
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxygen-tolerant, eosin Y mediated synthesis of protein–polymer biohybrids and protein-coated polymer nanoparticles†‡
Errika
Voutyritsa§
 abc,
Thomai
Lazou§
a,
Jonida
Bushi§
a,
Stavroula
Margaritaki
a,
Myrto
Charitaki
a,
Sune M.
Christensen
d,
Nikos S.
Hatzakis
bc and
Kelly
Velonia
*a
abc,
Thomai
Lazou§
a,
Jonida
Bushi§
a,
Stavroula
Margaritaki
a,
Myrto
Charitaki
a,
Sune M.
Christensen
d,
Nikos S.
Hatzakis
bc and
Kelly
Velonia
*a
aDepartment of Materials Science and Engineering, University of Crete, University Campus Voutes, 70013 Heraklion, Crete, Greece. E-mail: velonia@materials.uoc.gr
bDepartment of Chemistry, University of Copenhagen, Denmark
cNovo Nordisk Center for Optimised Oligo Escape, University of Copenhagen, Denmark
dNovonesis, Biologiens Vej 2, 2800 Kgs. Lyngby, Denmark
First published on 10th July 2024
Abstract
To avoid metal catalysts that are commonly used in conventional approaches for the synthesis of protein–polymer conjugates, an eosin Y/TEMED mediated, photoinduced polymerization of vinyl monomers was optimized. This oxygen tolerant, photoinduced approach allowed the grafting of a series of hydrophobic, hydrophilic and responsive polymers with quantitative protein macroinitiator consumption. CALB bioconjugates were also synthesized and found to retain part of the parent protein activity for extended periods of time. Notably, when BSA was used in the absence of an initiator, protein-coated nanoparticles were shown to form during emulsion polymerization.
Introduction
Protein–polymer conjugates are hybrid macromolecules designed to combine the unique functions and properties encoded in biomolecules with the limitless physicochemical and functional adaptability of synthetic polymers. As such, they have been systematically pursued over several decades.1–3 The vast range of potential applications of protein–polymer conjugates was first demonstrated with the synthesis of dextran–hemoglobin hybrids4 and the PEGylation of bovine serum albumin (BSA).5–7 The increased biocompatibility, stability and in vivo half-life of the first hybrid biomolecules sparked numerous investigations which have resulted in quite a few approved therapeutics by the United States Food and Drug Administration (FDA)8–10 and systems designed to address scientific goals in drug delivery,11–16 gated transport,17 sensing and detection,18 and catalysis.19–21Recent advances in polymer synthesis have also fostered significant progress in polymer bioconjugate synthesis. In the early synthesis of protein–polymer conjugates, most reports employed grafting-to approaches involving amino acid-specific or random couplings of prefunctionalized polymers to the protein22–24 or bioaffinity couplings.25–27 More recently, by capitalizing on the ability to synthesize polymers in rapid, efficient, and precise manners,28 numerous new methodologies have emerged for the synthesis of protein–polymer conjugates.2,29–33 These are mostly grafting-from methodologies involving controlled radical polymerization (CRP) approaches such as atom transfer radical polymerization (ATRP),34–41 Cu(0)-mediated radical polymerization,42,43 and ring-opening polymerization,44,45 as well as reversible addition–fragmentation chain transfer (RAFT)46–48 polymerization approaches and chain growth polymerization techniques.
Very recent reports on the synthesis of protein–polymer conjugates have focused on oxygen tolerant49–52 and photomediated metal catalyzed approaches.39,51,53 A significant advantage of photochemical approaches is that they offer temporal and spatial control under mild reaction conditions while oxygen tolerance is fundamental for the development of sustainable applications.54–57 However, several of these approaches require metal-based catalysts and metal contamination is a limiting factor when aiming at biomedical applications. For this reason, the metal-free organocatalyzed ATRP (O-ATRP)58–61 mediated synthesis of protein–polymer conjugates recently reported in seminal works by the groups of Sumerlin,62,63 Matyjaszewski,64 and Boyer65 provides a new, powerful tool in the realm of oxygen tolerant bioconjugation. In this respect, the commonly used xanthene electron acceptor derivative eosin Y (EY) has been used as the organocatalyst as it is known to mediate photoinduced polymerization of several families of monomers in conjunction with alkyl halides and amines.66–68 EY is cheap and commercially available, displays excellent biocompatibility and has therefore been widely used in biological applications.69,70 Taking these beneficial characteristics into account, Sumerlin and collaborators were the first to employ EY-catalyzed PET-RAFT for the synthesis of polymer–protein conjugates in the presence of a tertiary amine and under visible-light irradiation.62,63 Following this grafting-from approach, rapid synthesis of water soluble biohybrids over a range of targeted molecular weights became possible. In a more recent contribution, eosin Y acrylate was copolymerized with N-isopropylacrylamide (NIPAM) to afford a polymeric photocatalyst with temperature dependent hydrophilic-to-hydrophobic transition which enabled easy purification of the bioconjugates post polymerization and recovery of the catalyst.63 Importantly, ascorbic acid (AscA) was shown to efficiently deoxygenate the polymerization solution. Matyjaszewski and collaborators developed green-light-induced dual catalysis ATRP, i.e., using EY in combination with a copper complex which enabled rapid and well-controlled polymerization in water without the need for deoxygenation.71,72 Following this approach, hydrophilic acrylate-based protein–polymer hybrids were also synthesized under ambient conditions.64 Finally, Boyer and collaborators65 developed a photo-RAFT system allowing the synthesis of protein–polymer conjugates with excellent oxygen tolerance. During this study, the photocatalyst EY was combined with the reactive oxygen species (ROS), generating cocatalysts AscA and triethanolamine (TEOA). Evaluation of the impact of the ROS on model proteins led to the selection of EY/TEOA as the optimal photo-RAFT initiating system for preserving enzymatic activity.
Inspired by these reports, we developed an oxygen tolerant, EY photocatalyzed, grafting-from approach for the synthesis of protein–polymer conjugates in the absence of metal cocatalysts with targeted quantitative macroinitiator consumption. The latter is essential to avoid tedious purification that might render the approach difficult to scale-up for applications. We further reasoned that the strong adsorption of EY on proteins73 and/or its potential entrapment in the assemblies would enable direct imaging of the bioconjugates via fluorescence, offering in addition one pot synthesis and labeling for studies using advanced microscopy. Following this approach, we present herein the synthesis of hydrophilic, amphiphilic, and responsive bioconjugates under mild blue light irradiation and with quantitative macroinitiator consumption attained in the presence of a tertiary amine cocatalyst.65 To this end, BSA was selected as a model protein since it offers valuable characteristics to biohybrid systems including a lack of cytotoxicity, high stability, and the ability to evade interactions with blood serum components.74 Styrene, methacrylates, acrylates, and acrylamides were used as monomers. Expanding the biohybrid scope to enzymes, catalytically active biohybrids were also synthesized using the lipase B from Candida antarctica and were imaged with fluorescence microscopy. Considering that the tertiary amines act as co-initiators,60,75 the polymerization was studied in depth, revealing tandem formation of polymeric by-products. To valorize these polymers, emulsion polymerization was employed in the presence of native proteins, yielding protein-coated polymer nanoparticles.
Results and discussion
To achieve oxygen tolerant, photoinduced, organocatalyzed grafting of hydrophobic polymers from ATRP protein macroinitiators, we implemented an emulsion-based polymerization protocol.39 EY was used as the photoredox catalyst (Fig. S1‡) and styrene as the model monomer for the synthesis of amphiphilic bioconjugates. The use of BSA is widespread in bioconjugation studies due to its accessible free thiol, enabling specific functionalization, and, on this basis, it was selected as the model protein. The biomacroinitiator BSA-Br (Io) was synthesized following an established protocol, i.e., Michael addition of 2-bromo-2-methyl-propionic acid 2-(2,5-dioxo-2,5-dihydro-pyrrol-1-yl)-ethyl ester to the accessible cysteine residue (Cys-34) of the protein (Scheme S1 and Fig. S2‡). To evaluate the feasibility of this metal-free photoinduced approach, all reactions were studied at physiological pH (7.4) and ambient temperature under blue LED irradiation unless otherwise stated. A feed molar ratio of styrene/BSA-Br (Io) = 4000/1, ensuring the formation of a stable emulsion, was initially evaluated in the presence of 0.02 to 1 molar equivalent of EY (Table 1, entries 1–4) without adding either an oxygen scavenger or a sacrificial electron donor. It is important to note that all reactions were performed by simply eliminating the headspace from the reaction vessel without applying any deoxygenation76–79 means and that a ventilator was used to avoid temperature increase during the reaction (near ambient temperatures varying between 25 and 35 °C were measured in the reactor). Polyacrylamide gel electrophoresis (PAGE) and aqueous size exclusion chromatography (SEC) were employed to monitor the consumption of the macroinitiator and detect the formation of products. The formation of amphiphilic bioconjugates was observed for all feed molar ratios used under these conditions, yet, without quantitative macroinitiator consumption (Table 1, entries 1–3 and Fig. 1, Fig. S3, Fig. S4‡). A new band, not migrating past the stacking gel, was observed and attributed to the amphiphilic BSA-poly(styrene) biohybrid nanoparticles which were eluted with lower retention times than the macroinitiator in aqueous SEC.35,39 All biohybrids were characterized by infrared spectroscopy (FT-IR, Fig. S3‡). Lower or no macroinitiator consumption was observed when decreasing the catalyst concentration to as low as 0.02 molar equivalent (Table 1, entry 4 and Fig. S3, Fig. S4‡).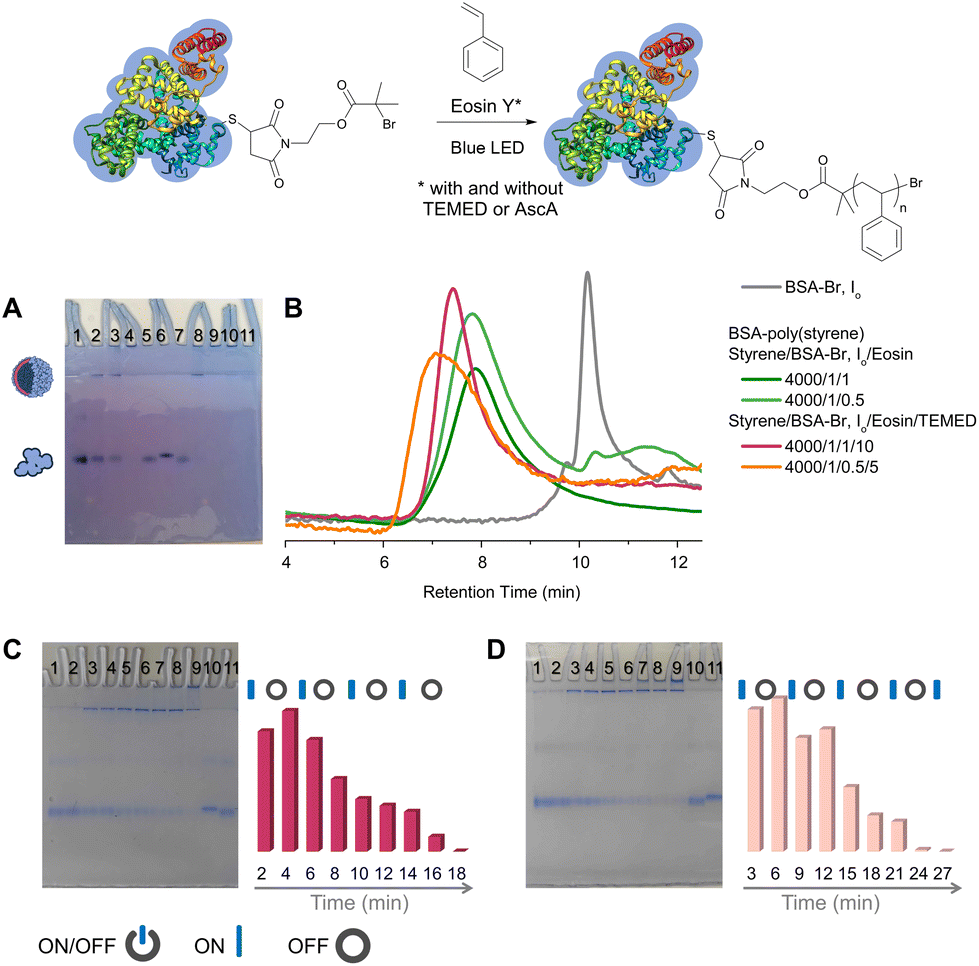 | ||
| Fig. 1 Synthesis of BSA-poly(styrene) via oxygen tolerant, organocatalyzed ATRP (top scheme). (A) Native PAGE, lanes 1–4 and 7–10: BSA-poly(styrene) lane 1: styrene/BSA-Br, Io/EY = 4000/1/0.02, lane 2: styrene/BSA-Br, Io/EY = 4000/1/0.2, lane 3: styrene/BSA-Br, Io/EY = 4000/1/0.5, lane 4: styrene/BSA-Br, Io/EY = 4000/1/1, lane 5: BSA-Br (Io), lane 6: native BSA, lane 7: styrene/BSA-Br, Io/EY/TEMED = 4000/1/0.02/0.2, lane 8: styrene/BSA-Br, Io/EY/TEMED = 4000/1/0.2/2, lane 9: styrene/BSA-Br, Io/EY/TEMED = 4000/1/0.5/5, lane 10: styrene/BSA-Br, Io/EY/TEMED = 4000/1/1/10, and lane 11: polystyrene. (B) SEC chromatographs of BSA-Br (Io) and BSA-poly(styrene) synthesized under different conditions. (C) ON/OFF time course experiment using a feed molar ratio of styrene/BSA-Br, Io/EY/TEMED = 4000/1/1/10. Left: native PAGE lanes 1–9: samples were withdrawn every 2 minutes of alternating blue LED ON and OFF periods, lane 10: native BSA, and lane 11: BSA-Br (Io). Right: semiquantitative analysis plot of BSA-Br (Io) consumption during the course of the reaction.39 (D) ON/OFF time course experiment using a feed molar ratio of styrene/BSA-Br, Io/EY/TEMED = 4000/1/0.5/5. Left: native PAGE lanes 1–9: samples were withdrawn every 3 minutes of alternating blue LED ON and OFF periods, lane 10: BSA-Br (Io), and lane 11: native BSA. Right: semiquantitative analysis plot of BSA-Br (Io) consumption during the course of the reaction.39 | ||
| Styrene/BSA-Br, Io/EY/AscA | Reaction time (min) | BSA-Br (Io) consumption | |
|---|---|---|---|
| 8 | 4000/1/1/0.5 | 120 | High |
| Styrene/BSA-Br, Io/EY/TEMED | Reaction time (min) | BSA-Br (Io) consumption | |
|---|---|---|---|
| a Without irradiation. b Formation of polystyrene nanoparticles. | |||
| 9 | 4000/1/1/10 | 10–15 | Quantitative |
| 10 | 4000/1/0.5/5 | 30–45 | Quantitative |
| 11 | 4000/1/0.2/2 | 120 | Near quantitative |
| 12 | 4000/1/0.02/0.2 | 240 or 480 | Low |
| 13 | 4000/0/1/10 | 240 | n.a.b |
| 14 | 4000/0/0.2/2 | 240 | n.a.b |
When control experiments were performed in the absence of a selected reaction component such as the catalyst (Table 1, entry 5), the monomer or irradiation (Table 1, entry 6 and Fig. S3‡), no biohybrid formation could be detected while in all cases, the macroinitiator was recovered unaffected. On the other hand, polystyrene nanoparticles were formed when styrene was subjected to emulsion polymerization conditions in the absence of the protein macroinitiator BSA-Br (Io) (Table 1, entry 7 and Fig. 2). The produced polystyrene was isolated and characterized with 1H-NMR spectroscopy (Fig. S5‡).
 | ||
| Fig. 2 (A) FE-SEM micrographs and (B) TEM micrographs of BSA-poly(styrene) (Table 1, entry 9) observed as two distinct populations of spherical nanoparticles with diameters between 100 and 130 nm and between 20 and 40 nm; (C) FE-SEM micrographs of poly(styrene) (Table 1, entry 13) observed as spherical nanoparticles with diameters between 10 and 40 nm; and (D) FE-SEM micrographs of BSA coated poly(styrene) nanoparticles with diameters between 10 and 50 nm. | ||
Throughout this study, quantitative macroinitiator consumption was targeted as it minimizes the effort required to isolate the biohybrids by rendering only a simple dialysis step necessary. We evaluated grafting in the presence of AscA since it was elegantly employed in recent protocols as a means to deoxygenate the reaction mixtures.63,65 Under the conditions used herein, grafting of styrene was found to proceed yet without quantitative macroinitiator consumption (Table 1, entry 8 and Fig. S3, S7‡). For this reason, we proceeded to investigate the effect of a tertiary amine sacrificial electron donor since it has been previously shown in EY mediated PET RAFT polymerizations that the stability of the generated amine radical cation enhanced both the efficiency of the reduction of excited-state EY and oxygen tolerance.80–82 Indeed, when N,N,N′,N′-tetramethylethylenediamine (TEMED) was added as a sacrificial electron donor,48 quantitative macroinitiator consumption could be attained after merely 10 to 15 minutes of blue LED irradiation at a feed molar ratio of styrene/BSA-Br, Io/EY/TEMED = 4000/1/1/10 (Fig. 1, Table 1, entry 9, Fig. S3 and S8‡). When a reduced EY feed molar ratio was used, quantitative macroinitiator consumption could again be achieved, albeit at increased irradiation times (between 30 minutes and 2 hours depending on the photoredox catalyst loading, Table 1, entries 10–12, Fig. S8‡).
Imaging of the products (Table 1, entry 9) with FE-SEM revealed two distinct populations of spherical nanoparticles with defined diameters between 100 and 130 nm and between 20 and 40 nm (Fig. 2A and B). Poly(styrene) formed in the absence of a macroinitiator appeared as spherical nanoparticles with significantly smaller diameters between 10 and 40 nm (Fig. 2C, Table 1, entries 13 and 14, Fig. S5‡). Hence, the spherical assemblies observed using FE-SEM can be most possibly attributed to hybrid polymer/bioconjugate nanoparticles (Fig. 2A and B). The nature of the nanoparticles was further elucidated through the synthesis of BSA-responsive polymer bioconjugates (vide infra). Taking into account the amphiphilicity of the bioconjugates and thus the lack of a solvent that would both dissolve them and preserve the conformation of the protein, the free polymer could not be efficiently removed from the product assemblies.
Next, intermittent light exposure was investigated to assess the possibility of activating and deactivating polymerization. Rapid macroinitiator consumption was observed after 2–3 minutes of irradiation, as can be observed in Fig. 1C (styrene/BSA-Br, Io/EY/TEMED = 4000/1/1/10, Table 1, entry 9 and Fig. S9‡). This short induction period can be ascribed to the time required to in situ remove the oxygen from the polymerization solution and the time required for the EY radical anion to form and in turn interact with the amine co-initiator to kick-start the polymerization.83–85 Under these conditions, the polymerization could be activated and deactivated by switching on and off the irradiation source until fully consuming BSA-Br (after 10 minutes of total ON irradiation time); nevertheless, temporal control was poor. We reasoned that the concentrations of EY and the tertiary amine would influence both the induction period and polymerization control, and therefore, to attain better temporal control, we lowered the concentration of the catalyst, i.e., styrene/BSA-Br, Io/EY/TEMED = 4000/1/0.5/5 (Table 1, entry 10 and Fig. 1C). The reaction could be again triggered or halted by turning the blue LEDs on and off with improved temporal control.
To further exploit this photoinduced methodology, we synthesized BSA-poly(styrene) on a larger scale using the same experimental setup. SEC verified that the macroinitiator consumption was quantitative on a 6 times larger scale without the need for further optimization (Fig. S9‡).
Monomer scope
To expand the monomer scope, the EY mediated, photoinduced ATRP grafting of a series of acrylates, methacrylates and acrylamides from BSA-Br (Io) was evaluated under the oxygen tolerant conditions established for styrene. For all hydrophobic monomers, emulsion polymerization conditions were pursued, i.e., formation of stable monomer emulsions prior to subjecting the monomer to polymerization. As seen in Fig. 3 (Table 2), hydrophobic acrylates, methacrylates and acrylamides could be successfully grafted from BSA-Br (Io), and for most monomers, quantitative macroinitiator consumption could be achieved through optimization of the feed molar ratio of the reactants. More specifically, the optimal feed molar ratio for the grafting of methyl acrylate (MA) was MA/BSA-Br/EY/TEMED = 4000/1/1/10, i.e., the same as the optimum feed ratio identified for styrene (Fig. S10 and S11‡). In the case of methyl methacrylate (MMA), quantitative macroinitiator consumption at the same feed molar ratio could only be achieved in the presence of 5% v/v toluene, presumably stabilizing the monomer emulsion (Fig. S12 and S13‡). In all cases, the amphiphilic bioconjugates were found to assemble into hybrid polymer/biopolymer spherical nanostructures (Fig. 3E).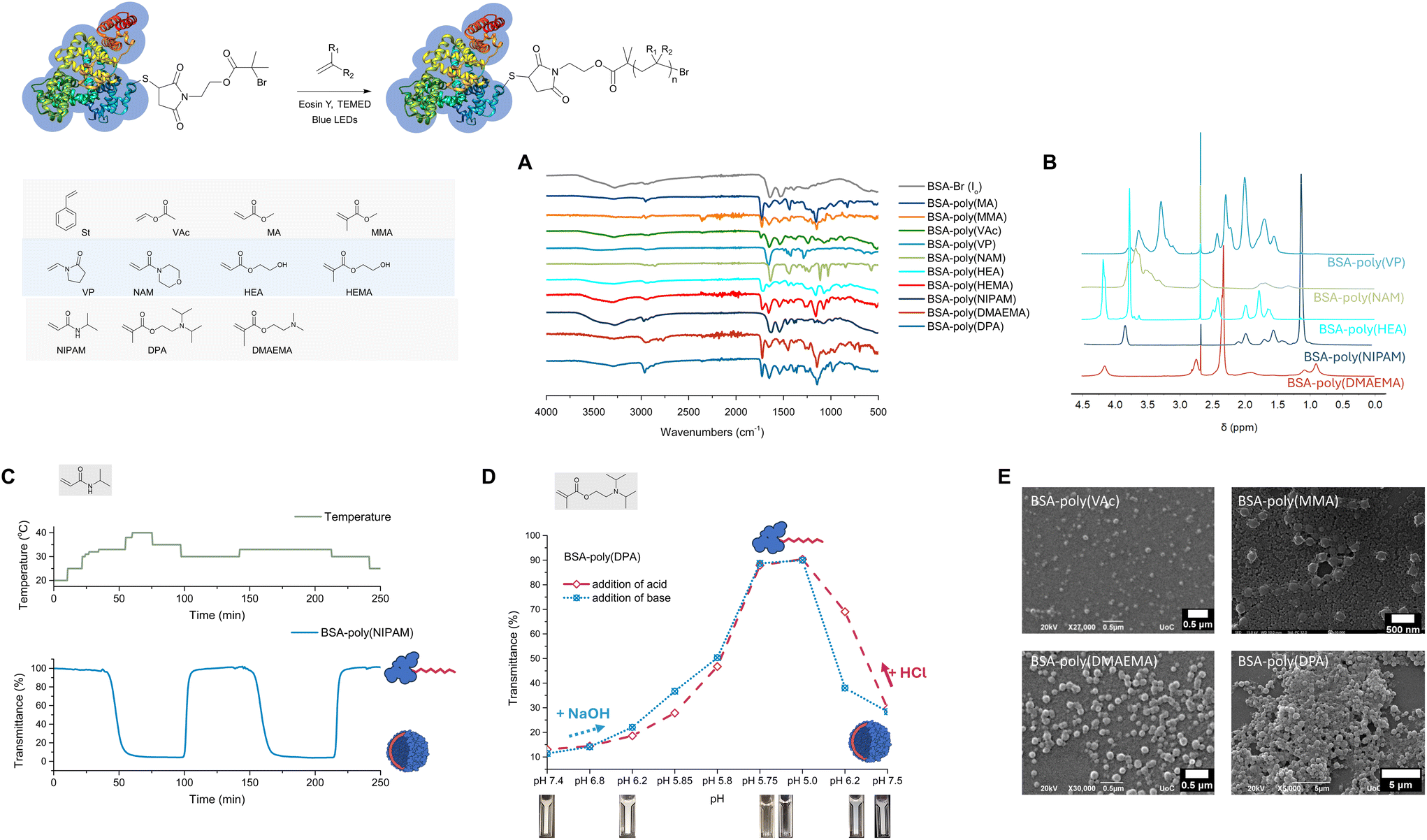 | ||
| Fig. 3 Characterization of amphiphilic, hydrophilic, and responsive protein–polymer conjugates. (A) IR spectra of the bioconjugates. (B) 1H-NMR spectra acquired for hydrophilic BSA-polymer conjugates. (C) Transmittance vs. time curve at different temperatures showing the rapid response of BSA-poly(NIPAM). (D) Transmittance vs. pH curves of BSA-poly(DPA). Two cycles are shown for the same sample in which the response was induced by changing the pH with the addition of HCl (pH decrease) or NaOH (pH increase). (E) SEM and FE-SEM micrographs of amphiphilic protein–polymer conjugate nanoparticles. | ||
| Entry | Monomer | Monomer/BSA-Br, Io/EY | BSA-Br (Io) consumption |
|---|---|---|---|
| a Addition of 5% v/v toluene. | |||
| 1 | MA | 4000/1/1/10 | Quantitative |
| 2 | MMA | 4000/1/1/10a | Quantitative |
| 3 | VAc | 8000/1/5/50 | High |
| 4 | VP | 4000/1/0.5/5 | Quantitative |
| 5 | NAM | 4000/1/1/10 | Near quantitative |
| 6 | HEA | 4000/1/1/10 | Quantitative |
| 7 | HEMA | 4000/1/1/10 | Quantitative |
| 8 | NIPAM | 1000/1/1/10 | Quantitative |
| 9 | NIPAM | 1000/1/0.2/10 | Quantitative |
| 10 | NIPAM | 100/1/0.2/10 | Quantitative |
| 11 | DMAEMA | 4000/1/0.2/10 | Quantitative |
| 12 | DPA | 4000/1/0.2/10 | Quantitative |
As seen with styrene, several other monomers used in this study (MMA, DPA and NIPAM vide infra) were also shown to polymerize in the absence of a macroinitiator (Fig. S6‡).
Grafting of the less activated monomer vinyl acetate (VAc) proved to be more demanding. In general, ATRP of VAc is considered highly challenging because the homolytic bond dissociation energy of the dormant poly(VAc) chains makes reactivation difficult while at the same time the VAc propagating radical is not stabilized either.86,87 Indeed, neither addition of an organic cosolvent nor increased catalyst loadings or grafting times could significantly increase biomacroinitiator consumption. The lowest amount of unreacted macroinitiator was detected at a feed molar ratio of VAc/BSA-Br, Io/EY/TEMED = 8000/1/5/10 (Fig. S14‡ and Fig. 3E).
We also sought to graft hydrophilic monomers from BSA-Br (Io), and for this reason, vinyl pyrrolidone (VP), N-acryloyl morpholine (NAM) and 2-hydroxyethyl acrylate (HEA) were selected since all produce polymers useful in a variety of pharmaceutical and biomedical applications (Fig. 3).88,891H-NMR spectroscopy provided an additional means to characterize hydrophilic protein–polymer conjugates while dialysis was sufficient to remove both unreacted monomers and the produced polymers from the bioconjugate solution. It should be noted that for hydrophilic monomers, the optimum conditions of emulsion polymerization did not result in macroinitiator consumption which was more difficult to attain. A feed molar ratio of VP/BSA-Br, Io/EY/TEMED = 4000/1/0.5/5 was found to be sufficient to yield BSA-poly(vinyl pyrrolidone) biohybrids (Fig. 3 and Fig. S15‡). For the synthesis of BSA-poly(N-acryloyl morpholine), near quantitative macroinitiator consumption was observed after optimization with NAM/BSA-Br/EY/TEMED = 4000/1/1/10 (Fig. 3 and Fig. S16‡). At the same molar loading, both 2-hydroxyethyl acrylate (HEA) and 2-hydroxyethyl methacrylate (HEMA) led to the formation of BSA-poly(HEA) (Fig. 3 and Fig. S187‡) and BSA-poly(HEMA) (Fig. 3 and Fig. S18‡), respectively.
Targeting the synthesis of responsive bioconjugates, N-isopropylacrylamide (NIPAM), 2-(dimethylamino)ethyl methacrylate (DMAEMA) and 2-(diisopropylamino)ethyl methacrylate (DPA) were grafted from the protein macroinitiator BSA-Br (Fig. 3). The conditions identified for full macroinitiator consumption are summarized in Table 2 (Fig. S19–S24‡). To get insight into the kinetics of this oxygen-tolerant approach, the photoinduced grafting of NIPAM from BSA-Br (Io) was further studied. In time course experiments performed under the conditions identified to be optimal (NIPAM/BSA-Br, Io/EY/TEMED = 2000/1/0.2/10, Fig. S19‡), the formation of biohybrids was apparent within the first 5 minutes of irradiation and full macroinitiator consumption could be achieved within 30 minutes. Importantly, when samples of the reaction mixture were withdrawn at fixed time points and studied with 1H-NMR spectroscopy without purification, full monomer consumption was also seen after 60 minutes (Fig. S19‡). The lower critical solution temperature (LCST) of BSA-poly(NIPAM) was determined to be between 32.8 and 33 °C at sufficiently dilute concentrations and was found to be reversible (Fig. 3 and Fig. S20‡). The spherical assemblies formed at temperatures higher than the LCST were imaged by SEM (Fig. S20‡). The response of BSA-poly(DPA) was also found to be reversible with the turning point determined to be at pH 5.8 (Fig. 3 and Fig. S21‡).90 Taking advantage of their response, both BSA-poly(NIPAM) and BSA-poly(DPA) could be effectively isolated from independently formed polymer chains, i.e., by performing dialysis after phase transition while retaining the conditions required for the biopolymer to be hydrophilic (at 20 °C for BSA-poly(NIPAM) and at pH below 5.8 for BSA-poly(DPA)). BSA-poly(DPA) samples were collected before and after dialysis performed at pH 5.5 and analyzed with native PAGE (Fig. S22†). The dialysate was collected and the released polymer was isolated and characterized with 1H-NMR spectroscopy (Fig. S22‡). Similar trends were observed in the synthesis of BSA-poly(DMAEMA) (Fig. S23 and S24‡).
Grafting multiple monomers
To capitalize on the unlimited chemical versatility of polymers, we investigated the possibility of grafting two different monomers from the protein initiator via this oxygen tolerant, metal-free, photocatalysis. A random BSA-poly(NIPAM-co-DPA) was synthesized by grafting both monomers together (Fig. S25‡). To explore the livingness of this approach, we then proceeded to consecutively graft two different monomers. To achieve this, full consumption of both the macroinitiator and the monomer was targeted for the first step. Quantitative consumption of the macroinitiator was confirmed by PAGE electrophoresis, and full monomer consumption was confirmed by 1H-NMR spectroscopy for the synthesis of BSA-poly(NIPAM) at a feed molar ratio of NIPAM/BSA-Br, Io/EY/TEMED = 100/1/0.2/10. After the first monomer, NIPAM, was fully consumed, styrene, EY and TEMED were added to the reaction mixture (styrene/BSA-Br, Io/EY/TEMED = 4000/1/0.2/10) and the reaction was allowed to proceed for another hour under blue LED irradiation. The formation of BSA-poly(NIPAM)-b-poly(styrene) was verified through the FT-IR spectrum of the biohybrid exhibiting all the characteristic vibrations of the protein and both polymer moieties (Fig. S26‡). Under the same conditions, the responsive BSA-poly(NIPAM)-b-poly(VP) was also synthesized (Fig. S27‡).Protein-coated polymer nanoparticles
An underlying goal of this study was to develop a facile, easy to implement and rapid protocol that would allow for easy tailoring of hybrid nanocarriers for diverse imaging applications given that EY strongly attaches to proteins. The key element in implementing such nanocarriers as drug or signal delivery systems is to attain mechanistic insights into internalization pathways and membrane/organelle interactions using fluorescent microscopy.91–93 Since several of the monomers used in this study polymerize under the conditions of this oxygen tolerant photoinduced polymerization to form polymer nanoparticles (vide supra), we envisioned that this approach could be used to construct protein-coated polymer nanoparticles. We reasoned that during emulsion polymerization, a native protein could act as a surfactant, further stabilizing the monomer emulsion. In such a case, protein-coated nanoparticles would be formed during EY mediated photopolymerization of styrene and stabilized through hydrophobic interactions.We therefore performed styrene polymerization in the presence of native BSA without adding the ATRP initiator BSA-Br, Io. In PAGE electrophoresis, a band not migrating past the stacking gel front was predominant upon completion of the reaction while native BSA could also be detected (Fig. S28‡). After the dialysis step, the presence of both BSA and poly(styrene) was confirmed in the product mixture with FT-IR. The nanoparticles were visualized via FE-SEM imaging to be spherical with diameters varying between 10 and 50 nm (Fig. 2D). Despite numerous efforts, the protein could not be fully detached from the nanoparticles by simple means that would allow further characterization post polymerization. We therefore proceeded to synthesize responsive poly(DPA) nanoparticles in the presence of native BSA (Fig. 4 and Fig. S23‡). To determine the nature of the produced nanoparticles, the product was characterized after synthesis and was then subjected to dialysis against phosphate buffer, pH 5.0, i.e., below the turning point (5.8, Fig. 3). Both the product and the dialysate were characterized. As seen in native PAGE analyses of the samples collected before and after the phase transition, native BSA (pI 4.8–5.0) was liberated, leaving no trace of the band attributed to the nanocarrier (Fig. S22‡). Our initial assumption was further supported through the detection of poly(DPA) obtained by acquiring a 1H-NMR spectrum of the dialysate (Fig. S22‡).
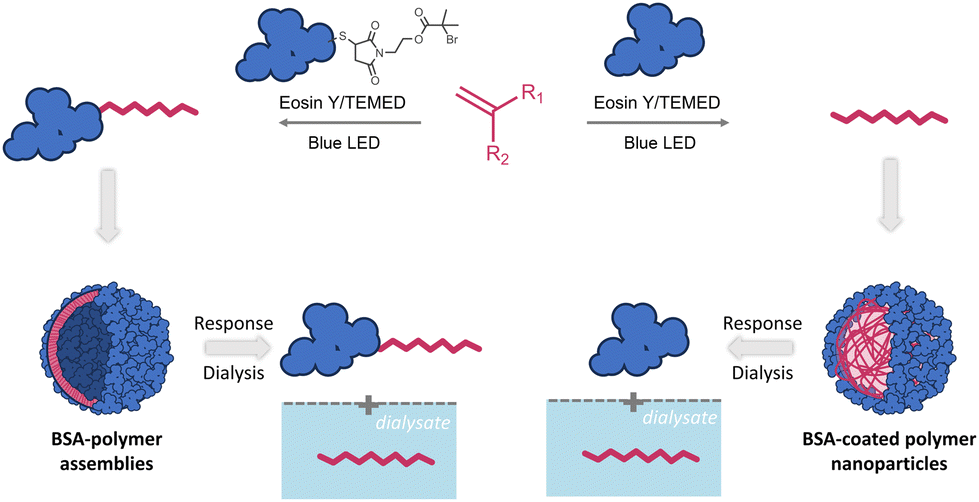 | ||
| Fig. 4 Proposed pathways to produce BSA-polymer and BSA-coated polymer nanoparticles. | ||
Imaging with fluorescence microscopy
EY has an emission peak at 516.5 nm and is detectable in fluorescence microscopy. As previously reported, EY strongly attaches to proteins which could be advantageous for the purposes of biomedical research imaging since EY is biocompatible and non-toxic. The protein-coated poly(styrene) nanoparticles were imaged with total internal reflection fluorescence (TIRF) microscopy and detected in the green channel (Fig. 5, Fig. S28 and S29‡).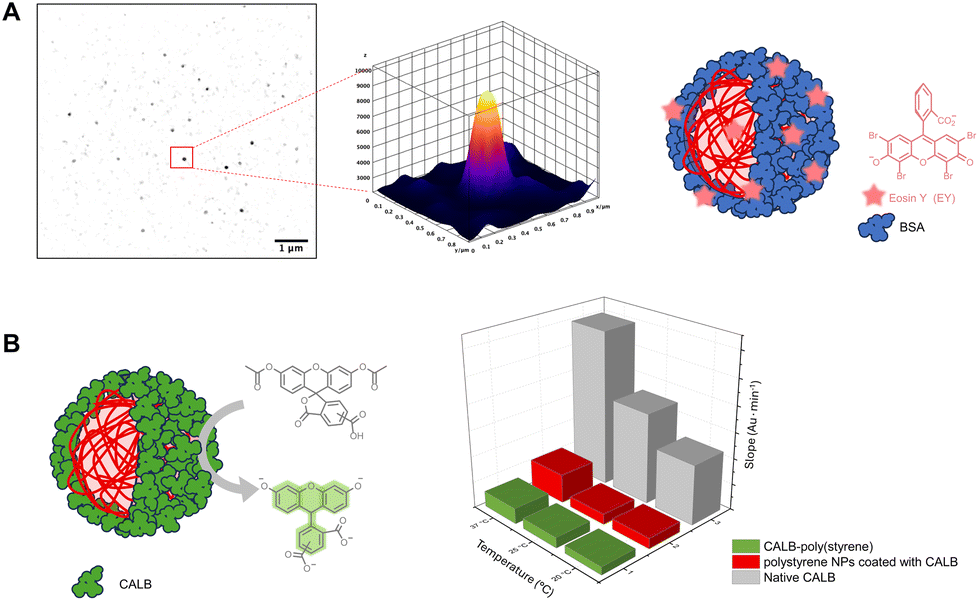 | ||
| Fig. 5 (A) Left: imaging of BSA coated poly(styrene) with internal reflection fluorescence (TIRF) microscopy. Right: 3D intensity plot of one nanoparticle. (B) Activity of CALB-poly(styrene), polystyrene nanoparticles coated with CALB and native CALB. The graph depicts the slopes of the activity kinetics recorded at 20, 25 and 37 °C. | ||
Protein scope – synthesis of CALB-poly(styrene)
To further explore the versatility of this oxygen tolerant photoinduced organocatalysis, the lipase B from Candida antarctica (CALB) was employed. To synthesize a CALB initiator, N-hydroxysuccinimide-2-bromo-2-methylpropionate was non-specifically linked via NHS-ester coupling to the exposed primary amines of the protein (Fig. S30‡). Upon isolating and characterizing the macroinitiator, the grafting of styrene was endeavored at a feed molar ratio of styrene/CALB-Br, Io/EY/TEMED = 4000/1/1/10. The formation of CALB-poly(styrene) was verified via SDS PAGE and FT-IR (Fig. S31‡). More specifically, a new non-migrating band was observed for the product in SDS PAGE which was pleasingly accompanied by the absence of the band corresponding to the macroinitiator. Consistent with what was seen with BSA, two populations of assembled nanoparticles were observed in SEM with the ones larger in diameter (100–150 nm) having spherical and distorted spherical architectures (Fig. S32‡). The FT-IR spectrum of the product verified the presence of all the characteristic vibrations of CALB-Br and poly(styrene) (Fig. S31‡).CALB catalyzes the hydrolysis of esters, converting triglycerides into glycerol and fatty acids, while being also one of the most used enzymes in biocatalysis with widespread applications.94 5-(6)-Carboxyfluorescein diacetate (CFDA) was used to test in vitro the catalytic activity of the CALB-poly(styrene) biohybrids by monitoring the formation of the hydrolysis product carboxyl fluorescein (CF) at 453 nm. The biohybrids were shown to retain part of the catalytic activity of the parent enzyme.
CALB-coated polystyrene nanoparticles
Poly(styrene) nanoparticles were synthesized in the presence of native CALB (1 equiv.) using a feed molar ratio of styrene/EY/TEMED = 4000/1/10 under blue LED irradiation for one hour. The nanoparticles were imaged with FE-SEM while the product was characterized with PAGE electrophoresis and FT-IR (Fig. S5‡).The CALB-coated polystyrene nanoparticles were also found to retain part of the esterase activity of native CALB (Fig. 5). Notably, the coated nanoparticles were proven to be remarkably stable as they retained their activity after storing for one year at 4 °C (Fig. S33,‡ activity data not shown). To the best of our knowledge, this is the first time that such significant stability of such nanocarriers is being reported.
Experimental
Materials and methods
Chemicals were purchased from commercial sources and were used as received, unless otherwise stated. Bovine serum albumin (BSA) was purchased from Sigma (>99%). The purification of CALB was performed according to the literature.95 Dialysis bags (Spectra/Por® Biotech Regenerated Cellulose Dialysis Membranes, MWCO 10, 25, and 50 kDa) were purchased from Spectrum Labs. The synthesis of the biomacroinitiators was performed using established protocols.35,39 Full experimental details and characterization are included in the ESI.‡General polymerization protocol for the oxygen tolerant, EY/TEMED mediated grafting of monomers from protein macroinitiators
A solution of EY was prepared by dissolving 1 mg of EY (1.54 μmol) in 1 mL of 20 mM phosphate buffer, pH 7.4, with the aid of sonication. 0.21 M and 0.1 M TEMED stock solutions were prepared by dissolving 3.3 μL (22 μmol) in 100 μL of nanopure water or 1.5 μL (10 μmol) in 98.5 μL of nanopure water. 2.8 μL–141.5 μL (0.044–0.218 μmol) of the stock solution of EY and the corresponding volume of the appropriate TEMED stock solution were dissolved in nanopure water to afford a solution with a fixed total volume (460 μL). The emulsion of the monomer was formed by adding the hydrophobic monomer (872 μmol, 4000 equiv.) and sonicating for ca. 5 minutes. The volume of water was adjusted for each feed molar ratio to retain a stable volume. The resulting emulsion was immediately transferred to a 6 mL polypropylene syringe equipped with a stirring bar, containing 0.625 mL of a 0.35 mM solution of the protein macroinitiator (Io) in 20 mM phosphate buffer, pH 7.4 (0.218 μmol, 1 equiv.). The headspace was eliminated to avoid the presence of undissolved oxygen and the reaction syringe was capped and placed under blue LED irradiation for specified amounts of time (varying from 5 minutes to 9 hours) with moderate stirring. A ventilator was used to avoid temperature increase, maintaining the temperature between 25 and 35 °C. The reaction mixture was then dialyzed using a 10 kDa MWCO regenerated cellulose dialysis membrane initially against a mixture of 5 mM phosphate buffer, pH 7.4, and 1% DMSO, then against 5 mM phosphate buffer, pH 7.4, and finally against 20 mM phosphate buffer, pH 7.4. The product solutions were analyzed by means of native or SDS PAGE electrophoresis, SEC, and FT-IR spectroscopy. 1H-NMR spectra were acquired for hydrophilic products. Dilute suspensions of the products in nanopure water were imaged with SEM or FE-SEM. All products were stored at 7 °C until further use.Conclusions
A comparative study of a robust EY/TEMED photocatalyzed, oxygen tolerant protocol that enables the synthesis of protein–polymer conjugates is presented. For the purposes of this study, we optimized photocatalysis and used it as a rapid and easy to implement protocol to graft diverse monomers from a protein macroinitiator while targeting at complete macroinitiator consumption in order to attain at the same time easy isolation means. In this manner, amphiphilic, hydrophilic and responsive bioconjugates were synthesized and characterized. The photoinduced EY/TEMED mediated grafting was found to be easily scalable. Expanding the scope of this protocol, triblock biohybrids could also be easily obtained. The EY mediated oxygen tolerant ATRP was also successfully applied to CALB, yielding biohybrids that retained part of the parent enzyme activity. Taking advantage of the ability of EY/TEMED to mediate vinyl monomer polymerization in emulsion, we studied the in situ formation and protein-coating of polymer nanoparticles. Both BSA and CALB were used to coat poly(styrene) and poly(DPA) nanoparticles. Interestingly, the CALB-coated poly(styrene) nanoparticles were found to be active after being stored for over a year at 4 °C. Nanoparticle protein coronas are spontaneously formed when they enter biological systems as part of their defence mechanisms. Our current studies are focusing on this extremely interesting feature of the EY mediated approach which provides the means to synthesize polymer nanoparticles coated with a defined protein corona to explore whether this can in turn dictate the biophysical and chemical identity of the nanoparticle.Author contributions
E. V., Th. L., J. B., S. M., M. Ch., and K. V.: investigation and data curation; S. M. C., N. S. H., and K. V.: supervision and resources; K. V.: conceptualization and methodology; E. V. and Th. L.: writing of the original draft; and K. V.: writing, review & editing. The final manuscript was reviewed by all authors.Data availability
The data supporting this article have been included as part of the ESI.‡Conflicts of interest
Sune M. Christensen is employed by Novonesis.Acknowledgements
We acknowledge Jeannette de Sparra Lundin, Gustav Hammerich Hansen and Christian Isak Jørgensen for assisting with CalB expression, purification, and characterization. The authors acknowledge the Hellenic Foundation for Research and Innovation (HFRI) EΛIΔEK program for funding under the 4th Call for HFRI PhD Fellowships (Fellowship Number: 11118). The authors acknowledge funding from the Independent Research Fund Denmark (1127-00432B), Villum Foundation (40801) and the NNF Challenge Center for Optimised Oligo Escape and Control of Disease (NNF23OC0081287).References
- R. A. Olson, A. B. Korpusik and B. S. Sumerlin, Chem. Sci., 2020, 11, 5142 RSC.
- M. S. Messina, K. M. Messina, A. Bhattacharya, H. R. Montgomery and H. D. Maynard, Prog. Polym. Sci., 2020, 100, 101186 CrossRef CAS.
- A. J. Russell, S. L. Baker, C. M. Colina, C. A. Figg, J. L. Kaar, K. Matyjaszewski, A. Simakova and B. S. Sumerlin, AIChE J., 2018, 64, 3230 CrossRef CAS.
- S. C. Tam, J. Blumenstein and K. Wong, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 2128 CrossRef CAS PubMed.
- A. Abuchowski, T. Van Es, N. C. Palczuk and F. F. Davis, J. Biol. Chem., 1977, 252, 3578 CrossRef CAS.
- F. M. Veronese, R. Largajolli, E. Boccù, C. A. Benassi and O. Schiavon, Appl. Biochem. Biotechnol., 1985, 11, 141 CrossRef CAS.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214 CrossRef CAS.
- E. M. Pelegri-O'Day, E. W. Lin and H. D. Maynard, J. Am. Chem. Soc., 2014, 136, 14323 CrossRef.
- S. N. Alconcel, A. S. Baas and H. D. Maynard, Polym. Chem., 2011, 2, 1442 RSC.
- A. M. Ramos-de-la-Peña and O. Aguilar, Int. J. Pept. Res. Ther., 2019, 26, 333 CrossRef.
- C. A. Stevens, K. Kaur and H.-A. Klok, Adv. Drug Delivery Rev., 2021, 174, 447 CrossRef CAS.
- P. Kiran, A. Khan, S. Neekhra, S. Pallod and R. Srivastava, Front. Med. Technol., 2021, 3, 676025 CrossRef PubMed.
- S. Bhattacharjee, W. G. Liu, W. H. Wang, I. Weitzhandler, X. H. Li, Y. Z. Qi, J. Y. Liu, Y. Pang, D. F. Hunt and A. Chilkoti, ChemBioChem, 2015, 16, 2451 CrossRef CAS PubMed.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2013, 65, 10 CrossRef CAS PubMed.
- Z. Liu, C. Dong, X. Wang, H. Wang, W. Li, J. Tan and J. Chang, ACS Appl. Mater. Interfaces, 2014, 6, 2393 CrossRef CAS.
- F. Duan, W. Jin, T. Zhang, Y. Sun, X. Deng and W. Gao, Adv. Mater., 2023, 2209765 CrossRef CAS PubMed.
- X. Huang, M. Li, D. Green, D. S. Williams, A. J. Patil and S. Mann, Nat. Commun., 2013, 4, 2239 CrossRef PubMed.
- J. M. Hoffman, P. S. Stayton, A. S. Hoffman and J. J. Lai, Bioconjugate Chem., 2015, 26, 29–38 CrossRef CAS PubMed.
- K. Velonia, A. E. Rowan and R. J. M. Nolte, J. Am. Chem. Soc., 2002, 124, 4224 CrossRef CAS PubMed.
- M. J. Boerakker, J. M. Hannink, P. H. H. Bomans, P. M. Frederik, R. J. M. Nolte, E. M. Meijer and N. A. J. M. Sommerdijk, Angew. Chem., Int. Ed., 2002, 41, 4239 CrossRef CAS.
- C. Bao, Y. Yin and Q. Zhang, Biomacromolecules, 2018, 19, 1539 CrossRef CAS; C. Y. Bao and Q. Zhang, Eur. Polym. J., 2019, 112, 263–272 CrossRef; C. W. Chiang, X. Liu, J. Sun, J. Guo, L. Tao and W. Gao, Nano Lett., 2020, 20, 1383 CrossRef PubMed.
- G. Mantovani, F. Lecolley, L. Tao, D. M. Haddleton, C. Clerx, J. J. L. M. Cornelissen and K. Velonia, J. Am. Chem. Soc., 2005, 127, 2966 CrossRef CAS.
- Y. Wang and C. Wu, Biomacromolecules, 2018, 19, 1804 CrossRef CAS.
- K. Velonia, Polym. Chem., 2010, 1, 944 RSC.
- C. A. Lackey, N. Murthy, O. W. Press, D. A. Tirrell, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 1999, 10, 401 CrossRef CAS PubMed.
- S. Kulkarni, C. Schilli, A. Müller, A. Hoffman and P. Stayton, Bioconjugate Chem., 2004, 15, 747 CrossRef CAS PubMed.
- X. Wan, G. Zhang, Z. Ge, R. Narain and S. Liu, Chem. – Asian J., 2011, 6, 2835 CrossRef CAS.
- K. Parkatzidis, H. S. Wang, N. P. Truong and A. Anastasaki, Chem, 2020, 7, 1575 Search PubMed.
- R. A. Olson, A. B. Korpusik and B. S. Sumerlin, Chem. Sci., 2020, 11, 5142 RSC.
- B. Kaupbayeva and A. J. Russell, Prog. Polym. Sci., 2020, 101, 101194 CrossRef CAS.
- X. Liu and W. Gao, Angew. Chem., Int. Ed., 2021, 60, 2 CrossRef.
- M. Heredero and A. Beloqui, ChemBioChem, 2023, 24, e202200611 CrossRef CAS.
- M. J. Tamasi, R. A. Patel, C. H. Borca, S. Kosuri, H. Mugnier, R. Upadhya, N. S. Murthy, M. A. Webb and A. J. Gormley, Adv. Mater., 2022, 34, 2201809 CrossRef CAS.
- K. L. Heredia, D. Bontempo, T. Ly, J. T. Byers, S. Halstenberg and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 16955 CrossRef CAS PubMed.
- B. Le Droumaguet and K. Velonia, Angew. Chem., Int. Ed., 2008, 47, 6263 CrossRef CAS.
- H. Murata, C. S. Cummings, R. R. Koepsel and A. J. Russell, Biomacromolecules, 2014, 15, 2817 CrossRef CAS PubMed.
- D. Cohen-Karni, M. Kovaliov, T. Ramelot, D. Konkolewicz, S. Graner and S. Averick, Polym. Chem., 2017, 8, 3992 RSC.
- C. Bao, J. Chen, D. Li, A. Zhang and Q. Zhang, Polym. Chem., 2020, 11, 1386 RSC.
- A. Theodorou, E. Liarou, D. M. Haddleton, I. G. Stavrakaki, P. Skordalidis, R. Whitfield, A. Anastasaki and K. Velonia, Nat. Commun., 2020, 11, 1486 CrossRef CAS PubMed.
- A. Theodorou, D. Gounaris, E. Voutyritsa, N. Andrikopoulos, C. I. M. Baltzaki, A. Anastasaki and K. Velonia, Biomacromolecules, 2022, 23, 4241 CrossRef CAS PubMed.
- E. Voutyritsa, C. Gryparis, A. Theodorou and K. Velonia, Macromol. Rapid Commun., 2023, 2200976 CrossRef CAS PubMed.
- Q. Zhang, M. Li, C. Zhu, G. Nurumbetov, Z. Li, P. Wilson, K. Kempe and D. M. Haddleton, J. Am. Chem. Soc., 2015, 137, 9344 CrossRef CAS.
- Y. Liu, T. K. Nevanen, A. Paananen, K. Kempe, P. Wilson, L. S. Johansson, J. J. Joensuu, M. B. Linder, D. M. Haddleton and R. Milani, ACS Appl. Mater. Interfaces, 2019, 11, 3599 CrossRef CAS.
- J. Lu, H. Wang, Z. Tian, Y. Hou and H. Lu, J. Am. Chem. Soc., 2020, 142, 1217 CrossRef CAS.
- C. Bao, X. Xu, J. Chenvand and Q. Zhang, Polym. Chem., 2020, 11, 682 RSC.
- J. Liu, V. Bulmus, D. L. Herlambang, C. Barner-Kowollik, M. H. Stenzel and T. P. Davis, Angew. Chem., Int. Ed., 2007, 46, 3099 CrossRef CAS.
- J. Xu, K. Jung, N. A. Corrigan and C. Boyer, Chem. Sci., 2014, 5, 3568 RSC.
- M. Kovaliov, M. L. Allegrezza, B. Richter, D. Konkolewicz and S. Averick, Polymer, 2018, 137, 338 CrossRef CAS.
- A. E. Enciso, L. Fu, A. J. Russell and K. Matyjaszewski, Angew. Chem., Int. Ed., 2018, 57, 933 CrossRef CAS PubMed.
- J. Yeow, R. Chapman, A. J. Gormley and C. Boyer, Chem. Soc. Rev., 2018, 47, 4357 RSC.
- L. Fu, Z. Wang, S. Lathwal, A. E. Enciso, A. Simakova, S. R. Das, A. J. Russell and K. Matyjaszewski, ACS Macro Lett., 2018, 7, 1248 CrossRef CAS PubMed.
- Y. Sun, S. Lathwal, Y. Wang, L. Fu, M. Olszewski, M. Fantin, A. E. Enciso, G. Szczepaniak, S. Das and K. Matyjaszewski, ACS Macro Lett., 2019, 8, 603 CrossRef CAS.
- A. Theodorou, P. Mandriotis, A. Anastasaki and K. Velonia, Polym. Chem., 2021, 12, 2228 RSC.
- X. Pan, M. Fantin, F. Yuan and K. Matyjaszewski, Chem. Soc. Rev., 2018, 47, 5457 RSC.
- A. Anastasaki, V. Nikolaou, Q. Zhang, J. Burns, S. R. Samanta, C. Waldron, A. J. Haddleton, R. McHale, D. Fox, V. Percec, P. Wilson and D. M. Haddleton, J. Am. Chem. Soc., 2014, 136, 1141 CrossRef CAS.
- N. D. Dolinski, Z. A. Page, E. H. Discekici, D. Meis, I. H. Lee, G. R. Jones, R. Whitfield, X. Pan, B. G. McCarthy, S. Shanmugam, V. Kottisch, B. P. Fors, C. Boyer, G. M. Miyake, K. Matyjaszewski, D. M. Haddleton, J. R. de Alaniz, A. Anastasaki and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 268 CrossRef CAS PubMed.
- S. Dadashi-Silab, I.-H. Lee, A. Anastasaki, F. Lorandi, B. Narupai, N. D. Dolinski, M. L. Allegrezza, M. Fantin, D. Konkolewicz, C. J. Hawker and K. Matyjaszewski, Macromolecules, 2020, 53, 5280 CrossRef CAS.
- D. A. Corbin and G. M. Miyake, Chem. Rev., 2022, 122, 1830 CrossRef CAS PubMed.
- C. Kutahya, F. S. Aykac, G. Yilmaz and Y. Yagci, Polym. Chem., 2016, 7, 6094 RSC.
- G. Yilmaz and Y. Yagci, Polym. Chem., 2018, 9, 1757 RSC.
- S. de Ávila Gonçalves, P. R. Rodrigues and R. Pioli Vieira, Macromol. Rapid Commun., 2021, 42, e2100221 CrossRef PubMed.
- B. S. Tucker, M. L. Coughlin, C. A. Figg and B. S. Sumerlin, ACS Macro Lett., 2017, 6, 452 CrossRef CAS PubMed.
- R. A. Olson, J. S. Levi, G. M. Scheutz, J. J. Lessard, C. A. Figg, M. N. Kamat, K. B. Basso and B. S. Sumerlin, Macromolecules, 2021, 54, 4880 CrossRef CAS.
- K. Kapil, A. M. Jazani, G. Szczepaniak, H. Murata, M. Olszewski and K. Matyjaszewski, Macromolecules, 2023, 56, 2017 CrossRef CAS PubMed.
- T. Zhang, Z. Wu, G. Ng and C. A. Boyer, Angew. Chem., Int. Ed., 2023, e202309582 CAS.
- C. Kutahya, F. C. Aykac, G. Yilmaz and Y. Yagci, Polym. Chem., 2016, 7, 6094 RSC.
- K. Parkatzidis, N. P. Truong, M. N. Antonopoulou, R. Whitfield, D. Konkolewicz and A. Anastasaki, Polym. Chem., 2020, 11, 4968 RSC.
- V. Bellotti, K. Parkatzidis, H. S. Wang, N. D. A. Watuthanthrige, M. Orfano, A. Monguzzi, N. P. Truong, R. Simonutti and A. Anastasaki, Polym. Chem., 2023, 14, 253 RSC.
- V. Srivastava and P. P. Singh, RSC Adv., 2017, 7, 31377 RSC.
- S. Shanmugam, S. Xu, N. N. M. Adnan and C. Boyer, Macromolecules, 2018, 51, 779 CrossRef CAS.
- G. Szczepaniak, J. Jeong, K. Kapil, S. Dadashi-Silab, S. S. Yerneni, P. Ratajczyk, S. Lathwal, D. J. Schild, S. R. Das and K. Matyjaszewski, Chem. Sci., 2022, 13, 11540 RSC.
- K. Kapil, G. Szczepaniak, M. R. Martinez, H. Murata, A. M. Jazani, J. Jeong, S. R. Das and K. Matyjaszewski, Angew. Chem., Int. Ed., 2023, e202217658 CAS.
- A. Cvetkovic, A. J. J. Straathof, R. Krishna and L. A. M. van der Wielen, Langmuir, 2005, 21, 1475 CrossRef CAS PubMed.
- J. Mariam, S. Sivakami and P. M. Dongre, Drug Delivery, 2016, 23, 2668 CrossRef CAS.
- J.-P. Fouassier, F. Morlet-Savary, J. Lalevée, X. Allonas and C. Ley, Materials, 2010, 3, 5130 CrossRef CAS PubMed.
- E. Liarou, R. Whitfield, A. Anastasaki, N. G. Engelis, G. R. Jones, K. Velonia and D. M. Haddleton, Angew. Chem., Int. Ed., 2018, 57, 8998 CrossRef CAS.
- E. Liarou, A. Anastasaki, R. Whitfield, C. E. Iacono, G. Patias, N. G. Engelis, A. Marathianos, G. R. Jones and D. M. Haddleton, Polym. Chem., 2019, 10, 963 RSC.
- E. Liarou, Y. Han, A. M. Sanche, M. Walker and D. M. Haddleton, Chem. Sci., 2020, 11, 5257 RSC.
- N. A. Swisher, D. A. Corbin and G. M. Miyake, ACS Macro Lett., 2021, 10, 453 CrossRef CAS PubMed.
- Y. Lee, C. Boyer and M. S. Kwon, Chem. Soc. Rev., 2023, 52, 3035 RSC.
- B. Nomeir, O. Fabre and K. Ferji, Macromolecules, 2019, 52, 6898 CrossRef CAS.
- Z. Liang, S. Xu, W. Tian and R. Zhang, Beilstein J. Org. Chem., 2015, 11, 425 CrossRef CAS PubMed.
- B. Cornils, W. A. Herrmann, J.-H. Xu and H.-W. Zanthoff, Catalysis from A to Z: a concise encyclopedia, John Wiley and Sons, Weinheim, 2020. DOI:10.1002/9783527809080.
- N. El Achi, Y. Bakkour, W. Adhami, J. Molina, M. Penhoat, N. Azaroual, L. Chausset-Boissarie and C. Rolando, Front. Chem., 2020, 8, 740 CrossRef CAS.
- D. Bondarev, K. Borská, M. Šoral, D. Moravčíková and J. Mosnáček, Polymer, 2019, 161, 122 CrossRef CAS.
- F. Lorandi and K. Matyjaszewski, Isr. J. Chem., 2020, 60, 108 CrossRef CAS.
- P. G. Falireas, V. Ladmiral, A. Debuigne, C. Detrembleur, R. Poli and B. Ameduri, Macromolecules, 2019, 52, 1266 CrossRef CAS.
- M. Teodorescu and M. Bercea, Polym.-Plast. Technol. Eng., 2015, 54, 923 CrossRef CAS.
- A. Oucif, N. Haddadine, D. Zakia, N. Bouslah, A. Benaboura, K. Beyaz, B. Guedouar and M. S. El-Shall, Polym. Bull., 2022, 79, 153 CrossRef.
- L. Papadimitriou, A. Theodorou, M. Papageorgiou, E. Voutyritsa, A. Papagiannaki, K. Velonia and A. Ranella, J. Drug Delivery Sci. Technol., 2022, 75, 103591 CrossRef CAS.
- H. D. Pinholt, S. S.-R. Bohr, J. F. Iversen, W. Boomsma and N. S. Hatzakis, Proc. Natl. Acad. Sci. U. S. A., 2021, 31, 118 Search PubMed.
- S. S.-R. Bohr, P. M. Lund, A. S. Kallenbach, H. Pinholt, J. Thomsen, L. Iversen, A. Svendsen, S. M. Christensen and N. S. Hatzakis, Sci. Rep., 2019, 9, 2654 CrossRef.
- H. Zhao, E. Ibarboure, V. Ibrahimova, Y. Xiao, E. Garanger and S. Lecommandoux, Adv. Sci., 2021, 8, 2102508 CrossRef CAS PubMed.
- F. Akram, A. S. Mir, I. u. Haq and A. Roohi, Mol. Biotechnol., 2022, 65, 521 Search PubMed.
- M. Skjøt, L. De Maria, R. Chatterjee, A. Svendsen, S. A. Patkar, P. R. Østergaard and J. Brask, ChemBioChem, 2009, 10, 520 CrossRef.
Footnotes |
| † Dedicated to the memory of Prof. R. J. M. Nolte, whose seminal contributions to the field and mentorship shaped our academic journeys. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00407h. Dedicated to the memory of Prof. R. J. M. Nolte, whose seminal contributions to the field and mentorship shaped our academic journeys. |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |