
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning network structures of hydrophobic hydrogels by controlling polymerization solvent†
Hailong
Fan
 *a,
Daito
Naohara
b,
Wei
Li
*a,
Xiang
Li
c and
Jian Ping
Gong
*ac
*a,
Daito
Naohara
b,
Wei
Li
*a,
Xiang
Li
c and
Jian Ping
Gong
*ac
aInstitute for Chemical Reaction Design and Discovery (WPI-ICReDD), Hokkaido University, Sapporo 001–0021, Japan. E-mail: fanhl@icredd.hokudai.ac.jp; wel208@icredd.hokudai.ac.jp; gong@sci.hokudai.ac.jp
bGraduate School of Life Science, Hokkaido University, Sapporo 001-0021, Japan
cFaculty of Advanced Life Science, Hokkaido University, Sapporo 001-0021, Japan
First published on 2nd May 2024
Abstract
Hydrophobic copolymer hydrogels hold great promise for various functional applications; however, the influence of monomer distribution and the resulting network structures on hydrogel properties remains largely unexplored. Here, hydrophobic hydrogels were synthesized through free-radical copolymerization using equimolar cationic and fluorous monomers. By employing precursor solutions with different solvent qualities, hydrogels with diverse copolymer network phase behaviors, varying from homogeneous networks to phase-separated structures, were obtained. The hydrophilicity disparity between monomers, coupled with their distribution in networks, dictates the hydrogel properties. Statistical copolymer hydrogels demonstrated swelling behaviors in both water and organic solvents, whereas phase-separated hydrogels exhibited anti-swelling behaviors. Moreover, statistical copolymer hydrogels equilibrated in saltwater showed superior mechanical strength and fracture energy compared to the phase-separated hydrogels. These findings underscore the pivotal role of monomer distribution in controlling copolymer hydrogel properties, providing a promising avenue for the design of hydrogel materials.
Introduction
Hydrophobic hydrogels present a progressively important branch within the extensive domain of hydrogel materials.1–3 At the center of this evolution lies the development of hydrophobization, which significantly expands the properties and applications of hydrogels. Hydrophobization is a process that involves the incorporation of hydrophobic species into the gel network, which assume roles as integral components of the structure or immobilized entities.4–13 This integration can give rise to hydrophobic domains within the matrix of hydrogels. Thus, it not only bolsters the solubilization capacity for hydrophobic drugs but also profoundly influences the overall characteristics of the hydrogels.14–19 The effects of hydrophobization extend to swelling behaviour, mechanical properties, and even the emergence of shape-memory characteristics.20–27 Moreover, hydrophobization enables hydrogel adhesion to a large array of surfaces, including hydrophilic substrates that traditional hydrogels fail to adhere to because of their highly swollen network structure.28–33A common approach to hydrophobization involves the one-step copolymerization of hydrophilic and hydrophobic monomers. While this approach is simple and efficient, it hinges on a comprehensive understanding of the interplay between hydrophilic and hydrophobic components during the polymerization process.5,34–38 Their distribution and arrangement within the polymer strand determine the hydrophilicity or hydrophobicity of the material at the molecular level, thereby dictating its overall interactions with water and other substances. For example, fine-tuned sequences endow hydrogels with enhanced underwater adhesion and superior mechanical strength.28 Furthermore, the phase separation between hydrophilic and hydrophobic components within the hydrogel, leading to the formation of distinct microdomains, also affects the hydrogel's macroscopic properties. Nevertheless, the effect of the copolymerization process on the overall properties of hydrophobic hydrogels remains largely unexplored.
Solvent properties play an important role in free radical copolymerization.39–42 The difference in interactions between reactant monomers and solvent molecules would induce local concentration variations, consequently leading to sequence changes in the resulting copolymers. Harnessing this effect presents an effective path to fabricate hydrogels with the same monomer compositions but different distributions and overall network structures.5
In this investigation, we synthesized hydrophobic gels through free-radical copolymerization, utilizing precursor solutions that transitioned from homogeneous to heterogeneous monomer phases (Fig. 1). This approach resulted in gels with identical monomer compositions but diverse network structures arising from phase separation. Specifically, cationic and fluorous monomers with equimolar ratio were chosen for copolymerization. In a homogeneous precursor solution, the two monomers underwent ideal copolymerization, forming statistical copolymer networks. The introduction of a poor solvent for the cationic monomer induced heterogeneity, leading to networks with phase-separated structures. By solvent exchange process, hydrogels were obtained. We systematically examined the swelling behaviour and mechanical properties of these hydrogels to elucidate the structure–property relationship. These findings are expected to have implications for the development of design principles that facilitate the fabrication of copolymer hydrogels with tailored properties for various functional applications.
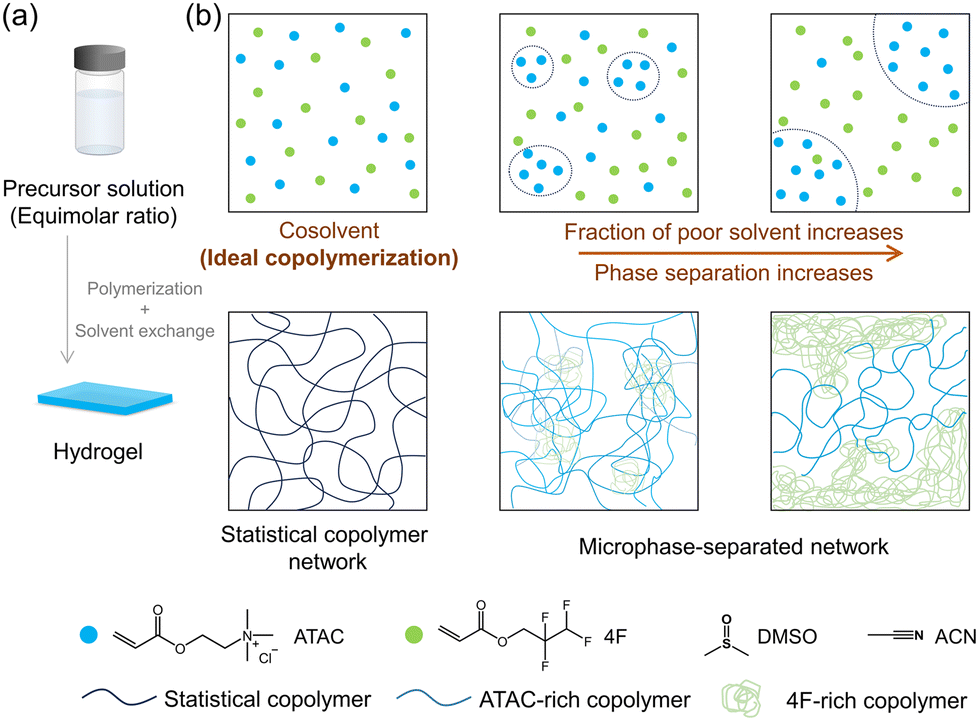 | ||
| Fig. 1 Schematic illustration of design strategy to synthesize hydrophobic copolymer hydrogels with different monomer distributions and network structures by tuning the solvent condition in reaction solutions. (a) Hydrogel fabrication by using free-radical copolymerization and solvent exchange processes. (b) Illustrations of local monomer distributions in precursor solutions with different solvents (upper row) and the resultant network structures in hydrogels (lower row). DMSO: dimethyl sulfoxide; ACN: acetonitrile; ATAC: 2-(acryloyloxy)ethyl trimethyl ammonium chloride; 4F: 2,2,3,3-tetrafluoropropyl acrylate. For simplicity, solvents, initiators, and crosslinkers are omitted in the illustration. | ||
Experimental
Materials
2-(Acryloyloxy)ethyl trimethyl ammonium chloride (ATAC, 79.4% in water), 2,2,3,3-tetrafluoropropyl acrylate (4F) were provided by Osaka Organic Chemical Co., Ltd, Japan. N,N′-Methylenebis(acrylamide) (MBAA), 2-oxoglutaric acid, NaCl, dimethyl sulfoxide (DMSO), and acetonitrile (ACN) were purchased from Wako Pure Chemical Industries, Ltd. All chemicals were used as purchased without further purification. Millipore deionized water was used in the experiments.Copolymerization kinetics in different solvents
To analyse the free-radical copolymerization kinetics of cationic and fluorous monomers, we utilized 1H-NMR spectroscopy (Agilent 500 MHz). The reaction solution, which comprised monomers at a total concentration of 1.0 M with varying monomer ratios, and 2-oxoglutaric acid (UV initiator, 2.5 mM), was polymerized in glass vials under UV light (3.9 mW cm−2) in a glove box. DMSO, ACN, and their mixtures were employed as solvents. For mixtures, the solvent was denoted as DxAy, where x and y are the weight percentage of DMSO and ACN, respectively. For example, the notation D70A30 represents a solution prepared by combining 70 wt% DMSO and 30 wt% ACN.Before polymerization, the reaction solution was pre-packaged into small vials, with each vial containing 200 μL of the solution. At different reaction times, we sampled one vial from the glove box and immediately exposed it to air to quench the reaction. Then, DMSO-d6 solvent was added to the sample to obtain a fully dissolved solution for NMR measurement. To determine the concentration of remaining monomers, we compared the integration of the double bond protons of the residual monomer to the integration of methylene protons of both residual monomer and formed copolymer.
In the monomer conversion measurement, the maximum polymerization time was set to 30 minutes. Because white precipitations showed up along with the copolymerization in the precursor solutions containing ACN, the determination of monomer conversion was performed till the precipitate became hardly soluble in DMSO-d6 for NMR measurements.
Hydrogel synthesis
To fabricate the gels, we utilized the free radical copolymerization of cationic and fluorous monomers in the presence of a chemical crosslinker, using DMSO, ACN, and their mixtures as the organic solvent. We dissolved the monomers (ATAC and 4F at a total concentration of 2.4 M with an equimolar ratio), chemical crosslinker (MBAA, 2.4 mM), and UV initiator (2-oxoglutaric acid, 6 mM) in the solvent. We then poured the resulting mixture into a reaction cell consisting of a pair of glass plates with a 1 mm spacing and irradiated it with 365 nm UV light at an intensity of 4 mW cm−2 for 11 hours in a glovebox. The NMR measurement revealed that the total monomer conversion (including both ATAC and 4F) was greater than 99% in all systems after 11 hours of UV exposure. To fabricate the homopolymer hydrogel P(ATAC), we dissolved the ATAC monomer (2.4 M), the chemical crosslinker (MBAA, 2.4 mM), and the UV initiator (2-oxoglutaric acid, 6 mM) in water. Subsequently, the resulting solution was poured into a reaction cell and exposed to UV irradiation within a glovebox for 11 hours.Swelling measurement
To prepare the samples, the 1 mm thick as-prepared organogel (with polymer content of gel ∼41 vol%) was cut into disk shapes with a diameter of 10 mm. Due to the stickiness of the as-prepared organogels, the margin of samples may appear irregular caused by peeling from the cutter. These samples were then immersed in various solutions, including organic solvents and salt solutions, to replace the solvent in the gel network. The solutions were changed every 12 hours for at least a week until the samples reached equilibrium. The swelling ratio Q was calculated as the ratio of the sample volume at swelling equilibrium V to that in the as-prepared state V0, Q = V/V0. As the swelling is isotropic, we measured the thickness of the gel in its as-prepared state as d0 and at equilibrium swelling as d. We then calculated the swelling ratio using the relationship Q = (d/d0)3.Mechanical test
Mechanical tests were conducted on hydrogel samples that had been equilibrated either in deionized water or a 0.7 M NaCl aqueous solution. To prepare the samples, the organogel was first immersed in an excess amount of either deionized or salt water to remove the solvent and residual chemicals. The deionized or salt water was changed every 12 hours for at least one week until the samples reached equilibrium. The hydrogels were then stored in either deionized or salt water until the mechanical tests were performed.For the uniaxial tensile stress–strain measurements, samples were cut into a dumbbell shape with the standard JIS-K6251-7 size (gauge length: 12 mm, width: 2 mm). The measurements were conducted using a universal testing machine (UTM, INSTRON 5965) at a steady velocity of 100 mm min−1 in air. The Young's modulus (E) was calculated from the slope of the stress–strain curve over 4–8% of the strain. The nominal tensile stress was calculated from the tensile force divided by the cross-sectional area of the virgin sample, and the tensile strain was calculated from the displacement of the cross-head of the testing machine divided by the initial gauge length.
For the tearing test, samples were cut into a trouser shape (width: 20 mm, length: 60 mm, notch length: 20 mm). The rate of extension was fixed at 100 mm min−1, and the fracture energy (Γ) was calculated from the average loading force (Fave) using the equation Γ = 2Fave/d, where d = 1.3–1.5 mm is the sample thickness.43
For the pure shear test, two different samples, one with a single edge notch and one without a notch, were employed to measure the fracture energy (Γ).44 The samples were cut into a rectangular shape with a width of 12 mm and a length of 50 mm. Then, the samples were clamped along their long edges at a height (H) of 10 mm. A notch (15 mm) was intentionally introduced into one of the samples, which was then gradually extended to a critical stretch value (λc) at which the crack began to propagate. The rate of extension was fixed at 100 mm min−1. Subsequently, an unnotched sample was stretched to the same stretch value (λc), with nominal stress (σ) recorded as a function of the stretch (λ). Based on the stress–stretch curve, the fracture energy of the gel can be calculated as 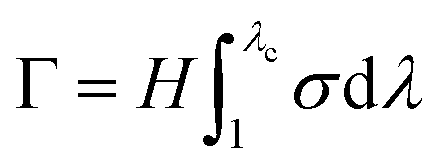 .
.
Results and discussion
Fabrication of gels under different solvent conditions
In DMSO, both ATAC and 4F monomers are completely dissolved, yielding a transparent true solution (Fig. S1†). With an increased proportion of ACN (a poor solvent for ATAC) in the precursor solution, the solubility of ATAC decreases, leading to a liquid–liquid phase separation of ATAC. The formation of emulsions, with separated domains rich in ATAC, causes the precursor solution to gradually turn translucent (Fig. S1†). We note that the emulsion state of the solution is relatively stable for hours and independent of the mixing condition. For consistency, both the characterization and the initiation of copolymerization were conducted within 10 minutes after preparing the precursor solutions in the experiment.By utilizing the solvent condition difference in the copolymerization, we proceeded to fabricate gels via one-pot free-radical copolymerization of equimolar ATAC and 4F monomers at a high total monomer concentration (2.4 M) with a small amount of chemical crosslinkers (MBAA 2.4 mM, i.e., 0.1 mol% in comparison with the monomers) in above solvents. The initiator and crosslinker are soluble in all solvents. The effect of their distributions in the precursor solution on network structures and properties of gels is neglected in the discussion.
Six gels were prepared in solvents varied from DMSO to DMSO/ACN mixtures and then ACN, which were labelled accordingly from Gel-1 to Gel-6 (Fig. 2). The appearance of the as-prepared organogels varies across this series. Gel-1, prepared in DMSO, is transparent, indicating that a randomly distributed sequence prevents ATAC aggregation—worth noting that a neat ATAC gel fabricated in DMSO is opaque. This observed phenomenon is similar to our previous studies, where gels with a statistical distribution of cationic and aromatic monomers exhibited transparency in their cosolvent.5,28 From Gel-2 to Gel-6, the as-prepared organogels turn from translucent to opaque, reflecting the precipitation of ATAC-rich phase in the poor solvent.
 | ||
| Fig. 2 Photographic images of as-prepare organogels and hydrogels equilibrated in deionized water, 0.154 M NaCl, and 0.7 M NaCl solution, respectively. Six gels fabricated in different solvents are labelled accordingly from Gel-1 to Gel-6. | ||
Hydrogels were prepared by submerging the organogels in a large volume of deionized or salt water for a sufficient time to reach swelling equilibrium. The monomer distribution difference also affects the appearance of the hydrogels, for which the aggregation of hydrophobic 4F monomers causes the transparency change while ATAC is soluble.
To better understand the monomer distributions within the networks, we examined the copolymerization kinetics of cationic and fluorous monomers in different solvents without adding the crosslinker. Fig. 3a–f plots the corresponding temporal evolution of monomer conversion in different systems with an equimolar ratio of two monomers at a total concentration of 1.0 M. In DMSO, the conversion curves of ATAC and 4F in DMSO almost completely overlapped with each other, suggesting that the two monomers were incorporated into the copolymer at the same rate (Fig. 3a). The discrepancy between the two monomer conversion curves emerged when incorporating ACN in the solution. With increasing the fraction of ACN, the increase in density inhomogeneity caused the monomer conversion curves to separate from each other with the reaction rate of ATAC being greater than that of 4F (Fig. 3b–f). The acceleration of the polymerization kinetics of ATAC in the poor solvent could be understood by the high local concentration of ATAC due to phase separation. At large ACN fractions, white precipitations showed up in the precursor solution within minutes from the beginning of the copolymerization (upon exposure to UV light), indicating the formation of non-soluble copolymers rich in consecutive ATAC segments (Fig. S2†).
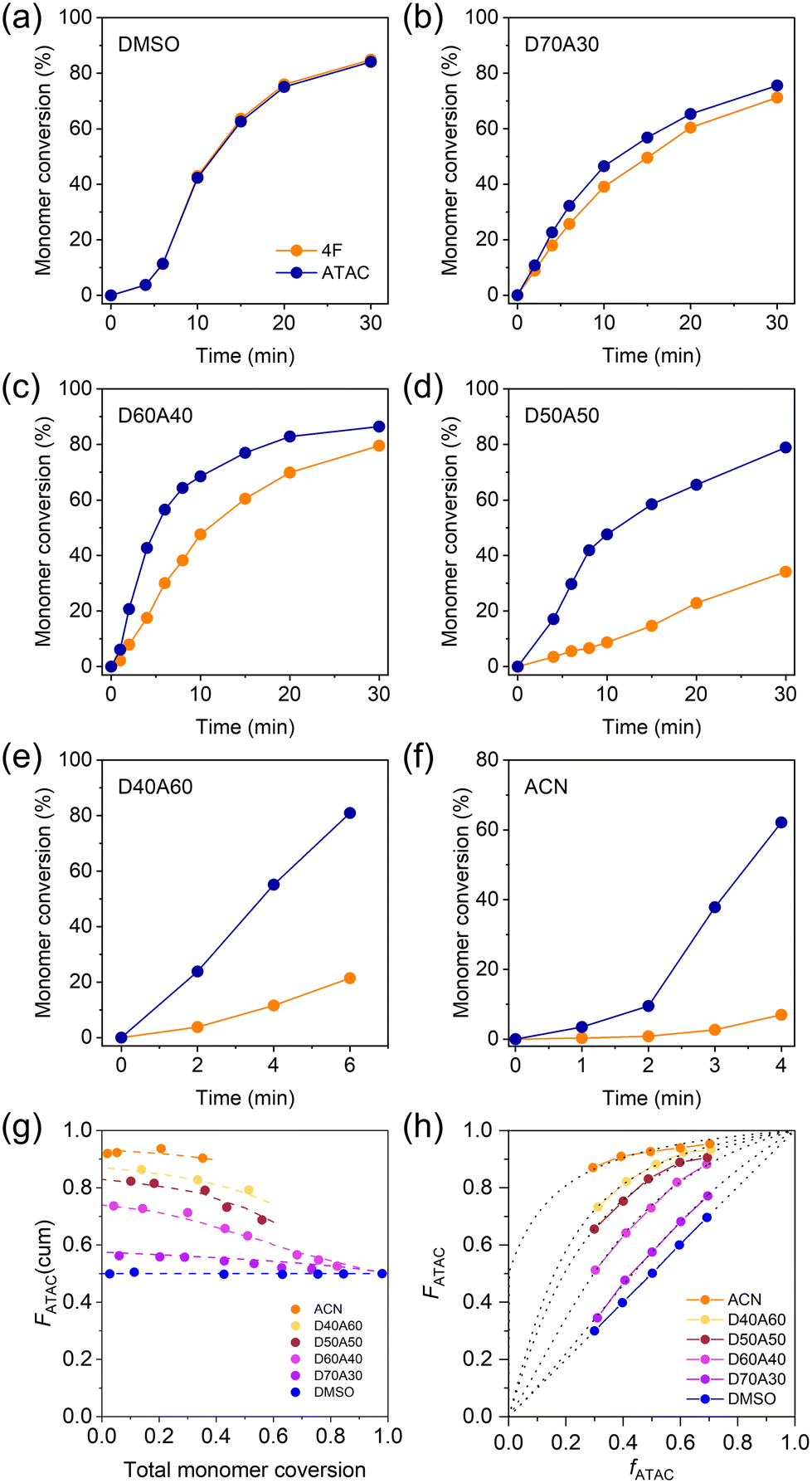 | ||
| Fig. 3 (a–g) Monomer conversion of cationic ATAC and fluorous 4F as a function of time in solutions with equimolar ratio and total concentration of 1.0 M of the two monomers. The solvent compositions as noted: (a) 100% DMSO, (b) 70% DMSO + 30% ACN, (c) 60% DMSO + 40% ACN, (d) 50% DMSO + 50% ACN, (e) 40% DMSO + 60% ACN, and (f) 100% ACN. (g) Cumulative ATAC mole fraction in copolymers against the total monomer conversion for equimolar systems (1.0 M in total) determined by 1H NMR. The dashed lines are a guide to the eyes. (h) ATAC mole fraction in copolymers, FATAC, as a function of ATAC mole fraction in feed, fATAC, in systems of different reaction solvents. The dashed lines are fitted curves by using the Mayo–Lewis equation. | ||
Utilizing the conversion data, Fig. 3g illustrates the cumulative ATAC composition in the copolymers plotted against the total monomer conversion. In the case of DMSO, the curve remains flat, indicating a consistent monomer composition in copolymers. In contrast, for other solvents, the curves initiate with a high ATAC composition exceeding 0.5, gradually decreasing to 0.5 with ongoing monomer conversion, indicating the obvious composition shift during copolymerization.
To gain insight into the monomer sequence from copolymerization, we varied ATAC mole fraction in feed, fATAC, in systems of different reaction solvents and measured ATAC mole fraction in copolymers, FATAC, at low monomer conversion (<10%). The results are shown in Fig. 3h. One finds that in DMSO, FATAC and fATAC were always identical, while in mixing solvents, FATAC was always larger than fATAC, consistent with the results in Fig. 3g. In the mixed solvents, in which phase separation of ATAC occurred, both FATAC and fATAC are average of the entire system.
By fitting the F–f curves to the Mayo–Lewis equation (dotted lines in Fig. 3h), we estimated the reactivity ratios (r) for ATAC and 4F (Table S1†). In DMSO, the two types of monomers exhibit similar reactivity ratios of ∼1, i.e., a typical feature of ideal copolymerization, which results in a statistical distribution of the two monomers in the chain sequence.5,45 This observed phenomenon aligns with our previous studies, where ideal copolymerization of cationic and aromatic monomers with the same double bond was identified in their cosolvent.5 However, upon the introduction of ACN into the precursor solution, copolymerization occurred in a heterogeneous system with obvious phase separation. In such cases, although reactivity ratios can be calculated by fitting from experimental data (Table S1†), they represent apparent values, which originated from a combination of two local reaction phases, thus they should not be suitable for sequence analysis.
Swelling behavior of hydrogels
Given that cationic ATAC is hydrophilic and responsive to the ionic strength of the medium while 4F is hydrophobic, the monomer distributions within the polymer strand play a crucial role in the hydrophilicity or hydrophobicity of the network, thereby dictating its overall interactions with water and organic solvent and subsequently determining the phase separations of the network. Therefore, the swelling behaviours of gels in water, saltwater, and organic solvents with different dielectric constants were studied. | ||
| Fig. 4 (a) Comparisons of swelling ratio Q for different gels in water and 0.7 M NaCl solution. (b) Volume swelling ratio Q of equilibrated hydrogels (equimolar in ATAC and 4F) with respect to the as-prepared organogels as functions of NaCl salt concentration Cs in the aqueous solution. A control study of homo-polycationic P(ATAC) hydrogel is shown for comparison. (c) Laser microscopy images of the fresh cut cross-section of hydrogels equilibrated in 0.7 M NaCl solution. | ||
Fig. 4b plots the swelling ratio Q of hydrogels with respect to the as-prepared organogel states as a function of salt concentration Cs. The differences between the swelling ratio Q curves indicate the structural difference of the gel networks. At high ionic strength (e.g., Cs = 0.7 M), the electrostatic repulsion is totally screened, and all six gels showed similar swelling ratios. Laser microscopy was applied to probe the hydrogel morphologies, and typical cross-section images of hydrogels equilibrated in 0.7 M NaCl solution are shown in Fig. 4c. From the images, Gel-1 and Gel-2 are mostly homogeneous. Heterogeneous aggregate structures show up in Gel-3, while the aggregation size and range increase from Gel-3 to Gel-6 with the large-scale phase separation enhancing the gel opaqueness in Fig. 2.
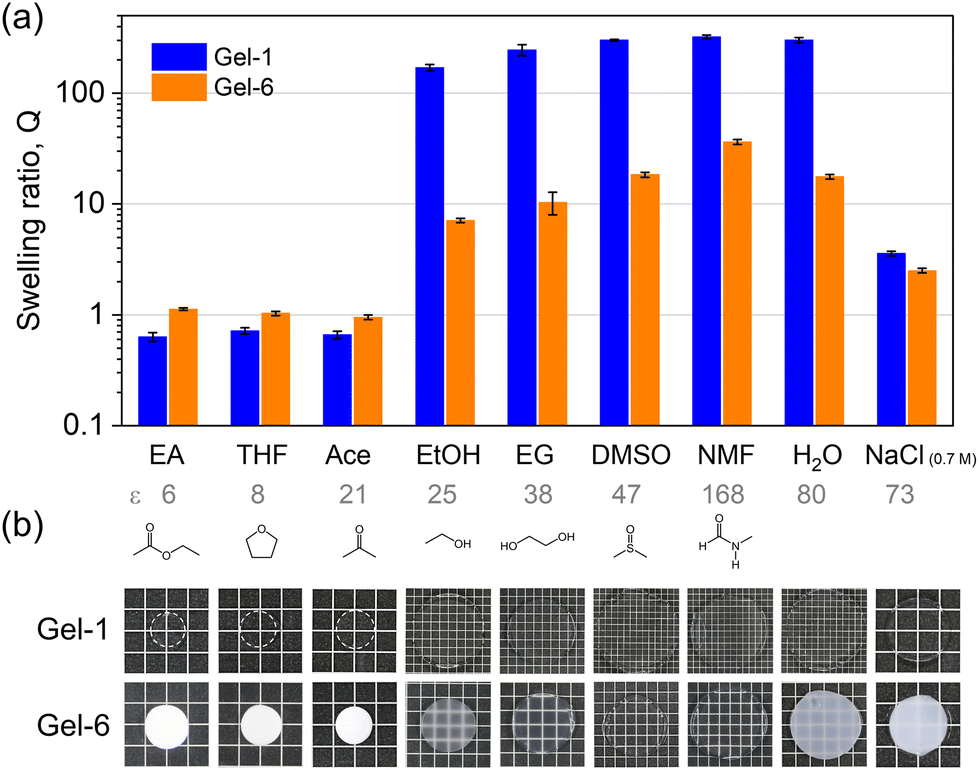 | ||
| Fig. 5 (a) Volume swelling ratio Q of Gel-1 and Gel-6 with respect to their as-prepared states in different solvents. The values of the solvent dielectric constant ε along with the molecular formula are shown below. (b) Photographic images of the gels equilibrated in these solvents. | ||
In general, the swelling behaviour of Gel-1 (with statistical sequence) is similar to that of the P(ATAC) hydrogel (Fig. S3†) and hence can be understood by the polyelectrolyte effect. Specifically, the shrinkage at low ε is attributed to the association of charged strands with counter-anions such that the strong ionic correlation condenses the gel.46 At high ε, the dissociation of the counterions produces high osmotic pressure and induces significant swelling, which is counter-balanced by the elasticity of the polymer network.
In contrast to Gel-1, Gel-6 presents a larger swelling ratio in the solvents with low ε but a smaller swelling ratio in the solvents with high ε. This suggests that the large 4F-rich phase in Gel-6 enlarges their contribution to the swelling behaviour (as reflected in the case of neat 4F gel in Fig. S3†), which is dictated by the affinity of 4F with individual solvents.
Mechanical properties of copolymer hydrogels
A uniaxial tensile test was conducted to examine the mechanical properties of the prepared hydrogels in deionized and salt water. When equilibrated in deionized water, Gel-1, Gel-2, and Gel-3 displayed mechanical weakness and fragility due to the highly swollen state (Fig. S4†). By contrast, Gel-4, Gel-5, and Gel-6 exhibited relatively larger strength with higher elastic modulus. Specifically, Gel-1 showed Young's modulus of approximately 6 kPa, whereas Gel-6 reached up to 60 kPa.When equilibrated in solution with high ionic strength that screens the electrostatic interactions (0.7 M NaCl solution), the mechanical performance of the hydrogels exhibited notable differences from their counterpart in water, highlighting the effects of deswelling and phase separation. Fig. 6a plots the stress–strain curves obtained at a strain rate of 0.14 s−1, and Fig. 6b presents extracted Young's modulus, E, for the gels. Gel-1 shows soft and stretchable characteristics, which has E ≈ 18 kPa. By increasing the phase separation domains from Gel-1 to Gel-6, E slightly decreases first and then undergoes a significant increase. Meanwhile, the gels become increasingly fragile. Particularly, Gel-6, exhibiting the strongest phase separation, has a Young's modulus of around 100 kPa but can only be elongated by 120% (with a maximum tensile strength of approximately 150 kPa).
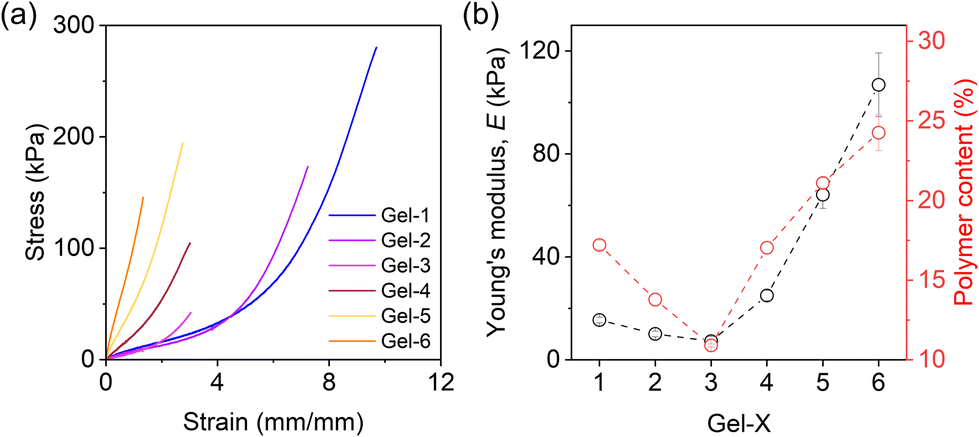 | ||
| Fig. 6 (a) Tensile stress–strain curves of hydrogels equilibrated in 0.7 M NaCl solution (obtained at a strain rate of 0.14 s−1). (b) Young's modulus and polymer content of the hydrogels equilibrated in 0.7 M NaCl solution. | ||
The intriguing observation of a non-monotonic trend in E is noteworthy, as we initially anticipated that an increase in phase separation would lead to a monotonic trend in E. To address this phenomenon, we measured the water content of the hydrogels in 0.7 M NaCl solution (Fig. S5†) and Young's modulus of the as-prepared organogels (Fig. S6†) in experiments. Both measurements showed nonmonotonic behaviours, while the corresponding polymer content in the hydrogel decreased first from Gel-1 and then started to increase at Gel-3, consistent with the behaviour of E (Fig. 6b).
The decrease of polymer content in Gel-2 and Gel-3 suggests that there might be more structural defects in their network formation processes which caused the relative weakness in mechanical properties. We attribute this feature to the fact that ACN has a higher radical transfer ability than DMSO (Fig. S7†). Thus, it is relatively harder for reactant monomers to form a gel in ACN, which reduces the connectivity and fraction of elastically effective strands of the formed networks that decrease E. Nevertheless, such an effect can be offset by the strong phase separation that significantly increases E (Fig. 6b).
Next, we proceeded to evaluate the fracture energy of the hydrogels using a trouser-shape tearing test and a pure shear test (Fig. 7), which quantifies the energy per unit area (J m−2) required to propagate a crack. Fig. 7a plots the tear force–distance curves of the gels. Gel-1 exhibits a remarkable loading force and a high tearing fracture energy of ∼110 J m−2. Its tear resistance ability is evident from the pronounced “zig-zag” curve with substantial fluctuations. From Gel-1 to Gel-6, the tear force initially decreases and then slightly increases. Accordingly, the fracture energy from the tearing test follows the same trend as Young's modulus, with Gel-3 exhibiting the lowest fracture energy among the hydrogels in Fig. 7b. A similar behaviour is also observed for the fracture energy from the pure shear test in Fig. 7d. These results indicate that Gel-1, having a statistical strand sequence and disordered network structure, performs the best in crack resistance, while the hydrogels with phase-separated structures (e.g., Gel-6), altering stress concentration and crack propagation, reduces crack resistance.
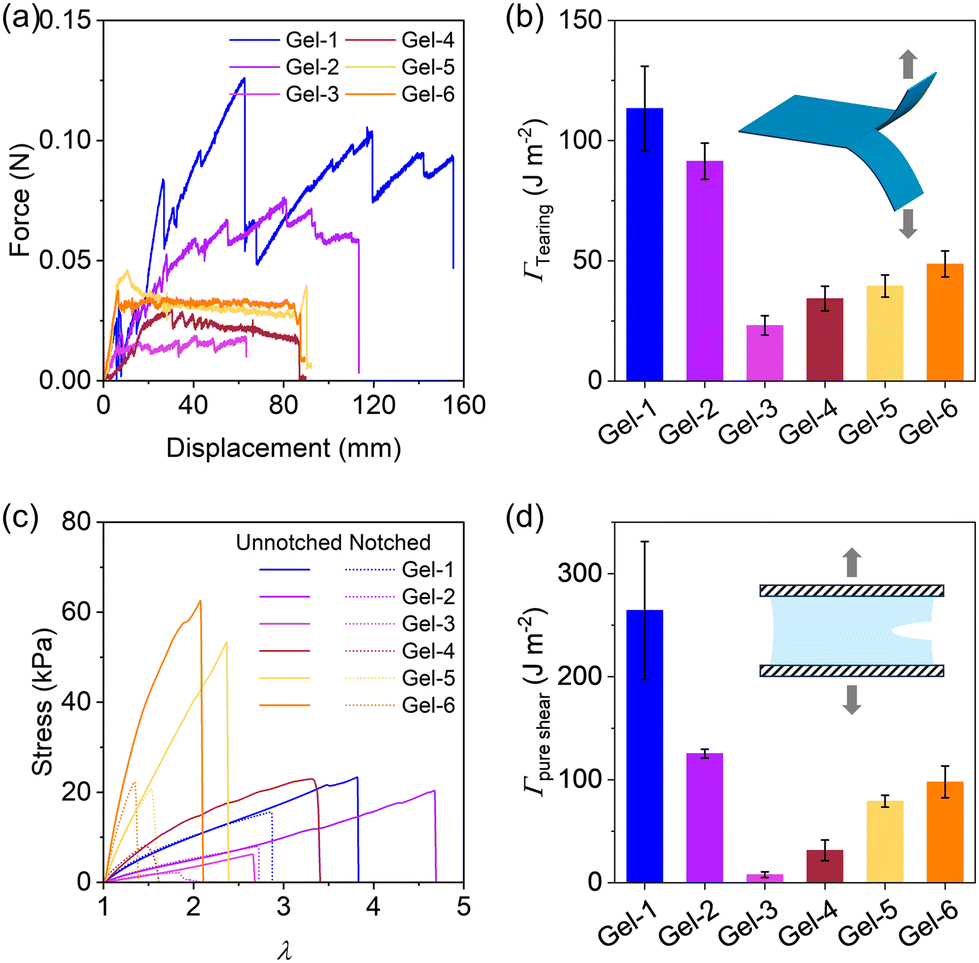 | ||
| Fig. 7 Fracture energy of hydrogels equilibrated in 0.7 M NaCl solution. (a) Tear force–distance curves of hydrogels. (b) Fracture energy determined by the tearing test. (c) Stress–stretch curves of hydrogels with and without notch. (d) Fracture energy determined by the pure shear test. Insets show the experimental setup. | ||
Conclusions
In summary, this study revealed that the monomer distribution and resulting network structures significantly impact the properties of hydrogels. By manipulating precursor solutions transitioning from homogeneous to heterogeneous monomer phases, we successfully obtained hydrogels with identical monomer compositions but diverse structures, ranging from statistical copolymer networks to various phase-separated configurations. The hydrophilicity disparity among monomers, combined with distinct phase-separated network structures, imparted unique properties to the resulting hydrogels. Statistical copolymer hydrogels exhibited hydrophobic swelling behaviour, while phase-separated hydrogels demonstrated anti-swelling characteristics. Notably, statistical copolymer hydrogels equilibrated in saltwater exhibited superior mechanical strength and fracture energy compared to their phase-separated counterparts. Overall, this work not only demonstrates a novel approach to control phase separate structures within hydrogels but also underscores the substantial influence of monomer distribution on the network structures and hydrogel properties, an aspect that has been relatively underexplored in previous studies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the JSPS KAKENHI Grant (Grant No. JP22H04968, JP22K21342, JP24K17728). The Institute for Chemical Reaction Design and Discovery (WPI-ICReDD) was established by the World Premier International Research Initiative (WPI), MEXT, Japan. The authors thank Mr Yoshiyuki Saruwatari at Osaka Organic Chemistry Co., Ltd, Japan for providing chemicals. Prof. Micheal Rubinstein in Duke University is highly acknowledged for useful suggestions and discussions.References
- M. Gosecka, M. Gosecki and D. Jaworska-Krych, Adv. Funct. Mater., 2023, 33, 2212302 CrossRef CAS.
- N. M. Oliveira, Y. S. Zhang, J. Ju, A.-Z. Chen, Y. Chen, S. R. Sonkusale, M. R. Dokmeci, R. L. Reis, J. F. Mano and A. Khademhosseini, Chem. Mater., 2016, 28, 3641–3648 CrossRef CAS.
- X. Liu, X. He, B. Yang, L. Lai, N. Chen, J. Hu and Q. Lu, Adv. Funct. Mater., 2021, 31, 2008187 CrossRef CAS.
- X. Yang, M. Cui, J. Zhou, L. Zhang, H. Zhou, Z. Luo, L. Zhou and H. Hu, ACS Appl. Bio Mater., 2021, 4, 523–532 CrossRef CAS PubMed.
- H. L. Fan, Y. R. Cai and J. P. Gong, Sci. China: Chem., 2021, 64, 1560–1568 CrossRef CAS.
- H. Guo, T. Nakajima, D. Hourdet, A. Marcellan, C. Creton, W. Hong, T. Kurokawa and J. P. Gong, Adv. Mater., 2019, 31, 1900702 CrossRef PubMed.
- I. Jeon, J. Cui, W. R. K. Illeperuma, J. Aizenberg and J. J. Vlassak, Adv. Mater., 2016, 28, 4678–4683 CrossRef CAS PubMed.
- M. Mo, S. Du, Y. Gao, B. Peng, L. Zhang and J. Zhu, J. Colloid Interface Sci., 2022, 616, 93–100 CrossRef CAS PubMed.
- A. Ghosh, S. Pandit, S. Kumar, D. Ganguly, S. Chattopadhyay, D. Pradhan and R. K. Das, Chem. Eng. J., 2023, 475, 146160 CrossRef CAS.
- Y. Wang, D. Yuan, L. Sun, S. Xu and S. Wan, Chem. Eng. J., 2023, 461, 142005 CrossRef CAS.
- M. Mansouri, S. Beemer, C. R. Kothapalli, T. Rhoades, P. S. Fodor, D. Das and N. D. Leipzig, ACS Appl. Mater. Interfaces, 2022, 14, 4899–4913 CrossRef CAS PubMed.
- L. Yang, S. Li, Z. Zhao, J. Wang, H. Lv and X. Yang, Polym. Chem., 2023, 14, 2212–2219 RSC.
- X. Li, H. Mutlu, C. Fengler, M. Wilhelm and P. Theato, Polym. Chem., 2021, 12, 361–369 RSC.
- C. Ma, Y. Shi, D. A. Pena, L. Peng and G. Yu, Angew. Chem., Int. Ed., 2015, 54, 7376–7380 CrossRef CAS PubMed.
- G. Huang, Z. Tang, S. Peng, P. Zhang, T. Sun, W. Wei, L. Zeng, H. Guo, H. Guo and G. Meng, Macromolecules, 2022, 55, 156–165 CrossRef CAS.
- X. Song, Z. Zhang, J. Zhu, Y. Wen, F. Zhao, L. Lei, N. Phan-Thien, B. C. Khoo and J. Li, Biomacromolecules, 2020, 21, 1516–1527 CrossRef CAS PubMed.
- E. Kumarasamy, I. M. Manning, L. B. Collins, O. Coronell and F. A. Leibfarth, ACS Cent. Sci., 2020, 6, 487–492 CrossRef CAS PubMed.
- H. Zhang, A. Patel, A. K. Gaharwar, S. M. Mihaila, G. Iviglia, S. Mukundan, H. Bae, H. Yang and A. Khademhosseini, Biomacromolecules, 2013, 14, 1299–1310 CrossRef CAS PubMed.
- P. Ravarino, D. Giuri, D. Faccio and C. Tomasini, Gels, 2021, 7, 43 CrossRef CAS PubMed.
- D. C. Tuncaboylu, M. Sari, W. Oppermann and O. Okay, Macromolecules, 2011, 44, 4997–5005 CrossRef CAS.
- G. Huang, H. Guo, Z. Tang, S. Peng, H. Liang, G. Meng and P. Zhang, Chem. Mater., 2023, 35, 5953–5962 CrossRef CAS.
- R. Liang, H. Yu, L. Wang, L. Lin, N. Wang and K.-u.-R. Naveed, ACS Appl. Mater. Interfaces, 2019, 11, 43563–43572 CrossRef CAS PubMed.
- T. Ono, T. Sugimoto, S. Shinkai and K. Sada, Nat. Mater., 2007, 6, 429–433 CrossRef CAS PubMed.
- J. Yang, F.-K. Shi, C. Gong and X.-M. Xie, J. Colloid Interface Sci., 2012, 381, 107–115 CrossRef CAS PubMed.
- D. Tan, F. Meng, Y. Ni, W. Sun, Q. Liu, X. Wang, Z. Shi, Q. Zhao, Y. Lei, S. Luan and L. Xue, Chem. Eng. J., 2023, 471, 144625 CrossRef CAS.
- L. Yang, S. Li, Z. Zhao, J. Wang, X. Yang and H. Lv, Chem. Eng. J., 2024, 482, 148808 CrossRef CAS.
- H. Chen, B. Hao, P. Ge and S. Chen, Polym. Chem., 2020, 11, 4741–4748 RSC.
- H. L. Fan, J. H. Wang, Z. Tao, J. C. Huang, P. Rao, T. Kurokawa and J. P. Gong, Nat. Commun., 2019, 10, 5127 CrossRef PubMed.
- H. L. Fan, J. H. Wang and J. P. Gong, Adv. Funct. Mater., 2021, 31, 2009334 CrossRef CAS.
- L. Han, M. Wang, L. O. Prieto-López, X. Deng and J. Cui, Adv. Funct. Mater., 2020, 30, 1907064 CrossRef CAS.
- S. Wang, L. Wang, X. Qu, B. Lei, Y. Zhao, Q. Wang, W. Wang, J. Shao and X. Dong, ACS Appl. Mater. Interfaces, 2022, 14, 50256–50265 CrossRef CAS PubMed.
- X. Fan, W. Zhou, Y. Chen, L. Yan, Y. Fang and H. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 32031–32040 CrossRef CAS PubMed.
- S. Huang, Y. Wan, X. Ming, J. Zhou, M. Zhou, H. Chen, Q. Zhang and S. Zhu, ACS Appl. Mater. Interfaces, 2021, 13, 41112–41119 CrossRef CAS PubMed.
- M. Mihajlovic, M. Staropoli, M.-S. Appavou, H. M. Wyss, W. Pyckhout-Hintzen and R. P. Sijbesma, Macromolecules, 2017, 50, 3333–3346 CrossRef CAS PubMed.
- S. Ida, D. Nishisako, A. Fujiseki and S. Kanaoka, Soft Matter, 2021, 17, 6063–6072 RSC.
- S. Ida, Polym. J., 2019, 51, 803–812 CrossRef CAS.
- S. Ida, T. Kawahara, H. Kawabata, T. Ishikawa and Y. Hirokawa, Gels, 2018, 4, 22 CrossRef PubMed.
- N. M. B. Smeets, M. Patenaude, D. Kinio, F. M. Yavitt, E. Bakaic, F.-C. Yang, M. Rheinstädter and T. Hoare, Polym. Chem., 2014, 5, 6811–6823 RSC.
- P. Deglmann, K.-D. Hungenberg and H. M. Vale, Ind. Eng. Chem. Res., 2021, 60, 10566–10583 CrossRef CAS.
- J. Hao, F. An, C. Lu and Y. Liu, J. Macromol. Sci., Part A: Pure Appl.Chem., 2019, 56, 1012–1021 CrossRef CAS.
- K. Liang and R. A. Hutchinson, Macromolecules, 2010, 43, 6311–6320 CrossRef CAS.
- M. L. Coote, T. P. Davis, B. Klumperman and M. J. Monteiro, J. Macromol. Sci., Part C, 1998, 38, 567–593 CrossRef.
- S. Y. Zheng, H. Ding, J. Qian, J. Yin, Z. L. Wu, Y. Song and Q. Zheng, Macromolecules, 2016, 49, 9637–9646 CrossRef CAS.
- X. H. Zhao, Soft Matter, 2014, 10, 672–687 RSC.
- S. Koltzenburg, M. Maskos and O. Nuyken, Polymer Chemistry. Springer, 2017 Search PubMed.
- W. Li, J.-M. Y. Carrillo, B. G. Sumpter and R. Kumar, ACS Macro Lett., 2020, 9, 1667–1673 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00256c |
| This journal is © The Royal Society of Chemistry 2024 |