols15,16 and carboxylic acids17–19 or function as initiating sites for cationic ring-opening polymerization (CROP),12,20–22 enabling diverse applications.13,14IPOx has been polymerized via both uncontrolled12,17,19,23–25 and controlled polymerization methods, e.g., anionic polymerization,20,26 group transfer polymerization,21 and reversible-deactivation radical polymerization (RDRP).27,28 The successful implementation of RDRP, and particularly copper-mediated RDRP (Cu-RDRP), is important since surface-initiated (SI) Cu-RDRP represents a major method of polymer brush synthesis, utilizing the copper catalyst both in the Cu(I) form (typically denoted as atom transfer radical polymerization, ATRP) or in the metallic Cu form (denoted here as Cu(0)-RDRP).29,30 Surprisingly enough, PIPOx brushes have been so far prepared mainly through uncontrolled radical polymerization processes,12,23,31 with two notable exceptions. Cu(0)-RDRP, utilizing a copper plate as the catalyst source,32,33 enabled preparation of PIPOx brushes of a defined thickness, even though comprehensive data on kinetic control were not reported. Unfortunately, this method is limited to planar surfaces as the polymer brush thickness is dependent on the Cu plate distance from the modified surface.33 Very recently, ATRP performed in organic solvents, was applied to SI grafting of IPOx from carbonyl iron particles.28 However, when applied in solution, this protocol was generally less well-controlled than the previously reported aqueous Cu(0)-RDRP of IPOx,27 and a precise control over the brush thickness was not attempted.28 Therefore, the field still lacks a robust and universal synthetic method for the preparation of PIPOx brushes with controlled brush thickness via RDRP. In this respect, the translation of the abovementioned aqueous Cu(0)-RDRP method, previously successfully employed in solution,27 to SI grafting could represent an ideal scenario. The applied exclusively chlorine-based initiation/catalytic system has recently showed promise in controlled polymerization of functional monomers in both aqueous34 and organic35 media, significantly enhancing the polymerization control as compared to traditional bromine-based systems. In this context, it is worth noticing that an overwhelming majority of literature protocols for SI Cu-RDRP grafting make use of brominated initiators, with chlorinated variants employed only in a handful of reports and mostly in organic solvents.36–41 It can be reasonably expected that the comparatively higher polymerization control attained in the all-chlorine aqueous Cu(0)-RDRP of IPOx27 could yield superior control over the brush growth when applied in an SI Cu(0)-RDRP setting. Moreover, the employed aqueous polymerization medium is highly convenient from the perspective of polymer brush synthesis due to both environmental concerns and suitability for organic solvent-sensitive substrates (e.g. plastics).42
It is expected that post-modification of PIPOx brushes can serve as a powerful tool for controlling the brush properties and introducing new functionality. For example, CROP of 2-oxazolines, initiated from PIPOx side groups, afforded highly dense bottle-brush brushes on various substrates.12,23 Further, Raus et al. have recently demonstrated that PIPOx can be easily transformed into ATRP macroinitiators through the reaction of its 2-oxazoline rings with suitable 2-halocarboxylic acids under relatively mild conditions.27 The obtained PIPOx-based ATRP macroinitiators were then successfully used for solution grafting of styrene and methyl methacrylate. Presumably, when applied to PIPOx brushes, similar transformation would significantly expand the range of monomers introducible into the side chains of the bottle-brush brushes/graft copolymer brushes, allowing for fine-tuning of PIPOx brush properties. Nevertheless, the prospects for PIPOx brush post-modification are much wider, particularly when considering the immense number of commercially available (functional) carboxylic acids and thiols.
Importantly, PIPOx is increasingly considered as a promising platform for designing materials for biomedical applications, which is facilitated by the hydrophilic character of the 2-oxazoline side groups.18 Kroneková et al. studied the toxicological profile of PIPOx, revealing that PIPOx is biocompatible and possesses immunomodulative properties.17 Considering the application potential of PIPOx brushes as interfaces in complex biological environments (e.g. blood plasma), it appears desirable to establish the PIPOx brush resistance to non-specific protein adsorption. Unfortunately, such data are currently missing in literature.
In this study, we develop conditions for the preparation of PIPOx brushes via an aqueous all-chlorine Cu-RDRP protocol, exercising a high degree of control over brush thickness. Additionally, we demonstrate, for the first time, the high resistance of PIPOx brushes to non-specific protein adsorption from several single protein solutions and complex biological media. Further, we highlight the utility of PIPOx brushes in post-modification reactions by transforming them, via reactions with azido- and halo-carboxylic acids, into substrates for click reactions and macroinitiators for Cu-RDRP, respectively. Successful model CuAAC click reaction and Cu-RDRP synthesis of graft copolymer brushes are then demonstrated.
Results and discussion
Synthesis and characterization of PIPOx brushes
The major objective of the current study was to develop a synthetic protocol affording a high degree of temporal control over the growth of PIPOx brushes in a wide thickness range under mild experimental conditions. To this end, we decided to utilize the all-chlorine Cu(0)-RDRP system that has been recently successfully used for the controlled polymerization of IPOx in aqueous solutions at r.t.27 In that report, various chlorinated initiators were tested, with best results obtained using 2-chloropropionitrile. However, this initiator's structure does not lend itself to the transformation into a SI Cu-RDRP initiator. Therefore, we focused on another initiator variant, the structurally similar methyl 2-chloropropionate (MCP), that provided rapid polymerization in the original report, particularly when combined with 1,1,4,7,10,10-hexamethyltriethylenetetramine (HMTETA) as a ligand. For the purpose of SI grafting, we synthesized an MCP analogue, 11-(trichlorosilyl)undecyl-2-chloropropionate (TUCP), via a modified literature procedure,43 and used it to modify Si chips to obtain a Cu-RDRP initiator layer (Scheme 1 and Fig. S1†). | ||
| Scheme 1 Modification of the silicon chip surface with a 2-chloropropionate group-bearing initiator and subsequent grafting of IPOx under Cu(0)-RDRP conditions. | ||
We suspected that the experimental conditions developed for solution polymerization of IPOx might not be directly transferable to SI grafting due to the differences in kinetics of these processes.42 In addition, the solution Cu(0)-RDRP of IPOx tended to be highly sensitive to the stoichiometry of the catalytic/initiation system.27 This parameter is markedly different in the SI grafting process due to the comparatively low initiator concentration (unless a sacrificial initiator is used). For this reason, we screened here several ligands as this component has a major impact on the Cu-RDRP equilibrium that determines the overall polymerization control.27 Besides HMTETA, we tested also the following common Cu-RDRP ligands: 1,1,4,7,7-pentamethyldiethylenetriamine (PMDETA), tris[2-(dimethylamino)ethyl]amine (Me6TREN), tris(2-pyridylmethyl)amine (TPMA), and 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane (Me4cyclam).
As can be seen from the development of PIPOx brush thickness with time displayed in Fig. 1, the ligand selection was indeed instrumental from the perspective of brush growth rate and the overall control of the process. TPMA, which was the preferred ligand in the solution Cu(0)-RDRP of IPOx,27 afforded quickly a 10 nm brush the thickness of which increased further only extremely slowly during the experiment duration (120 min). Another active ligand, Me6TREN, showed a rather similar kinetic profile, only the initial rapid hike in brush thickness was more pronounced (ca. 20 nm), with an approximately 30 nm brush reached within 120 min. The high initial rate of brush growth followed by a slow growth period could be ascribed to poor polymerization control in the initial polymerization period, leading to pronounced radical termination. The decreased overall concentration of the remaining “living” chain ends on the surface is expected to suppress further termination events. Additionally, termination leads to the generation of the Cu(II) deactivator44 whose increased (local)45 concentration could further contribute to improving the polymerization control. The slower but controlled development of brush thickness in later polymerization stages appears to be consistent with these assumptions. In contrast, the remaining tested ligands provided a linear increase in brush thickness directly from the polymerization start, which indicates faster establishment of the controlling Cu-RDRP equilibrium. The use of PMDETA led the slowest brush growth observed in our screening, attaining a 11 nm brush in 120 min. However, this process was extremely well controlled as documented by the R2 value (0.996) of the corresponding linear regression fit. This ligand could thus be beneficial in applications where thin but perfectly defined brush layers are required. HMTETA proved to be the most universal option of all ligands tested, affording rather rapid (δd = 58 nm h−1) and well-controlled brush growth throughout the polymerization course, ultimately reaching a ca. 120 nm thick brush layer. This observation appears to confirm the good match between the 2-chloropropionate initiator (MCP) and HMTETA revealed previously in the solution Cu(0)-RDRP of IPOx.27 Finally, we found that Me4cyclam provides even faster IPOx polymerization than HMTETA that, however, appears to be characterized by two distinct kinetic domains (the first domain is visualized by a linear fit in Fig. 1) within the 120 min experiment duration. First, a fast (δd = 132 nm h−1) linear brush growth period was observed for ca. 80 min within which an approximately 180 nm thick brush was reached. Afterwards, the brush growth decelerated (δd = 33 nm h−1), ultimately yielding a 200 nm brush in 120 min. Taken together, these values correspond to the average brush growth rate of δd = 100 nm h−1. To the best of our knowledge, this is the highest growth rate of a PIPOx brush reported, with the Cu plate-mediated Cu(0)-RDRP utilized by Zhang et al. affording δd = 40 nm h−1 only.32
 | ||
| Fig. 1 Influence of the Cu(0)-RDRP ligand on the PIPOx brush growth rate, as measured by spectroscopic ellipsometry. Linear regression fitting curves are used as a visual aid. | ||
It is important to note that, similarly as in the solution Cu(0)-RDRP of IPOx,27 we employed here a disproportionation step during which the Cu(0) catalyst and the Cu(II) deactivator are supposedly generated from a Cu(I) salt in the presence of a ligand.46 In practical terms, we allowed the catalytic system (CuCl/CuCl2 and a ligand) to react in water for 30 min prior to the IPOx addition and polymerization start by transferring the obtained mixture to the flask containing an initiator-modified Si chip. The disproportionation technique, introduced by Haddleton and coworkers, has been previously successfully employed in the polymerization of a range of monomers in aqueous media.46–48 It is of note that the omission of the disproportionation period in the solution polymerization of PIPOx was found to impair the polymerization control.27 Importantly, when under otherwise identical experimental conditions (HMTETA as a ligand) we added IPOx to the mixture prior to the 30 min disproportionation period, emulating thus some typical SI Cu-RDRP protocols,49–51 the brush thickness still grew linearly with time, only at a much lower rate (Fig. 2). This finding is interesting also in the context of the results obtained by Tsarevsky et al. who revealed that the Cu(I)/HMTETA complex shows much lower tendency toward disproportionation in water than complexes with other standard Cu-RDRP ligands.52 Therefore, it is still not completely clear which Cu species is the major Cu-RDRP activator under the conditions used in the present study. Nevertheless, our data clearly highlight the utility of performing the disproportionation step in neat water when attempting the SI Cu(0)-RDRP grafting of IPOx.
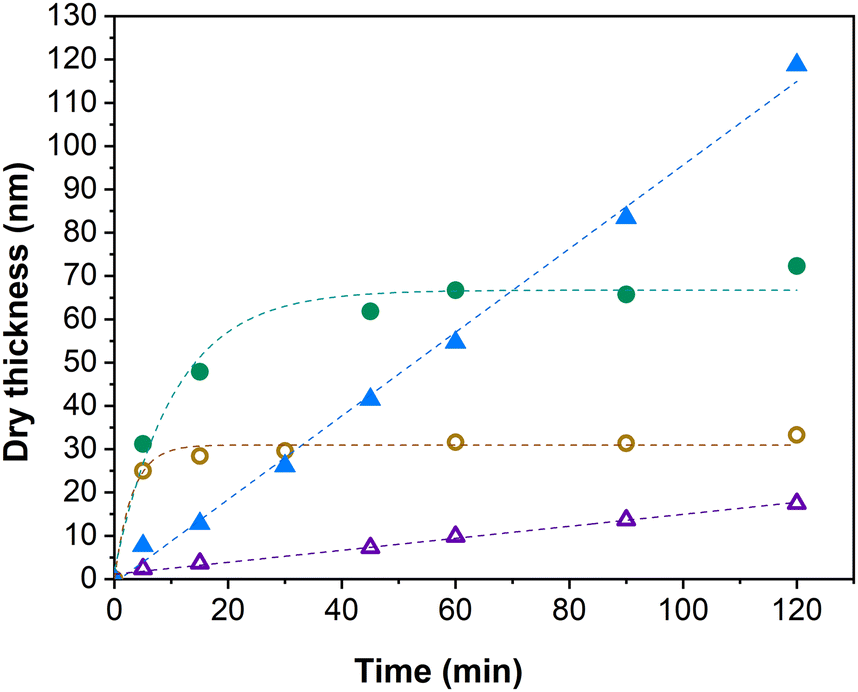 | ||
| Fig. 2 Influence of the initiation group and the medium used in the disproportionation step on the PIPOx brushes growth rate; triangles: TUCP initiator, circles: TUBiB initiator; closed symbols: disproportionation in water, open symbols: disproportionation in the water/IPOX mixture. Standard experimental conditions were used (HMTETA as a ligand). Fitting curves are used as a visual aid. | ||
To demonstrate the advantage of using the all-chlorine initiation/catalytic system in aqueous SI Cu(0)-RDRP grafting of IPOx, we conducted comparison experiments with a typical brominated initiator group. To this end, we modified Si chips with 11-(trichlorosilyl)undecyl-2-bromoisobutyrate, introducing the 2-bromoisobutyrate initiation group that is probably the most extensively used initiator in both solution and SI Cu-RDRP protocols.29,30,53,54 For this comparison, we used HMTETA as a ligand and CuCl/CuCl2 as the catalyst source, performing the polymerization using both the disproportionation regimes detailed above. The obtained kinetic data revealed that PIPOx brushes were grown in an uncontrolled fashion with the Br-based system (Fig. 2). In the experiment where the disproportionation step was performed in the water/IPOx mixture, we observed a rapid increase in brush thickness, reaching an average of 25 nm within the first 5 min. This initial phase was then followed by noticeable deceleration of the growth rate, resulting in the final brush thickness of approximately 33 nm after 120 min. Such a distinctive drop in the polymer brush growth rate is indicative of extensive chain termination events. When disproportionation was performed in neat water, the kinetic profile remained qualitatively similar, only the brush growth was significantly more rapid, reaching the brush thickness of approximately 70 nm in 120 min. The in-water disproportionation thus accelerated the polymerization similarly as observed previously for the chlorinated system. Overall, the data in Fig. 2 demonstrate the superior polymerization control obtained with the all-chlorine system, corroborating some previous observations made in solution Cu(0)-RDRP.27,34,47
The composition of the Cl-based ATRP initiator self-assembled monolayer (SAM) and the synthesized PIPOx brushes was confirmed by an XPS analysis. The XPS spectrum of the TUCP initiator SAM taken in the C 1s region shows the predominance of the ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) –C and –H component at 285.0 eV, attributed to the alkane backbone of the TUCP initiator (Fig. 3). In addition, the spectrum envelope could be resolved with contributions of the –Si bond, the carbon atoms of –O–(C
–C and –H component at 285.0 eV, attributed to the alkane backbone of the TUCP initiator (Fig. 3). In addition, the spectrum envelope could be resolved with contributions of the –Si bond, the carbon atoms of –O–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and Cl––(CO)–O, and the ester group ((O)–O) centered at 284.2, 286.8, and 289.5 eV, respectively. The presence of C–
O) and Cl––(CO)–O, and the ester group ((O)–O) centered at 284.2, 286.8, and 289.5 eV, respectively. The presence of C–![[l with combining low line]](https://www.rsc.org/images/entities/char_006c_0332.gif) groups (spin-split Cl 2p3/2 → Cl 2p1/2 doublet, main contribution centered at 200.6 eV, separation between contributions of 1.7 eV) is further confirmed by the high-resolution spectrum of the Cl 2p region (Fig. S2†).
groups (spin-split Cl 2p3/2 → Cl 2p1/2 doublet, main contribution centered at 200.6 eV, separation between contributions of 1.7 eV) is further confirmed by the high-resolution spectrum of the Cl 2p region (Fig. S2†).
 | ||
| Fig. 3 Representative high-resolution core-level spectra of the Cl-based TUCP initiator layer and of a PIPOx brush taken in the C 1s and N 1s regions. Measured spectra are shown in open circles whereas their fittings are shown in red lines. The individual contributions of different functional groups are displayed in blue lines. | ||
The high-resolution XPS spectrum of a representative PIPOx brush (Fig. 3), grown from the TUCP initiator layer, taken in the C 1s region, displays predominant C–H bonding carbon signals centered at 285.0 eV, corresponding to the methacrylic backbone. The tertiary carbon signal of C*-oxazoline can be identified as a secondary shift centered at 285.5 eV. The carbon atoms present in the 2-oxazoline rings give rise to contributions at 285.9, 286.8, and 287.5 eV due to their C–N and C–O, –O–N– bonds, respectively. Furthermore, in the high-resolution spectra taken in the N 1s region, a single species of nitrogen from the –O–C![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) oxazoline structure can be identified at 398.9 eV. The obtained XPS data indicate that the oxazoline groups are well-preserved following Cu(0)-RDRP, affording PIPOx brushes with the expected chemical structure. Additional confirmation of the chemical structure of the PIPOx brushes was achieved through grazing angle attenuated total reflection Fourier-transform infrared (GAATR-FTIR) spectroscopy measurements (Fig. S3†). The following absorption bands, characteristic to the 2-oxazoline ring, were found in the spectra of PIPOx brushes: 1656 cm−1 (CN stretching), 1130 cm−1 (C–O stretching), and 985, 954, and 925 cm−1 (ring skeletal vibrations).20
oxazoline structure can be identified at 398.9 eV. The obtained XPS data indicate that the oxazoline groups are well-preserved following Cu(0)-RDRP, affording PIPOx brushes with the expected chemical structure. Additional confirmation of the chemical structure of the PIPOx brushes was achieved through grazing angle attenuated total reflection Fourier-transform infrared (GAATR-FTIR) spectroscopy measurements (Fig. S3†). The following absorption bands, characteristic to the 2-oxazoline ring, were found in the spectra of PIPOx brushes: 1656 cm−1 (CN stretching), 1130 cm−1 (C–O stretching), and 985, 954, and 925 cm−1 (ring skeletal vibrations).20
Resistance of PIPOx brushes to non-specific protein adsorption
The ability to resist non-specific protein adsorption, also denoted as fouling, is a prerequisite for surfaces which are to be utilized in biomedical applications or medical diagnostics.55–57 For the first time, we evaluated here the resistance of PIPOx brushes against adsorption from several single-protein solutions and, importantly, also from undiluted blood plasma, which is one of the most challenging biological media.55,56,58–60 Protein fouling onto solid surfaces is a dynamic process involving multiple steps. These include, but are not limited to, initial adsorption of protein molecules, desorption of reversibly bound proteins, and conformational changes of irreversibly adsorbed proteins.61,62 The surface plasmon resonance (SPR) method enables the quantification of the proteins deposited irreversibly on the surface of an SPR chip.In order to determine the resistance of PIPOx brushes against protein fouling, gold-coated SPR chips with a 15 nm thick SiO2 top layer were modified with PIPOx brushes, using the same conditions as for silicon substrates (i.e., the all-chlorine initiation/catalytic system, HMTETA, 30 min in-water disproportionation). Subsequently, the PIPOx-modified SPR chips were exposed to the selected biological fluid for 15 min: undiluted human blood plasma (BP), fibrinogen (Fbg, 1 mg mL−1 in PBS), fetal bovine serum (FBS, 10% in PBS), and bovine serum albumin (BSA, 5 mg mL−1 in PBS). For the PIPOx brushes employed in the fouling measurements, a dry thickness of 15 nm was selected based on results reported for other antifouling coatings, which suffered from increased fouling at thicknesses lower than 10–15 nm.63,64 At the same time, the swelling ratio of the PIPOx brushes (defined as dswollen/ddry) was determined to be 4.0. We therefore did not attempt to measure the fouling resistance of thicker coatings, as the SPR technique is sensitive to changes in refractive index in the vicinity of the interface, and a high thickness can lead to the loss of sensitivity.65
From the obtained SPR sensograms, we calculated the amount of irreversibly bound proteins on the surface of both the bare and PIPOx-modified SPR chips as the difference in the detected baselines before and after the surface exposure to the respective protein solution or blood plasma. The obtained results were compared with the adsorption on the non-modified SiO2-coated SPR sensor that served as a control.
As follows from the data collected in Fig. 4, the modification of SPR sensors with PIPOx brushes effectively prevented fouling from all single protein solutions, whereby the surface mass coverage was below the limit of detection (LOD) of the used SPR instrument. High resistance of the PIPOx brushes to fibrinogen fouling is of a particular importance as this protein is one of the most abundant proteins found in blood plasma and, once adsorbed on a surface, it can unfold and promote adhesion of platelets (involved in blood coagulation), monocytes, and macrophages (associated with the foreign body reaction to biomaterials).66 Moreover, PIPOx brushes also resisted adsorption from diluted FBS, a complex biological medium commonly used in cell culture experiments. Most importantly, however, the surfaces modified with PIPOx brushes led to a significant reduction in the non-specific protein adsorption from undiluted blood plasma, as documented by the observed 96% decrease in protein mass deposition in comparison with an unmodified SPR chip. Overall, the obtained values compare favorably to other well-established fouling-resistant polymer brushes reported in literature, showing a similar ability to resist non-specific protein adsorption from undiluted blood plasma as poly[oligo(ethylene glycol) methacrylate] and poly(2-hydroxyethyl methacrylate) brushes.67,68
 | ||
| Fig. 4 Non-specific protein adsorption after 15 min contact time on bare SPR chips with a SiO2 layer and analogous chips modified with PIPOx brushes. The presented fouling values are the average values ± standard deviation from three independent experiments. An asterisk represents a value below the LOD. | ||
Modification of PIPOx brushes
In order to highlight the vast opportunities for PIPOx brush post-polymerization modification, we focused here on two relevant scenarios: controlled grafting from PIPOx brushes and the PIPOx brush modification via the copper-catalyzed azide–alkyne cycloaddition (CuAAC) click reaction.69–73 For this purpose, specific functionalities, i.e., Cu(0)-RDRP initiators and azide groups, had to be first installed onto PIPOx chains. To this end, the straightforward, byproduct-free reaction of 2-oxazoline rings with carboxylic acids can be conveniently used.13 While PIPOx reactions with (non-functional) carboxylic acids are frequently performed at relatively harsh conditions involving high temperatures (up to 160 °C),17,74,75 it has been shown that milder conditions (e.g., 60 °C) can also be employed, particularly when functional carboxylic acids alpha-substituted with electron-withdrawing groups are used.15,27 These groups increase the carboxylic function acidity, which appears to be the main driving force increasing the reactivity toward the 2-oxazoline ring.11,15,27First, we focused on Cu(0)-RDRP grafting from PIPOx brushes. Surface-immobilized graft copolymers, referred to as graft copolymer brushes or ‘bottle-brush’ brushes (when grafting density is high), feature unique interfacial properties76 that endow them with exceptional flexibility, fast responsiveness to environmental stimuli, and a high degree of mechanical stability.77,78 As substrates, we used here PIPOx brushes with the thickness of 20 nm. In order to ensure grafting exclusively along the PIPOx backbone and not from the terminal units retained after PIPOx synthesis, we substituted the chlorine atoms at chain ends using sodium azide prior to the introduction of Cu-RDRP initiation sites (Scheme 2).70,79 In the next step, we conducted the reaction of the 2-oxazoline side groups with a large excess of 2-chloropropionic acid (CPA) at 60 °C for 24 h (Scheme 2).27
 | ||
| Scheme 2 The synthetic pathway for the fabrication of PIPOx-g-poly(HPMA) brushes from the underlying CPA-functionalized PIPOx layer serving as a Cu(0)-RDRP macroinitiator. | ||
The desired introduction of the 2-chloropropionate groups was verified by GAATR-FTIR (Fig. S3†). Apparently, the PIPOx brush modification with CPA proceeded to a great extent, as evidenced by (i) the almost negligible intensities of bands in the region corresponding to vibrations of 2-oxazoline rings (985–925 cm−1), (ii) the appearance of the sharp signal at 1743 cm−1 corresponding to carbonyl stretching CO vibrations in the newly formed ester bond, and (iii) the amide bond formation, visible as the broadening of the bands at 1656 cm−1 (amide I) and 1525 cm−1 (amide II). Moreover, the presence of 2-chloropropionate moieties in the PIPOx brush macroinitiators was confirmed by an XPS analysis (Fig. 5), where the contribution of chlorine atoms in the corresponding XPS spectra was clearly detected. In addition to the C–Cl group observed after the modification with CPA (spin-split Cl 2p3/2 → Cl 2p1/2 doublet, main contribution centered at 200.7 eV, separation between contributions of 1.6 eV), the 2-chloropropionate group presence was evidenced by the amide and ester species appearing in the high-resolution spectra taken in the C 1s at 288.4 and 289.2 eV. The change in the chemical state of nitrogen upon ring opening is evident in the N 1s spectrum due to the appearance of an amide contribution at 400.31 eV.
 | ||
| Fig. 5 Comparison of high-resolution core-level spectra of the parent PIPOx brush, the CPA-modified PIPOx brush macroinitiator (PIPOx-CPA), and the PIPOx-g-poly(HPMA) brush taken in the Cl 2p, C 1s, and N 1s regions. Measured spectra are shown in open circles whereas their fittings are shown in red lines. The individual contributions of different functional groups are displayed in blue lines. | ||
Having successfully transformed PIPOx brushes to macroinitiators decorated with the 2-chloropropionate Cu-RDRP initiation sites, we proceeded to the Cu(0)-RDRP grafting of poly[N-(2-hydroxypropyl)methacrylamide] (poly(HPMA)) to prepare the PIPOx-g-poly(HPMA) brushes. HPMA was chosen here as a model monomer because it is extensively used in SI grafting to fabricate non-fouling surfaces.60,80,81 The grafting polymerization was initiated by PIPOx macroinitiator brushes (thickness of 20 nm) under similar conditions as those employed for the synthesis of original PIPOx brushes (Me4Cyclam, CuCl/CuCl2, 30 °C, water). We opted for the Me4cyclam ligand here because it has been previously successfully used in SI Cu-RDRP of HPMA.60,67
Fig. 6 presents the evolution of the PIPOx-g-poly(HPMA) brush thickness with time as determined by spectroscopic ellipsometry. Because of the potentially complex, non-linear topology of the brush grafts, the obtained thickness values should be considered only as apparent. In the initial 15 min period, the apparent thickness of the PIPOx-g-poly(HPMA) brush increased rapidly and linearly from the original value of 20 nm (PIPOx macroinitiator brush) to 35 nm. However, following this initial stage, the growth rate was reduced dramatically, with no apparent brush thickness increase registered from 30 min onwards. In standard SI grafting, such a deceleration would indicate the presence of extensive chain termination; however, we cannot disregard here the limited space available for the growth of the poly(HPMA) grafts within the already dense PIPOx brush environment. Consequently, as the polymerization progresses, the chain ends of the growing poly(HPMA) grafts will become less accessible due to steric hindrance, which could contribute to the observed chain growth rate reduction. Note that we also confirmed that the original PIPOx end-chain initiation sites were successfully eliminated in the abovementioned reaction with NaN3 by performing a control experiment in which HPMA grafting was attempted from a (CPA-nonmodified) NaN3-treated PIPOx brush. Indeed, no increase in thickness was observed in this case, confirming that HPMA chains grow exclusively from the newly introduced 2-chloropropionate initiation sites during the PIPOx-g-poly(HPMA) synthesis.
 | ||
| Fig. 6 Kinetics of PIPOx-g-poly(HPMA) brush growth initiated from 20 nm thick CPA-modified PIPOx brushes. | ||
The surfaces of prepared PIPOx-g-poly(HPMA) brushes were analyzed by XPS (Fig. 5). An analysis of the C 1s spectra revealed that grafting of poly(HPMA) chains from the CPA-modified PIPOx macroinitiator brush afforded distinct signals at 285.0, 285.4, 286.1, 286.6, and 287.9 eV, corresponding to –C and –H, *–CO, –N, –O, and N–O carbons, respectively. These signals are characteristic of poly(HPMA) brushes, confirming the successful graft growth from the underlying PIPOx layer.67,68
Next, we studied the possibility of PIPOx brush modification through the CuAAC click reaction. The CuAAC click reaction enables highly effective, straightforward, and selective functionalization of polymer brushes.82 For instance, we have recently used CuAAC click for polymer brush modification with cell-adhesive peptides, which facilitated subsequent adhesion and proliferation of mammalian cells.69,70 Importantly, Weber et al. showed that 4-azidobenzoic acid-modified PIPOx can be readily employed for CuAAC click reaction with an alkyne-substituted substrate.15 Considering the possible vast applications, we explored the possibility of employing this technique also in the modification of our well-defined PIPOx brushes. To this end, we first introduced azide moieties into the parent 20 nm thick PIPOx brushes by the reaction with 11-azido-3,6,9-trioxaundecanoic acid (ADA) conducted in DMF at 60 °C for 24 h (Scheme 3).
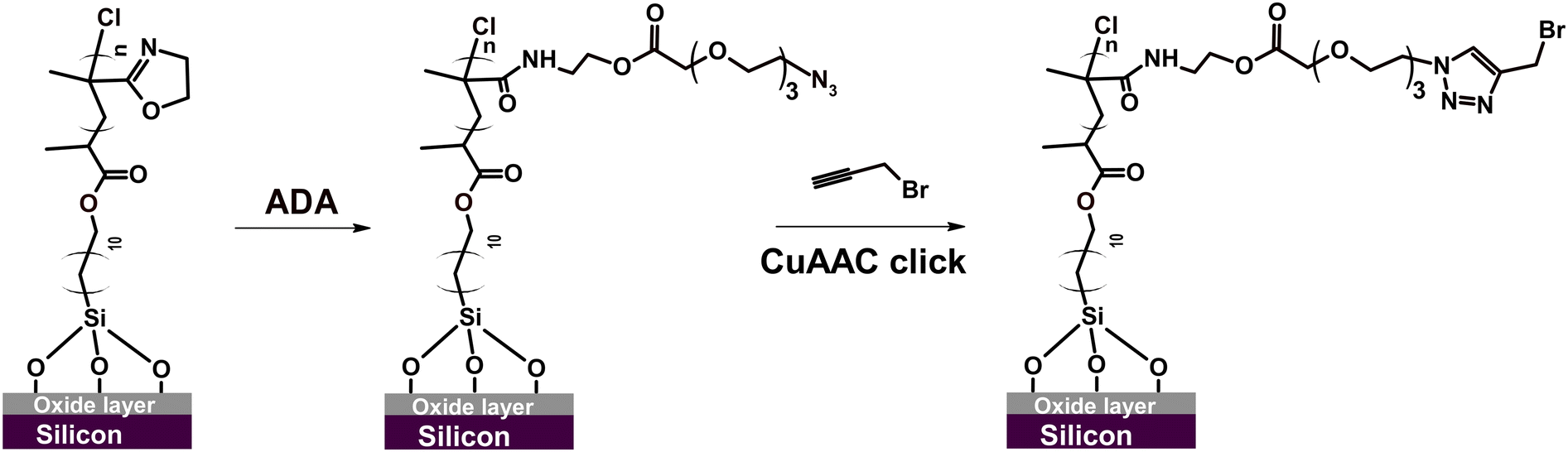 | ||
| Scheme 3 CuAAc click reaction of propargyl bromide with ADA-modified PIPOx brushes. | ||
GAATR-FTIR analysis of the obtained ADA-modified PIPOx brushes (Fig. S3†) indicated that the extent of 2-oxazoline ring modification was somewhat lower in this case as compared to the modification with CPA. This may be partially attributed to the bulkiness of ADA that may limit the extent of modification inside the brush.83 Nevertheless, the bands corresponding to the formed ester carbonyl CO (1743 cm−1) and to amide I and amide II group vibrations are clearly observable. Importantly, the presence of azide groups in the modified brushes was unequivocally confirmed by the appearance of the characteristic band at 2107 cm−1. The incorporation of ADA into the PIPOx brushes was further verified through an XPS analysis. As follows from the C 1s spectra presented in Fig. 7, the modification of PIPOx brushes is accompanied by the increase in the intensity of C–O and O–CO signals. Additionally, three distinct new peaks corresponding to H–CO and N− and N+ nitrogen of azide group are observed at 399.9, 401.0, and 404.9 eV, in N 1s spectrum, respectively.
 | ||
| Fig. 7 High-resolution core-level XPS spectra of the parent PIPOx brush, the ADA-modified PIPOx brush, and the PIPOx brush functionalized through the CuAAC click reaction in the (A) C 1s, (B) N1s and (C) Br 3d regions. Measured spectra are shown in open circles whereas their fittings are shown in red lines. The individual contributions of different functional groups are displayed in blue lines. | ||
As a model alkyne-bearing substrate for the click reaction, we selected propargyl bromide because it contains a primary bromine atom that is readily detectable by XPS. The modification was performed under standard reaction conditions as detailed in Scheme 3. The success of the click reaction was confirmed by the appearance of a new signal in the Br 3d XPS spectrum, as shown in Fig. 7C. Moreover, a decrease in the intensity of the azide signal in N 1s spectra and the appearance of a new signal corresponding to the newly formed triazole ring at 73.1 eV (Fig. 7B) were also observed.
Conclusions
In this study, we developed an aqueous Cu(0)-RDRP protocol yielding PIPOx brushes with precisely controlled thickness via SI grafting. We achieved this by taking advantage of the all-chlorine initiation/catalytic system and the catalyst generation in an in situ disproportionation step, building on our previous results on IPOx polymerization in solution. We demonstrated that the ligand choice has a prominent impact on the polymerization rate and control, highlighting the feasibility of temporal control over brush thickness. In this respect, HMTETA was identified as the most versatile ligand option, providing fast and controlled brush growth throughout the whole evaluated time period. Additionally, PMDETA afforded slow but exceptionally well-controlled brush growth, which may be beneficial when the high-precision control over brush thickness is desirable. Furthermore, we showed that the all-chlorine Cu(0)-RDRP protocol provides superior polymerization control as compared to the traditional systems based on a brominated (2-bromoisobutyrate-bearing) initiator. Additionally, we also revealed that using neat water as the medium during the catalyst disproportionation step leads to a considerable acceleration of the PIPOx brush growth as compared to protocols where the disproportionation step is performed in the water/monomer mixture. Altogether, the developed conditions represent the most universal SI RDRP method for controlled IPOx grafting, avoiding the need of external stimuli such as UV light and suitable for substrates of different geometry. The considerable versatility of the highly-functional PIPOx brushes in terms of post-polymerization modification was successfully demonstrated on two relevant scenarios building upon the facile and byproduct-free PIPOx reaction with functional carboxylic acids: the straightforward synthesis of PIPOx-g-poly(HPMA) graft copolymer brushes via aqueous Cu(0)-RDRP and the CuAAC click reaction on a PIPOx brush decorated with azido groups. Importantly, we also showed for the first time that PIPOx brushes exhibit high resistance to non-specific protein adsorption from complex biological media, which highlights their potential in biomedical and diagnostic applications where minimizing biofouling is essential. This resistance to protein fouling, combined with the possibility of direct functionalization under mild reaction conditions without activation steps, renders PIPOx brushes an attractive choice for the fabrication of advanced multifunctional surfaces. We envision that this approach may be used to functionalize not only Si substrates, but also other substrates amenable to silanization (i.e., glass, metal oxides) by applying the initiator presented herein, or any other material surfaces by employing suitable initiator anchoring ad-layers such as polydopamine.84Author contributions
Conceptualization: R.P., V.R.; data curation: M.S., L.P., A.S.P., O.P.G., J.S., V.R., R.P.; funding acquisition: A.S.P. and T.R.; investigation: M.S., A.S.P., O.P.G., J.S., T.R., S.G., Z.S. and R.P.; methodology: R.P.; resources: O.P.G., Z.S. and R.P.; supervision: R.P.; validation: M.S. and R.P.; visualization: M.S., L.P., J.S., V.R., R.P.; writing – original draft: M.S., V.R., R.P.; writing – review & editing: L.P., A.S.P., O.P.G., J.S., V.R., and R.P.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Czech Science Foundation (Project No. 22-27329S). O.P.G. and J.S. acknowledge funding from the Czech Science Foundation (Project No. 21-16729K). S.G. is a PhD student at Charles University, Prague, Czech Republic.References
- L. Chen, C. Yan and Z. Zheng, Mater. Today, 2018, 21, 38–59 CrossRef CAS.
- I. Lilge and H. Schönherr, Polymer, 2016, 98, 409–420 CrossRef CAS.
- K. A. Günay, N. Schüwer and H.-A. Klok, Polym. Chem., 2012, 3, 2186–2192 RSC.
- J. Slabý, M. Bocková and J. Homola, Sens. Actuators, B, 2021, 347, 130629 CrossRef.
- M. Forinova, A. Pilipenco, I. Visova, N. S. Lynn Jr, J. Dostalek, H. Maskova, V. Honig, M. Palus, M. Selinger, P. Kocova, F. Dycka, J. Sterba, M. Houska, M. Vrabcova, P. Horak, J. Anthi, C. P. Tung, C. M. Yu, C. Y. Chen, Y. C. Huang, P. H. Tsai, S. Y. Lin, H. J. Hsu, A. S. Yang, A. Dejneka and H. Vaisocherova-Lisalova, ACS Appl. Mater. Interfaces, 2021, 13, 60612–60624 CrossRef CAS PubMed.
- M. Badoux, M. Billing and H.-A. Klok, Polym. Chem., 2019, 10, 2925–2951 RSC.
- W. Hu, X. Li, G. He, Z. Zhang, X. Zheng, P. Li and C. M. Li, Biosens. Bioelectron., 2013, 50, 338–344 CrossRef CAS PubMed.
- I. Lilge and H. Schonherr, Angew. Chem., Int. Ed., 2016, 55, 13114–13117 CrossRef CAS PubMed.
- I. Lilge and H. Schonherr, Langmuir, 2016, 32, 838–847 CrossRef CAS PubMed.
- T. Shimizu, M. Yamato, A. Kikuchi and T. Okano, Biomaterials, 2003, 24, 2309–2316 CrossRef CAS PubMed.
- M. N. Leiske, A. M. Mahmoud, N. M. Warne, J. A. C. M. Goos, S. Pascual, V. Montembault, L. Fontaine, T. P. Davis, M. R. Whittaker and K. Kempe, Polym. Chem., 2020, 11, 5681–5692 RSC.
- N. Zhang, M. Steenackers, R. Luxenhofer and R. Jordan, Macromolecules, 2009, 42, 5345–5351 CrossRef CAS.
- B. Kopka, B. Kost and M. Basko, Polym. Chem., 2022, 13, 4736–4746 RSC.
- R. Merckx, J. Becelaere, E. Schoolaert, O. Frateur, M. N. Leiske, D. Peeters, F. A. Jerca, V. V. Jerca, K. De Clerck and R. Hoogenboom, Chem. Mater., 2023, 35, 7079–7093 CrossRef CAS.
- C. Weber, T. Neuwirth, K. Kempe, B. Ozkahraman, E. Tamahkar, H. Mert, C. R. Becer and U. S. Schubert, Macromolecules, 2011, 45, 20–27 CrossRef.
- T. Nishikubo, A. Kameyama and H. Tokai, Polym. J., 1996, 28, 134–138 CrossRef CAS.
- Z. Kronekova, M. Mikulec, N. Petrencikova, E. Paulovicova, L. Paulovicova, V. Jancinova, R. Nosal, P. S. Reddy, G. D. Shimoga, D. Chorvat Jr and J. Kronek, Macromol. Biosci., 2016, 16, 1200–1211 CrossRef CAS PubMed.
- F. A. Jerca, A. M. Anghelache, E. Ghibu, S. Cecoltan, I.-C. Stancu, R. Trusca, E. Vasile, M. Teodorescu, D. M. Vuluga, R. Hoogenboom and V. V. Jerca, Chem. Mater., 2018, 30, 7938–7949 CrossRef CAS.
- V. V. Jerca, F. A. Nicolescu, A. Baran, D. F. Anghel, D. S. Vasilescu and D. M. Vuluga, React. Funct. Polym., 2010, 70, 827–835 CrossRef CAS.
- N. Zhang, S. Huber, A. Schulz, R. Luxenhofer and R. Jordan, Macromolecules, 2009, 42, 2215–2221 CrossRef CAS.
- N. Zhang, S. Salzinger, B. S. Soller and B. Rieger, J. Am. Chem. Soc., 2013, 135, 8810–8813 CrossRef CAS PubMed.
- J.-J. Kang, K. Shehu, C. Sachse, F. A. Jung, C.-H. Ko, L. C. Barnsley, R. Jordan and C. M. Papadakis, Colloid Polym. Sci., 2020, 299, 193–203 CrossRef.
- N. Zhang, T. Pompe, I. Amin, R. Luxenhofer, C. Werner and R. Jordan, Macromol. Biosci., 2012, 12, 926–936 CrossRef CAS PubMed.
- T. Kagiya, T. Matsuda and K. Zushi, J. Macromol. Sci., Part A, 1972, 6, 1349–1372 CrossRef CAS.
- T. Kagiya and T. Matsuda, Polym. J., 1972, 3, 307–314 CrossRef CAS.
- H. Feng, M. Changez, K. Hong, J. W. Mays and N.-G. Kang, Macromolecules, 2016, 50, 54–62 CrossRef.
- V. Raus, A. Hološ, J. Kronek and J. Mosnáček, Macromolecules, 2020, 53, 2077–2087 CrossRef CAS.
- M. Ilčíková, M. Mrlík, M. Cvek, D. Bondarev, Z. Kroneková, J. Kronek, P. Kasák and J. Mosnáček, Macromolecules, 2023, 56, 3904–3912 CrossRef.
- R. Barbey, L. Lavanant, D. Paripovic, N. Schuwer, C. Sugnaux, S. Tugulu and H. A. Klok, Chem. Rev., 2009, 109, 5437–5527 CrossRef CAS PubMed.
- J. O. Zoppe, N. C. Ataman, P. Mocny, J. Wang, J. Moraes and H. A. Klok, Chem. Rev., 2017, 117, 1105–1318 CrossRef CAS PubMed.
- Y. Du, T. Zhang, D. Gieseler, M. Schneider, D. Hafner, W. Sheng, W. Li, F. Lange, E. Wegener, I. Amin and R. Jordan, Chemistry, 2020, 26, 2749–2753 CrossRef CAS PubMed.
- T. Zhang, Y. Du, F. Müller, I. Amin and R. Jordan, Polym. Chem., 2015, 6, 2726–2733 RSC.
- T. Zhang, E. M. Benetti and R. Jordan, ACS Macro Lett., 2019, 8, 145–153 CrossRef CAS PubMed.
- V. Raus and L. Kostka, Polym. Chem., 2019, 10, 564–568 RSC.
- S. Gupta and V. Raus, React. Funct. Polym., 2023, 183, 105509 CrossRef CAS.
- S. Sant and H.-A. Klok, Eur. Polym. J., 2024, 205, 112706 CrossRef CAS.
- M. M. Mecwan, M. J. Taylor, D. J. Graham and B. D. Ratner, Biointerphases, 2019, 14, 041006 CrossRef PubMed.
- Y. Gotoh, H. Suzuki, N. Kumano, T. Seki, K. Katagiri and Y. Takeoka, New J. Chem., 2012, 36, 2171–2175 RSC.
- J. T. Park, J. H. Koh, J. K. Koh and J. H. Kim, Appl. Surf. Sci., 2009, 255, 3739–3744 CrossRef CAS.
- C. L. Chochos, A. A. Stefopoulos, S. Campidelli, M. Prato, V. G. Gregoriou and J. K. Kallitsis, Macromolecules, 2008, 41, 1825–1830 CrossRef CAS.
- M.-J. Chang, J.-Y. Tsai, C.-W. Chang, H.-M. Chang and G. J. Jiang, J. Appl. Polym. Sci., 2007, 103, 3680–3687 CrossRef CAS.
- C. W. Pester, H.-A. Klok and E. M. Benetti, Macromolecules, 2023, 56, 9915–9938 CrossRef CAS.
- C. Rodriguez-Emmenegger, S. Janel, A. de los Santos Pereira, M. Bruns and F. Lafont, Polym. Chem., 2015, 6, 5740–5751 RSC.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- R. E. Behling, B. A. Williams, B. L. Staade, L. M. Wolf and E. W. Cochran, Macromolecules, 2009, 42, 1867–1872 CrossRef CAS.
- Q. Zhang, P. Wilson, Z. Li, R. McHale, J. Godfrey, A. Anastasaki, C. Waldron and D. M. Haddleton, J. Am. Chem. Soc., 2013, 135, 7355–7363 CrossRef CAS PubMed.
- G. R. Jones, A. Anastasaki, R. Whitfield, N. Engelis, E. Liarou and D. M. Haddleton, Angew. Chem., Int. Ed., 2018, 57, 10468–10482 CrossRef CAS PubMed.
- A. Anastasaki, V. Nikolaou, G. Nurumbetov, P. Wilson, K. Kempe, J. F. Quinn, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Chem. Rev., 2016, 116, 835–877 CrossRef CAS PubMed.
- A. de los Santos Pereira, T. Riedel, E. Brynda and C. Rodriguez-Emmenegger, Sens. Actuators, B, 2014, 202, 1313–1321 CrossRef CAS.
- C. Rodriguez-Emmenegger, O. A. Avramenko, E. Brynda, J. Skvor and A. B. Alles, Biosens. Bioelectron., 2011, 26, 4545–4551 CrossRef CAS PubMed.
- C. Rodriguez-Emmenegger, O. Kylian, M. Houska, E. Brynda, A. Artemenko, J. Kousal, A. B. Alles and H. Biederman, Biomacromolecules, 2011, 12, 1058–1066 CrossRef CAS PubMed.
- N. V. Tsarevsky, W. A. Braunecker and K. Matyjaszewski, J. Organomet. Chem., 2007, 692, 3212–3222 CrossRef CAS.
- K. Matyjaszewski and N. V. Tsarevsky, Nat. Chem., 2009, 1, 276–288 CrossRef CAS PubMed.
- C. Boyer, N. A. Corrigan, K. Jung, D. Nguyen, T. K. Nguyen, N. N. Adnan, S. Oliver, S. Shanmugam and J. Yeow, Chem. Rev., 2016, 116, 1803–1949 CrossRef CAS PubMed.
- C. Rodriguez Emmenegger, E. Brynda, T. Riedel, Z. Sedlakova, M. Houska and A. B. Alles, Langmuir, 2009, 25, 6328–6333 CrossRef CAS PubMed.
- C. Blaszykowski, S. Sheikh and M. Thompson, Biomater. Sci., 2015, 3, 1335–1370 RSC.
- J. Homola, Chem. Rev., 2008, 108, 462–493 CrossRef CAS PubMed.
- Z. Riedelová, A. de los Santos Pereira, J. Svoboda, O. Pop-Georgievski, P. Májek, K. Pecánková, F. Dycka, C. Rodriguez-Emmenegger and T. Riedel, Macromol. Biosci., 2022, 22, e2200247 CrossRef PubMed.
- T. Riedel, Z. Riedelova-Reicheltova, P. Majek, C. Rodriguez-Emmenegger, M. Houska, J. E. Dyr and E. Brynda, Langmuir, 2013, 29, 3388–3397 CrossRef CAS PubMed.
- C. Rodriguez-Emmenegger, E. Brynda, T. Riedel, M. Houska, V. Subr, A. B. Alles, E. Hasan, J. E. Gautrot and W. T. Huck, Macromol. Rapid Commun., 2011, 32, 952–957 CrossRef CAS PubMed.
- Q. Wei, T. Becherer, S. Angioletti-Uberti, J. Dzubiella, C. Wischke, A. T. Neffe, A. Lendlein, M. Ballauff and R. Haag, Angew. Chem., Int. Ed., 2014, 53, 8004–8031 CrossRef CAS PubMed.
- R. E. Baier and R. C. Dutton, J. Biomed. Mater. Res., 1969, 3, 191–206 CrossRef CAS PubMed.
- C. Zhao, L. Li, Q. Wang, Q. Yu and J. Zheng, Langmuir, 2011, 27, 4906–4913 CrossRef CAS PubMed.
- M. Vorobii, O. Pop-Georgievski, A. de los Santos Pereira, N. Y. Kostina, R. Jezorek, Z. Sedláková, V. Percec and C. Rodriguez-Emmenegger, Polym. Chem., 2016, 7, 6934–6945 RSC.
- J. Homola, Surface Plasmon Resonance Based Sensors, Springer-Verlag Berlin Heidelberg, 2006 Search PubMed.
- T. A. Horbett, J. Biomed. Mater. Res., Part A, 2018, 106, 2777–2788 CrossRef CAS PubMed.
- F. Surman, T. Riedel, M. Bruns, N. Y. Kostina, Z. Sedlakova and C. Rodriguez-Emmenegger, Macromol. Biosci., 2015, 15, 636–646 CrossRef CAS PubMed.
- M. Vorobii, A. de los Santos Pereira, O. Pop-Georgievski, N. Y. Kostina, C. Rodriguez-Emmenegger and V. Percec, Polym. Chem., 2015, 6, 4210–4220 RSC.
- R. Poreba, A. de Los Santos Pereira, R. Pola, S. Jiang, O. Pop-Georgievski, Z. Sedlakova and H. Schonherr, Macromol. Biosci., 2020, 20, e1900354 CrossRef PubMed.
- A. Schulte, A. de los Santos Pereira, R. Pola, O. Pop-Georgievski, S. Y. Jiang, I. Romanenko, M. Singh, Z. Sedláková, H. Schönherr and R. Poreba, Macromol. Biosci., 2023, 23, e2200472 CrossRef PubMed.
- P. V. Mendonça, A. C. Serra, A. V. Popov, T. Guliashvili and J. F. J. Coelho, React. Funct. Polym., 2014, 81, 1–7 CrossRef.
- M. Barbosa, N. Vale, F. M. Costa, M. C. Martins and P. Gomes, Carbohydr. Polym., 2017, 165, 384–393 CrossRef CAS PubMed.
- M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- V. V. Jerca, F. A. Nicolescu, R. Trusca, E. Vasile, A. Baran, D. F. Anghel, D. S. Vasilescu and D. M. Vuluga, React. Funct. Polym., 2011, 71, 373–379 CrossRef CAS.
- F. A. Jerca, V. V. Jerca, A. M. Anghelache, D. M. Vuluga and R. Hoogenboom, Polym. Chem., 2018, 9, 3473–3478 RSC.
- A. L. Liberman-Martin, C. K. Chu and R. H. Grubbs, Macromol. Rapid Commun., 2017, 38, 1700058 CrossRef PubMed.
- B. Zhao, J. Phys. Chem. B, 2021, 125, 6373–6389 CrossRef CAS PubMed.
- R. Wang, Q. Wei, W. Sheng, B. Yu, F. Zhou and B. Li, Angew. Chem., Int. Ed., 2023, 62, e202219312 CrossRef CAS PubMed.
- V. Parrillo, A. de Los Santos Pereira, T. Riedel and C. Rodriguez-Emmenegger, Anal. Chim. Acta, 2017, 971, 78–87 CrossRef CAS PubMed.
- A. R. Kuzmyn, A. T. Nguyen, L. W. Teunissen, H. Zuilhof and J. Baggerman, Langmuir, 2020, 36, 4439–4446 CrossRef CAS PubMed.
- E. Roeven, A. R. Kuzmyn, L. Scheres, J. Baggerman, M. M. J. Smulders and H. Zuilhof, Langmuir, 2020, 36, 10187–10199 CrossRef CAS PubMed.
- S. V. Orski, G. R. Sheppard, S. Arumugam, R. M. Arnold, V. V. Popik and J. Locklin, Langmuir, 2012, 28, 14693–14702 CrossRef CAS PubMed.
- N. Schüwer, T. Geue, J. P. Hinestrosa and H.-A. Klok, Macromolecules, 2011, 44, 6868–6874 CrossRef.
- O. Pop-Georgievski, C. Rodriguez-Emmenegger, A. de los Santos Pereira, V. Proks, E. Brynda and F. Rypacek, J. Mater. Chem. B, 2013, 1, 2859–2867 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00424h |
| This journal is © The Royal Society of Chemistry 2024 |