
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploiting a branched radical polymerization strategy to enhance the degree of grafting onto graphene oxide†
Wai Hin
Lee
 and
Stefan A. F.
Bon
*
and
Stefan A. F.
Bon
*
Department of Chemistry, The University of Warwick, Coventry CV4 7AL, UK. E-mail: s.bon@warwick.ac.uk; Web: https://bonlab.info
First published on 3rd May 2024
Abstract
The synthesis of polymer-grafted graphene oxide (GO) using a branched radical polymerization strategy is described. A methacrylate-based macromonomer is used as a chain-transfer agent to enhance the degree of grafting and prevent macroscopic gel formation. A methacrylic acid-based macromonomer was first synthesized via aqueous solution catalytic chain transfer polymerization. The resultant macromonomer was used as a chain transfer agent in the radical polymerization of poly(ethylene glycol) methacrylate (PEGMA) in the presence of a dispersion of GO in a water/DMF mixture (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 w/w). The degree of grafting was determined using thermogravimetric analyses and infrared spectroscopy, and the geometrical evolution was investigated using atomic force microscopy. The grafting efficiency increased with the degree of branching.
:50 w/w). The degree of grafting was determined using thermogravimetric analyses and infrared spectroscopy, and the geometrical evolution was investigated using atomic force microscopy. The grafting efficiency increased with the degree of branching.
Introduction
Graphene is a high aspect ratio 2D macromolecule composed of single-layered sp2-hybridized carbon with limited defects at its outer edges and holes.1 Graphene tends to stack into the naturally occurring graphite. Its exfoliation and fabrication of single sheet dispersions in liquids is, therefore, not straightforward. Liquid-phase exfoliation methods require high energy inputs, such as sonication, which, besides the desired exfoliation of sheets, causes ruptures and fragmentation.2 In addition, graphene's colloidal characteristics such as its low surface charge density make it hard for sheets not to coagulate once dispersed, especially in water.As a compromising solution, graphite is commonly oxidised into graphene oxide (GO) hereby introducing oxygen-containing functional groups, such as hydroxy, ketone, epoxy, and carboxylic acid moieties (Scheme 1). Its synthetic method can be traced back to 1859 by Brodie who used perchlorate and nitric acid as oxidizing agents.3 Hummers improved upon this method using less precarious oxidizing agents such as potassium permanganate, concentrated sulfuric acid and/or nitric acid.4 The molecular structure of GO was not clearly understood at that time.5–7 Currently, the most accepted model was proposed by Klinowski in 1998, which describes GO as a single sheet with domains of conjugated sp2 carbon in a hexagonal arrangement, oxygen functional groups at the edges, and the presence of defects between domains.8 Recently, Thickett et al. further used the deconvolution of 2D 13C{1H} NMR spectroscopy of GO to elucidate both local and long-range order of the functional groups, from which they reconstructed the GO structure in domains of approximately 20 nm.9
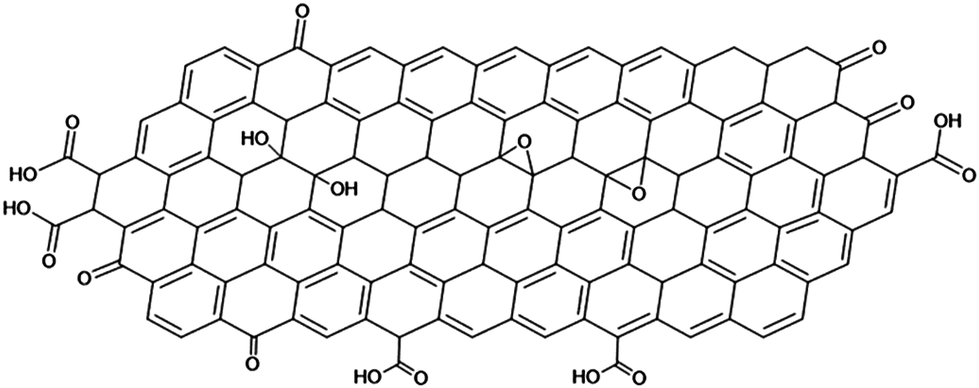 | ||
| Scheme 1 Klinowski's model of graphene oxide (GO). | ||
Owing to the enhanced hydrophilicity, surface charge, and loss of conjugated domains introducing non-planarity, GO can be exfoliated into large single sheets in water and kept colloidally stable with a prolonged shelf-life.10–13 Dispersions of GO can be further modified. One modification is to reduce the GO back to recover part of the original graphene features. The synthetic route of chemical reduction of GO was proposed far before a proper structural understanding. Hofmann and König investigated the efficiency of FeCl2 and hydrazine to reduce GO (known as graphite oxide in the literature then). They found a reduction in the C/O ratio up to 82.3% C with the lamellar spacing of 3.5 Å in 1937.14 This indicated the restoration of graphitic crystallite with scarce structural defects and oxygen residues on GO. Ruoff et al. explored the hydrazine reduction and used a polymer stabilizer, that is, poly(styrene sulfonate), to warrant colloidal stability of the GO sheets in water and prevent restacking upon reaction due to the loss of hydroxy and carboxylic acid groups.15–17
The task of keeping GO and reduced GO (rGO) dispersed in liquids as single sheets is not an easy one, especially at higher concentrations.18 Chemical approaches such as the introduction of functional groups, for example, nucleophilic addition or substitution at the epoxy or carboxylic acid moieties,19,20 together with physical approaches that include π-stacking onto the conjugated domain,21,22 and cationic exchange at the carboxylate group23,24 are employed. With our particular interest in radical polymerization, we would like to focus on grafting polymer chains onto GO through the polymerization process. The covalent attachment of polymer chains to GO can occur either through radical addition to a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C of the GO and further propagation (grafting through), initiation of a polymer chain from the position of a generated radical on the GO by means of an earlier chain transfer event (grafting from), and attachment of a growing polymer chain onto GO through bimolecular termination (grafting to).
C of the GO and further propagation (grafting through), initiation of a polymer chain from the position of a generated radical on the GO by means of an earlier chain transfer event (grafting from), and attachment of a growing polymer chain onto GO through bimolecular termination (grafting to).
Numerous reversible deactivation radical polymerization approaches have been reported to modify GO with polymer chains, in which GO was pre-modified chemically to serve as an initiator and/or mediator. Examples include reversible-addition fragmentation chain transfer (RAFT) processes,19,25–27 atom transfer radical polymerizations (ATRP),28–32 and nitroxide-mediated polymerizations (NMP).33 The more straightforward approach, carrying out a radical polymerization in the presence of unmodified GO is often overlooked despite less complicated experimental procedures. Examples, where GO serves as an ordinary free radical initiator, include using cerium(IV) nitrate redox chemistry,34 thermal reduction and self-initiation of GO35,36 and γ-radiated initiation.37 We must be mindful that GO is not inert towards radical species and that the grafting of polymer chains to, from, and through GO sheets readily occurs. These pathways have been long proposed in the free radical polymerization with other analogous carbon materials such as carbon black,38–40 C6041–44 and carbon nanotubes.45,46 An excellent study by Kan and coworkers demonstrated that polymer-grafted GO could be obtained by simple free radical polymerization.47 They demonstrated the polymerization of twelve different monomers and proposed that polymeric radicals were capable of grafting through the non-aromatic CC groups. Macroscopically homogeneous dispersions of modified GO were obtained in good solvents for the graft polymer. This strategy can be further extended to heterogeneous polymerization systems. For example, Zetterlund et al. demonstrated the Pickering mini-emulsion polymerization using GO as stabilizer.48 Herein, we would like to study the grafting mechanism in the case of branched polymers and if the pendant vinyl groups act as secondary graphing points.
Kinetic studies of free radical polymerization in the presence of crosslinker can be traced back to the pioneering work by Walling in 1945.49 He investigated the polymerization of ethylene glycol dimethacrylate and its copolymerization with methyl methacrylate in both bulk and solution systems, and compared the results with the gel formation theory by Flory and Stockmayer.50,51 He observed a delay in the gel point from its theoretical value, and such discrepancy was magnified when a chain transfer prone solvent was used. Despite the lack of accurate kinetics of polymerization in the presence of crosslinker, perplexed by the concurrent cyclization, multiple crosslinking and nanoscale segregation,52–54 it was generally accepted that a linear polymer chain was generated in the first instance. At higher vinyl group conversion, the pendant vinyl groups on the pre-formed polymer chains would react, leading to branching and eventually macroscopic crosslinking.55,56
Later, Sherrington used this approach with thiol-based chain transfer agent in various ratio to monomer and crosslinker to limit the crosslinking and thus prevent macroscopic gelation.57–60Via this route, a higher content of crosslinker (2% mol/mol in a solution of 40% v/v total monomer) can be incorporated than by radical polymerization in absence of chain transfer agent (gelation even at <0.5% mol/mol). Despite a less defined branched structure, this class of polymers is often regarded as an alternative to dendrimers because of the simpler and more versatile synthetic route. This strategy has also recently been explored by Rannard et al. to synthesize hyperbranched telomers using crosslinker and chain transfer agents only.61–64
Herein, we would like to exploit the advantages of free radical polymerization as a means to synthesize polymer-grafted GO and combine this with the addition fragmentation chain-transfer (AFCT) mediated branched polymer synthesis to enhance the degree of grafting. Poly(methacrylic acid) macromonomer was first synthesized via cobalt-mediated catalytic chain transfer polymerization (CCTP) in an aqueous solution. The resulting macromonomers were used as AFCT-agents in synthesising branched poly(ethylene glycol) methacrylate in water/DMF dispersions of GO (Scheme 2). Note that using thiols as chain-transfer agent needs to be avoided as these compounds can react directly with GO in both basic and acidic conditions.65 The product was characterized in detail to quantify the grafting efficiency, its grafting density, and layer thickness on the GO.
 | ||
| Scheme 2 Reaction schemes for the synthesis of PMAA macromonomer via aqueous CCTP mediated by CoEtBF (top); and in situ grafting branched copolymer of PEGMA onto GO via PMAA macromonomer-mediated radical polymerization in water/DMF dispersion of GO. | ||
Experimental
Materials
The aqueous dispersion of GO (solids content: 1.062 wt%) was kindly provided by William Blythe Ltd. Prior to use, impurities such as residual ions during synthesis were removed by dialysis. Cellulose acetate dialysis tubing with a molecular weight cut-off of 14000 g mol−1 was purchased from Sigma-Aldrich. Prior to use the dialysis tubing was placed in a large beaker filled with de-ionized water at room temperature for 30 min, and subsequently rinsed out with more de-ionized water. In a typical procedure, the tubing was filled with an aqueous GO dispersion (150 mL). This was dialyzed against deionized water (2 L). The dialysate was exchanged daily until the conductivity of the wastewater was below 1 μS cm−1.
After dialysis, the GO dispersion was centrifugated at 5000 rpm for 30 min. Centrifugation was performed on an Eppendorf centrifuge equipped with Falcon 50 mL centrifuge tubes. This was necessary to remove residual unexfoliated graphite flakes and other colloidal impurities. The supernatant was stored as a dispersion of exfoliated GO and used in all subsequent experiments. Two batches of aqueous GO dispersion were prepared following the same procedures. The solids content of the two batches of purified GO dispersion were 0.3687 and 0.4020% w/w respectively.
Poly(ethylene glycol) methacrylate (PEGMA 500; number average molecular weight = 500 g mol−1), methacrylic acid (MAA), diethylene glycol dimethacrylate (DEGDMA), tetraethyleneglycol dimethacrylate (TEGDMA), and N,N-dimethylformamide (DMF) were purchased from Sigma-Aldrich and used as received. Methanol and diethyl ether were purchased from Fischer Scientific. 4,4′-Azobis(4-cyanovaleric acid) (ACVA) initiator was supplied by Alfa Chemicals and used as purchased. Cobalt(II) catalyst (CoEtBF) was synthesized following a procedure by Espenson and coworkers.66
Instrumentation
Poly(methacrylic acid) (PMAA) macromonomer via solution catalytic chain transfer polymerization (CCTP)
The synthesis of PMAA macromonomer was carried out following a modified procedure based on the one reported by Haddleton et al.67 In a typical experiment, ACVA (0.1890 g, 0.67 mmol) and CoEtBF (2.69 mg, 6.0 mmol) were put in to a 250 mL round bottom flask (flask 1). CoEtBF (1.53 mg, 3.4 mmol) was placed into a separate flask (flask 2). Water (100 g, 100 mL) and MAA (45.9 g, 45 mL) were also put into two separate flasks (flasks 3 and 4). All flasks were deoxygenated by purging with nitrogen gas for 1 h. Water (90 mL) was transferred from flask 3 into flask 1 with a nitrogen filled syringe and heated to 80 °C oil bath. MAA (40 mL) was transferred from flask 4 into flask 2 with a nitrogen filled syringe. Mild sonication in a water bath was applied to flask 2 to facilitate the dissolution of CoEtBF in MAA. The CoEtBF solution in MAA (36.8 mL, 37.5 g, 0.436 mol) was fed from flask 2 into flask 1 over 1 h using a syringe pump whilst maintaining flask 1 at 80 °C. The polymerization was allowed to continue for a further 2 h to reach near complete monomer conversion.Grafting of branched polymer onto graphene oxide
Two series of reactions were performed using DEGDMA and TEGDMA as crosslinker, respectively. The experimental details were summarized in Tables S.1 and S.2.† The nomenclature of the experiment was designated as L, HB or HBT x–y–z where L represented linear architecture without addition of crosslinker; HB or HBT represented branched architecture using DEGDMA or TEGDMA as crosslinker respectively; x:y:z represented the molar ratio of [PEGMA 500]:[crosslinker]:[PMAA MM] in the initial reaction mixture.
In a typical experiment (HB 10-1-1), DEGDMA (0.0485 g, 2.0 mmol), PEGMA 500 (1.00 g, 0.02 mol), DMF (4.20 g), water (0.18 g), GO dispersion (2.7122 g, 0.01 g GO, SC: 0.3687% w/w) and PMAA macromonomer solution (1.8552 g, 0.55 g PMAA, 2.0 mmol, SC: 29.7% w/w) were mixed. DMF was added to the mixture in advance to prevent permanent complexation of carboxylic acid and PEGMA. The reaction mixture was deoxygenated by purging with nitrogen gas for 0.5 h. It was heated and thermostated at 70 °C overnight under a slight overpressure of nitrogen gas in the absence of agitation to avoid shear-induced aggregation of the GO.
The non-grafted polymer was separated from the reaction mixture for analysis as follows. The crude final product (3 mL) was diluted with methanol (9 mL) and centrifuged at 7500 rpm for 120 min using the Eppendorf centrifuge. The clear supernatant was discarded, and the black GO-sediment was redispersed with fresh methanol (12 mL). This process was repeated three times to fully remove all soluble non-grafted polymer. The absence of precipitation of an aliquot of the supernatant in diethyl ether confirmed this. Next, the same centrifugation process was repeated on the now methanol-based dispersion of GO, using deionized water as a replacement solvent to fabricate the purified aqueous dispersion of polymer-grafted GO.
Results
PMAA macromonomer synthesis via CCTP in water
PMAA macromonomers were synthesized to serve as addition–fragmentation chain-transfer (AFCT) agents. To avoid side reactions between GO and sulfur/halogen in the subsequent modification, reversible deactivation radical polymerization methods of sulfur-based RAFT and atom transfer radical polymerization (ATRP) or related analogues were excluded. The AFCT agents regulate the molecular weight, as in they reduce it, which is needed to control branching and to avoid macroscopic crosslinking, resulting in permanent gelation of the entire system. The PMAA macromonomers were made by semi-batch catalytic chain transfer polymerization of MAA in water mediated by CoEtBF as chain transfer agent. The monomer conversion (p) and number degree of polymerization (DPn) were determined to be 98% and 33 by 1H NMR (Fig. S.1 and Table S.3†) respectively according to eqn (S.1) and (S.2).† In this step, low molecular weight was preferred to ensure the PMAA solution was in the dilute regime below polymer chain overlap to prevent macroscopic gelation via intermolecular hydrogen bonding.68Synthesis of branched polymer
The PMAA macromonomers are used as reversible addition fragmentation chain transfer (AFCT) agents. It is important to note that the chain transfer constant of these ω-unsaturated methacrylate-based macromonomers is estimated to be around 0.2 for radical polymerization of methacrylate monomers.56 Batch solution polymerizations of PEGMA 500 (monomer content: 10% w/w) were carried out in water DMF mixture (50:50 w/w) at 70 °C in presence (L10-0-1) and absence (L-10-0-0) of 10% mol/mol of macromonomer-to-monomer, to demonstrate its effect as AFCT agent. The conversion of L 10–0–1 was monitored over 8 h was 94% by 1H NMR (Fig. S.2 and Table S.4†). SEC analysis of the non-grafted fraction of polymer was obtained by filtration through a 0.2 mm membrane filter.
In the next set of reactions, DEGDMA was introduced as a crosslinker at a molar ratio of 10:1 of [PEGMA 500]:[DEGDMA] while maintaining the monomer content at 10% w/w. The polymerization was performed in the same conditions as aforementioned, in water/DMF mixture (50:50 w/w) at 70 °C for 24 h. The polymerization was carried out in the presence (HB 10-1-1) and absence (HB 10-1-0) of the AFCT agent. Macroscopic gelation was observed at 20% mol/mol DEGDMA-to-PEGMA or higher crosslinker concentration. The molecular weight distributions of the polymer are shown in Fig. 1. The SEC results showed that the incorporation of PMAA macromonomer reduced the molecular weight considerably from 368 kg mol−1 (DP = 736) to 8.0 kg mol−1 (DP = 10; excluded 5.1 kg mol−1 from PMAA33 macromonomer) for the linear polymer (Table 1). This indicated the change from termination (theoretical DP = 892 for kp = 8375 M−1 s−1, [M] = 0.2 M, kt = 5 × 107 M−2 s−1 [II] = 1 mM and f = 0.7 at 10% w/w PEGMA and 70 °C)69 to chain transfer (theoretical DP = 10) as the major pathway. The incorporation of 0.1 molar equivalent of DEGDMA crosslinker to PEGMA 500 only increased the molecular weight slightly to 383 and 11.6 kg mol−1 in the absence and presence of PMAA macromonomer respectively. However, comparison at higher [DEGDMA]:[PEGMA 500] was not possible due to macroscopic gelation in the absence of macromonomer. The effect of [DEGDMA]:[PEGMA 500] were discussed in later section.
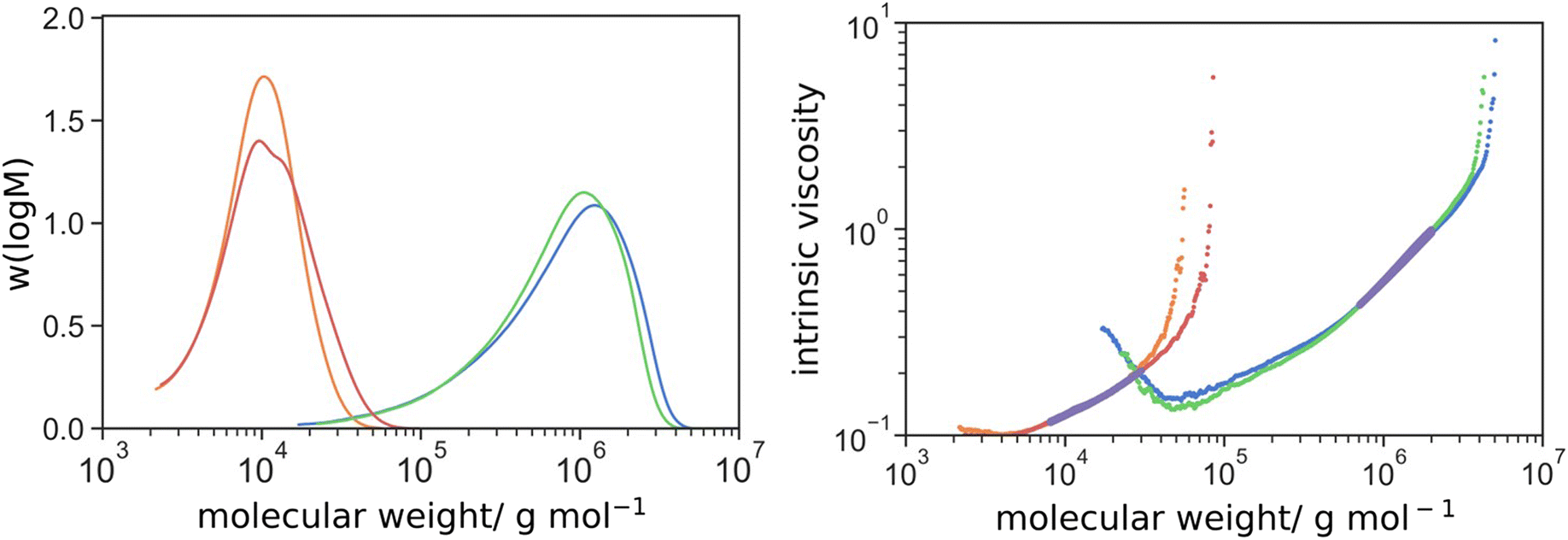 | ||
| Fig. 1 Molecular weight distributions of L10-0-0 (green), L10-0-1 (orange), HB10-1-0 (blue) and HB 10-1-1 (red) determined by triple detection SEC and their intrinsic viscosity against molecular weight; the magenta lines indicate the selected linear regions for triple detection analyses. | ||
|
|
|
α/— | |
|---|---|---|---|
| L 10-0-0 | 368.0 | 934.0 | 0.78 |
| L 10-0-1 | 8.0 | 11.0 | 0.51 |
| HB 10-1-0 | 383.0 | 1,039 | 0.75 |
| HB 10-1-1 | 11.6 | 20.3 | 0.47 |


The Mark–Houwink–Sakurada parameter (α) was determined from triple detection (refractive index, viscosity and light scattering) according to the slope of selected linear region of intrinsic viscosity vs. molecular weight. It is an index to evaluate the compactness of the molecular topology. The more extended the structure, the larger the α value or vice versa (α > 0.7 for linear polymer in a good solvent; = 0.5 in a theta solvent; <0.5 in a poor solvent). Branched polymers have smaller α values than their linear counterparts because of a higher density molecular architecture. Indeed, the α values reduced significantly from 0.78 to 0.51 by the addition of macromonomer as a CTA, even in the absence of crosslinker, suggested that the topology deviated from linear. This was rationalized by the fact that PEGMA 500 in use had a relatively long side chain, with respect to the degree of polymerization. In the presence of CTA, the DP of PEGMA 500 calculated from  was only 10. This is comparable to the length of the PEG side chain and, therefore, the polymer chain had a bottlebrush-like topology instead of linear. In the initial experiments incorporating DEGDMA crosslinker at [DEGDMA]:[PEGMA 500] = 1:10, the drop in α value was minuscule. This indicates that not all the polymer chains are branched. This is logical, as at a low overall degree of polymerization and small amounts of crosslinker, statistically, many chains remain linear.
was only 10. This is comparable to the length of the PEG side chain and, therefore, the polymer chain had a bottlebrush-like topology instead of linear. In the initial experiments incorporating DEGDMA crosslinker at [DEGDMA]:[PEGMA 500] = 1:10, the drop in α value was minuscule. This indicates that not all the polymer chains are branched. This is logical, as at a low overall degree of polymerization and small amounts of crosslinker, statistically, many chains remain linear.
Effect of crosslinker concentration
The crosslinker concentration played an important role in controlling the molecular weight, degree of branching and crosslinking and the pendant vinyl groups as grafting sites. Due to the AFCT activity of macromonomer, unlike thiol, the chain transfer agent was not consumed throughout the reaction, while the monomer concentration continuously reduced in the course of polymerization, [CTA]:[monomer] remained sufficiently high to control the molecular weight and avoid gelation throughout the reaction.
The molar ratio of crosslinker to macromonomer from 1:1 to 5:1 has been chosen. The average molecular weights and the α values (Mark–Houwink–Sakurada parameter) are summarized in Table 2. Upon the increasing crosslinker concentration, the  was increasing from 18.0 to 56.1 kg mol−1 and the α value decreased from 0.48 to 0.35, at a 10:1 ratio of PEGMA 500:DEGDMA and a 2:1 ratio of PEGMA 500:DEGDMA, respectively (Fig. 2). This increase of weight average molecular weight suggested that more polymer chains were bundled into a branched structure, and the decline in the α value also indicated that the molecules were more compact due to a more crosslinked environment.
was increasing from 18.0 to 56.1 kg mol−1 and the α value decreased from 0.48 to 0.35, at a 10:1 ratio of PEGMA 500:DEGDMA and a 2:1 ratio of PEGMA 500:DEGDMA, respectively (Fig. 2). This increase of weight average molecular weight suggested that more polymer chains were bundled into a branched structure, and the decline in the α value also indicated that the molecules were more compact due to a more crosslinked environment.
 | ||
| Fig. 2 Molecular weight distributions of HB 10-1-1 ([DEGDMA]:[CTA] = 1:1 mol/mol, blue), HB 10-2-1 ([DEGDMA]:[CTA] = 2:1 mol/mol, orange), HB 10-3-1 ([DEGDMA]:[CTA] = 3:1 mol/mol, green), HB 10-4-1 ([DEGDMA]:[CTA] = 4:1 mol/mol, purple) and HB 10-5-1 ([DEGDMA]:[CTA] = 5:1 mol/mol, red), determined by triple detection SEC. | ||
|
|
|
α/— | |
|---|---|---|---|
| HB 10-1-1 | 9.8 | 18.0 | 0.48 |
| HB 10-2-1 | 10.1 | 22.6 | 0.45 |
| HB 10-3-1 | 10.7 | 29.9 | 0.42 |
| HB 10-4-1 | 10.7 | 38.1 | 0.39 |
| HB 10-5-1 | 10.2 | 56.1 | 0.35 |


Recently, Rannard et al. reported that the length of the spacer between polymerizable groups in the crosslinker molecule played an important role in the degree of crosslinking.61,64,70 The vinyl groups on a crosslinker molecule with a longer spacer are less sterically hindered by the polymer backbone which facilitates a separate propagating radical to react via intermolecular crosslinking. Moreover, the extended spacer locally screened the second vinyl group from the propagating radical, statistically favouring intermolecular crosslinking over intramolecular cyclization.71
Herein, we adopted this concept into our system and investigated the possibility to further enhance the degree of grafting using a crosslinker with a longer spacer. Similar experiments were performed using tetraethylene glycol dimethacrylate (4 ethylene glycol units) instead of DEGDMA (2 ethylene glycol units). The molecular weight distributions were shown in Fig. 3.  ,
,  and the α values were summarized in Table 3.
and the α values were summarized in Table 3.
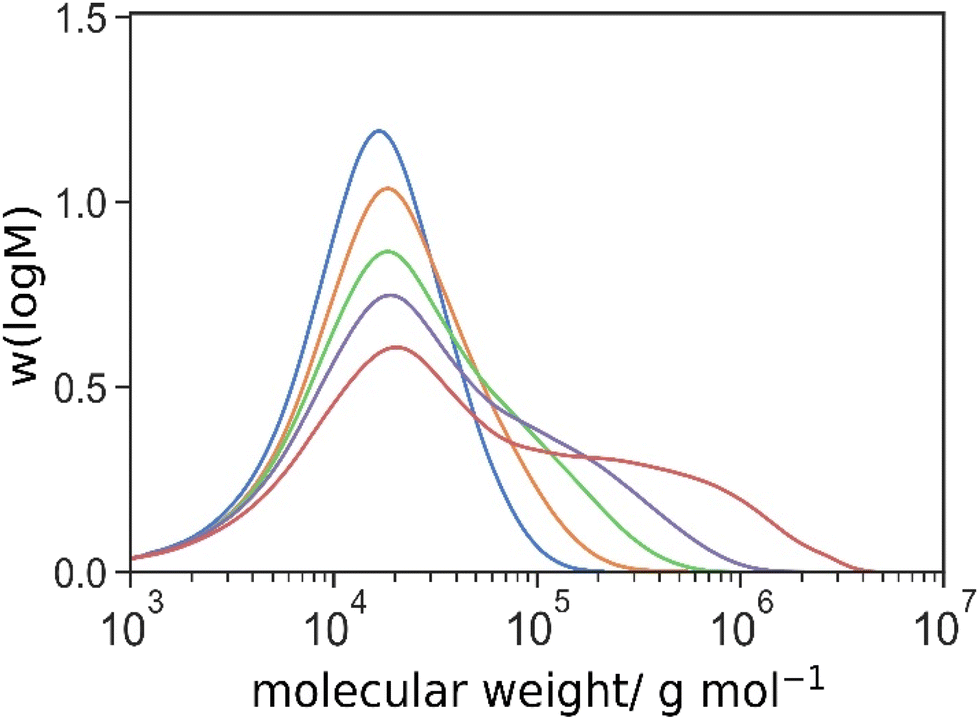 | ||
| Fig. 3 Molecular weight distributions of HBT 10-1-1 ([TEGDMA]:[CTA] = 1:1 mol/mol, blue), HBT 10-2-1 ([TEGDMA]:[CTA] = 2:1 mol/mol, orange), HBT 10-3-1 ([TEGDMA]:[CTA] = 3:1 mol/mol, green), HBT 10-4-1 ([TEGDMA]:[CTA] = 4:1 mol/mol, purple) and HBT 10-5-1 ([TEGDMA]:[CTA] = 5:1 mol/mol, red), determined by triple detection SEC. | ||
|
|
|
α/— | |
|---|---|---|---|
| HBT 10-1-1 | 10.6 | 21.2 | 0.46 |
| HBT 10-2-1 | 11.7 | 29.5 | 0.44 |
| HBT 10-3-1 | 12.5 | 45.5 | 0.40 |
| HBT 10-4-1 | 13.3 | 75.9 | 0.37 |
| HBT 10-5-1 | 14.3 | 205.8 | 0.34 |


Higher molecular weights and smaller α values of the branched polymers from TEGDMA were found in all experiments, compared to its equivalence using DEGDMA in the same molar ratio of [crosslinker]:[macromonomer] (Fig. 3, note: molar ratio of molecules not vinyl groups). This suggested the TEGDMA crosslinker resulted into a more branched and compact architecture. Focusing on the experiments using the highest amount of crosslinker HB(T) 10-5-1 ([crosslinker]:[macromonomer] = 5:1), the 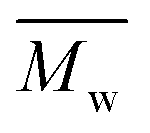 using TEGDMA increasing from 56.1 to 205.8 kg mol−1 by 3.7 times. This substantial increment ruled out a possible explanation that the difference in
using TEGDMA increasing from 56.1 to 205.8 kg mol−1 by 3.7 times. This substantial increment ruled out a possible explanation that the difference in  was simply due to the higher molar mass of TEGDMA only, where a molecular weight of 60.1 kg mol−1 would be expected if in this case. Based on the
was simply due to the higher molar mass of TEGDMA only, where a molecular weight of 60.1 kg mol−1 would be expected if in this case. Based on the  determined from the linear analogue (L 10-0-1;
determined from the linear analogue (L 10-0-1;  : 11.0 kg mol−1), the average number of branches of HB 10-5-1 and HBT 10-5-1 was 5.1 and 18.6 respectively. This demonstrated the enhanced crosslinking efficiency using a crosslinker with an extended spacer length.
: 11.0 kg mol−1), the average number of branches of HB 10-5-1 and HBT 10-5-1 was 5.1 and 18.6 respectively. This demonstrated the enhanced crosslinking efficiency using a crosslinker with an extended spacer length.
In situ grafting polymer on to GO via radical polymerization
Radical attack to GO and its use to graft polymer onto GO have been reported previously,47,72 and it was proposed that carbon-centred radical was capable to couple with GO at the conjugated CC (Scheme 3). The radical species could rearrange within the conjugated domain and cause reduction, then further react with monomers (grafting through) or form inactive species (grafting to). We expect this mechanism was undertaken, however the exact mechanism, or the combination of other pathways such as chain transfer, are not able to be deduced from our characterization and beyond the scope of current study. The topological conformation of the grafted polymer will be discussed in later section.
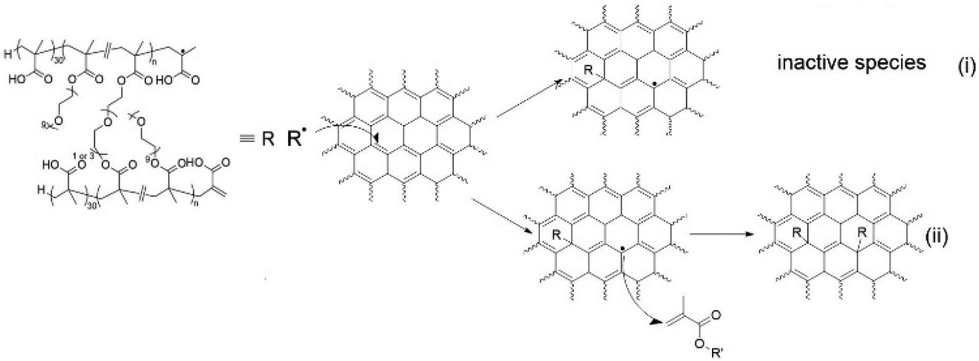 | ||
| Scheme 3 Proposed mechanism of propagating polymeric radical to graft to GO at conjugated CC. | ||
FTIR was initially used to determine the grafted polymer content on GO after purification by comparing the characteristic band of GO and the grafted polymer. This, however, was perplexed by the combination of intrinsic IR absorption and scattering effect of GO (Fig. S.3 and Table S.5†). It only semi-quantitatively demonstrated that the ratio of the CC stretching band at 1626 cm−1 from GO to the CO stretching band from the grafted polymer at 1699 cm−1 reduced with increasing crosslinker concentration, indicating that a higher grafted polymer content was attained at larger crosslinker concentration.
TGA was used to determine the degree of grafting of polymer onto GO quantitatively. To interpret the thermal decomposition profile, the blank samples of the initial PMAA macromonomer, linear and branched PEGMA were first characterized. Herein, it was found that the decomposition profile of polymer-grafted GO (HB 10-1-1) differs from pristine GO and the corresponding polymers at high temperature (ca. 300 °C). Instead of well-defined steps, HB 10-1-1 showed gradual decomposition between 250 °C and 450 °C, followed by the sharp decline ending at 550 °C (Fig. 4). This step appeared at a higher temperature than the complete degradation of GO and the corresponding polymer themselves. This was also observed by Grácio et al. in the modification of GO using ATRP, in which they suggested the synergistic enhancement in the thermal stability resulted from the vicinal radical entrapment by GO.31
 | ||
| Fig. 4 Thermal degradation profiles (left) of HB 10-1-1 (blue), HB 10-2-1 (orange), HB 10-3-1 (green), HB 10-4-1 (red), HB 10-5-1 (purple) grafted GO, GO (magneta) and HB 10-1-1 branched polymer itself (grey), and its calculated mass fraction of grafted polymer onto GO (right). | ||
Due to this distinct degradation profile at high temperature, we calculated the mass fractions of polymer and GO from the degradation step from 125 °C to 250 °C only, and assumed that a linear combination of GO and the polymers as the only components using eqn (1). The first derivatives were shown in Fig. S.4† to demonstrate its significance.
| m = fGOmGO + fpmp | (1) |
:[CTA] was increasing from 1:1 to 5:1 (Fig. 6). This indicated that the polymer content on GO was higher when more crosslinker was used. The calculated degree of grafting increased with the [DEGDMA]:[CTA] ratio from 0.328 at L 10-0-1 ([DEGDMA]:[CTA] = 0:1) to 0.495 at HB 10-5-1 ([DEGDMA]:[CTA] = 5:1), indicating that the more branched topology promoted the degree of grafting onto GO. The density of grafting (ρgr) can also be calculated from eqn (2) using the number average molecular weight of the polymers and compositional data of GO by XPS (Fig. S.5 and Table S.6†):73 | (2) |
 | (3) |
| f p | ρ gr/nm−2 | A gr/nm2 | R g,n /nm | A g,n/nm2 | l/nm | |
|---|---|---|---|---|---|---|
| L 10-0-1 | 0.328 | 0.0210 | 47.6 | 2.44 | 18.7 | 2.91 |
| HB 10-1-1 | 0.380 | 0.0216 | 46.4 | 2.50 | 19.7 | 2.68 |
| HB 10-2-1 | 0.438 | 0.0266 | 37.6 | 2.58 | 20.9 | 1.77 |
| HB 10-3-1 | 0.478 | 0.0295 | 33.9 | 2.68 | 22.5 | 1.22 |
| HB 10-4-1 | 0.483 | 0.0301 | 33.2 | 2.77 | 24.1 | 0.96 |
| HB 10-5-1 | 0.495 | 0.0331 | 30.2 | 2.79 | 24.5 | 0.61 |
It was observed that the average spacing between grafted polymer was smaller than the radius of gyration when [DEGDMA]:[CTA] above 2, which means that the GO surfaces were in a crowded condition. In this crowded condition, the direct grafting of polymer onto GO is less probable. Then to the HBT series where TEGDMA was used as crosslinker with a longer spacer, we would like to investigate whether the extended crosslinker will enhance the accessibility of the pendent vinyl group, and promote a more branched polymer architecture, thus the overall degree of grafting.
The thermal degradation profiles by TGA showed an identical pattern as the HB series, and therefore, the same calculation method was adopted to determine the polymer content in the product (Fig. 5). The effect of [crosslinker]:[CTA] on the grafted polymer content was even more pronounced in the series of TEGDMA. fp increased from 0.385 in HBT 10-1-1 ([TEGDMA]:[CTA] = 1:1) to 0.665 in HBT 10-1-1 ([TEGDMA]:[CTA] = 5:1). These values were higher than the counterpart using DEGDMA as crosslinker (fp = 0.380 and 0.495 for HB 10-1-1 and HB 10-5-1, [DEGDMA]:[CTA] = 1:1 and [DEGDMA]:[CTA] = 5:1, respectively) suggesting that the strategy to enhance the grafted polymer content onto GO by enhancing intermolecular crosslink was successful.
 | ||
| Fig. 5 Thermal degradation profiles (left) of HBT 10-1-1 (blue), HB 10T-2-1 (orange), HBT 10-3-1 (green), HBT 10-4-1 (red), HBT 10-5-1 (purple) grafted GO, and their calculated mass fraction of grafted polymer onto GO (right). | ||
Strikingly, opposite to the DEGDMA series, the series of TEGDMA deviated from the predicted maximum grafted polymer fraction based on the radius of gyration (Rg) of the branched copolymer (Table 5). In fact, the spacing between grafted polymers was always smaller than the radius of gyration of the branched polymer in all reactions. This was more significant in HBT 10-4-1 ([TEGDMA]:[CTA] = 4:1) and HBT 10-5-1 ([TEGDMA]:[CTA] = 5:1) where negative statistical spacing was calculated, suggesting the graft polymers were in the overlapping regime. This was attributed to the longer spacer length of TEGDMA than DEGDMA such that the pendant vinyl groups were more extended and mitigate their burial in the grafted polymer layer. Regarding the polymer conformation, a mechanism (Scheme 4) is proposed: initially polymeric radicals (red) graft onto GO (black) directly. At higher conversion when GO surface is saturated, the pendant CC on the as-grafted polymers serve as secondary grafting site, which is not achievable for linear polymer. For low branching system, this secondary grafting is limited and the entire grafted polymers remain separated. In highly branched system, more pendant vinyl groups are accessible to the later polymeric radicals (blue) to achieve higher grafting density at overlapping regime. While the exact mechanism cannot be determined from the experimental data, it is postulated that the grafted polymer would overlap and slightly penetrate to the neighbouring one, analogous to branched polymer melt. However, owing to the low molecular weight and high crosslink density, segments between two crosslinks would be below the entanglement molecular weight, and entanglement was unlikely to occur (Scheme 4).
 | ||
| Scheme 4 Proposed conformation of branched polymer grafted GO. Red: primary polymer grafted on to GO directly; green: secondary grafted polymer at the pendent vinyl group. | ||
| f p | ρ gr/nm−2 | A gr/nm2 | R g,n/nm | A g,n/nm2 | l/nm | |
|---|---|---|---|---|---|---|
| HBT 10-1-1 | 0.385 | 0.0204 | 49.1 | 2.63 | 21.7 | 1.32 |
| HBT 10-2-1 | 0.435 | 0.0227 | 44.1 | 2.74 | 23.6 | 1.01 |
| HBT 10-3-1 | 0.529 | 0.0310 | 32.3 | 2.95 | 27.3 | 0.26 |
| HBT 10-4-1 | 0.584 | 0.0364 | 27.5 | 3.09 | 30.0 | −0.13 |
| HBT 10-5-1 | 0.665 | 0.0478 | 20.9 | 3.24 | 33.0 | −0.66 |
Finally, to achieve a better understanding of the growth of the grafted polymer layer on GO, the kinetics of HBT 10-5-1 ([PEGMA]:[TEGDMA]:[CTA] = 10:5:1) were studied by taking samples over the first 6 h of the reaction at 70 °C, which has the highest degree of grafting, the conversion was monitored by 1H NMR following the decline of the vinyl C–H in the monomer (Fig. S.2†). TGA was performed to determine fp at every hour over 6 h (Fig. 6). It showed that fpversus the reaction time and conversion. It revealed that instead of a constant build-up of polymer layer throughout the entire reaction, a significant portion of polymers were grafted onto GO in a short period at the early stage of the reaction, fp = 0.587 at 1 h (34% monomers conversion only), followed by a moderate and steady growth of the grafted polymer to fp = 0.631 at 6 h (94% monomers conversion). The discrepancy between the bulk monomer conversion and fp was also noticed by Kan et al.47 and we postulate that the drastic increment at the very beginning is due to the initial grafting onto blank GO. At the later stages of polymerization, the GO surface is plausibly shielded by the grafted polymer, hindering direct grafting onto the GO sheet. Instead, the pendant vinyl groups on the crosslinker and the AFCT activity of the ω-vinyl macromonomer served as secondary grafting sites to allow further attachment of polymer chains. Note that transfer to polymer potentially also occurs as a grafting mechanism. For reference, a similar fp (≈0.5) but much lower ρgr (0.016 chain nm−2) was reported in Kan's result in FRP. However, it will be nonsensical to further compare the strategy due to excessive variables in both the materials (e.g. graphite source, GO compositions and functionalities, GO synthesis repeatability) and reaction conditions (e.g. targeted DP, radical flux, monomer choice).
 | ||
| Fig. 6 Thermal degradation profiles HB 10-5-1 grafted GO at the reaction time of 1 (blue), 2 (orange), 3 (red), 4 (purple), 5 (brown) and 6 (green) h, and the mass fraction by the reaction (middle) and equivalent monomer conversion (right). | ||
Thickness measurement by atomic force microscopy
Owing to the 2D structure of GO, one can expect that polymer will be grafted onto the basal plane predominately, and less onto the edge. Therefore, the increase in the thickness of GO will provide additional data related to the geometry and quantity of the grafted polymer. It should be noted that the thickness measured here are specific to the material sources and reaction conditions, and thus the topology of the derived grafted polymer, and the sole comparison in the absolute values in thickness between literatures would not reflect any significance. The thickness of the final product after washing by centrifugation was measured using AFM. First, from the HB series (Fig. S.5† and Fig. 7), it is clear that after the polymerization, the GO became thicker than the pristine GO (h = 1.14 nm). Then with the increasing crosslinker concentration, the polymer layer was also thicker, from 1.87 nm of HB 10-1-1 to 3.27 nm of HB 10-5-1. Despite the similar fp of HB 10-3-1, HB 10-4-1 and HB 10-5-1, the thickness increased with the crosslinker concentration (Table 6). Here we postulated that while the total polymer contents were similar, the size of an individual more branched polymer molecule in HB 10-5-1 is larger than in HB 10-3-1. As a result, a thicker but less densely packed polymer layer was grafted in the case of more crosslinker, or vice versa when less crosslinker.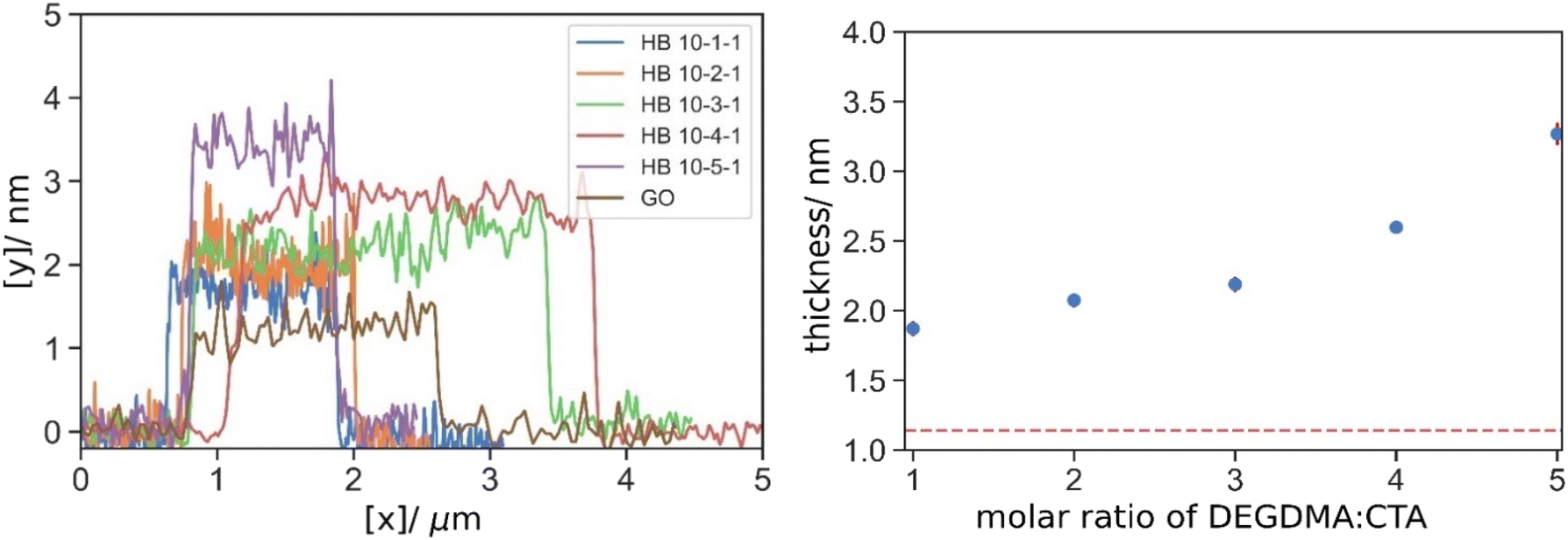 | ||
| Fig. 7 Typical linear AFM depth profiles of GO (brown), HB 10-1-1 (blue), HB 10-2-1 (orange), HB 10-3-1 (green), HB10-4-1 (red), HB 10-5-1 (brown) grafted GO, and their mean thickness; red dotted line indicated the thickness of pristine GO prior to the reaction. | ||
| Thickness/nm | Thickness/nm | ||
|---|---|---|---|
| Pristine GO | 1.14 | ||
| HB 10-1-1 | 1.87 | HBT 10-1-1 | 1.87 |
| HB 10-2-1 | 2.07 | HBT 10-2-1 | 2.13 |
| HB 10-3-1 | 2.19 | HBT 10-3-1 | 2.80 |
| HB 10-4-1 | 2.60 | HBT 10-4-1 | 3.29 |
| HB 10-5-1 | 3.27 | HBT 10-5-1 | 3.56 |
Next we would like to compare the difference between TEGDMA and DEGDMA on the thickness of the grafted polymer layer on GO. The typical AFM images and depth profiles were also demonstrated in Fig. S.6† and Fig. 8. The thickness of polymer grafted GO at various [TEGDMA]:[CTA] was summarized in Table 5 and Fig. 8. At the same [Crosslinker]: [CTA], the branched polymers with TEGDMA as crosslinker have a higher molecular weight than its analogous product with DEGDMA. Due to both the larger individual polymer molecule and higher degree of grafting, one can expect a more significant increase in the thickness of GO by using TEGDMA. When [crosslinker]:[CTA] = 1:1, linear polymers were still the predominated species with very limited branching, and thus there is no difference in the thickness of 1.87 nm. As [crosslinker]:[CTA] becomes larger, the effect of TEGDMA on the polymer layer thickness is more pronounced. When [crosslinker]:[CTA] = 2:1, HBT 10-2-1 was thicker than HB 10-2-1 by 0.06 nm only; however, when [crosslinker]:[CTA] = 3:1 and 4:1, there was a clear increment of 0.60 nm and 0.69 nm respectively when using TEGDMA. For reference only, the thickness was comparable to surface-initiated ATRP by Fang et al. (3.6 nm),28 where low molecular weight polymer was targeted, and much lower than that in FRP by Kan et al. (12.8 nm).47 Again, it is ill-judged to evaluate the effectiveness based on the value itself between methods owing to the numerous other factors.
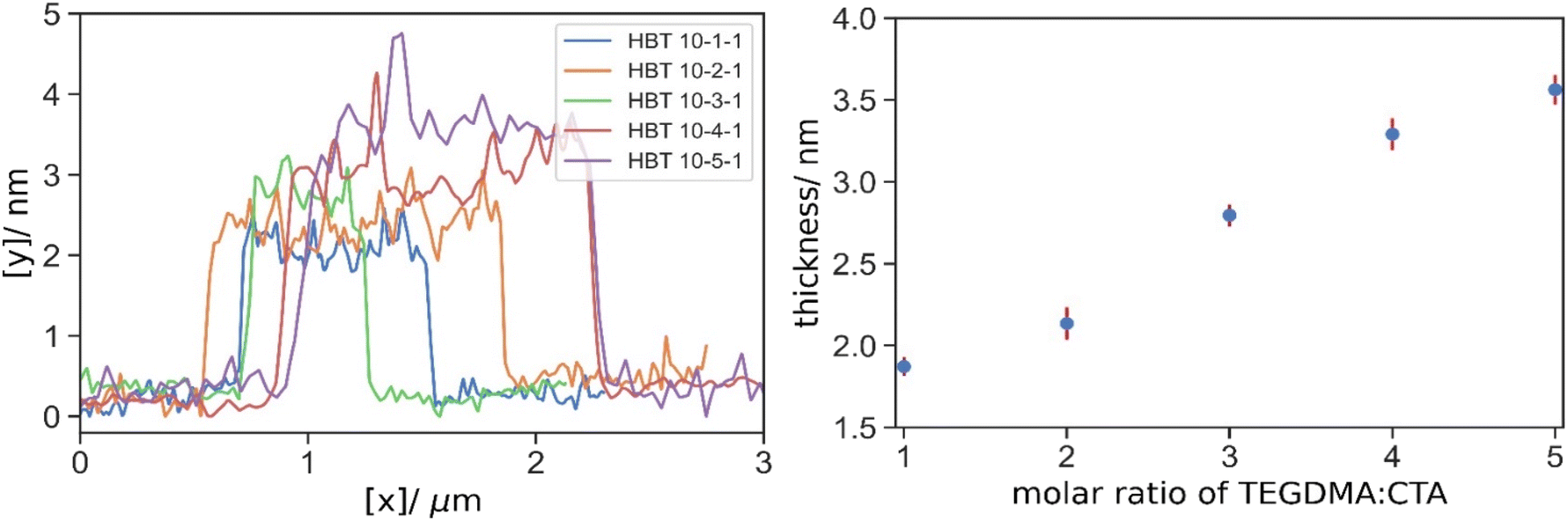 | ||
| Fig. 8 Typical depth profiles of HBT 10-1-1 (blue), HBT 10-2-1 (orange), HBT 10-3-1 (green), HBT10-4-1 (red), HBT 10-5-1 (purple) grafted GO and their mean thickness. | ||
Lastly, the kinetics of the evolution of the grafted polymer layer was studied. HBT 10-5-1 was chosen here because it has the highest degree of grafting and the thickest polymer layer which minimized the error due to the limit on depth resolution in the early stage of grafting. The typical AFM images and depth profiles of HBT 10-5-1 grafted GO over the polymerization time are shown in Fig. 9 and S.8.†
 | ||
| Fig. 9 Typical AFM depth profiles of HBT 10-5-1 grafted onto GO at time of 1, 2, 3, 4, 5 and 6 h, and their mean thickness by reaction time and equivalent conversion. | ||
Herein, it was found that the thickness increased in two regimes. The thickness increased drastically from 1.14 nm to 2.32 nm at the early stage of the reaction within 1 h (Fig. 9). Then, the thickness increased gently and steadily throughout the rest of the reactions over the 6 h sampling time. To further illustrate this two-step grafting, the thickness evolution was rescaled by conversion, and again, a non-linear growth of the grafted polymer layer versus conversion was observed. It should be noted that the evolution of the thickness cannot be estimated simply from the conversion and bulk density of the polymer. This is because the radius of gyration of the individual molecules by GPC was already in the nanoscale, and the excluded volume and molecular rigidity have to be considered in such a length scale.
Complementary to the previous TGA results on the grafting mechanism. The initial drastic increment in the thickness was caused by polymer grafting onto pristine GO, whereas the later steady and moderate growth corresponded to the continuous grafting of branched-polymer at the pendant vinyl groups or chain extension from the ω-vinyl terminated macromonomer. Oppositely, demonstrated by Kan et al., a plateau would be expected for linear non-functionalized polymer in conventional radical polymerization without crosslinker and AFCT moiety.47 Moreover, with the regulation of molecular weight by CTA and the branched architecture, a homogeneous and dense grafted polymer layer was obtained, in contrast to distinct patches scattering on GO in the case of linear high MW polymer.
Conclusion
In this work, a water soluble PMAA macromonomer was synthesized via aqueous solution CCTP. Grafting of branched PEGMA 500 onto GO was performed using PMAA macromonomer-mediated radical polymerization of PEGMA 500 in the presence of crosslinkers in aqueous GO dispersion. With the aid of a PMAA macromonomer, the branched polymer was successfully synthesized without macroscopic gelation. The effects of the crosslinker's concentration and spacer length on the resultant molecular weight and the polymer topology were examined using triple detection SEC.On the polymer grafted GO, the grafted polymer content was determined by TGA and FTIR. It was found that the grafting efficiency can be improved by enhancing the degree of branching. At a high [crosslinker]:[CTA] using a crosslinker with an extended spacer ([TEGDMA]:[CTA] = 5:1 mol/mol in this study), the grafted polymer content was as high as 66.5% of the product (2:1 w/w of grafted polymer to GO). The mechanism of grafting was further investigated in the combination of morphological study using AFM. It was postulated that the grafting process was divided into two stages. Initially, polymeric radicals directly grafted onto the intrinsic reactive sites of GO. This was indicated by the rapid increment in the grafted polymer content and evolution in the thickness within a short reaction time (1 h), or low conversion (34%). As the surface of GO was crowded, the pendent vinyl groups of the crosslinker and/or the ω-vinyl terminated macromonomer became predominant grafting sites. At this stage, a moderate and steady rise of the grafted polymer content and its thickness on GO was observed during the remaining progress of the reaction. This work opened a facile route to enhance the grafting efficiency by incorporating a small quantity of crosslinker, instead of excessive monomers. The kinetic study also offered a better understanding of the grafting mechanism of polymeric radicals onto GO.
Author contributions
Conceptualization (SAFB), data curation (WHL, SAFB), formal analysis (WHL, SAFB), funding acquisition (SAFB), investigation (WHL), methodology (WHL, SAFB), project administration (WHL, SAFB), supervision (SAFB), validation (WHL, SAFB), visualisation (WHL), writing – original draft (WHL, SAFB) writing – review & editing (WHL, SAFB).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Polymer Characterization Research Technology Platform (RTP) at the University of Warwick for providing the use of their Size Exclusion Chromatography and Thermal Gravimetric Analysis equipment. We also thank Synthomer and William Blythe for funding and supplying crude graphene oxide dispersion.References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- Z. Li, R. J. Young, C. Backes, W. Zhao, X. Zhang, A. A. Zhukov, E. Tillotson, A. P. Conlan, F. Ding, S. J. Haigh, K. S. Novoselov and J. N. Coleman, ACS Nano, 2020, 14, 10976–10985 CrossRef CAS PubMed.
- B. C. Brodie, Philos. Trans. R. Soc. London, 1859, 149, 249–259 CrossRef.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339–1339 CrossRef CAS.
- F. A. De La Cruz and J. M. Cowley, Nature, 1962, 196, 468–469 CrossRef CAS.
- W. Scholz and H. P. Boehm, Z. Anorg. Allg. Chem., 1969, 369, 327–340 CrossRef CAS.
- A. Clause, R. Plass, H.-P. Boehm and U. Hofmann, Z. Anorg. Allg. Chem., 1957, 291, 205–220 CrossRef.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- A. Rawal, S. H. C. Man, V. Agarwal, Y. Yao, S. C. Thickett and P. B. Zetterlund, ACS Appl. Mater. Interfaces, 2021, 13(5), 18255–18263 CrossRef CAS PubMed.
- I. Chowdhury, N. D. Mansukhani, L. M. Guiney, M. C. Hersam and D. Bouchard, Environ. Sci. Technol., 2015, 49, 10886–10893 CrossRef CAS PubMed.
- M. Wang, Y. Niu, J. Zhou, H. Wen, Z. Zhang, D. Luo, D. Gao, J. Yang, D. Liang and Y. Li, Nanoscale, 2016, 8, 14587–14592 RSC.
- Y. Jiang, R. Raliya, J. D. Fortner and P. Biswas, Environ. Sci. Technol., 2016, 50, 6964–6973 CrossRef CAS PubMed.
- A. S. Adeleye, K. T. Ho, M. Zhang, Y. Li and R. M. Burgess, Environ. Sci. Technol., 2019, 53, 5858–5867 CrossRef CAS PubMed.
- U. Hofmann and E. König, Z. Anorg. Allg. Chem., 1937, 234, 311–336 CrossRef CAS.
- S. Stankovich, R. D. Piner, X. Chen, N. Wu, S. T. Nguyen and R. S. Ruoff, J. Mater. Chem., 2006, 16, 155–158 RSC.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- S. Park, J. An, J. R. Potts, A. Velamakanni, S. Murali and R. S. Ruoff, Carbon, 2011, 49, 3019–3023 CrossRef CAS.
- J. I. Paredes, S. Villar-Rodil, A. Martínez-Alonso and J. M. D. Tascón, Langmuir, 2008, 24, 10560–10564 CrossRef CAS PubMed.
- H. R. Thomas, D. J. Phillips, N. R. Wilson, M. I. Gibson and J. P. Rourke, Polym. Chem., 2015, 6, 8270–8274 RSC.
- H. Yang, J.-S. Li and X. Zeng, ACS Appl. Nano Mater., 2018, 1, 2763–2773 CrossRef CAS.
- S. Song, C. Wan and Y. Zhang, RSC Adv., 2015, 5, 79947–79955 RSC.
- Q. Wei, X. Wang and F. Zhou, Polym. Chem., 2012, 3, 2129–2137 RSC.
- C. F. Matos, F. Galembeck and A. J. G. Zarbin, Carbon, 2014, 78, 469–479 CrossRef CAS.
- W. Meng, E. Gall, F. Ke, Z. Zeng, B. Kopchick, R. Timsina and X. Qiu, J. Phys. Chem. C, 2015, 119, 21135–21140 CrossRef CAS.
- H. M. Etmimi, M. P. Tonge and R. D. Sanderson, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1621–1632 CrossRef CAS.
- K. Jiang, C. Ye, P. Zhang, X. Wang and Y. Zhao, Macromolecules, 2012, 45, 1346–1355 CrossRef CAS.
- V. T. Huynh, D. Nguyen, C. H. Such and B. S. Hawkett, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1413–1421 CrossRef CAS.
- M. Fang, K. Wang, H. Lu, Y. Yang and S. Nutt, J. Mater. Chem., 2010, 20, 1982–1992 RSC.
- K. Ohno, C. Zhao and Y. Nishina, Langmuir, 2019, 35, 10900–10909 CrossRef CAS PubMed.
- S. H. Lee, D. R. Dreyer, J. An, A. Velamakanni, R. D. Piner, S. Park, Y. Zhu, S. O. Kim, C. W. Bielawski and R. S. Ruoff, Macromol. Rapid Commun., 2010, 31, 281–288 CrossRef CAS PubMed.
- G. Gonçalves, P. A. A. P. Marques, A. Barros-Timmons, I. Bdkin, M. K. Singh, N. Emami and J. Grácio, J. Mater. Chem., 2010, 20, 9927–9934 RSC.
- A. Kumar, B. Behera, G. D. Thakre and S. S. Ray, Ind. Eng. Chem. Res., 2016, 55, 8491–8500 CrossRef CAS.
- D. Vuluga, J.-M. Thomassin, I. Molenberg, I. Huynen, B. Gilbert, C. Jérôme, M. Alexandre and C. Detrembleur, Chem. Commun., 2011, 47, 2544–2546 RSC.
- B. Wang, D. Yang, J. Z. Zhang, C. Xi and J. Hu, J. Phys. Chem. C, 2011, 115, 24636–24641 CrossRef CAS.
- D. Voylov, T. Saito, B. Lokitz, D. Uhrig, Y. Wang, A. Agapov, A. Holt, V. Bocharova, A. Kisliuk and A. P. Sokolov, ACS Macro Lett., 2016, 5, 199–202 CrossRef CAS PubMed.
- F. Beckert, A. M. Rostas, R. Thomann, S. Weber, E. Schleicher, C. Friedrich and R. Mülhaupt, Macromolecules, 2013, 46, 5488–5496 CrossRef CAS.
- B. Zhang, Y. Zhang, C. Peng, M. Yu, L. Li, B. Deng, P. Hu, C. Fan, J. Li and Q. Huang, Nanoscale, 2012, 4, 1742 RSC.
- S. Hayashi, S. Handa, Y. Oshibe, T. Yamamoto, N. Tsubokawa, S. Handa, Y. Oshibe, T. Yamamoto and N. Tsubokawa, Polym. J., 1995, 27, 623–630 CrossRef CAS.
- K. Ohkita, N. Tsubokawa, E. Saitoh, M. Noda and N. Takashina, Carbon, 1975, 13, 443–448 CrossRef CAS.
- J. Ueda, H. Yamaguchi, K. Shirai, T. Yamauchi and N. Tsubokawa, J. Appl. Polym. Sci., 2008, 107, 3300–3305 CrossRef CAS.
- C. E. Bunker, G. E. Lawson and Y.-P. Sun, Macromolecules, 1995, 28, 3744–3746 CrossRef CAS.
- D. Stewart and C. T. Imrie, Chem. Commun., 1996, 1383–1384 RSC.
- K. Kirkwood, D. Stewart and C. T. Imrie, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 3323–3325 CrossRef CAS.
- A. G. Camp, A. Lary and W. T. Ford, Macromolecules, 1995, 28, 7959–7961 CrossRef CAS.
- M. S. P. Shaffer and K. Koziol, Chem. Commun., 2002, 2074–2075 RSC.
- S. Qin, D. Qin, W. T. Ford, J. E. Herrera, D. E. Resasco, S. M. Bachilo and R. B. Weisman, Macromolecules, 2004, 37, 3965–3967 CrossRef CAS.
- L. Kan, Z. Xu and C. Gao, Macromolecules, 2011, 44, 444–452 CrossRef CAS.
- Y. Cai, Y. Fadil, F. Jasinski, S. C. Thickett, V. Agarwal and P. B. Zetterlund, Carbon, 2019, 149, 445–451 CrossRef CAS.
- C. Walling, J. Am. Chem. Soc., 1945, 67, 441–447 CrossRef.
- P. J. Flory, J. Am. Chem. Soc., 1941, 63, 3083–3090 CrossRef CAS.
- W. H. Stockmayer, J. Chem. Phys., 1944, 12, 125–131 CrossRef CAS.
- D. T. Landin and C. W. Macosko, Macromolecules, 1988, 21, 846–851 CrossRef CAS.
- W.-H. Li, A. E. Hamielec and C. M. Crowe, Polymer, 1989, 30, 1513–1517 CrossRef CAS.
- W.-H. Li, A. E. Hamielec and C. M. Crowe, Polymer, 1989, 30, 1518–1523 CrossRef CAS.
- O. Okay, Polymer, 1994, 35, 2613–2618 CrossRef CAS.
- O. Okay and H. J. Naghash, Polym. Bull., 1994, 33, 665–672 CrossRef CAS.
- N. O'Brien, A. McKee, D. C. Sherrington, A. T. Slark and A. Titterton, Polymer, 2000, 41, 6027–6031 CrossRef.
- P. A. Costello, I. K. Martin, A. T. Slark, D. C. Sherrington and A. Titterton, Polymer, 2002, 43, 245–254 CrossRef CAS.
- F. Isaure, P. A. G. Cormack and D. C. Sherrington, Macromolecules, 2004, 37, 2096–2105 CrossRef CAS.
- S. Camerlynck, P. A. G. Cormack, D. C. Sherrington and G. Saunders, J. Macromol. Sci., Part B: Phys., 2005, 44, 881–895 CrossRef CAS.
- S. R. Cassin, S. Flynn, P. Chambon and S. P. Rannard, Polym. Chem., 2022, 13, 2295–2306 RSC.
- S. R. Cassin, S. Wright, S. Mckeating, O. B. Penrhyn-Lowe, S. Flynn, S. Lomas, P. Chambon and S. P. Rannard, Polym. Chem., 2023, 14, 1905–1914 RSC.
- O. B. Penrhyn-Lowe, S. Flynn, S. R. Cassin, S. Mckeating, S. Lomas, S. Wright, P. Chambon and S. P. Rannard, Polym. Chem., 2021, 12, 6472–6483 RSC.
- S. Flynn, O. B. Penrhyn-Lowe, S. Mckeating, S. Wright, S. Lomas, S. R. Cassin, P. Chambon and S. P. Rannard, RSC Adv., 2022, 12, 31424–31431 RSC.
- A. de Leon, M. Mellon, J. Mangadlao, R. Advincula and E. Pentzer, RSC Adv., 2018, 8, 18388–18395 RSC.
- A. Bakač, M. E. Brynildson and J. H. Espenson, Inorg. Chem., 1986, 25, 4108–4114 CrossRef.
- D. M. Haddleton, E. Depaquis, E. J. Kelly, D. Kukulj, S. R. Morsley, S. A. F. Bon, M. D. Eason and A. G. Steward, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2378–2384 CrossRef CAS.
- C. Robin, C. Lorthioir, C. Amiel, A. Fall, G. Ovarlez and C. Le Cœur, Macromolecules, 2017, 50, 700–710 CrossRef CAS.
- S. Smolne, S. Weber and M. Buback, Macromol. Chem. Phys., 2016, 217, 2391–2401 CrossRef CAS.
- O. B. Penrhyn-Lowe, S. Flynn, S. R. Cassin, S. Mckeating, S. Lomas, S. Wright, P. Chambon and S. P. Rannard, Polym. Chem., 2021, 12, 6472–6483 RSC.
- J. Ikeda, Y. Hasei, Y. Yasuda, H. Aota and A. Matsumoto, J. Appl. Polym. Sci., 2004, 94, 1086–1093 CrossRef CAS.
- A. Bonanni, C. K. Chua and M. Pumera, Chem. – Eur. J., 2014, 20, 217–222 CrossRef CAS PubMed.
- S. Zhang, H. Wang, J. Liu and C. Bao, Mater. Lett., 2020, 261, 127098 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR and FTIR characterization, XPS analysis, AFM analysis. See DOI: https://doi.org/10.1039/d4py00300d |
| This journal is © The Royal Society of Chemistry 2024 |