
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFast and switchable ring-opening polymerization of biorenewable omega-substituted lactones towards sustainable copolymers with facile control over monomer sequences†
Jiayun
Jiang
a,
Xue
Liang
a,
Jiewen
Wang
 a,
Hongru
Qiang
a,
Jianrui
Li
a,
Jianzhong
Du
*bc and
Yunqing
Zhu
*a
a,
Hongru
Qiang
a,
Jianrui
Li
a,
Jianzhong
Du
*bc and
Yunqing
Zhu
*a
aDepartment of Polymeric Materials, School of Materials Science and Engineering, Tongji University, Shanghai 201804, China. E-mail: 1019zhuyq@tongji.edu.cn
bDepartment of Gynaecology and Obstetrics, Shanghai Key Laboratory of Anesthesiology and Brain Functional Modulation, Clinical Research Center for Anesthesiology and Perioperative Medicine, Translational Research Institute of Brain and Brain-Like Intelligence, Shanghai Fourth People's Hospital, School of Medicine, Tongji University, Shanghai 200434, China. E-mail: jzdu@tongji.edu.cn
cKey Laboratory of Advanced Civil Engineering Materials of Ministry of Education, School of Materials Science and Engineering, Tongji University, 4800 Caoan Road, Shanghai 201804, China
First published on 21st May 2024
Abstract
Highly active catalysts for the ring-opening polymerization (ROP) of biorenewable monomers, especially lactones, are essential for sustainable polymers. (Thio)urea/base catalyst systems have been found to be active for unsubstituted lactones. However, while omega-substituted lactones are an important class of biorenewable monomers, the ROP of such lactones suffers from low polymerization rates, and (thio)ureas usually show low catalytic activity for omega-substituted lactones due to their high steric hindrance. Herein, fast and controlled ROPs of omega-substituted lactones, including ε-decalactone (ε-DL), δ-caprolactone (δ-CL) and δ-decalactone (δ-DL), were achieved using cheap and commercially available (thio)urea/base catalyst systems, whose activities were significantly improved by pairing the pKas of (thio)ureas with bases. (Thio)urea/IMes pairs exhibited superior activity, with the most active TU3/IMes showing turnover frequencies of 270 h−1, 996 h−1 and 331 h−1 in the ROP of ε-DL, δ-CL and δ-DL, respectively. Moreover, the block, gradient and random copolymer chain sequences can be facilely modulated by different catalyst pairs during the copolymerization of ε-caprolactone (ε-CL) and ε-DL to match different application scenarios. Therefore, this work provides fresh ideas for the transformation of similar low-activity catalysts to high-activity ones and contributes to the efficient construction of structurally and functionally diverse biorenewable polymer materials.
Introduction
With increasing global environmental awareness, biorenewable aliphatic polyesters have been an effective alternative to traditional petroleum-derived plastics due to their renewability, biodegradability, and biocompatibility. With these properties, they have been extensively studied for potential applications in electronics, biomedical, packaging and other fields.1–5 The ring-opening polymerization (ROP) of lactones has been extensively studied as a promising method for synthesizing aliphatic polyesters. An increasing number of new bioderived lactones are now emerging, among which omega-substituted lactones should be of particular interest. This is because the production of biorenewable lactones from hydroxy fatty acids is a commonly used process,6,7 leading to the structural characteristic of many biorenewable lactones with omega substituents on the ring. Thus, it can be found that many biorenewable lactones, such as δ-decalactone,8–10 δ-caprolactone,11–13 γ-decalactone, γ-valerolactone, carvomenthide,14 dihydrocarvide,14 and menthide,15,16 are monomers with an omega-substituted structure, and have been used for the synthesis of biorenewable polymers. These biorenewable polymers usually have low Tg, low crystallinity, good flexibility, and the potential for post-modification on the substituents.12,16–18 Despite these advantages, the ring-opening polymerization of omega-substituted lactones also suffers from low polymerization rates, high reaction temperatures or rather elaborate catalysts. Therefore, achieving fast and controlled ring-opening polymerization of omega-substituted lactones is of great significance for the synthesis of functional biorenewable polyesters and deserves further research.In addition to exploring how to efficiently polymerize biorenewable monomers (e.g. omega-substituted lactones), achieving greener and more efficient modulation of the copolymer sequence is one of the key challenges in the field of polymer synthesis. Homopolymers or copolymers with no sequence-controlled features usually do not have structural and functional complexity. Developing simple and efficient methods for monomer sequence modulation is of great value as it could afford copolymers with block, gradient and random structures to achieve different properties.19–21 For example, the main method to prepare block copolymers at present is the sequential addition of monomers, which is relatively complicated and difficult to operate compared to the emerging chemoselective copolymerization method, which can build block copolymers from a mixed monomer feedstock.19 Since Coates's group reported the one-pot polymerization of epoxide, cyclic anhydride, and CO2 using a single catalyst,22 chemoselective polymerization has been widely studied.23–27 However, it is difficult to tune the chemoselectivity of such catalytic systems to freely build random, gradient, and block copolymers according to the application scenarios. Therefore, it is necessary to develop a catalytic system that can facilely modulate copolymer sequences to afford copolymers with structural diversity.
Currently, various catalysts have been developed for the ring-opening polymerization of lactones. Organocatalysts, including Lewis acids, phosphazene bases, N-heterocylic carbenes (NHCs) and H-bond bifunctional compounds, have received the most attention since they can avoid metal residues in polyesters while still providing a high level of control over the polymerization compared to metal-based catalysts.1,3,28–30 Among them, (thio)urea [(T)U], a hydrogen-bond organocatalyst, stands out for its advantages of low cost, commercial availability, and applicability in switchable polymerization.31–33 Most studies have focused on the applications of (thio)urea catalysts for the ROP of unsubstituted lactones such as lactide (LA) and caprolactone (CL), which have shown high activity. However, like other types of catalysts,18,34–37 (thio)ureas tend to be much less active for the ROP of omega-substituted lactones, which could be due to the large steric hindrance contributed by the substituents. This problem limits the development and transformation of bio-based omega-substituted lactones.
In Prof. Waymouth's work, it has been found that the pairing of (thio)ureas with bases of different pKa values greatly affects the activity of the catalyst systems for the ROP of δ-valerolactone (VL).38 However, whether this phenomenon can be generally applied to other lactones, especially to omega-substituted lactones, remains unclear, as the ROP of omega-substituted lactones is greatly affected by their substituents and is rather different from that of unsubstituted lactones. Therefore, in this work, a series of (thio)urea/base catalyst systems were carefully selected and paired to evaluate their catalytic activity in search of efficient catalysts for the ROP of omega-substituted lactones. ε-Decalactone (ε-DL), a biologically derived lactone that can be prepared from castor oil,36,39 was first selected as the monomer for the screening of the catalyst systems. To further verify the activity and trends of the efficient catalysts, the monomer scope was extended to other omega-substituted lactones, including δ-caprolactone (δ-CL) and δ-decalactone (δ-DL), which are naturally derived from coconut water and Cryptocarya massoy, respectively.40,41 In addition, the sequence of polymer chains can be modulated by simply using different (thio)urea/base pairs in the copolymerization of ε-DL and ε-CL to construct block, gradient and random copolymers.
Results and discussion
Ring-opening polymerization of ε-DL using different (thio)urea/base pairs
According to previous studies, pairing (thio)ureas and bases with different pKa values usually leads to different catalytic mechanisms, including anionic and cooperative mechanisms, leading to varied catalytic activities.38 Therefore, to explore the best catalytic system for omega-substituted lactones, a series of (thio)ureas with different substituents (Fig. 1a) combined with three organic bases (1,8-diazabicyclo[5.4.0]undec-7-ene, DBU; 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine, BEMP; and 1,3-dimesitylimidazol-2-ylidene, IMes) (Fig. 1b) were screened for the ring-opening polymerization of ε-DL, one of the commonly used biobased omega-substituted lactones (Fig. 1c). The choice of (thio)urea/base pairs covers a wide range of pKa values and features both mechanisms. The pKa values in DMSO of (thio)ureas and bases are labelled in Fig. 1.18,42–45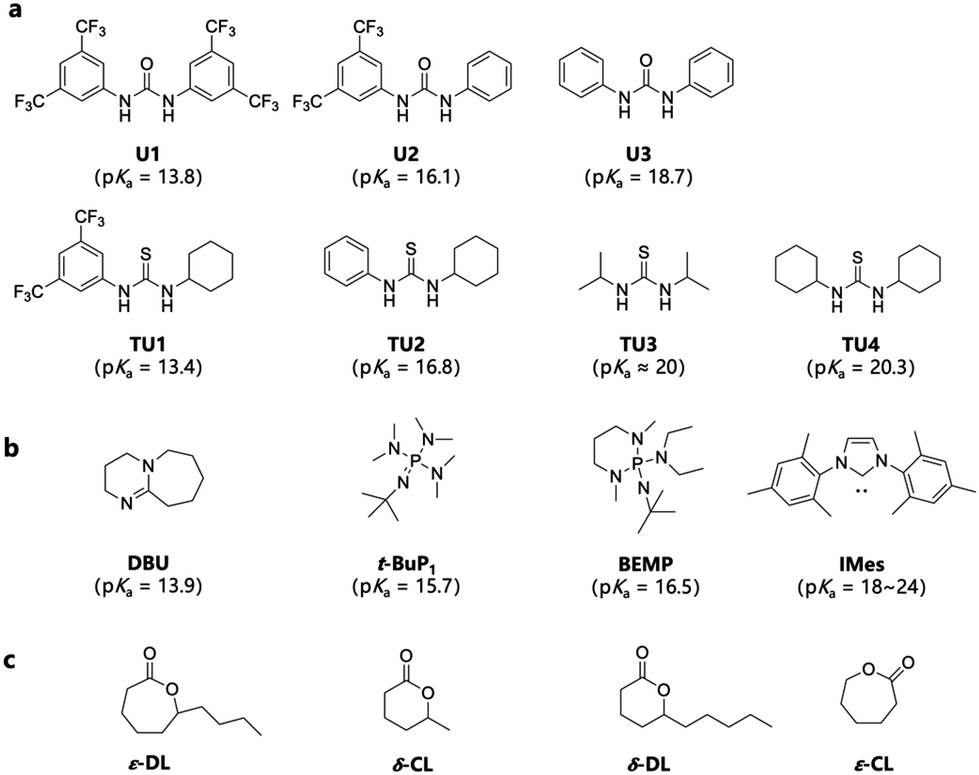 | ||
| Fig. 1 (a) Ureas and thioureas with their pKa values in DMSO.30,42 (b) Organic bases with their pKa values in DMSO.43–45 (c) Monomers studied. | ||
All of the ROPs of ε-DL were carried out in toluene with BnOH as the initiator at 25 °C and the reactions were terminated at predetermined time durations (Table 1). Instead of a high polarity solvent namely DMSO, toluene with medium polarity was selected as the solvent for the ROPs to simultaneously ensure the occurrence of different catalytic mechanisms and maintain good control over the polymerization process.46 The 1H NMR spectrum for the ROP of ε-DL is shown in Fig. S1.† All the polymers displayed unimodal SEC traces with narrow distributions (Fig. S2†). Significant differences in the catalytic effects of different catalyst systems can be observed in the ROP of ε-DL. For the weakest DBU, no considerable conversion of ε-DL was observed in combination with either urea or thiourea, even when the reaction time was extended to 120 h. U1 and TU1, with pKa values close to that of DBU, can lead to 18% and 4% conversion of ε-DL, respectively. This could be due to the high activity in the ‘matching’ case, where the highest catalytic activity of (thio)urea/base catalyst systems towards lactide and unsubstituted lactones was usually observed when the pKa value of the (thio)urea closely matched that of the protonated base.38 This could be the result of the competition between two mechanisms, the anionic mechanism and the cooperation mechanism, since in the ‘matching’ case, the deprotonation of (thio)urea by the base and the formation of a hydrogen-bonded complex with the base will proceed simultaneously, leading to equilibrium.47,48
| # | (Thio)ureas | Bases | t (h) | Conv.b (%) | TOFc (h−1) |
M
n,theo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) d (kDa) d (kDa) |
M
n,SECe (kDa) |
Đ |
|---|---|---|---|---|---|---|---|---|
| a Polymerizations were performed in dry toluene at 25 °C with [ε-DL]0 = 5 M; [ε-DL]/[(T)U]/[base]/[I] = 100/2.5/2.5/1; I = BnOH. b Monomer conversion determined by 1H NMR in CDCl3 using integrals of the characteristic signals. c Turnover frequency (TOF) = mole of the consumed monomer/mole of the (thio)urea catalyst per hour. d M n,theo = M(ε-DL) × ([ε-DL]0/[I]0) × conv. + M(BnOH). e Determined by SEC in THF at a flow rate of 1.0 mL min−1 at 40 °C, calibrated with polystyrene standards. | ||||||||
| 1 | U1 | DBU | 120 | 18 | — | — | — | — |
| 2 | BEMP | 144 | 79 | 0.2 | 13.6 | 11.4 | 1.03 | |
| 3 | IMes | 72 | 49 | 0.3 | 8.5 | 8.3 | 1.12 | |
| 4 | U2 | DBU | 120 | 12 | — | — | — | — |
| 5 | BEMP | 144 | 46 | 0.1 | 7.9 | 5.8 | 1.06 | |
| 6 | IMes | 48 | 85 | 0.7 | 14.6 | 11.8 | 1.03 | |
| 7 | t-BuP1 | 48 | 0 | — | — | — | — | |
| 8 | U3 | DBU | 120 | 0 | — | — | — | — |
| 9 | BEMP | 120 | 0 | — | — | — | — | |
| 10 | IMes | 2 | 86 | 17 | 14.7 | 10.1 | 1.10 | |
| 11 | t-BuP1 | 48 | 0 | — | — | — | — | |
| 12 | TU1 | DBU | 120 | 4 | — | — | — | — |
| 13 | BEMP | 120 | 0 | — | — | — | — | |
| 14 | IMes | 72 | 24 | 0.1 | — | — | — | |
| 15 | TU2 | DBU | 120 | 0 | — | — | — | — |
| 16 | BEMP | 120 | 0 | — | — | — | — | |
| 17 | IMes | 24 | 93 | 2 | 15.9 | 11.3 | 1.13 | |
| 18 | t-BuP1 | 48 | 0 | — | — | — | — | |
| 19 | TU3 | DBU | 120 | 0 | — | — | — | — |
| 20 | BEMP | 120 | 0 | — | — | — | — | |
| 21 | IMes | 8 min | 90 | 270 | 15.4 | 11.3 | 1.20 | |
| 22 | t-BuP1 | 48 | 0 | — | — | — | — | |
| 23 | TU4 | IMes | 3 | 92 | 12 | 15.7 | 10.3 | 1.15 |
When DBU is not strong enough to deprotonate the (thio)urea, i.e. when its pKa value is relatively smaller, the polymerization is dominated by the cooperative mechanism. In this case, almost all combinations involved with DBU resulted in failed polymerization. Similarly, when pairing the moderate BEMP with (thio)ureas of greater pKa values (Table 1, entries 9 and 20), none of the cases of the cooperative mechanism can lead to the ROP of ε-DL. This is in contrast to the results of previous studies involving lactide and unsubstituted lactones,38 as all catalyst systems featuring the cooperative mechanism (Table 1, entries 4, 8, 9, 15, 19 and 20) led to failures in the ROP of ε-DL. In addition, N′-tert-butyl-N,N,N′,N′,N′′,N′′-hexamethylphosphorimidic triamide (t-BuP1) with a pKa value of 15.7 was selected in combination with U2, U3, TU2, and TU3 (pKa > 16.1), respectively, to further validate the low activity of the cooperative mechanism towards omega-substituted lactones (Table 1, entries 7, 11, 18 and 22). Consistent with the results of DBU and BEMP, none of these catalyst pairs of the cooperative mechanism could initiate the ring-opening polymerization of ε-DL, suggesting that the cooperative mechanism may not work for omega-substituted lactones. This may be due to the following reasons: first, the hydroxyl group at the propagating chain end formed by the ROP of omega-substituted lactones is a secondary alcohol. It has been noted that secondary alcohols are less reactive than primary alcohols,49,50 making it more difficult to activate secondary alcohols at the propagating chain end. In addition, compared to the anionic mechanism in which the activations of the propagating chain end and the monomer occur on the same (thio)urea molecule, the nucleophilic attack on the monomer in the cooperative mechanism would be more difficult due to the steric hindrance of the substituents.
BEMP led to successful polymerization only in combination with U1 and U2. It is noteworthy that, in contrast to the previous findings, BEMP/U1 (anionic mechanism) showed higher activity than BEMP/U2 (‘matching’ mechanism) in the ROP of ε-DL. As it is out of the scope of this work, this unusual phenomenon will be investigated in the future. In contrast, TU1 and TU2, which have similar pKa values, did not initiate the ROP of ε-DL. Similarly, when combined with DBU, U1 and U2 also showed better activity than TU1 and TU2. This could be due to the fact that ureas are stronger hydrogen bond donors than thioureas of similar acidity, which has also been reported by Kozlowski.46,51,52 In addition, the urea anion is a stronger base than the thiourea anion due to the large sulfur atom that increased the stability of the conjugate base.
When the strongest IMes was used, ε-DL ROP occurred in all cases. The activity of the catalyst systems reached its maximum in the ‘matching’ case, as the TOF value of TU3/IMes was as high as 270 h−1. In contrast to the observations about U1/TU1 and U2/TU2, TU3 exhibited higher activity than U3 in the ROP of ε-DL when combined with IMes. It is hypothesized that this unusual phenomenon could be due to the steric hindrance since the nitrogen atom of TU3 is bonded to an isopropyl group, instead of a bulky benzene ring of U3. This steric hindrance effect may not be profound in the ROP of unsubstituted lactones but could pose a significant impact on the ROP of omega-substituted lactones due to their large substituents. To verify this hypothesis, another thiourea, TU4, with higher steric hindrance but almost identical pKa to TU3, was employed to pair with IMes for ε-DL ROP (Table 1, entry 23). The TOF value of TU4/IMes was found to be 12 h−1, which is much lower than that of TU3/IMes. This indicates that steric hindrance indeed has a great impact on the (thio)urea/base catalyzed ROP of substituted lactones. Low steric hindrance of the (thio)ureas facilitates the simultaneous activation of the propagating chain end and the monomer on the same (thio)urea molecule, leading to efficient nucleophilic attack.
Due to the particularly high activity of TU3/IMes, the reaction rate is too high for aliquoting and monitoring, as the monomer was almost completely consumed as soon as the reaction mixture was prepared in a glove box. Therefore, entry 6 was chosen as the model reaction for kinetic studies due to its moderate activity. As shown in Fig. 2a and b, the linear increase in molecular weight with monomer conversion indicates the characteristics of controlled living polymerization. However, the SEC traces in Fig. 2b are not perfectly symmetrical, showing a slight ‘tail’ at a shorter retention time due to initiation from a trace amount of water present in the monomers. This phenomenon has been discussed in detail in previous works by other groups.29,53 Furthermore, semi-logarithmic plot analysis demonstrated the first-order feature, which is also consistent with the living polymerization features (Fig. 2c). A chain extension experiment was later performed to further confirm the living features of the ROP of ε-DL mediated by the (thio)urea/base catalyst systems. After 100 equivalents of ε-DL were completely converted, another 100 equivalents of ε-DL were added to the reaction mixture. The SEC traces in Fig. S3† demonstrate an increase in the molecular weight of the polymer from 13.6 kDa to 25.7 kDa that occurred after the sequential addition of monomers while still maintaining a narrow molecular weight distribution (<1.08).
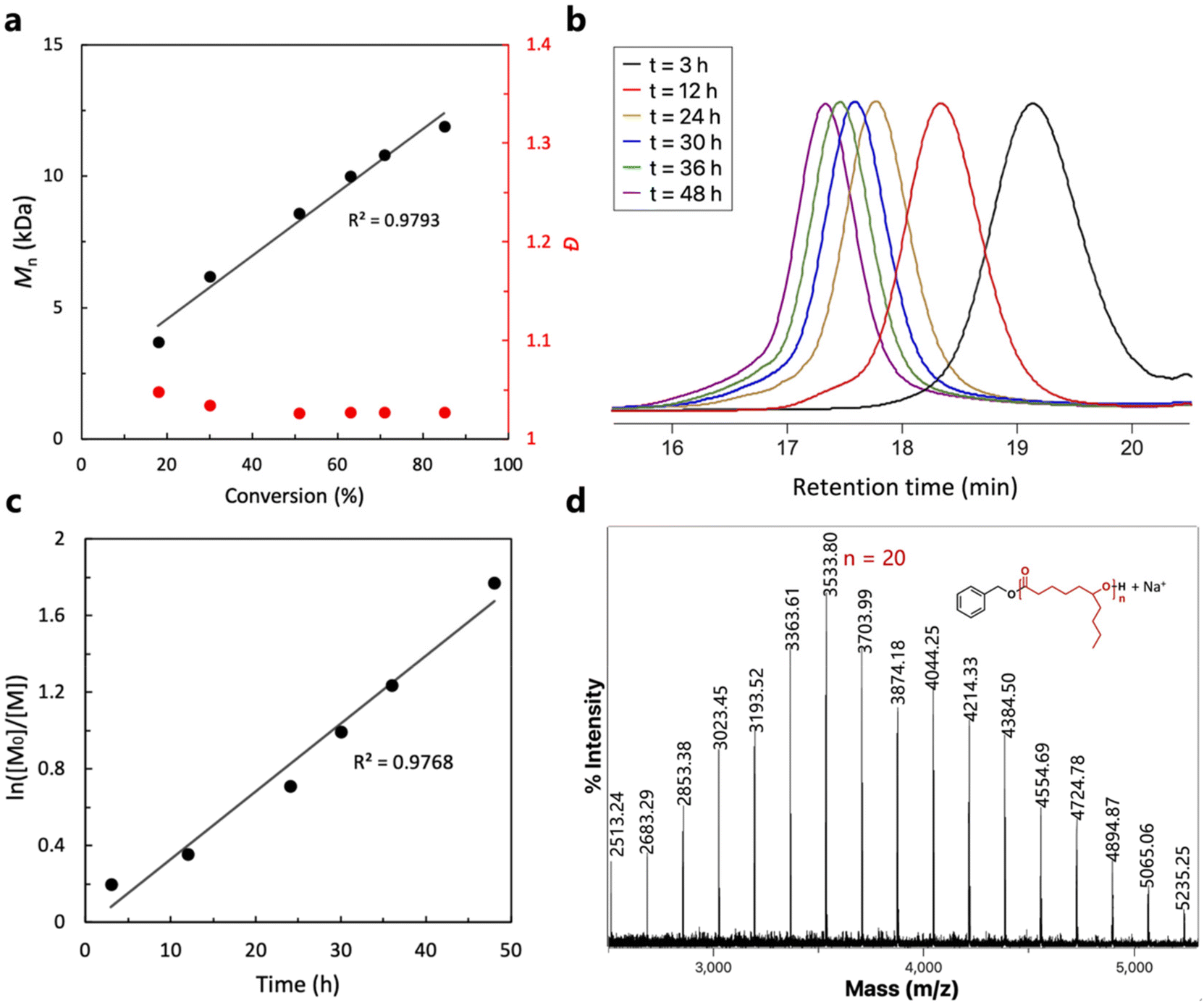 | ||
| Fig. 2 (a) Mn,SEC and dispersity (Đ) vs. monomer conversion, (b) typical SEC curves of aliquots taken at different time intervals during the ROP of ε-DL, (c) semi-logarithmic plot of monomer concentration over time and (d) the MALDI-TOF mass spectrum of P(ε-DL) produced with U2/IMes as the catalyst (Mn = 6.0 kDa, Đ = 1.04, Table 1, entry 6). | ||
(Thio)urea catalysts have previously been found to initiate polymer chains, making it difficult to predict molecular weights from the amount of initiator, compromising the controllable nature of living polymerization.34 Therefore, in order to further identify the polymer structure and initiation mechanism, the matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) technique was used to analyze the structure of the obtained P(ε-DL). The MALDI-TOF mass spectrum reveals only one series of molecular ion peaks with an interval of ca. 170.13 Da, corresponding to the exact molar mass of the ε-DL repeat unit (Fig. 2d). The molar mass M = n × 170.13 (MDL) + 108.14 (MBnOH) + 22.99 (MNa+) of each peak corresponds to linear P(ε-DL) chains initiated solely by BnOH and terminated by the hydroxyl group, which is also consistent with the results of the 1H NMR spectrum (Fig. S1†), suggesting that the catalyst system does not initiate polymer chains and has excellent control over the molecular weight and structure of the polymer. The chain structure of the polymer catalyzed by U2/BEMP was also characterized by MALDI-TOF (Fig. S4†) and the results are also in agreement with the previous ones.
The conversions at different times during the ROP of ε-DL by different catalyst systems were plotted into graphs (Fig. S5†), followed by the discussion on the activity trends and reaction mechanisms for different catalyst pairs. In conclusion, in the ROP of ε-DL, the (thio)urea/IMes pairs exhibited superior activity in all catalyst systems. In particular, TU3/IMes was used for the ROP of ε-DL at room temperature with a TOF value of 270 h−1, exhibiting activity far exceeding that of any other catalysts at the same or higher polymerization temperatures (Fig. S6†).18,34,36
Ring-opening polymerization of other omega-substituted lactones using different (thio)urea/IMes pairs
In the screening of the (thio)urea/base catalytic system carried out in the ROP of ε-DL, the (thio)urea/IMes catalyst pairs showed high activity superior to the other groups. To further verify the high activity and activity trends of the (thio)urea/IMes catalyst pairs by a wider range of omega-substituted lactones, these catalyst systems were subsequently used for the ring-opening polymerization of δ-CL and δ-DL (Tables S1 and S2†). The 1H NMR spectra for the ROP of δ-CL and δ-DL are shown in Fig. S7 and S8.†The (thio)urea/IMes catalyst systems showed particularly high activity when screened by the ring-opening polymerization of δ-CL (Table S1†), compared to the performance in the ROP of ε-DL under the same conditions. All (thio)urea/IMes systems had much higher TOF values in the ROP of δ-CL than ε-DL. This difference may be due to the greater steric hindrance of the substitution of ε-DL, which confines its polymerization rate. This can be further confirmed by the results of the ring-opening polymerization of δ-DL catalyzed by(thio)urea/IMes (Table S2†). The TOF values in the ROP of δ-DL were smaller than those observed in the ROP of δ-CL, which can be attributed to the substitution of the n-amyl group on the ring. Moreover, SEC analyses of polymers obtained by the ROPs of δ-CL and δ-DL mediated by (thio)urea/IMes all exhibited monomodal curves and narrow molecular weight distributions, confirming that the polymerization remains highly controlled (Fig. S9 and S10†). The living behaviour of the polymerization of δ-CL and δ-DL is subsequently demonstrated by the first-order features shown by the plots of ln[([M]0 − [M]eq)/([M]t − [M]eq)] vs. time, the linear increase in molecular weight with conversion and the consistently low molecular weight distribution (Fig. 3). Here, neither the polymerization of δ-CL nor that of δ-DL can achieve a conversion greater than 90%, which is probably caused by the equilibrium of the monomer and the polymer in the reaction system. As the thermodynamic driving force for the polymerization of cyclic monomers is closely related to ring size, relatively low equilibrium conversion is commonly observed in the polymerization of six-membered lactones with low ring strain.54 Additionally, some reports indicated that the equilibrium conversion of substituted δ-lactones decreases with larger substituents on the ring,55 which is consistent with the observation that the equilibrium conversion of δ-DL (ca. 70%) is generally lower than that of δ-CL (ca. 80%) under the same conditions.
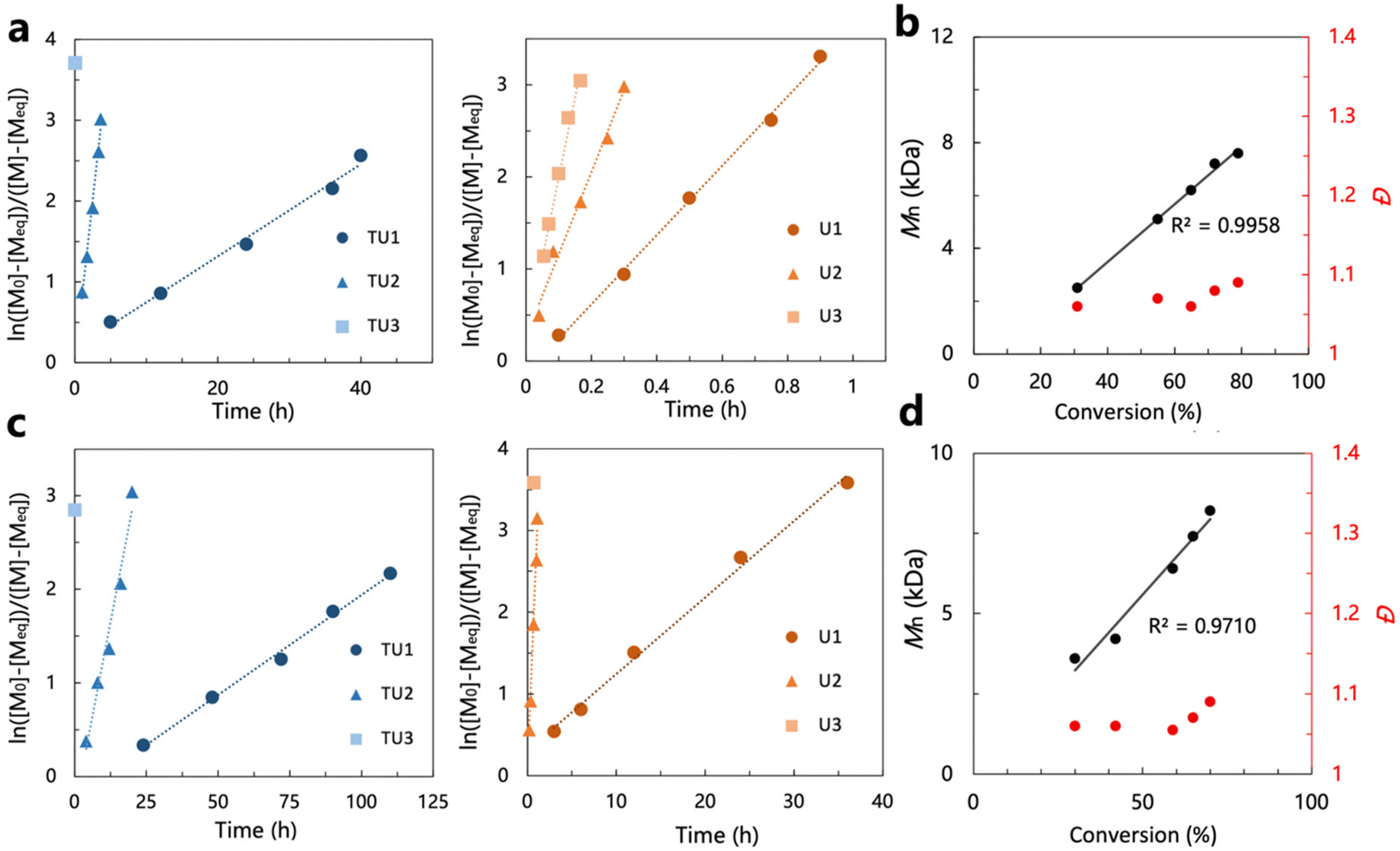 | ||
| Fig. 3 (a) Kinetic plots of ln[([M]0 − [M]eq)/([M]t − [M]eq)] vs. time for the ROP of δ-CL and (b) Mn,SEC and dispersity (Đ) vs. monomer conversion (U2/IMes), (c) kinetic plots of ln[([M]0 − [M]eq)/([M]t − [M]eq)] vs. time for the ROP of δ-DL and (d) Mn,SEC and dispersity (Đ) vs. monomer conversion (U2/IMes). | ||
The process of the ring-opening polymerization of δ-CL and δ-DL using the (thio)urea/IMes catalyst systems was sampled for monitoring and the results are presented in plots to visualize the trends in activity (Fig. 3a and c). The activity trends of the (thio)urea/IMes catalyst systems are consistent with the previous findings in ε-DL ROP. All the activities increased with decreasing acidity of the urea or thiourea, which corresponds to the shift from the anionic mechanism to the ‘matching’ mechanism. In the ring-opening polymerizations of δ-CL and δ-DL, considerable monomer conversion could be achieved in minutes using IMes and TU3/U3. In particular, the TOF values of TU3/IMes in the ROP of δ-CL and δ-DL were as high as 996 h−1 and 331 h−1, respectively, which were much higher than those of the catalysts (e.g. DPP and TBD) commonly used in the ROP of δ-CL and δ-DL at room temperature (Fig. S11†).9,13,56,57 Although the TOF values of a strontium catalyst (TOF = 360 h−1 and 228 h−1 for δ-CL and δ-DL, respectively) are reluctantly close to our system, its application may still be limited by the high cost, pyrophoric feature, and concerns regarding the toxicity of metal residues in polymers. Therefore, the higher activity exhibited by (thio)urea/IMes in the ROP of δ-CL and δ-DL further verified that the anionic and ‘matching’ mechanisms are more applicable to omega-substituted lactones.
Ring-opening copolymerization of ε-DL and ε-CL using (thio)urea/IMes pairs
From the previous experimental results, it is clear that the (thio)urea/base catalyst systems are much more active for lactones with smaller side chains, such as δ-CL. Hence, given the reactivity difference of these catalyst systems in the homopolymerization of different monomers, it may be possible to achieve well-defined polymer sequences by the ROPs of lactone mixtures in one pot. We believe that the modulation of the copolymer structure may be achieved by selecting catalyst systems with different activities for the synthesis of random, gradient and block copolymers (Scheme 1b). To ensure a high reactivity difference, unsubstituted lactone ε-caprolactone(ε-CL) was chosen as the comonomer for copolymerization with ε-DL (Table 2). Although ε-CL is usually prepared industrially from petrol, more and more studies are now focused on developing green and efficient methods for the preparation of ε-CL,58 which makes it potentially sustainable. U2/IMes, which exhibits moderate activity in the homopolymerization of ε-DL, was first selected as the catalyst for copolymerization with ε-CL (Table 2, entry 1). The copolymerization was monitored by 1H NMR spectroscopy (Fig. S12 and S13†). Due to the high activity of U2/IMes towards ε-CL, ε-CL reached complete conversion within 30 min, followed by the ROP of ε-DL to produce an ε-CL/ε-DL copolymer. The SEC curve of the copolymer (Fig. S14†) showed a monomodal distribution and the molecular weight also matched the theoretical values (Mn = 16.6 kDa, Đ = 1.32), indicating that the copolymerization process is highly controlled.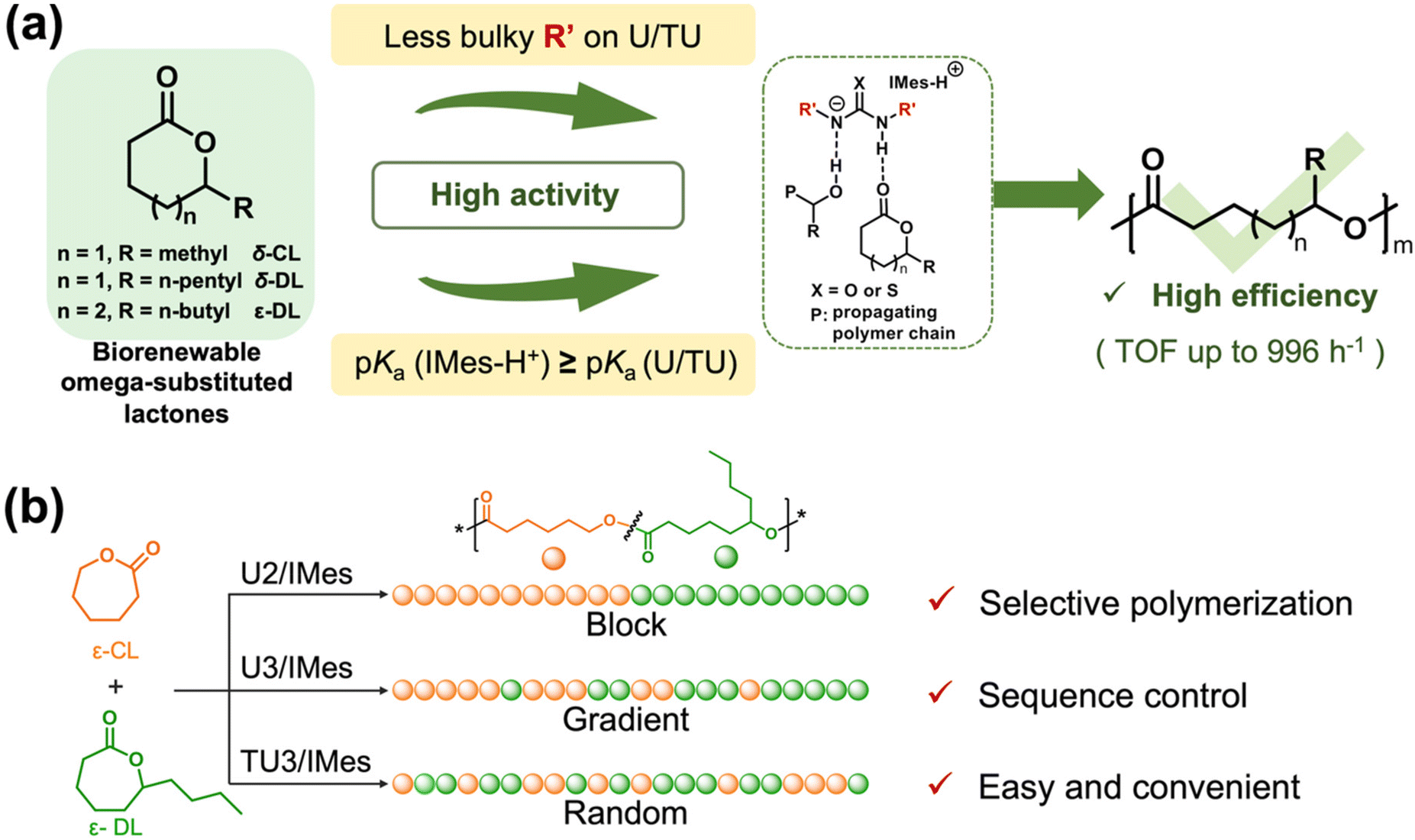 | ||
| Scheme 1 (a) Fast and efficient ring-opening polymerization of biorenewable omega-substituted lactones catalyzed by (thio)urea/base catalyst systems and (b) copolymerization of ε-DL and ε-CL to produce block, gradient and random copolymers using different (T)U/base catalyst systems. | ||
| Dyade | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst (s) | [ε-CL]/[ε-DL] | t (h) | Conv. ε-CLb (%) | Conv. ε-DLb (%) |
M
n,theoc (kDa) |
M
n,SECd (kDa) |
Đ | CL*–CL | CL*–DL | DL*–CL | DL*–DL |
| a Polymerizations were performed in dry toluene at 25 °C with [ε-CL]0 = 2.5 M; [(T)U]/[base]/[I] = 2.5/2.5/1; I = BnOH. b Monomer conversion determined by 1H NMR in CDCl3 using integrals of the characteristic signals. c M n,theo = M(ε-CL) × ([ε-CL]0/[I]0) × conv. + M(ε-DL) × ([ε-DL]0/[I]0) × conv. + M(BnOH). d Determined by SEC in THF at a flow rate of 1.0 mL min−1 at 40 °C, calibrated with polystyrene standards. e Determined by quantitative 13C NMR spectroscopy, with * defining the carbonyl analyzed. | ||||||||||||
| 1 | U2/IMes | 100/100 | 102 | 99 | 75 | 23.3 | 16.6 | 1.32 | 0.46 | 0.08 | 0.02 | 0.44 |
| 2 | U3/IMes | 100/100 | 4 | 99 | 86 | 26.0 | 22.1 | 1.33 | 0.39 | 0.12 | 0.09 | 0.40 |
| 3 | U3/IMes | 50/100 | 3 | 99 | 93 | 21.6 | 19.3 | 1.30 | 0.20 | 0.10 | 0.09 | 0.61 |
| 4 | TU3/IMes | 100/100 | 40 min | 99 | 92 | 27.1 | 28.8 | 1.35 | 0.31 | 0.20 | 0.19 | 0.30 |
To further confirm the selectivity of U2/IMes for ε-CL and ε-DL, the copolymerization of ε-CL/ε-DL by U2/IMes was also monitored using in situ IR spectroscopy (Fig. 4a). The FTIR spectra of ε-DL and ε-CL monomers are shown in Fig. S15† to justify the choice of bands for monitoring. It was found that ε-CL ROP occurred first, as evidenced by the decrease in the resonance attributed to ε-CL at 1051 cm−1. During this period, ε-DL did not undergo any conversion, as the corresponding resonance at 1530 cm−1 hardly decreased. A reduction of the resonance assigned to ε-DL was only observed after the complete consumption of ε-CL. This suggests that U2/IMes preferentially catalyzes the ROP of ε-CL and switches to the ROP of ε-DL once ε-CL is completely consumed. Therefore, the copolymerization of ε-CL and ε-DL by the U2/IMes catalytic system affords copolymers with a block structure.
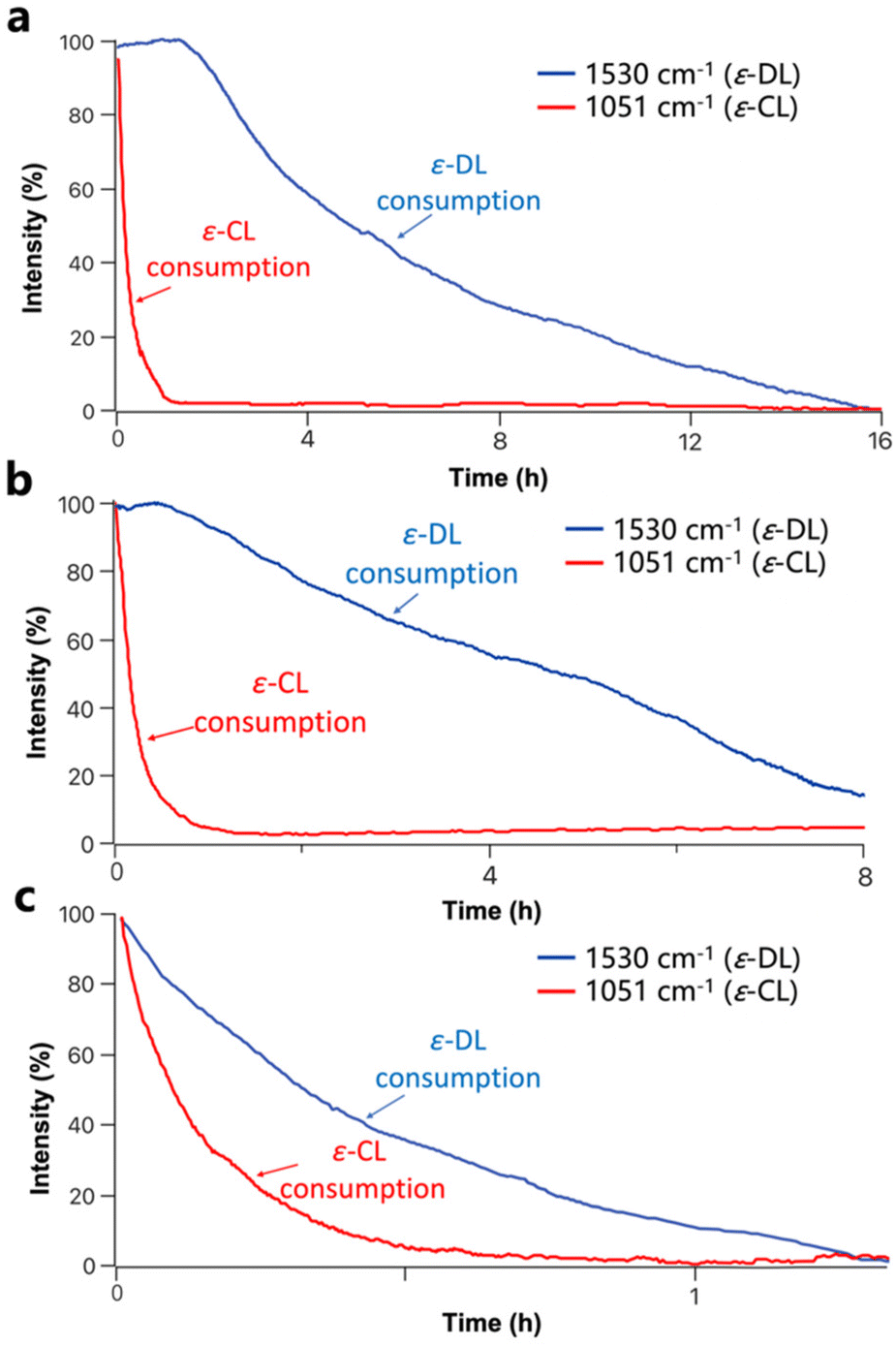 | ||
| Fig. 4 Formation of the ε-CL/ε-DL copolymer produced by (a) U2/IMes, (b) U3/IMes and (c) TU3/IMes monitored by in situ IR spectroscopy. | ||
Alternatively, the more reactive U3/IMes and the most reactive TU3/IMes were selected for the copolymerization of ε-CL and ε-DL. The monomers were able to reach considerable conversion (> 80%) within a few hours and copolymers with the corresponding molecular weights and narrow molecular weight distributions were synthesized (Table 2, entries 2–4). The ring-opening copolymerization of ε-CL and ε-DL mediated by U3/IMes and TU3/IMes was also monitored by in situ IR spectroscopy. For U3/IMes, the conversion of ε-DL occurred before the complete conversion of ε-CL, suggesting that a gradient copolymer structure was most likely formed (Fig. 4b). For TU3/IMes, a simultaneous decrease in the resonances attributed to ε-CL and ε-DL at 1051 cm−1 and 1530 cm−1 can be clearly observed, respectively. This suggests a mutual propagation of ε-DL and ε-CL, leading to the formation of a random copolymer (Fig. 4c). Besides, 1H DOSY NMR spectra of copolymers prepared by U2/IMes, U3/IMes and TU3/IMes all show a single diffusion coefficient, which confirms the formation of ε-CL/ε-DL copolymers (Fig. S16, S17 and S18†).
To verify the chain sequence, all ε-CL/ε-DL copolymers were analyzed by quantitative 13C NMR spectroscopy, and the carbonyl region was analyzed and integrated to reveal the chain structure (Fig. 5). The sequence of two (different) adjacent monomers in a polymer is characterized by the carbonyl dyad resonance. The carbonyl dyad resonance corresponding to an ε-CL carbonyl adjacent to an ε-CL repeat unit (CL*–CL, where the asterisk represents the observed carbonyl) can be observed at δ = 173.6 ppm, while the smaller shoulder peak on the left corresponds to the carbonyl dyad resonance of CL*–DL. Similarly, the three split peaks of the carbonyl dyad resonance observed at δ = 173.4 ppm can be attributed to DL*–DL and DL*–CL resonances, respectively. For copolymers produced by U2/IMes, the integration of the CL*–DL (0.08) and DL*–CL (0.02) resonances is particularly smaller than those of DL*–DL (0.44) and CL*–CL (0.46), indicating the high selectivity of U2/IMes and the block-like polymer chain sequence. Notably, some transesterification may occur due to the relatively long reaction time of entry 1, which could account for the presence of some CL*–DL and DL*–CL carbonyl dyad resonances.
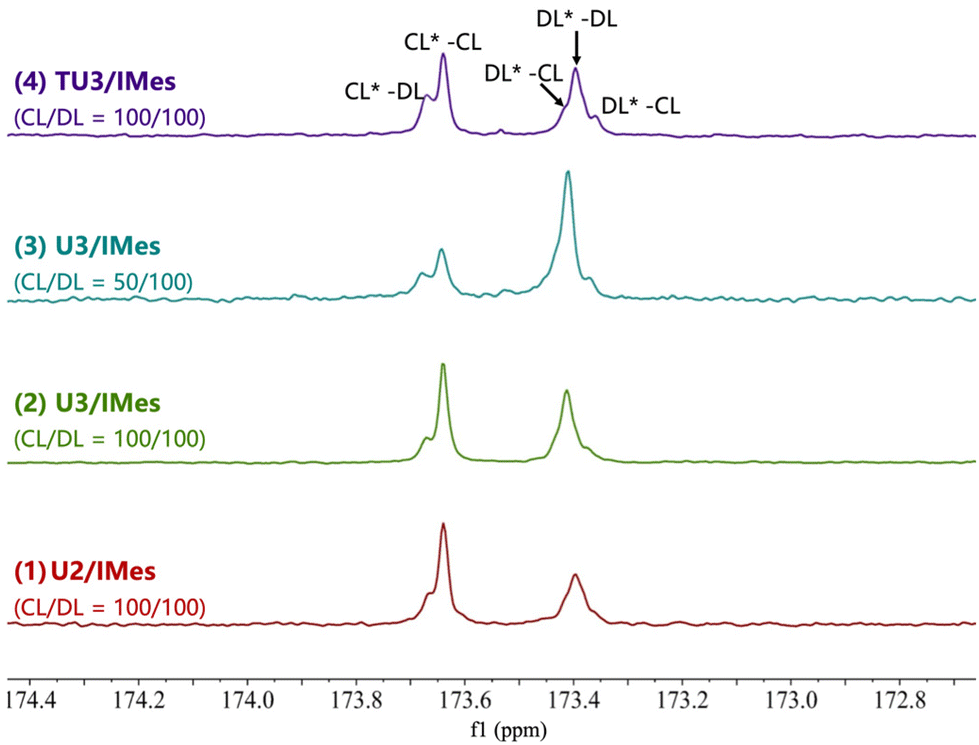 | ||
| Fig. 5 Quantitative 13C NMR spectra of the carbonyl region of the ε-DL/ε-CL copolymer (125 MHz, CDCl3, 298 K). | ||
When the U3/IMes with moderate activity was used, there was an increase in the integrals of the CL*–DL (from 0.08 to 0.12) and DL*–CL (from 0.02 to 0.09) resonances, but still much less than those of the DL*–DL and CL*–CL resonances. This phenomenon indicates that the selectivity for ε-CL and ε-DL is reduced in the U3/IMes system, thus favouring the formation of copolymers with a potential gradient structure. For comparison, the monomer ratio of ε-CL/ε-DL was then reduced to 50/100 with U3/IMes (Table 2, entry 3). The result of 13C NMR shows that the integrals of the major DL*–DL and CL*–CL dyad resonances in the carbonyl region of the copolymer have also changed to match the initial monomer feed ratio. There is a slight decrease in the integral of the CL*–DL resonance in the copolymer from 0.12 to 0.10 as the content of ε-CL decreases, suggesting a reduction of the gradation length in the copolymer. When it comes to the most active TU3/IMes, the relative integrals obtained by deconvolution of the carbonyl resonances corresponding to CL*–DL and CL*–CL became closer (0.20 vs. 0.31), indicating that a random copolymer was likely to be produced. This may be due to the very high activity of TU3/IMes that the reactivity difference between ε-CL and ε-DL became smaller, making the ε-DL monomer getting inserted into the polymer chain at a similar rate to that of ε-CL. It can also be evidenced by the fact that the least active U2/IMes has the highest selectivity for the copolymerization of monomer mixtures. Therefore, by simply changing the combination of (thio)ureas and bases, copolyesters with different structures (e.g. block, gradient and random structures) can be facilely and efficiently prepared in one-pot from a mixed monomer feedstock.
The thermal properties of the copolymers were investigated by DSC analysis to demonstrate the effect of block, gradient and random structures on thermal properties. In Fig. 6, the copolymer prepared from U2/IMes exhibits a glass transition temperature (Tg) of −56 °C, which is between that of the two homopolymers [−53 and −60 °C for P(ε-DL) and P(ε-CL), respectively], and a Tm at 41 °C, which can be assigned to the semi-crystalline PCL block. This suggests that the block structure of the copolymer is favourable for generating phase separation. This observation further evidenced the successful production of block copolymers by U2/IMes through its excellent selectivity. In contrast, the copolymers obtained from U3/IMes and TU3/IMes all showed only one Tg without melting transition, which corresponded to the gradient and random structures due to reduced selectivity. Furthermore, the disappearance of Tm indicates that the crystallinity of PCL can be simply and effectively reduced by the choice of catalyst pairs. This indicates that by simply changing the combination of catalyst pairs, the crystallinity of copolyesters can be easily tuned. These results confirmed the synthesis of polymers with different chain structures by selecting catalyst systems with different activities. This is of great value in terms of modulating the polymer chain sequence, which can further modulate its properties to match different application scenarios.
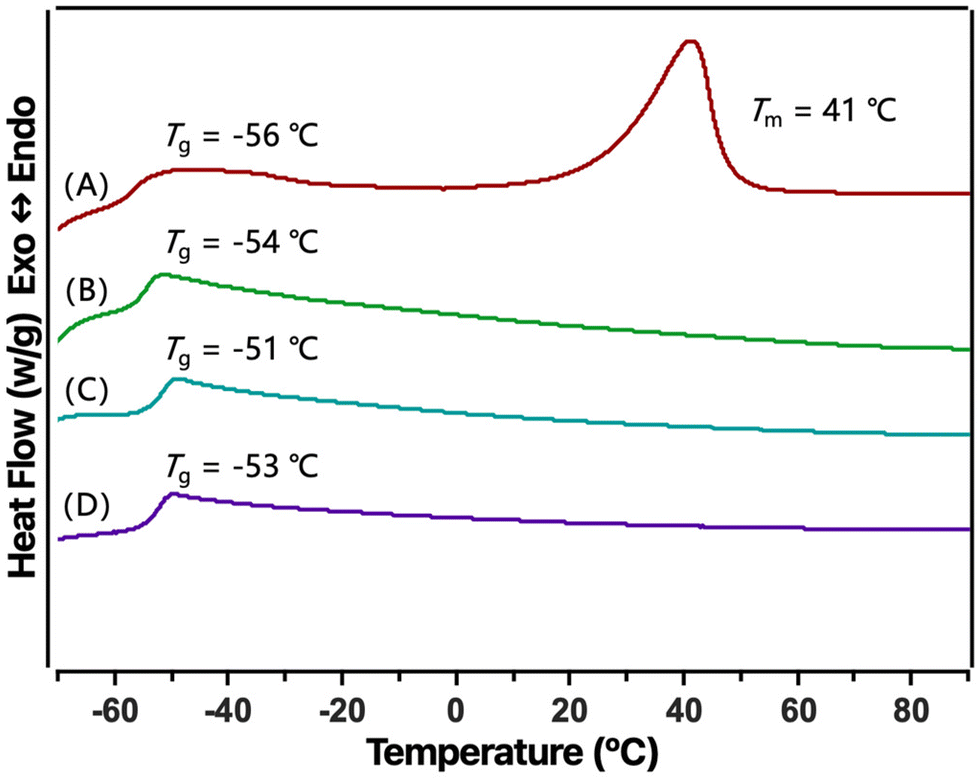 | ||
| Fig. 6 DSC thermograms for P(ε-CL-co-ε-DL) produced by different catalyst systems: (A) U2/IMes (Table 2, entry 1), (B) U3/IMes (Table 2, entry 2), (C) U3/IMes (Table 2, entry 3), and (D) TU3/IMes (Table 2, entry 4). | ||
Conclusions
In summary, a series of biorenewable omega-substituted lactones were successfully polymerized using inexpensive and readily commercially available catalysts and co-catalysts. The polymerization process is highly controlled and exhibits living features. The (thio)urea/base systems produce polymers with very narrow molecular weight distributions (<1.10 in most cases). The most active TU3/IMes demonstrated a TOF value of up to 270 h−1 in the ROP of ε-DL, far exceeding those of the previously reported catalysts, including TBD, Sn(Oct)2, zinc complex, etc. This indicates that for the (thio)urea/IMes catalytic pairs, the ‘matching’ mechanism features higher activity for the ROP of omega-substituted lactones, and this is further confirmed by the ROP of δ-CL and δ-DL with (thio)urea/IMes pairs, where the TOF values were up to 996 h−1 and 331 h−1, respectively. Unlike previous ROPs of LA and unsubstituted lactones, the cooperative mechanism showed hardly any activity in the ROPs of omega-substituted lactones. In addition to the catalytic mechanism, steric hindrance induced by the substituents of the omega-substituted lactones also has a great impact on the polymerization activity. By changing the structure of thiourea from TU3 to TU4 with greater steric hindrance, the catalytic activity was significantly reduced with a TOF of only 12 h−1. Moreover, by utilizing the selectivity differences of different catalyst systems for the copolymerization of ε-CL and ε-DL, it is feasible to modulate the chain structure of the copolymers to easily achieve block, gradient and random structures. The modulation of monomer sequences during copolymerization is of great importance in polymer synthesis, as it significantly affects thermal and mechanical properties, allowing the construction of materials suitable for different applications. Besides, this polyester is promising for applications in the adhesive, coating, and biomedical fields. The above results provide ideas for the design of similar H-bond donor/acceptor catalysts and contribute to the development of more structurally diverse polymers.Author contributions
Jiang, Du and Zhu conceived the idea, proposed the strategy, designed the experiments, evaluated the data and wrote the manuscript together. Jiang performed the majority of the experiments. Liang, Wang, Qiang and Li participated in the polymerization experiments and related characterization analysis. All the authors proofread the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2022YFC2402900), the National Natural Science Foundation of China (51903190, 22175131, 22335005, and 21925505), the Innovation Program of Shanghai Municipal Education Commission (2023ZKZD28), and the International Scientific Collaboration Fund of Science and Technology Commission of Shanghai Municipality (21520710100). J. D. is a recipient of the National Science Fund for Distinguished Young Scholars.References
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS.
- A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- A. Tardy, J. Nicolas, D. Gigmes, C. Lefay and Y. Guillaneuf, Chem. Rev., 2017, 117, 1319–1406 CrossRef CAS.
- C. A. R. Picken, O. Buensoz, P. D. Price, C. Fidge, L. Points and M. P. Shaver, Chem. Sci., 2023, 14, 12926–12940 RSC.
- W. Lu, J. E. Ness, W. Xie, X. Zhang, J. Minshull and R. A. Gross, J. Am. Chem. Soc., 2010, 132, 15451–15455 CrossRef CAS PubMed.
- R. Kourist and L. Hilterhaus, in Microorganisms in Biorefineries, ed. B. Kamm, Springer, Berlin, Heidelberg, 2015, vol. 26, pp. 275–301 Search PubMed.
- S. Katsuhara, Y. Takagi, N. Sunagawa, K. Igarashi, T. Yamamoto, K. Tajima, T. Isono and T. Satoh, ACS Sustainable Chem. Eng., 2021, 9, 9779–9788 CrossRef CAS.
- M. T. Martello, A. Burns and M. Hillmyer, ACS Macro Lett., 2012, 1, 131–135 CrossRef CAS PubMed.
- D. Bandelli, C. Helbing, C. Weber, M. Seifert, I. Muljajew, K. D. Jandt and U. S. Schubert, Macromolecules, 2018, 51, 5567–5576 CrossRef CAS.
- C. Li, L. Wang, Q. Yan, F. Liu, Y. Shen and Z. Li, Angew. Chem., Int. Ed., 2022, 61, e202201407 CrossRef CAS PubMed.
- Q. Yan, C. Li, T. Yan, Y. Shen and Z. Li, Macromolecules, 2022, 55, 3860–3868 CrossRef CAS.
- D. Bandelli, C. Weber and U. S. Schubert, Macromol. Rapid Commun., 2019, 40, 1900306 CrossRef CAS PubMed.
- J. R. Lowe, M. T. Martello, W. B. Tolman and M. A. Hillmyer, Polym. Chem., 2011, 2, 702–708 RSC.
- D. Zhang, M. A. Hillmyer and W. B. Tolman, Biomacromolecules, 2005, 6, 2091–2095 CrossRef CAS PubMed.
- J. A. Wilson, S. A. Hopkins, P. M. Wright and A. P. Dove, Biomacromolecules, 2015, 16, 3191–3200 CrossRef CAS.
- G. L. Gregory, G. S. Sulley, L. P. Carrodeguas, T. T. D. Chen, A. Santmarti, N. J. Terrill, K.-Y. Lee and C. K. Williams, Chem. Sci., 2020, 11, 6567–6581 RSC.
- S. Thongkham, J. Monot, B. Martin-Vaca and D. Bourissou, Macromolecules, 2019, 52, 8103–8113 CrossRef CAS.
- C. Romain, Y. Zhu, P. Dingwall, S. Paul, H. S. Rzepa, A. Buchard and C. K. Williams, J. Am. Chem. Soc., 2016, 138, 4120–4131 CrossRef CAS PubMed.
- J.-F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 1238149 CrossRef.
- S. Rupf, P. Pröhm and A. J. Plajer, Chem. Sci., 2022, 13, 6355–6365 RSC.
- R. C. Jeske, J. M. Rowley and G. W. Coates, Angew. Chem., Int. Ed., 2008, 47, 6041–6044 CrossRef CAS PubMed.
- C. Romain and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 1607–1610 CrossRef CAS PubMed.
- Z. Yang, C. Hu, F. Cui, X. Pang, Y. Huang, Y. Zhou and X. Chen, Angew. Chem., Int. Ed., 2022, 61, e202117533 CrossRef CAS PubMed.
- V. K. Chidara, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. S. Feng, Macromolecules, 2021, 54, 2711–2719 CrossRef CAS.
- J. B. Zhang, L. B. Wang, S. F. Liu, X. H. Kang and Z. B. Li, Macromolecules, 2021, 54, 763–772 CrossRef CAS.
- Y. K. Ma, X. X. You, J. B. Zhang, X. W. Wang, X. H. Kou, S. F. Liu, R. L. Zhong and Z. B. Li, Angew. Chem., Int. Ed., 2023, 62, e202303315 CrossRef CAS PubMed.
- M. Hong, J. Chen and E. Y. X. Chen, Chem. Rev., 2018, 118, 10551–10616 CrossRef CAS PubMed.
- A. P. Dove, ACS Macro Lett., 2012, 1, 1409–1412 CrossRef CAS PubMed.
- B. Lin and R. M. Waymouth, J. Am. Chem. Soc., 2017, 139, 1645–1652 CrossRef CAS.
- K. V. Fastnacht, S. S. Spink, N. U. Dharmaratne, J. U. Pothupitiya, P. P. Datta, E. T. Kiesewetter and M. K. Kiesewetter, ACS Macro Lett., 2016, 5, 982–986 CrossRef CAS PubMed.
- X. Zhang, G. O. Jones, J. L. Hedrick and R. M. Waymouth, Nat. Chem., 2016, 8, 1047–1053 CrossRef CAS PubMed.
- J. U. Pothupitiya, R. S. Hewawasam and M. K. Kiesewetter, Macromolecules, 2018, 51, 3203–3211 CrossRef CAS.
- P. Olsén, T. Borke, K. Odelius and A.-C. Albertsson, Biomacromolecules, 2013, 14, 2883–2890 CrossRef PubMed.
- L. Jasinska-Walc, M. Bouyahyi, A. Rozanski, R. Graf, M. R. Hansen and R. Duchateau, Macromolecules, 2015, 48, 502–510 CrossRef CAS.
- J.-O. Lin, W. Chen, Z. Shen and J. Ling, Macromolecules, 2013, 46, 7769–7776 CrossRef CAS.
- J. Bai, J. Wang, Y. Wang and L. Zhang, Polym. Chem., 2018, 9, 4875–4881 RSC.
- B. Lin and R. M. Waymouth, Macromolecules, 2018, 51, 2932–2938 CrossRef CAS.
- C. Romero-Guido, I. Belo, T. M. N. Ta, L. Cao-Hoang, M. Alchihab, N. Gomes, P. Thonart, J. A. Teixeira, J. Destain and Y. Waché, Appl. Microbiol. Biotechnol., 2011, 89, 535–547 CrossRef CAS PubMed.
- T. H. Parliment, W. W. Nawar and I. S. Fagerson, J. Dairy Sci., 1965, 48, 615–616 CrossRef CAS PubMed.
- T. Rali, S. W. Wossa and D. N. Leach, Molecules, 2007, 12, 149–154 CrossRef CAS PubMed.
- G. Jakab, C. Tancon, Z. Zhang, K. M. Lippert and P. R. Schreiner, Org. Lett., 2012, 14, 1724–1727 CrossRef CAS PubMed.
- T. Bug, T. Lemek and H. Mayr, J. Org. Chem., 2004, 69, 7565–7576 CrossRef CAS PubMed.
- D. Martin, O. Illa, A. Baceiredo, G. Bertrand, R. M. Ortuño and V. Branchadell, J. Org. Chem., 2005, 70, 5671–5677 CrossRef CAS PubMed.
- R. Schwesinger, H. Schlemper, C. Hasenfratz, J. Willaredt, T. Dambacher, T. Breuer, C. Ottaway, M. Fletschinger, J. Boele, H. Fritz, D. Putzas, H. W. Rotter, F. G. Bordwell, A. V. Satish, G. Z. Ji, E. M. Peters, K. Peters, H. G. vonSchnering and L. Walz, Liebigs Ann., 1996, 1055–1081 CrossRef CAS.
- R. M. Cywar, J.-B. Zhu and E. Y. X. Chen, Polym. Chem., 2019, 10, 3097–3106 RSC.
- D. E. Gomez, L. Fabbrizzi, M. Licchelli and E. Monzani, Org. Biomol. Chem., 2005, 3, 1495–1500 RSC.
- C. Perez-Casas and A. K. Yatsimirsky, J. Org. Chem., 2008, 73, 2275–2284 CrossRef CAS.
- G. Hua, J. Franzén and K. Odelius, Polym. Chem., 2016, 54, 1908–1918 CrossRef CAS PubMed.
- S. Penczek, Polym. Chem., 2000, 38, 1919–1933 CrossRef CAS.
- R. R. Walvoord, P. N. H. Huynh and M. C. Kozlowski, J. Am. Chem. Soc., 2014, 136, 16055–16065 CrossRef CAS PubMed.
- J. Ho, V. E. Zwicker, K. K. Y. Yuen and K. A. Jolliffe, J. Org. Chem., 2017, 82, 10732–10736 CrossRef CAS PubMed.
- M. R. Stühler, C. Gallizioli, S. M. Rupf and A. J. Plajer, Polym. Chem., 2023, 14, 4848–4855 RSC.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2016, 49, 2419–2428 CrossRef CAS.
- P. Olsén, K. Odelius and A.-C. Albertsson, Biomacromolecules, 2016, 17, 699–709 CrossRef PubMed.
- J. Zhao and N. Hadjichristidis, Polym. Chem., 2015, 6, 2659–2668 RSC.
- D. Bandelli, I. Muljajew, K. Scheuer, J. B. Max, C. Weber, F. H. Schacher, K. D. Jandt and U. S. Schubert, Macromolecules, 2020, 53, 5208–5217 CrossRef CAS.
- H. Chen, R. Liu, S. Cai, Y. Zhang, C. Zhu, H. Yu and S. Li, Biotechnol. J., 2024, 19, e2300210 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00404c |
| This journal is © The Royal Society of Chemistry 2024 |