
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Kinetic investigation of photoiniferter-RAFT polymerization in continuous flow using inline NMR analysis†
Magdalena A.
Bereś
 a,
Bo
Zhang
b,
Tanja
Junkers
*b and
Sébastien
Perrier
*acd
a,
Bo
Zhang
b,
Tanja
Junkers
*b and
Sébastien
Perrier
*acd
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: s.perrier@warwick.ac.uk
bPolymer Reaction Design Group, School of Chemistry, Monash University, Clayton, VIC 3800, Australia. E-mail: tanja.junkers@monash.edu
cWarwick Medical School, University of Warwick, Coventry, CV4 7AL, UK
dFaculty of Pharmacy and Pharmaceutical Sciences, Monash University, Parkville, VIC 3052, Australia
First published on 26th June 2024
Abstract
Photo reversible deactivation radical polymerization and, in particular, photoiniferter-reversible addition–fragmentation chain transfer (PI-RAFT) polymerization have become popular approaches to polymer synthesis in recent years. There is, however, a lack of fundamental investigations concerning the mechanism and kinetics of such reactions. Herein, we apply an automated continuous flow platform featuring inline NMR analysis that allows for rapid kinetic screening via transient timesweep experiments for detailed investigation of the PI-RAFT of two model monomers with different propagation rates and radical stabilities—methyl acrylate (MA) and methyl methacrylate (MMA). The effect of the structure of the RAFT agent on polymerization kinetics is studied. For the polymerization of MA, RAFT agents with a more stabilised R group lead to an induction period whose extent can be tuned by varying the light intensity. Faster polymerization of MA with xanthates than with trithiocarbonates suggests the important role of reversible termination in the PI-RAFT mechanism. The slower apparent rate of propagation for the polymerization of acrylates compared to polymerization of methacrylates, when mediated by trithiocarbonate RAFT agents, indicates that polymerization of MA is retarded due to the lower radical stability of the propagating radical compared to methacrylic radicals.
Introduction
Photoiniferter reversible addition–fragmentation chain transfer (PI-RAFT) polymerization is one of the simplest photopolymerization methods available as it does not require any exogenous initiator or catalyst. The RAFT agent itself acts as the initiator, chain transfer agent (CTA) and terminating agent, as in the iniferter system originally reported by Otsu and coworkers.1–3 However, in the PI-RAFT case, both chain transfer and termination are reversible, leading to a greater level of control. Upon activation with either UV4 or visible light,5,6 the RAFT agent undergoes homolytic cleavage at its C–S bond to release a C-centred radical (derived from the RAFT agent R group), which can act as an initiator, and a thiyl radical which can reversibly terminate with the propagating radical and enter chain transfer RAFT equilibrium. Interestingly, with this mechanism a persistent radical effect is established, and the dominant form of termination reforms a macroRAFT agent rather than producing dead polymer chains. For a detailed mechanism, the reader is directed to reviews on the topic.7,8 Albeit visible light is considered a mild energy source, it can lead to photodegradation of the RAFT agent and/or RAFT polymer end group, particularly for RAFT agents and monomers which give relatively stable radicals upon photodissociation.9 Care must be exercised not to oversaturate the system with light and often stopping the reaction at earlier conversions is necessary to maintain good livingness, as increased light intensity and exposure can lead to increased termination.10,11 Nevertheless, when appropriately tuned, PI-RAFT exhibits unprecedent livingness, as demonstrated by Carmean et al. in the synthesis of ultrahigh molecular weight polymers via PI-RAFT.12,13 Yet, higher light intensity leads to increased photodissociation of the RAFT agent and increased radical flux, improving the rate of polymerization.14,15 However, the rate of PI-RAFT can also be regulated by changing the wavelength of light to target the π to π* or n to π* electronic transitions of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond of the CTA, with the latter leading to faster polymerization despite a lower molar extinction coefficient.16,17 Moreover, within an electronic transition, the wavelength of light can be further optimised and often the most activating wavelength of light is red-shifted relative to the absorption maximum.18–20 An alternative approach to tune the rate of photopolymerization is temperature control due to the thermal nature of the propagation step, as neatly demonstrated by Junkers and co-workers, who reported increased monomer conversions for the PI-RAFT polymerization of isoprene when the reaction temperature was increased to 145 °C.21 Rubens et al. reported significant enhancement in the rate of photopolymerization of methacrylic monomers when polymerization was carried out at 90 °C in continuous flow.22 The reaction was completed in an hour and due to such a short reaction time, no notable degradation of trithiocarbonate was observed. Although designing a heated batch photoreactor is difficult due to limited light penetration and the necessity to keep the system under an inert atmosphere, in a flow set-up the reactor can be simply wrapped around a light source and submerged in an oil bath. The attractiveness of flow chemistry for photochemistry is reflected in the plethora of publications in the field of Reversible-Deactivation Radical Polymerizations (RDRP).11,23–28 The narrow channels in flow reactors and hence their high ratio of surface area to volume lead to improved heat transfer, which reduces the number of side reactions. More importantly, for photo reactions the short optical pathway results in uniform and complete light exposure, accelerating photoreactions sometimes by order of magnitudes compared to classical batch chemistry.29–35 Together these make flow photopolymerization both more reproducible and scalable. Further, flow set-ups allow for the easy incorporation of various online and inline analysis instruments.36–38 The entire reaction and analysis suite can be automated, enabling self-optimizing reactor set-ups by use of feedback loops and machine learning.39 Over the years, inline analysis featuring benchtop NMR has been developed for reaction monitoring for polymer chemistry and the specific targeting of monomer conversions from machine-learning algorithms.36,40 Low-field benchtop NMR is equipped with a permanent rather than a superconducting magnet and hence does not require extensive cryogenic cooling, leading to lower cost, maintenance and size than high-field NMR.41,42 Although it gives lower resolution than high-field NMR, it is perfectly suited for monitoring monomer conversion36 and bypasses the need for a special integrated flow tube as required for high-field NMR.43 While batch reactors can be continuously sampled or reaction streams looped through an NMR spectrometer, flow reactors are somewhat more dynamic and direct in measurement. Usually, inline NMR is performed under steady-state conditions. Steady-state kinetics require separate reaction conditions for each time point (residence time) unless a probe can be placed alongside the reactor.44 This leads to enormous waste of reagents and time. Mozharov et al. introduced a dynamic flow experiment – transient timesweep kinetics – in which the reaction stream can be treated as a series of mini batch reactors (reaction plugs) with individual reaction times and the reaction progress can be analysed at the outlet of the reactor, a concept that Haven et al. later adopted for the online mass spectrometric monitoring of polymerization reactions.45,46 In a single flow-experiment, the reactor is stabilised at a given flow rate and after a stabilisation period, a step change in flow rate is introduced. As a consequence, different reaction times are traversed and sampled until the reactor stabilises at a new flow rate. An alternative approach involves a controlled flow rate ramp which allows for fine tuning of the rate of change in residence time; however, such a set-up is more complicated to implement.47 Recently, Drelinkiewicz et al. reported a new method for timesweep kinetics, specifically concerning photochemical reactions in which no flow rate change is applied, but after stabilisation of the reactor the light is simply turned off, thus creating a gradient of effective reaction time (by choosing irradiation time relative to reactor residence time), assuming no reaction takes place when the light is turned off.48 Junkers and co-workers gave a recent overview of timesweep transient kinetic measurements in flow for polymer reaction monitoring49 and used this platform to study the PI-RAFT polymerization of various methacrylic monomers and the effect of light intensity and trithiocarbonate RAFT agent on polymerization kinetics.15 A lower target degree of polymerization and increased light intensity resulted in faster rates of polymerization of methyl methacrylate due to increased concentration and rate of photolysis of the RAFT agent, respectively. Further, an increased reaction rate with an oligomeric RAFT agent with two inserted methyl methacrylate units was observed, highlighting the importance of RAFT pre-equilibrium on polymerization kinetics.
S bond of the CTA, with the latter leading to faster polymerization despite a lower molar extinction coefficient.16,17 Moreover, within an electronic transition, the wavelength of light can be further optimised and often the most activating wavelength of light is red-shifted relative to the absorption maximum.18–20 An alternative approach to tune the rate of photopolymerization is temperature control due to the thermal nature of the propagation step, as neatly demonstrated by Junkers and co-workers, who reported increased monomer conversions for the PI-RAFT polymerization of isoprene when the reaction temperature was increased to 145 °C.21 Rubens et al. reported significant enhancement in the rate of photopolymerization of methacrylic monomers when polymerization was carried out at 90 °C in continuous flow.22 The reaction was completed in an hour and due to such a short reaction time, no notable degradation of trithiocarbonate was observed. Although designing a heated batch photoreactor is difficult due to limited light penetration and the necessity to keep the system under an inert atmosphere, in a flow set-up the reactor can be simply wrapped around a light source and submerged in an oil bath. The attractiveness of flow chemistry for photochemistry is reflected in the plethora of publications in the field of Reversible-Deactivation Radical Polymerizations (RDRP).11,23–28 The narrow channels in flow reactors and hence their high ratio of surface area to volume lead to improved heat transfer, which reduces the number of side reactions. More importantly, for photo reactions the short optical pathway results in uniform and complete light exposure, accelerating photoreactions sometimes by order of magnitudes compared to classical batch chemistry.29–35 Together these make flow photopolymerization both more reproducible and scalable. Further, flow set-ups allow for the easy incorporation of various online and inline analysis instruments.36–38 The entire reaction and analysis suite can be automated, enabling self-optimizing reactor set-ups by use of feedback loops and machine learning.39 Over the years, inline analysis featuring benchtop NMR has been developed for reaction monitoring for polymer chemistry and the specific targeting of monomer conversions from machine-learning algorithms.36,40 Low-field benchtop NMR is equipped with a permanent rather than a superconducting magnet and hence does not require extensive cryogenic cooling, leading to lower cost, maintenance and size than high-field NMR.41,42 Although it gives lower resolution than high-field NMR, it is perfectly suited for monitoring monomer conversion36 and bypasses the need for a special integrated flow tube as required for high-field NMR.43 While batch reactors can be continuously sampled or reaction streams looped through an NMR spectrometer, flow reactors are somewhat more dynamic and direct in measurement. Usually, inline NMR is performed under steady-state conditions. Steady-state kinetics require separate reaction conditions for each time point (residence time) unless a probe can be placed alongside the reactor.44 This leads to enormous waste of reagents and time. Mozharov et al. introduced a dynamic flow experiment – transient timesweep kinetics – in which the reaction stream can be treated as a series of mini batch reactors (reaction plugs) with individual reaction times and the reaction progress can be analysed at the outlet of the reactor, a concept that Haven et al. later adopted for the online mass spectrometric monitoring of polymerization reactions.45,46 In a single flow-experiment, the reactor is stabilised at a given flow rate and after a stabilisation period, a step change in flow rate is introduced. As a consequence, different reaction times are traversed and sampled until the reactor stabilises at a new flow rate. An alternative approach involves a controlled flow rate ramp which allows for fine tuning of the rate of change in residence time; however, such a set-up is more complicated to implement.47 Recently, Drelinkiewicz et al. reported a new method for timesweep kinetics, specifically concerning photochemical reactions in which no flow rate change is applied, but after stabilisation of the reactor the light is simply turned off, thus creating a gradient of effective reaction time (by choosing irradiation time relative to reactor residence time), assuming no reaction takes place when the light is turned off.48 Junkers and co-workers gave a recent overview of timesweep transient kinetic measurements in flow for polymer reaction monitoring49 and used this platform to study the PI-RAFT polymerization of various methacrylic monomers and the effect of light intensity and trithiocarbonate RAFT agent on polymerization kinetics.15 A lower target degree of polymerization and increased light intensity resulted in faster rates of polymerization of methyl methacrylate due to increased concentration and rate of photolysis of the RAFT agent, respectively. Further, an increased reaction rate with an oligomeric RAFT agent with two inserted methyl methacrylate units was observed, highlighting the importance of RAFT pre-equilibrium on polymerization kinetics.
Herein, we apply a previously developed transient sweep methodology to study the kinetics of photoiniferter RAFT polymerization in greater detail.15,50 The aforementioned simplicity of PI-RAFT works both for its advantage and its disadvantage. While PI-RAFT is usually easy to implement, optimisation is more cumbersome. It is further hindered by the lack of consistency in reporting experimental details. Within the published literature, various incomparable photoreactor setups are reported with light intensity values often lacking. Here, we use an automated continuous flow photoreactor platform at elevated temperature featuring inline NMR analysis and light intensity control to overcome some of the common sources of error. In this system, the light intensity is controlled with a variable power supply and quantified with a light meter. Further, automated inline NMR analysis dramatically increases the number of data points collected per experiment and reduces the error associated with sample preparation and handling.50 We apply this platform to study the effect of structure and reactivity of the RAFT agent on the control over and kinetics of photoiniferter polymerization. While control over RAFT polymerization is usually governed by a reactivity match between CTA and monomer,51 here we investigate photoiniferter RAFT specific subtleties over the control and kinetics of polymerization of two model monomers—methyl acrylate and methyl methacrylate—which have different radical stability of the corresponding propagating radicals and rates of propagation. We investigate how kinetics can be tuned by varying the stability of the CTA carbon-centred radical via systematic variation of the RAFT agent R group structure, which affects the rate of photolysis and reinitiation efficiency of the CTA. Xanthate and trithiocarbonate RAFT agents are compared to highlight importance of both reversible termination and degenerative chain transfer for the success of photoiniferter RAFT. Furthermore, we show how the monomer reactivity and stability affect the rate-determining step of the photoiniferter process.
Results and discussion
We designed a library of xanthate and trithiocarbonate iniferter/RAFT agents, with systematically varied R groups of different radical stabilities and the same alkyl chain on the Z group (Fig. 1). While xanthates are known to perform poorly with more activated monomers (MAMs) in conventional thermal RAFT,51,52 they provide significant advantages in photoiniferter RAFT. Due to their lower bond dissociation energy, the quantum yield for homolytic bond cleavage for xanthates is higher than that of trithiocarbonates, making them good photoinitiators.53 This was exploited by Lehnen et al., who demonstrated that doping a trithiocarbonate PI-RAFT process with small quantities of xanthate significantly increases the rate of polymerization.54 The same feature leads to high conversions for xanthate-mediated polymerization even at higher degrees of polymerization,55 while significant rate retardation is observed for trithiocarbonates.56 Further, fast photodissociation of xanthates increases the role of reversible deactivation in the photoiniferter process and this enabled Easterling et al. to invert the sequence of block copolymers.57 This library of RAFT agents was used with two model monomers characterised by different rates of propagation and stability of the propagating radicals – methyl acrylate (MA) and methyl methacrylate (MMA).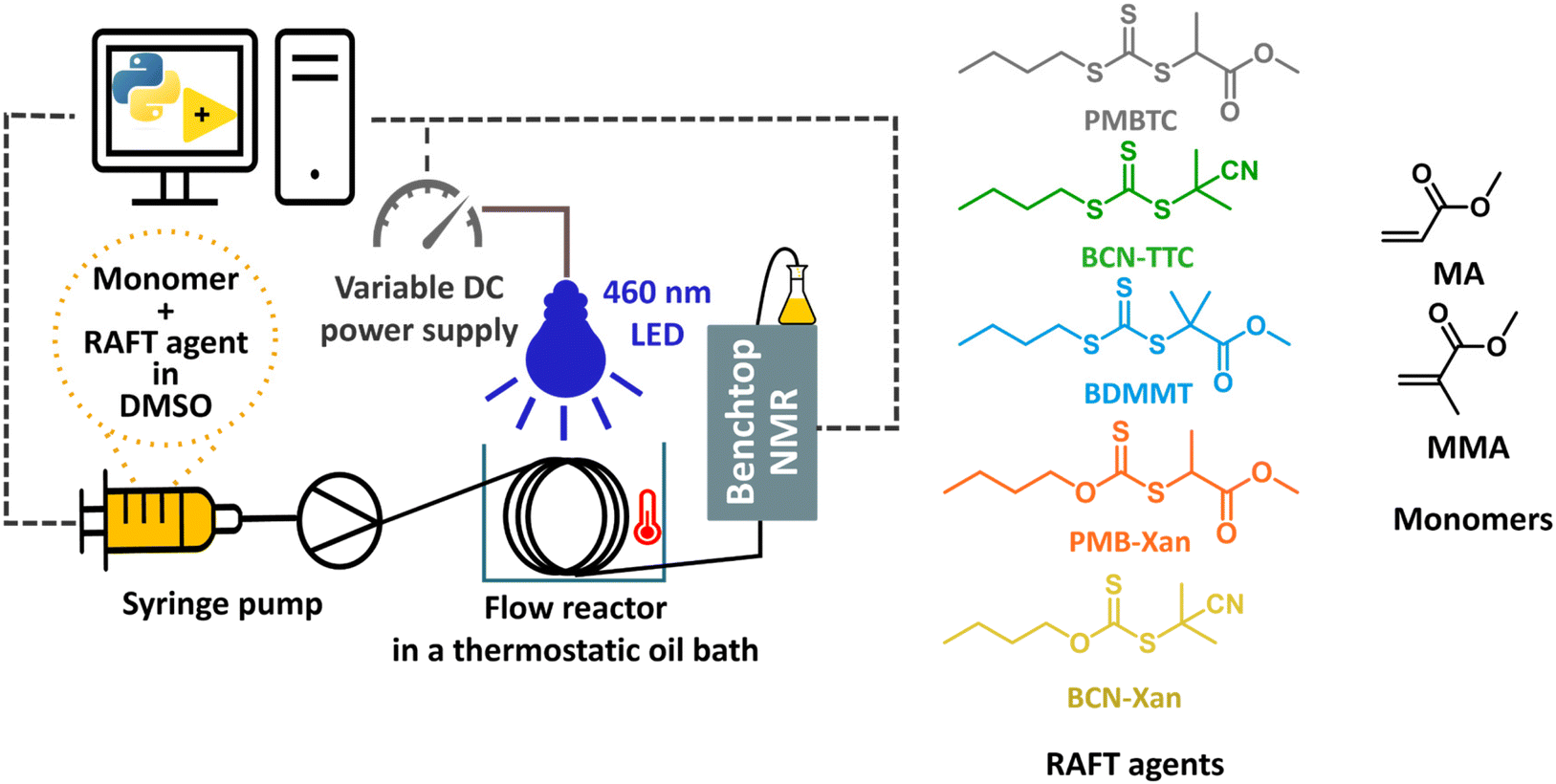 | ||
| Fig. 1 Left: the photo-flow reactor set-up consisting of syringe pump, flow reactor, blue light source (460 nm) with controller, benchtop NMR for inline analysis and computer. Right: structures of RAFT agents and monomers studied. | ||
The investigation started with methyl 2-(((butylthio)carbonothioyl)thio)propanoate (PMBTC) as RAFT agent for the polymerization of methyl acrylate (MA) in dimethyl sulfoxide (DMSO), target degree of polymerization 50, under blue light irradiation (460 nm) (Fig. 2) as this RAFT agent is well known to perform well in the PI-RAFT polymerization of acrylic monomers under blue light.5 The carbon-centred radical derived from PMBTC resembles a methyl acrylate propagating radical, so we anticipated the fast establishment of RAFT equilibrium, without an induction period. Polymerization was carried out at elevated temperature (70 °C) to ensure fast polymerization at lower light intensities to minimise photodegradation of the RAFT agent. MA polymerization kinetics were followed from 4 min onwards (flow rate 0.45 mL min−1) as shorter reaction times require faster flow rates, which can lead to deviations in NMR measurements.36,58,59 Indeed, when a control photoreaction in the absence of RAFT agent was run, significant data scatter was observed for shorter reaction times (ESI Fig. 8†). Interestingly, we found that the MMA signal stabilised faster and polymerizations could be followed from 2 min onwards (flow rate 0.225 mL min−1) without observing such a deviation which can be an effect of different relaxation times of both monomers.60 For molecules with longer T1 relaxation times, nuclei may not have enough time to align along the z axis, leading to an underestimated signal intensity, which will be exacerbated at a faster flow rate as the reaction mixture spends less time in the magnetic field.43 Light intensities from 5.3 mW cm−2 to 22.2 mW cm−2 were screened and it was found that the apparent rate of propagation increased linearly with light intensity, as reported previously for PI-RAFT polymerization of methyl methacrylate,15 which confirms that the RAFT agent plays the role of an initiator (Fig. 2). This is different from the square root relationship reported for PET-RAFT polymerization.61 For light intensities below 10 mW cm−2, an induction period, defined as a period of no or minimal monomer conversion without the formation of oligomers, was observed, which gradually decreased until it completely disappeared at 15 mW cm−2. It is important to note here that a small deviation in monomer conversion was observed—while no monomer conversion was detected in the first several minutes of polymerization, as confirmed with offline high-field NMR measurements, some monomer conversion (∼5%) was observed for online sweep experiment. This, however, does not affect the analysis of the length of the inhibition period or rate of polymerization, as the slope of the kinetic curve remains unaffected. Such deviations are inherent to continuous inline NMR monitoring and were also observed in control experiments with no RAFT agent (ESI, Fig. 8†). This can be further exacerbated by incorrect baseline correction and noisy signal integration.15 Increasing light intensity is analogous to increasing temperature in conventional radical polymerization to induce faster decomposition of a thermal initiator. RDRP techniques rely on the fast and uniform initiation of polymerization and hence the temperature of the reaction and the initiator concentration need to be carefully selected. In PI-RAFT a higher concentration of CTA indeed leads to a faster reaction,15 but it also affects the degree of polymerization. As too high light intensity can lead to photodegradation, we decided to use 15 mW cm−2 for the rest of our screenings.
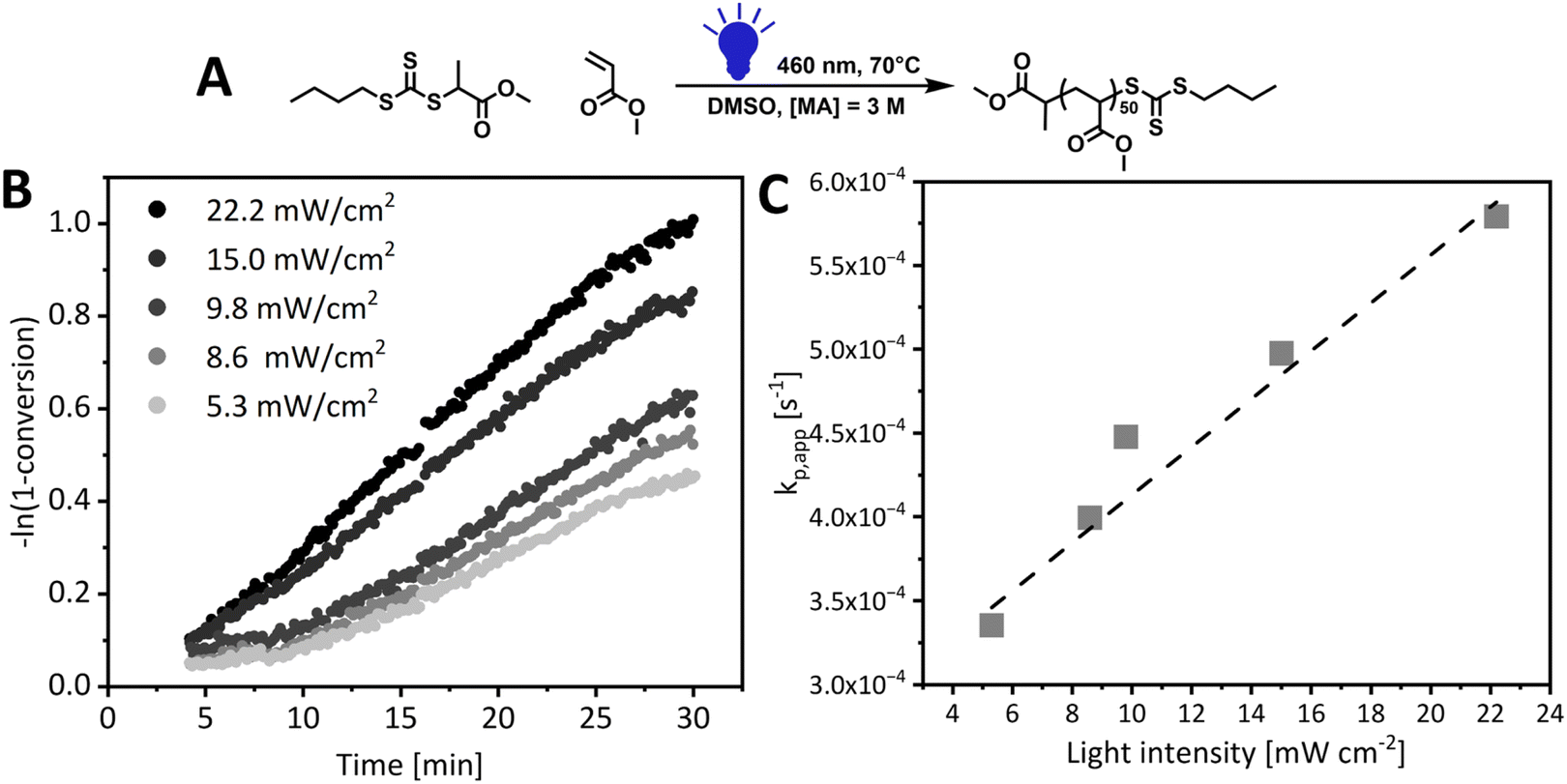 | ||
| Fig. 2 (A) Reaction scheme for DP50 methyl acrylate polymerization under blue light (460 nm) in DMSO ([MA] = 3 M, [MA]/[CTA] = 50) at 70 °C. (B) Pseudo-first-order kinetics plot. (C) Relation of light intensity and apparent rate of propagation (ka,app). | ||
With established standard photopolymerization conditions, we then examined the influence of the structure of the RAFT agent on the PI-RAFT polymerization of MA. Two most commonly used families of RAFT agents were studied: xanthates and trithiocarbonates, as these have dramatically different UV-vis absorption profiles. As both the stability and the reactivity of the R group (a carbon-centred radical) and the stabilising effect of the Z-group affect RAFT equilibrium, we kept the Z group alkyl chain the same to look only at the effect of the R group, which was systematically varied (2-propanoate, 2-2-methylpropanoate, 2-cyanopropan-2-yl, Fig. 1) within each family. From the UV-vis spectra of the RAFT agents (ESI, Fig. 9†), it is clear that the n–π* transition of xanthates is significantly blue-shifted, compared to trithiocarbonates and increased stability of the R group leads to a small red-shift of the maxima, both for xanthates (355 nm < 360 nm for PMB-Xan and BCN-Xan, respectively) and for trithiocarbonates (433 nm < 441 nm < 446 nm for PMBTC, BDMMT and BCN-TTC, respectively). Yet, despite possessing no significant absorbance in the blue light region, xanthates led to significantly faster MA polymerization compared to trithiocarbonates (Fig. 3A). The apparent polymerization rate coefficient (kp,app) for PMB-Xan was 1.2 × 10−3 s−1 while polymerization with PMB-TTC was 2.4 times slower, with kp,app 5.0 × 10−4 s−1.
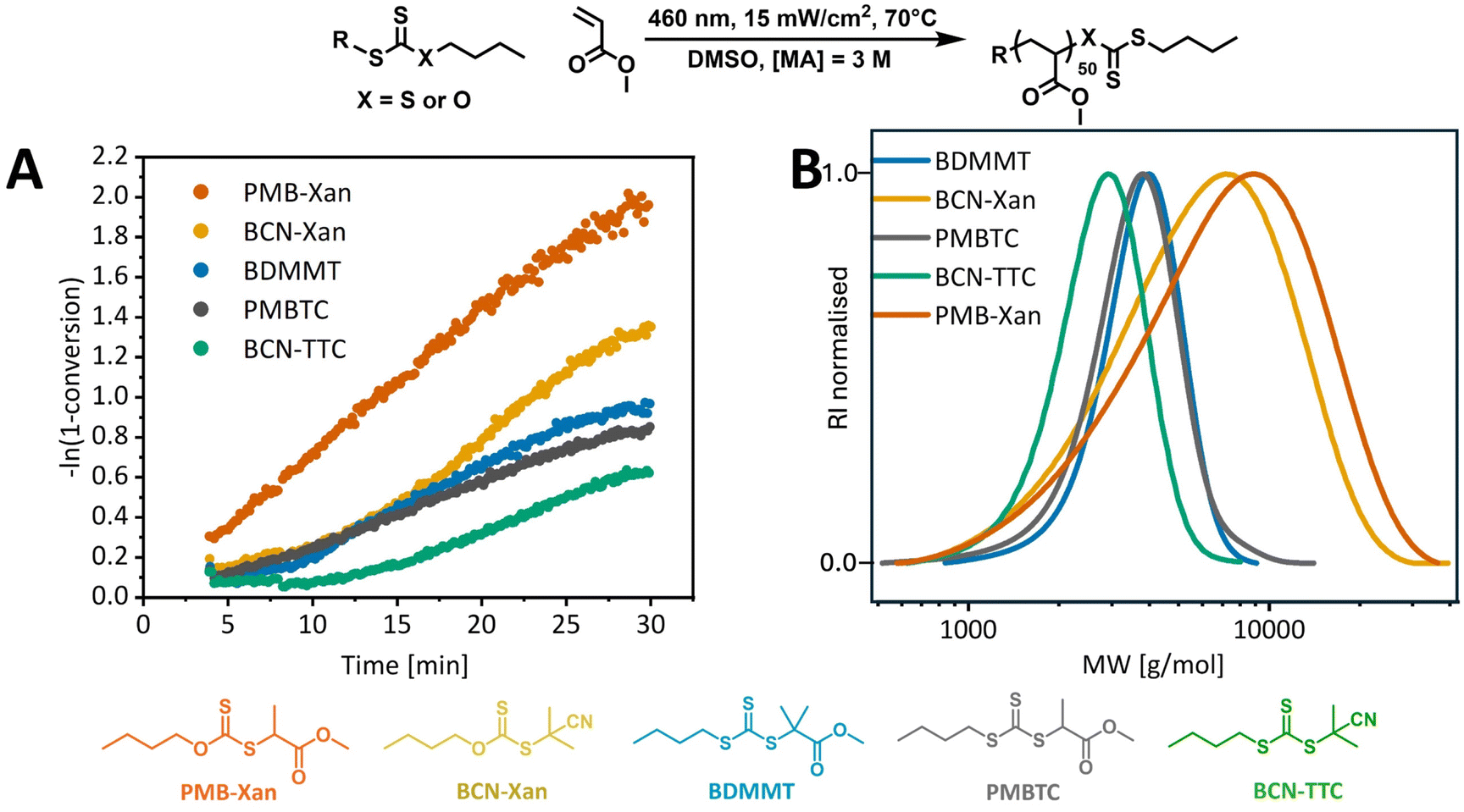 | ||
| Fig. 3 (A) Pseudo-first-order plot for MA PI-RAFT polymerization with different RAFT agents in DMSO ([MA] = 3 M, [MA]/[CTA] = 50) under blue light (460 nm, 15 mW cm−2) at 70 °C, with residence time from 4 to 30 min. (B) THF size exclusion chromatograms for corresponding photoiniferter RAFT polymerizations. | ||
While successful blue light polymerization with xanthates has been reported previously,56 this rate difference was still surprising. In fact, electron spin resonance spin-trapping experiments reported by Li et al. showed that, while a wavelength of light (λ) above 390 nm was more effective in the photolysis of ethyl 2-((ethoxycarbonothioyl)thio)propanoate xanthate than λ > 440 nm, even for the latter wavelength xanthate generated more radicals than trithiocarbonates.62 This highlights that a higher molar absorption coefficient at a given wavelength does not necessarily correspond to a higher quantum yield for homolytic bond dissociation and often the most activating wavelength for a photochemical process is actually red-shifted compared to the absorption spectrum.18,63
Within xanthates and trithiocarbonates, the R group has a significant effect on the kinetics of MA polymerization. For both families, more stable R groups (2-2-methylpropanoate and 2-cyanopropan-2-yl) led to induction periods, during which minimal or no monomer conversion was observed. This could be either an effect of the difference in photodissociation rates of the RAFT agents, or the time needed to establish RAFT equilibrium. A more stable R group should lead to increased rates of photolysis.9 While the molar absorption coefficients (ε) for the maxima of the n–π* transition follow a reverse trend to R group stability (ε = 46.7 > 41.8 > 34.4 M−1 cm−1 for PMBTCλ=433![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) nm, BDMMTλ=441nm and BCN-TTCλ=446nm, respectively (ESI, Fig. 10†)), at the wavelength of interest, 460 nm, PMBTC has the lowest coefficient (22.4 M−1 cm−1). Further, if the action plot for bond cleavage is red-shifted, as is often the case for wavelength-dependent processes,18,20 the photodissociation of PMBTC would be even further diminished compared to BCN-TTC and DDMAT. Yet, PMBTC is the only trithiocarbonate which did not result in an induction period, and photodissociation alone cannot explain this. This contrasts with previously results from McKenzie et al., who reported that only trithiocarbonates with secondary (but not tertiary) R groups resulted in an induction period in the polymerization of methyl acrylate under blue light (λmax = 460 nm, 1.5 mW cm−2) due to the slower rate of photolysis.9 This may be a result of the higher light intensity used in our study as well as an elevated temperature, which favours propagation and decreases the barrier in bond dissociation energy.
nm, BDMMTλ=441nm and BCN-TTCλ=446nm, respectively (ESI, Fig. 10†)), at the wavelength of interest, 460 nm, PMBTC has the lowest coefficient (22.4 M−1 cm−1). Further, if the action plot for bond cleavage is red-shifted, as is often the case for wavelength-dependent processes,18,20 the photodissociation of PMBTC would be even further diminished compared to BCN-TTC and DDMAT. Yet, PMBTC is the only trithiocarbonate which did not result in an induction period, and photodissociation alone cannot explain this. This contrasts with previously results from McKenzie et al., who reported that only trithiocarbonates with secondary (but not tertiary) R groups resulted in an induction period in the polymerization of methyl acrylate under blue light (λmax = 460 nm, 1.5 mW cm−2) due to the slower rate of photolysis.9 This may be a result of the higher light intensity used in our study as well as an elevated temperature, which favours propagation and decreases the barrier in bond dissociation energy.
As in conventional thermal RAFT polymerization, the R group needs to fragment efficiently to avoid induction periods (transition from pre to main equilibrium),64,65 so in PI-RAFT, the RAFT agent needs to undergo fast photolysis, followed by the equally fast addition of monomer to the R group radical. While the RAFT agents giving the most stable C-centred radicals fragment fastest, they are also the least reactive, precisely due to increased radical stability. As the R group becomes more electron withdrawing and/or stable, the addition rates decrease and fragmentation rates increase.64 If addition of R radicals to monomer is sufficiently low, it can cause an induction period, as observed for a tertiary cyanoalkyl group, which exhibits a very slow rate of addition to methyl acrylate.64 If on top of this, the R group fragments significantly faster than the propagating radical, an initialisation period can be observed during which the original RAFT agent is fully consumed before propagation and hence formation of oligomeric species occurs.66 Hence, both the xanthate and trithiocarbonate with a 2-propanoate R group that mimics an acrylic propagating radical perform best, while 2-2-methylpropanoate and 2-cyanopropan-2-yl groups lead to induction periods of about 8 and 10 min for the corresponding trithiocarbonates. Besides, no retardation is observed, and the rates of polymerization are the same within trithiocarbonate and xanthate families. The differences in the final monomer conversion (30 min residence time) as can be observed in Table 1 entries 1, 3, 4 and entries 2, 5 are the result of an induction period and an effectively delayed start of the polymerization. As a macroRAFT agent is generated, the effect of the α-end group diminishes and the reaction should follow the same kinetics after reaching RAFT equilibrium. Unsurprisingly, the dispersities for xanthate-mediated polymerization are broader as xanthates exhibit moderate activity for acrylates which are MAMs (More Activated Monomers).51,67
| Entry | Monomer | RAFT agent | Conversionb [%] |
M
n,SECc [g mol−1] |
M n,theo. [g mol−1] | Đ |
|---|---|---|---|---|---|---|
| a Polymerizations were conducted with [monomer] = 3 M in DMSO, target degree of polymerization 50, blue light (460 nm, 15 mW cm−2) irradiation at 70 °C for 30 min. b Monomer conversion was determined with 1H NMR spectroscopy. c Number-average molecular weights (Mn,SEC) were determined by THF SEC. | ||||||
| 1 | MA | BDMMT | 62% | 3450 | 2940 | 1.11 |
| 2 | MA | BCN-Xan | 74% | 4610 | 3400 | 1.53 |
| 3 | MA | PMBTC | 57% | 3230 | 2710 | 1.16 |
| 4 | MA | BCN-TTC | 47% | 2520 | 2260 | 1.13 |
| 5 | MA | PMB-Xan | 87% | 5260 | 3980 | 1.63 |
| 7 | MMA | BDMMT | 52% | 6460 | 2870 | 1.74 |
| 8 | MMA | BCN-Xan | 38% | 20900 |
2120 | 1.74 |
| 9 | MMA | PMBTC | 19% | 71000 |
1200 | 1.71 |
| 10 | MMA | BCN-TTC | 60% | 4570 | 3240 | 1.48 |
| 11 | MMA | PMB-Xan | 26% | 54400 |
1540 | 1.62 |
Different trends were observed for the polymerization of MMA. MMA is notoriously difficult to polymerize using photopolymerization, especially with PI-RAFT, and reactions rarely achieve full conversion and yield polymers with broad dispersities.4,13,22 The poorer control for methacrylates than acrylates or acrylamides can arise from increased photodegradation of the polymeric end group due to the increased stability of the propagating radicals, like in the case of the photolytic stability of RAFT agents.9 Sumerlin and co-workers hypothesised that thiyl radicals can terminate polymer chains by abstracting α-hydrogens from propagating methacrylic radicals.13 Common practices to improve control over the polymerization of MMA include the addition of a tertiary amine catalyst which acts as single-electron reductant that will reduce the thiyl radical to an anion to improve its photostability.68 Indeed, the levels of control we observed for methyl methacrylate polymerization in DMSO were significantly lower than for methyl acrylate, even for butyl(2-cyano-2-propyl)trithiocarbonate (Table 1, entries 1 and 10). None of the methyl methacrylate polymerizations exhibit a long induction period, as PMMA macroRAFT radicals have higher or similar stability to RAFT agent R groups. However, significant rate retardation for less stable R groups is observed (Fig. 4). This is likely due to slower fragmentation for RAFT agents with less stable R groups. Another potential reason for rate retardation is consumption of the intermediate radicals in reversible or irreversible side reactions,69 which will also be affected by the stability of the intermediate and so of the R group. PMBTC exhibits remarkably slow polymerization due to mismatch of reactivity with the more stabilised methacrylate monomer. As the 2-propanoate R group is a poorer leaving group than the monomer, methyl methacrylate fragments preferentially and high molecular weight polymers are produced.70 Indeed, at only 19% monomer conversion the number average molecular weight of PMMA chain is 71000 g mol−1 (Mn,theo. = 1200 g mol−1, Table 1, entry 9). Surprisingly, xanthates that led to fastest polymerization of MA led to slower polymerization of MMA than trithiocarbonates. Given the low chain transfer constant for xanthate, we anticipated that PI-RAFT of MMA mediated with xanthates will predominantly undergo reversible termination, which should be fast for xanthates, rather than degenerative chain transfer. However, slow kinetics and poor control suggest that RAFT equilibrium plays an important role and reduced fragmentation leads to the formation of high molecular weight polymers, similar to the PMBTC case. This equates to fewer carbonylthio moieties per propagating chain and hence slower photolysis and an overall reduced rate of polymerization. Indeed, this rate reduction is specific to PI-RAFT as in conventional thermal RAFT experiments, and the apparent rate of propagation was very similar for BCN-Xan and BCN-TTC (9.97 × 10−5 s−1 and 8.83 × 10−5 s−1, respectively, ESI Fig. 11†).
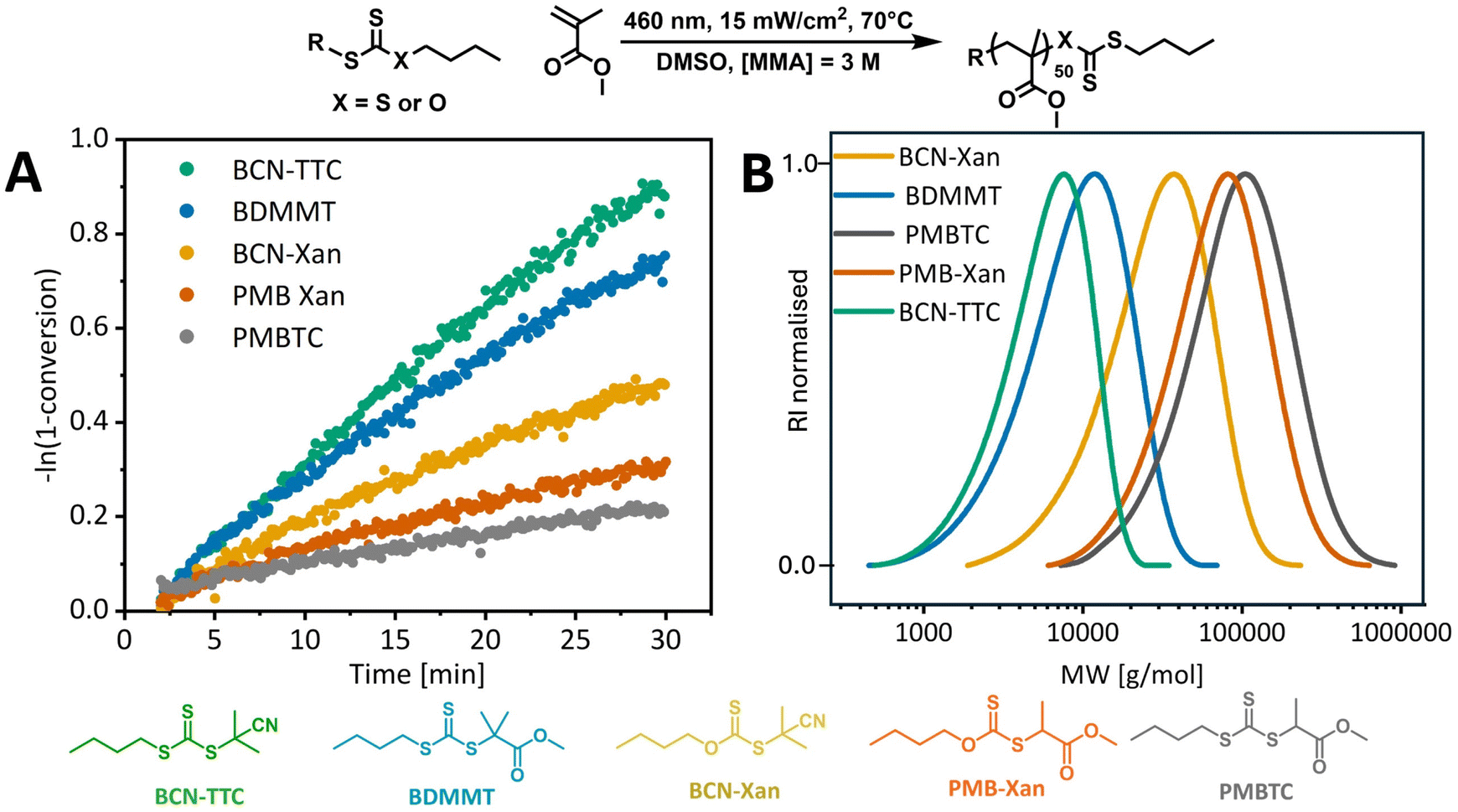 | ||
| Fig. 4 (A) Pseudo-first-order plot for MMA PI-RAFT polymerization with different RAFT agents in DMSO ([MMA] = 3 M, [MMA]/[CTA] = 50) under blue light (15 mW cm−2) at 70 °C, with residence time from 4 to 30 min. (B) THF size exclusion chromatograms for corresponding photoiniferter RAFT polymerizations. | ||
A comparison of MA and MMA polymerization kinetics clearly shows that a difference in reaction rate is apparent (Fig. 5), in addition to the absence of an induction period for MMA. MMA polymerization with tertiary R group RAFT agents exhibits similar polymerization rates to MA, while polymerization with xanthates is faster for MA. For further comparison, we focused on a tertiary alkyl cyano group RAFT agent as it does not cause significant retardation of MA or MMA polymerization in control thermal experiments (ESI Fig. 11†). For PI-RAFT with BCN-TTC, the apparent propagation rate coefficients for MA and MMA are 5.83 × 10−4 s−1 and 5.32 × 10−4 s−1, respectively (Fig. 5), suggesting retardation of MA polymerization in the PI-RAFT process, as at 70 °C the rate coefficient for free radical polymerization of MA is about 31 times faster than that of MMA (32798 L mol−1 s−1vs. 1055 L mol−1 s−1).71,72 In the case of PI-RAFT BCN-Xan polymerization, the apparent rate coefficient of MA polymerization is about 7 times faster than that of MMA (1.83 × 10−3 s−1 and 2.72 × 10−4 s−1). We hypothesise that this difference arises from a photodissociation/reversible termination process – the formation of a secondary propagating radical (MA) is less favourable than for a tertiary radical (MMA); hence the rate of polymerization of MA but not MMA is retarded. While xanthate undergoes photodissociation faster than trithiocarbonate due to lower bond dissociation energy,53 trithiocarbonate fragments less efficiently and photodissociation becomes the rate-determining step for the PI-RAFT polymerization of MA.
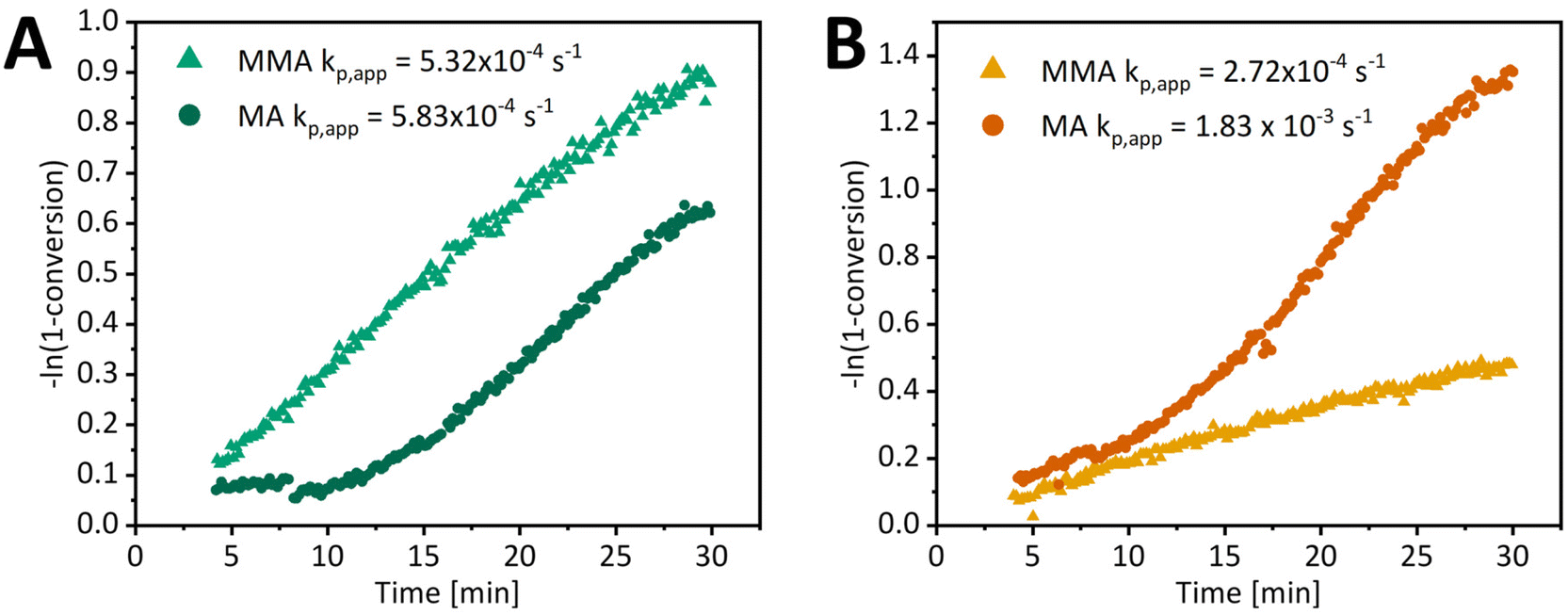 | ||
| Fig. 5 Pseudo-first-order plots for MMA and MA PI-RAFT polymerization in DMSO ([monomer] = 3 M, [monomer]/[CTA] = 50) under blue light (15 mW cm−2) at 70 °C with (A) butyl(2-cyano-2-propyl)trithiocarbonate as CTA and (B) O-butyl S-(2-cyanopropan-2-yl)xanthate as CTA. | ||
Conclusions
We have studied the effects of the structure and reactivity of the RAFT agent on the photoiniferter RAFT kinetics of two model monomers with different propagation rates and radical stabilities – methyl acrylate and methyl methacrylate. Significant induction periods, which can be eliminated by increasing light intensity, were observed for MA but not for MMA. Despite more efficient photodissociation, RAFT agents with more stable R groups lead to increased induction periods for the polymerization of MA, highlighting the importance of RAFT pre-equilibrium on polymerization kinetics. Further, photodissociation influences the rate of polymerization. Fast photodissociating xanthates, despite their poor absorption overlap with blue light, give rise to faster polymerization of MA while trithiocarbonates, which are characterised by a poorer quantum yield of photo bond cleavage, lead to retardation. Despite photodissociation and reversible termination playing important roles in the PI-RAFT mechanism, xanthates and trithiocarbonate with a secondary R group lead to the slow and uncontrolled polymerization of MMA. This study highlights that, besides the required activity matching between RAFT and monomer, other factors such as radical stability and photo flux significantly affect the kinetics of PI-RAFT. The automated transient kinetics flow polymerization platform gives increased resolution due to high data density, which allows us to look at small differences in induction periods, which may not be easily accessible otherwise, and a variable power supply allows for a reproducible study of polymerization kinetics.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from the Australian Research Council (ARC) via the provision of a LIEF grant (LE210100100) is gratefully acknowledged. M. A. B. thanks Lubrizol and the University of Warwick for the provision of a scholarship.References
- T. Otsu, Iniferter concept and living radical polymerization, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2121–2136 CrossRef CAS.
- T. Otsu and A. Kuriyama, Living Radical Polymerization in Homogeneous System by Using Iniferter: Design of Block Copolymers, J. Macromol. Sci., Chem., 1984, 21, 961–977 Search PubMed.
- T. Otsu and M. Yoshida, Role of Initiator-Transfer Agent-Terminator (Iniferter) in Radical Polymerizations: Polymer Design by Organic Disulfides as Iniferters, Makromol. Chem., Rapid Commun., 1982, 3, 127–132 CrossRef CAS.
- J. F. Quinn, L. Barner, C. Barner-Kowollik, E. Rizzardo and T. P. Davis, Reversible Addition–Fragmentation Chain Transfer Polymerization Initiated with Ultraviolet Radiation, Macromolecules, 2002, 35, 7620–7627 CrossRef CAS.
- T. G. McKenzie, Q. Fu, E. H. H. Wong, D. E. Dunstan and G. G. Qiao, Visible Light Mediated Controlled Radical Polymerization in the Absence of Exogenous Radical Sources or Catalysts, Macromolecules, 2015, 48, 3864–3872 CrossRef CAS.
- J. Xu, S. Shanmugam, N. A. Corrigan and C. Boyer, in Controlled Radical Polymerization: Mechanisms, ed. K. Matyjaszewski, B. S. Sumerlin, N. V. Tsarevsky and J. Chiefari, American Chemical Society, Washington, 1st edn, 2015, pp. 247–267 Search PubMed.
- M. Hartlieb, Photo-Iniferter RAFT Polymerization, Macromol. Rapid Commun., 2022, 43, 2100514 CrossRef CAS PubMed.
- M. Chen, M. Zhong and J. A. Johnson, Light-Controlled Radical Polymerization: Mechanisms, Methods, and Applications, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed.
- T. G. McKenzie, L. P. M. Da Costa, Q. Fu, D. E. Dunstan and G. G. Qiao, Investigation into the photolytic stability of RAFT agents and the implications for photopolymerization reactions, Polym. Chem., 2016, 7, 4246–4253 RSC.
- H. Zhou and J. A. Johnson, Photo-controlled growth of telechelic polymers and end-linked polymer gels, Angew. Chem., Int. Ed., 2013, 52, 2235–2238 CrossRef CAS.
- M. Chen and J. A. Johnson, Improving photo-controlled living radical polymerization from trithiocarbonates through the use of continuous-flow techniques, Chem. Commun., 2015, 51, 6742–6745 RSC.
- R. N. Carmean, T. E. Becker, M. B. Sims and B. S. Sumerlin, Ultra-High Molecular Weights via Aqueous Reversible-Deactivation Radical Polymerization, Chem, 2017, 2, 93–101 CAS.
- R. N. Carmean, M. B. Sims, C. A. Figg, P. J. Hurst, J. P. Patterson and B. S. Sumerlin, Ultrahigh Molecular Weight Hydrophobic Acrylic and Styrenic Polymers through Organic-Phase Photoiniferter-Mediated Polymerization, ACS Macro Lett., 2020, 9, 613–618 CrossRef CAS PubMed.
- R. W. Lewis, R. A. Evans, N. Malic, K. Saito and N. R. Cameron, Ultra-fast aqueous polymerisation of acrylamides by high power visible light direct photoactivation RAFT polymerisation, Polym. Chem., 2018, 9, 60–68 RSC.
- G. D. Ammini, J. P. Hooker, J. Van Herck, A. Kumar and T. Junkers, Comprehensive high-throughput screening of photopolymerization under light intensity variation using inline NMR monitoring, Polym. Chem., 2023, 14, 2708–2716 RSC.
- R. W. Hughes, M. E. Lott, J. I. Bowman and B. S. Sumerlin, Excitation Dependence in Photoiniferter Polymerization, ACS Macro Lett., 2022, 12, 14–19 CrossRef PubMed.
- J. D. Coyle, The photochemistry of thiocarbonyl compounds, Tetrahedron, 1985, 41, 5393–5425 CrossRef CAS.
- M. Nardi, E. Blasco and C. Barner-Kowollik, Wavelength-Resolved PhotoATRP, J. Am. Chem. Soc., 2022, 144, 1094–1098 CrossRef CAS.
- S. Gauci, F. Du Prez, J. Holloway, H. Houck and C. Barner-Kowollik, The Power of Action Plots: Unveiling Reaction Selectivity of Light-Stabilized Dynamic Covalent Chemistry, Angew. Chem., Int. Ed., 2023, 62, e202310274 CrossRef CAS PubMed.
- J. P. Menzel, B. B. Noble, J. P. Blinco and C. Barner-Kowollik, Predicting wavelength-dependent photochemical reactivity and selectivity, Nat. Commun., 2021, 12, 1–12 CrossRef.
- F. Lauterbach, M. Rubens, V. Abetz and T. Junkers, Ultrafast PhotoRAFT Block Copolymerization of Isoprene and Styrene Facilitated through Continuous-Flow Operation, Angew. Chem., Int. Ed., 2018, 57, 14260–14264 CrossRef CAS PubMed.
- M. Rubens, P. Latsrisaeng and T. Junkers, Visible light-induced iniferter polymerization of methacrylates enhanced by continuous flow, Polym. Chem., 2017, 8, 6496–6505 RSC.
- A. Sivokhin, D. Orekhov, O. Kazantsev, K. Otopkova, O. Sivokhina, Y. Chesnokov, M. Smirnov, A. Ovchinnikov and I. Makhov, High-molecular weight bottlebrushes via continuous flow photoiniferter polymerization of macromonomers, Polym. Chem., 2023, 13, 3186–3195 RSC.
- R. M. Myers, D. E. Fitzpatrick, R. M. Turner and S. V. Ley, Flow Chemistry Meets Advanced Functional Materials, Chem. – Eur. J., 2014, 20, 12348–12366 CrossRef CAS PubMed.
- B. Wenn, M. Conradi, A. D. Carreiras, D. M. Haddleton and T. Junkers, Photo-induced copper-mediated polymerization of methyl acrylate in continuous flow reactors, Polym. Chem., 2014, 5, 3053–3060 RSC.
- A. Melker, B. P. Fors, C. J. Hawker and J. E. Poelma, Continuous flow synthesis of poly(methyl methacrylate) via a light-mediated controlled radical polymerization, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2693–2698 CrossRef CAS.
- F. Zhong, Y. Zhou and M. Chen, The influence of mixing on chain extension by photo-controlled/living radical polymerization under continuous-flow conditions, Polym. Chem., 2019, 10, 4879–4886 RSC.
- J. Gardiner, C. H. Hornung, J. Tsanaktsidis and D. Guthrie, Continuous flow photo-initiated RAFT polymerisation using a tubular photochemical reactor, Eur. Polym. J., 2016, 80, 200–207 CrossRef CAS.
- V. Hessel, D. Kralisch, N. Kockmann, T. Noël and Q. Wang, Novel process windows for enabling, accelerating, and uplifting flow chemistry, ChemSusChem, 2013, 6, 746–789 CrossRef CAS.
- Y. Su, N. J. W. Straathof, V. Hessel and T. Noël, Photochemical transformations accelerated in continuous-flow reactors: Basic concepts and applications, Chem. – Eur. J., 2014, 20, 10562–10589 CrossRef CAS PubMed.
- G. Jas and A. Kirschning, Continuous Flow Techniques in Organic Synthesis, Chem. – Eur. J., 2003, 9, 5708–5723 CrossRef CAS.
- S. V. Ley, On being green: Can flow chemistry help?, Chem. Rec., 2012, 12, 378–390 CrossRef CAS.
- C. Sambiagio and T. Noël, Flow Photochemistry: Shine Some Light on Those Tubes!, Trends Chem., 2020, 2, 92–106 CrossRef CAS.
- K. S. Elvira, X. C. I. Solvas, R. C. R. Wootton and A. J. Demello, The past, present and potential for microfluidic reactor technology in chemical synthesis, Nat. Chem., 2013, 5, 905–915 CrossRef CAS.
- R. L. Hartman, J. P. McMullen and K. F. Jensen, Deciding whether to go with the flow: Evaluating the merits of flow reactors for synthesis, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef CAS.
- S. T. Knox, S. Parkinson, R. Stone and N. J. Warren, Benchtop flow-NMR for rapid online monitoring of RAFT and free radical polymerisation in batch and continuous reactors, Polym. Chem., 2019, 10, 4774–4778 RSC.
- T. Durand, C. Henry, D. Bolien, D. C. Harrowven, S. Bloodworth, X. Franck and R. J. Whitby, Thermolysis of 1,3-dioxin-4-ones: Fast generation of kinetic data using in-line analysis under flow, React. Chem. Eng., 2016, 1, 82–89 RSC.
- S. Schwolow, F. Braun, M. Rädle, N. Kockmann and T. Röder, Fast and Efficient Acquisition of Kinetic Data in Microreactors Using In-Line Raman Analysis, Org. Process Res. Dev., 2015, 19, 1286–1292 CrossRef CAS.
- S. T. Knox, S. J. Parkinson, C. Y. P. Wilding, R. A. Bourne and N. J. Warren, Autonomous polymer synthesis delivered by multi-objective closed-loop optimisation, Polym. Chem., 2022, 13, 1576–1585 RSC.
- M. Rubens, J. Van Herck and T. Junkers, Automated Polymer Synthesis Platform for Integrated Conversion Targeting Based on Inline Benchtop NMR, ACS Macro Lett., 2019, 8, 1437–1441 CrossRef CAS PubMed.
- J. Mitchell, L. F. Gladden, T. C. Chandrasekera and E. J. Fordham, Low-field permanent magnets for industrial process and quality control, Prog. Nucl. Magn. Reson. Spectrosc., 2014, 76, 1–60 CrossRef CAS PubMed.
- M. V. Gomez and A. De La Hoz, NMR reaction monitoring in flow synthesis, Beilstein J. Org. Chem., 2017, 13, 285–300 CrossRef CAS.
- A. M. R. Hall, J. C. Chouler, A. Codina, P. T. Gierth, J. P. Lowe and U. Hintermair, Practical aspects of real-time reaction monitoring using multi-nuclear high resolution FlowNMR spectroscopy, Catal. Sci. Technol., 2016, 6, 8406–8417 RSC.
- S. Mozharov, A. Nordon, J. M. Girkin and D. Littlejohn, Non-invasive analysis in micro-reactors using Raman spectrometry with a specially designed probe, Lab Chip, 2010, 10, 2101–2107 RSC.
- S. Mozharov, A. Nordon, D. Littlejohn, C. Wiles, P. Watts, P. Dallin and J. M. Girkin, Improved method for kinetic studies in microreactors using flow manipulation and noninvasive Raman spectrometry, J. Am. Chem. Soc., 2011, 133, 3601–3608 CrossRef CAS.
- J. J. Haven, J. Vandenbergh and T. Junkers, Watching polymers grow: real time monitoring of polymerizations via an on-line ESI-MS/microreactor coupling, Chem. Commun., 2015, 51, 4611–4614 RSC.
- J. S. Moore and K. F. Jensen, Batch kinetics in flow: Online IR analysis and continuous control, Angew. Chem., Int. Ed., 2014, 53, 470–473 CrossRef CAS.
- D. Drelinkiewicz, S. T. Alston, T. Durand and R. J. Whitby, The switch-off method: rapid investigation of flow photochemical reactions, React. Chem. Eng., 2023, 8, 2134–2140 RSC.
- J. Van Herck and T. Junkers, Rapid Kinetic Screening via Transient Timesweep Experiments in Continuous Flow Reactors, Chem.: Methods, 2022, 2, e202100090 CAS.
- J. Van Herck, I. Abeysekera, A.-L. Buckinx, K. Cai, J. Hooker, K. Thakur, E. Van de Reydt, P.-J. Voorter, D. Wyers and T. Junkers, Operator-independent high-throughput polymerization screening based on automated inline NMR and online SEC, Digital Discovery, 2022, 1, 519–526 RSC.
- S. Perrier, 50th Anniversary Perspective: RAFT Polymerization – A User Guide, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- A. Ajayaghosh and R. Francis, A xanthate-derived photoinitiator that recognizes controls the free radical polymerization pathways of methyl methacrylate and styrene, J. Am. Chem. Soc., 1999, 121, 6599–6606 CrossRef CAS.
- B. Zhao, J. Li, Y. Xiu, X. Pan, Z. Zhang and J. Zhu, Xanthate-Based Photoiniferter RAFT Polymerization toward Oxygen-Tolerant and Rapid Living 3D Printing, Macromolecules, 2022, 55, 1620–1628 CrossRef CAS.
- A.-C. Lehnen, J. Gurke, A. M. Bapolisi, M. Reifarth, M. Bekir and M. Hartlieb, Xanthate-supported photo-iniferter (XPI)-RAFT polymerization: Facile and rapid access to complex macromolecular architectures, Chem. Sci., 2023, 14, 593–603 RSC.
- A.-C. Lehnen, J. A. M. Kurki and M. Hartlieb, The difference between photo-iniferter and conventional RAFT polymerization: high livingness enables the straightforward synthesis of multiblock copolymers, Polym. Chem., 2022, 13, 1537–1546 RSC.
- C. Ding, C. Fan, G. Jiang, X. Pan, Z. Zhang, J. Zhu and X. Zhu, Photocatalyst-Free and Blue Light-Induced RAFT Polymerization of Vinyl Acetate at Ambient Temperature, Macromol. Rapid Commun., 2015, 36, 2181–2185 CrossRef CAS.
- C. P. Easterling, Y. Xia, J. Zhao, G. E. Fanucci and B. S. Sumerlin, Block Copolymer Sequence Inversion through Photoiniferter Polymerization, ACS Macro Lett., 2019, 8, 1461–1466 CrossRef CAS.
- F. Dalitz, M. Cudaj, M. Maiwald and G. Guthausen, Process and reaction monitoring by low-field NMR spectroscopy, Prog. Nucl. Magn. Reson. Spectrosc., 2012, 60, 52–70 CrossRef CAS.
- N. Zientek, K. Meyer, S. Kern and M. Maiwald, Quantitative Online NMR Spectroscopy in a Nutshell, Chem. Ing. Tech., 2016, 88, 698–709 CrossRef CAS.
- J. G. Powles, B. I. Hunt and D. J. H. Sandiford, Proton Spin Lattice Relaxation and Mechanical Loss in a Series of Acrylic Polymers, Polymer, 1964, 5, 505–515 CrossRef CAS.
- M. L. Allegrezza and D. Konkolewicz, PET-RAFT Polymerization: Mechanistic Perspectives for Future Materials, ACS Macro Lett., 2021, 10, 433–446 CrossRef CAS.
- J. Li, M. Zhang, J. Zhu and X. Zhu, Investigation into the Direct Photolysis Process of Photo-Induced RAFT Polymerization by ESR Spin Trapping, Polymers, 2019, 11, 1722 CrossRef CAS.
- I. M. Irshadeen, S. L. Walden, M. Wegener, V. X. Truong, H. Frisch, J. P. Blinco and C. Barner-Kowollik, Action Plots in Action: In-Depth Insights into Photochemical Reactivity, J. Am. Chem. Soc., 2021, 143, 21113–21126 CrossRef CAS PubMed.
- G. Moad, in RAFT Polymerization: Methods, Synthesis and Applications: Volume 1 and 2, ed. G. Moad and E. Rizzardo, Wiley, 1st edn, 2021, vol. 1–2, pp. 359–492 Search PubMed.
- S. Perrier, C. Barner-Kowollik, J. F. Quinn, P. Vana and T. P. Davis, Origin of inhibition effects in the reversible addition fragmentation chain transfer (RAFT) polymerization of methyl acrylate, Macromolecules, 2002, 35, 8300–8306 CrossRef CAS.
- J. B. McLeary, J. M. McKenzie, M. P. Tonge, R. D. Sanderson and B. Klumperman, Initialisation in RAFT-mediated polymerisation of methyl acrylate, Chem. Commun., 2004, 17, 1950–1951 RSC.
- M. Destarac, D. Matioszek, X. Vila, J. Ruchmann-Sternchuss and S. Z. Zard, in Reversible Deactivation Radical Polymerization: Mechanisms and Synthetic Methodologies, ed. K. Matyjaszewski, H. Gao, B. S. Sumerlin and N. V. Tsarevsky, American Chemical Society, Washington, 1st edn, 2018, vol. 1284, pp. 291–305 Search PubMed.
- Q. Fu, T. G. McKenzie, S. Tan, E. Nam and G. G. Qiao, Tertiary amine catalyzed photo-induced controlled radical polymerization of methacrylates, Polym. Chem., 2015, 6, 5362–5368 RSC.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Advances in RAFT polymerization: The synthesis of polymers with defined end-groups, Polymer, 2005, 46, 8458–8468 CrossRef CAS.
- G. Pound, J. B. McLeary, J. M. McKenzie, R. F. M. Lange and B. Klumperman, In-situ NMR spectroscopy for probing the efficiency of RAFT/MADIX agents, Macromolecules, 2006, 39, 7796–7797 CrossRef CAS.
- S. Beuermann, M. Buback, T. P. Davis, R. G. Gilbert, R. A. Hutchinson, O. F. Olaj, G. T. Russell, J. Schweer and A. M. Van Herk, Critically evaluated rate coefficients for free-radical polymerization, 2: Propagation rate coefficients for methyl methacrylate, Macromol. Chem. Phys., 1997, 198, 1545–1560 CrossRef CAS.
- C. Barner-Kowollik, S. Beuermann, M. Buback, P. Castignolles, B. Charleux, M. L. Coote, R. A. Hutchinson, T. Junkers, I. Lacík, G. T. Russell, M. Stach and A. M. Van Herk, Critically evaluated rate coefficients in radical polymerization – 7. Secondary-radical propagation rate coefficients for methyl acrylate in the bulk, Polym. Chem., 2014, 5, 204–212 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00409d |
| This journal is © The Royal Society of Chemistry 2024 |