
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent effects on surface-grafted and solution-born poly[N-(2-hydroxypropyl)methacrylamide] during surface-initiated RAFT polymerization†
Yu-Min
Wang
 *a,
Anna
Kálosi
bc,
Yuriy
Halahovets
c,
Hynek
Beneš
a,
Andres
de los Santos Pereira
a and
Ognen
Pop-Georgievski
*a
*a,
Anna
Kálosi
bc,
Yuriy
Halahovets
c,
Hynek
Beneš
a,
Andres
de los Santos Pereira
a and
Ognen
Pop-Georgievski
*a
aInstitute of Macromolecular Chemistry, Czech Academy of Sciences, Heyrovského nám. 2, 16200 Prague, Czech Republic. E-mail: yumin810229@gmail.com; georgievski@imc.cas.cz
bCentre for Advanced Materials Application, Slovak Academy of Sciences, Dúbravská cesta 9, 84511 Bratislava, Slovakia
cDepartment of Multilayers and Nanostructures, Institute of Physics, Slovak Academy of Sciences, Dúbravská cesta 9, 84511 Bratislava, Slovakia
First published on 26th April 2024
Abstract
The difference in the molar mass between surface-grafted and solution-born polymers grown during surface-initiated (SI) polymerization has caused controversy for years. To understand it, we study the solvent effects on the polymer formed on the surface and in the solution by investigating their macromolecular parameters. We utilized reversible addition fragmentation chain-transfer (RAFT) polymerization to grow surface-grafted and solution-born poly[N-(2-hydroxypropyl)methacrylamide] (p(HPMA)) under different solvent conditions. Changing the solvent proticity and/or polarity influences the solution propagation rate, leading to mass transfer limitations and a concomitant discrepancy in the molar masses of the polymer formed in solution and grafted from the surface. Moreover, the solvent effects were found to directly determine the grafting density of surface-grafted p(HPMA). These results highlight how decisive the solvent effects on the SI-RAFT polymerization of HPMA are and that they may be key to regulate the physical and macromolecular parameters of the obtained surface-grafted p(HPMA) brushes.
Introduction
Poly[N-(2-hydroxypropyl)methacrylamide] (p(HPMA)) represents the state of the art of biocompatible, non-toxic, and non-immunogenic polymers and has found extensive use in drug delivery.1–4 Its polymer brush coatings on surfaces inhibit non-specific protein binding (fouling) in various biomedical applications.5–8 P(HPMA) brushes showed outstanding hemocompatibility and prevented surface thrombogenicity under dynamic flow conditions.6,9 In our previous study,10 we reported that the antifouling performance of the p(HPMA) brushes increased with increasing grafting density of the polymer chains. Importantly, we showed how the selection of the grafting method directly affects the grafting density of the p(HPMA) coatings on the surface and that denser polymer brushes can be achieved by the grafting-from (GF) approach. Methods employed for GF typically rely on controlled radical polymerizations (CRP),11,12 such as nitroxide-mediated polymerization,13–15 atom-transfer radical polymerization (ATRP)16–21 and reversible addition fragmentation chain-transfer (RAFT) polymerization.22–28 In particular, surface-initiated (SI) RAFT polymerization is gaining increased attention due to its advantageous features, such as excellent monomer compatibility and lack of metal catalysts toxic to cells, which is beneficial for biomaterial applications.29–31Besides the grafting density of the polymer brush, the molar mass of the polymer chains on the surface is also an important physical parameter influencing the coating's antifouling ability32 but it is challenging to measure it directly.33–35 The difficulty in accessing the molar mass distribution of surface-grafted polymer generated during SI polymerizations comes from the fact that its mass is extremely low, making analysis by typical methods such as size-exclusion chromatography (SEC) problematic. While some authors have assumed that the molar mass of the surface-grafted polymer is comparable with that of the polymer grown concomitantly in solution during the SI-polymerization,36 experimental studies have demonstrated that the polymer molar mass on the surface and the solution-born polymers may be similar or differ greatly, depending on polymerization conditions.37–39 Genzer's and Spencer's groups attributed this discrepancy to a phenomenon they termed “crowding” during SI-ATRP polymerization at high grafting density and/or with a high propagation rate.40,41 Crowding limits mass transfer at the surface, slowing down the propagation rate in comparison to that in the solution. However, it remains an open question to which extent such effect is present in SI-RAFT polymerization and which factors may influence it. This is crucial to optimize the GF-method using SI-RAFT polymerization.
In order to study the SI-RAFT polymerization kinetics of the surface-grafted polymer, the group of Prof. Barner-Kowollik introduced single-molecule force spectroscopy (SMFS) as a powerful method.33 This technique harnesses the sensitivity of atomic force microscopy (AFM) to record force–distance curves as individual surface-grafted polymer chains are bound to the tip and extended during tip retraction to extract their contour length.42 To ensure that the whole contour length of the polymer is accessed during measurement, the chains must be bound to the AFM tip by their end group.33 For this purpose, after SI-RAFT polymerization the terminal chain transfer agent group capping the chains can be conveniently converted to thiol, allowing for chemical bonding between polymer chain and a gold-coated AFM tip.10,26,43 Thus, SMFS offers a practical and effective approach for characterizing the molar mass distribution of surface-grafted polymer brushes.33,44,45 Brooks and colleagues conducted a comprehensive evaluation of SMFS on polymer brushes that had varying grafting densities and showed excellent agreement between the molar masses obtained by SMFS and by SEC from de-grafted polymer chains.35 Several studies demonstrated the reliability of AFM and AFM-SMFS, establishing them as powerful alternatives for the molar mass determination of molecular brushes and surface-grafted polymer chains, which can bypass the difficulties of traditional methods.35,46–48
The choice of solvent plays a crucial role in the mechanism of CRP including their solution and surface-initiated polymerizations. Solvent effects can differently affect the propagation rate of various monomers.49 They are also critical when the solvent molecules determine the interactions between polymer chains or directly interact with them, for example in polyzwitterionic brushes.50,51 Moreover, in ATRP the solvent polarity affects the reaction kinetics by influencing the rates of activation and deactivation and the ATRP equilibrium constant.37,52,53 Similarly, in RAFT polymerization the solvent composition can influence the fragmentation rate of the RAFT agent-radical adduct and impact the attainable control and conversion, as shown by Thang and co-workers specifically for the RAFT polymerization of HPMA in solution.54,55 However, the effects of solvent composition on the SI-RAFT polymerization for the grafting of p(HPMA) brushes remain unexplored, even though such brushes are of great interest due to their excellent fouling resistance.
In the current study, we investigate the differences between surface-grafted and solution-born polymer during the RAFT polymerization of HPMA by manipulating the solvent composition. We characterize the conversion and molecular weight distribution in solution by NMR spectroscopy and SEC and assess the concomitant growth of surface-grafted p(HPMA) brushes by spectroscopic ellipsometry and SMFS. Additionally, we measured the dynamic viscosity of the polymerization solutions and its shear-rate dependence to address the possible mass transfer limitations occurring during the SI-RAFT polymerization, due to the effect of viscosity on diffusion. By leveraging solvent effects, we verify a discrepancy between surface-grafted and solution-born polymers in the context of SI-RAFT polymerization. The influence of solvent composition on the surface-grafted polymer is revealed to be complex and act by various simultaneous mechanisms, affecting propagation rate, mass transport and grafting density.
Experimental section
Materials
Silicon wafers (orientation 〈100〉) bearing native silicon oxide layer were purchased from Siegert Wafer GmbH (Germany). Copper(I) bromide (CuBr, 99.99%), 2,2′-bipyridyl (BiPy, 99%), 4-cyano-4-(phenylcarbonothioylthio) pentanoic acid (CTA), 2,2′-azobis(2-methylpropionitrile) (AIBN), triethylamine (TEA, 99.5%) and hexylamine (99%) were purchased from Sigma-Aldrich (Czech Republic). TEA was purified by distillation over CaH2 before use. All other reagents were used as received. Dimethyl sulfoxide (DMSO), dimethylformamide (DMF), 1,4-dioxane and toluene were purchased from Acros Organics (extra dry, kept over molecular sieves, and filtered using 0.22 μm syringe filter before use). The other organic solvents of analytical grade were from Lach-Ner (Czech Republic) at the highest available purity and used as received. Deionized (DI) water was obtained from a Milli-Q purification system (Milli-Q gradient A10, Merck-Millipore). [11-(2-Bromo-2-methyl)-propionyloxy]undecyltrichlorosilane (Br-silane) and N-(2-hydroxypropyl)methacrylamide (HPMA) were synthesized according to the procedures reported earlier.56,57Synthesis
Characterization
Single-molecule force spectroscopy (SMFS)
The SMFS measurements were performed using an atomic force microscopy setup microscope (Bruker JPK Nanowizard) working in force spectroscopy mode with gold-coated tips (MikroMasch, HQ:CSC38/Cr–Au, consisting of three cantilevers with a nominal tip radius of 35 nm). The SMFS measurements were carried out in DI water. After mounting the cantilever, calibration was done for the sensitivity and the spring constant. The tip separation from the sample was set to 500 nm and the movement of the force curves was performed with a frequency of 0.5 Hz. The data was analysed in the Data Processing software of Bruker JPK. From the measured curves, the ones with rupture events were selected and were fitted to the worm-like chain (WLC) model to obtain the contour length of the polymer chains (see ESI†).33Size exclusion chromatography equipped with multiple angle laser light scattering (SEC-MALS)
The molar mass distribution of the solution-born p(HPMA) was measured by using a Shimadzu HPLC system equipped with a Superose 6 column, online UV detector (Shimadzu), differential refractive index detector (Wyatt Optilab T-rEX) and multi-angle light scattering (Wyatt Dawn Heleos-II). The mobile phase used for the measurement was 0.3 M sodium acetate buffer (pH 6.5) with 1 g L−1 of sodium azide flowing at 0.5 mL min−1, and the specific refractive index increment (dn·dc−1) of p(HPMA) was taken to be as 0.167.Dynamic viscosity measurement
Dynamic viscosity was measured using a rotational rheometer ARES G2 (TA Instruments, USA). Shear flow measurements were performed at 23 °C in the range of shear rates from 1 to 10 s−1 using cone-plate geometry with a plate diameter of 25 mm and cone angle of 0.1 rad. Additionally, viscosity measurements were attempted at the polymerization temperature of 80 °C but could not be carried out due to solvent evaporation losses in the open measurement chamber.Results and discussion
In pursuit of SI-RAFT polymerization conditions providing poly(HPMA) brushes of controlled parameters, we investigated the discrepancy between surface and solution polymerization. We focused on the effect of the solvent composition, as this was previously shown by Thang and coworkers to have a critical influence on the propagation rate and attainable control in the conventional solution-based RAFT polymerization of HPMA.54 We adjust the solution propagation rate by the solvent effects and observe the discrepancy in the molar mass between polymers grown in solution and on the surface. The set of conditions employed for the SI-RAFT polymerization of HPMA differed only in composition of the solvent, which consisted of mixtures of water and either DMF (polar, aprotic) or 1,4-dioxane (non-polar, aprotic) (see Table 1, pure 1,4-dioxane was not used due to incomplete solubility of the monomer). P(HPMA) was grown simultaneously in solution and on the surface from sites of CTA attachment. Solution-phase RAFT polymerization follows a well-known mechanism,55,58–60 but on the surface, the process has a slightly different initiation (Scheme 1). In this study, the R-group of the CTA was bonded covalently to the surface (I). | ||
| Scheme 1 Illustration and chemical structure of propagation on the surface and in the solution during SI-RAFT polymerization of HPMA. | ||
| Entry | Solvent composition | Designation | k app (min−1) | |
|---|---|---|---|---|
| a AIBN and CTA are insoluble in pure water. b HPMA monomer is only partially soluble in pure 1,4-dioxane. | ||||
| 1 | DMF 100% | Water 0% | DMF 100% | 0.0013 |
| 2 | DMF 75% | Water 25% | DMF 75% | 0.0023 |
| 3 | DMF 50% | Water 50% | DMF 50% | 0.0026 |
| 4 | DMF 25% | Water 75% | DMF 25% | 0.0035 |
| 5 | 1,4-Dioxane 75% | Water 25% | 1,4-Dioxane 75% | 0.0028 |
| 6 | 1,4-Dioxane 50% | Water 50% | 1,4-Dioxane 50% | 0.0046 |
| 7 | 1,4-Dioxane 25% | Water 75% | 1,4-Dioxane 25% | 0.0039 |
The radicals are initially formed in solution (II) and transfer reversibly to the thiocarbonylthio (S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–S) moiety of surface-bound CTA, leaving behind a radical in the surface-anchored R-group (II). Subsequently, the surface propagation (III) starts, involving HPMA monomer diffusion to the radical-bearing R-group on the surface. In this way, the surface-grafted and solution-born polymers grow simultaneously (IV) via RAFT polymerization.10 The surface-attached CTA forms a monolayer. While its local concentration on the surface is high, its total amount is very low in comparison with the monomer and initiator concentrations in solution. Because of this, in SI-RAFT it is usually necessary to add “free” soluble CTA in the polymerization mixture in order to attain controlled RAFT polymerization in solution. This free CTA also aids in the exchange of radicals between surface and solution to limit termination, which can be increased on the surface due to the high local concentration of radicals. In the case of HPMA, a methacrylamide monomer, termination could occur by radical mechanisms as well as possibly by chain-end degradation, facilitated by a nucleophilic attack on the thiocarbonyl by the terminal methacrylamide unit.61–63 Addition of free CTA has also been shown to be beneficial in the synthesis of molecular brushes via RAFT using the R-group approach.64
C–S) moiety of surface-bound CTA, leaving behind a radical in the surface-anchored R-group (II). Subsequently, the surface propagation (III) starts, involving HPMA monomer diffusion to the radical-bearing R-group on the surface. In this way, the surface-grafted and solution-born polymers grow simultaneously (IV) via RAFT polymerization.10 The surface-attached CTA forms a monolayer. While its local concentration on the surface is high, its total amount is very low in comparison with the monomer and initiator concentrations in solution. Because of this, in SI-RAFT it is usually necessary to add “free” soluble CTA in the polymerization mixture in order to attain controlled RAFT polymerization in solution. This free CTA also aids in the exchange of radicals between surface and solution to limit termination, which can be increased on the surface due to the high local concentration of radicals. In the case of HPMA, a methacrylamide monomer, termination could occur by radical mechanisms as well as possibly by chain-end degradation, facilitated by a nucleophilic attack on the thiocarbonyl by the terminal methacrylamide unit.61–63 Addition of free CTA has also been shown to be beneficial in the synthesis of molecular brushes via RAFT using the R-group approach.64
Initially, we focused on changing mainly the proticity of the solvent by using mixtures of DMF and water (Table 1, entries 1–4). The molar mass and dispersity of the polymer grown in solution were monitored via SEC-MALS (see Fig. S1 and Table S1, ESI†). For each DMF/water composition, the solution polymerization showed a near-linear pseudo-first-order kinetic, pointing to a controlled radical polymerization (see Fig. 1a and b) with a narrow dispersity (see Fig. 1c). In comparison with pure DMF, the conversion increased by 1.6, 1.8 and 2.1 times and the molar mass increased by 1.5, 2.3 and 2.8 times in the solution when the water content changed to 25%, 50% and 75%, respectively (see detailed value at Table S1, ESI†). We performed a linear regression fit of the conversion plots using eqn (S2)† to obtain the apparent propagation rate constant kp, app. kp, app gradually increased with the increase in solvent proticity, i.e. higher percentage of water present in the polymerization mixture (Table 1). These results are comparable with Thang's study in that having more water in the solution is beneficial for the control of the RAFT polymerization of HPMA.54
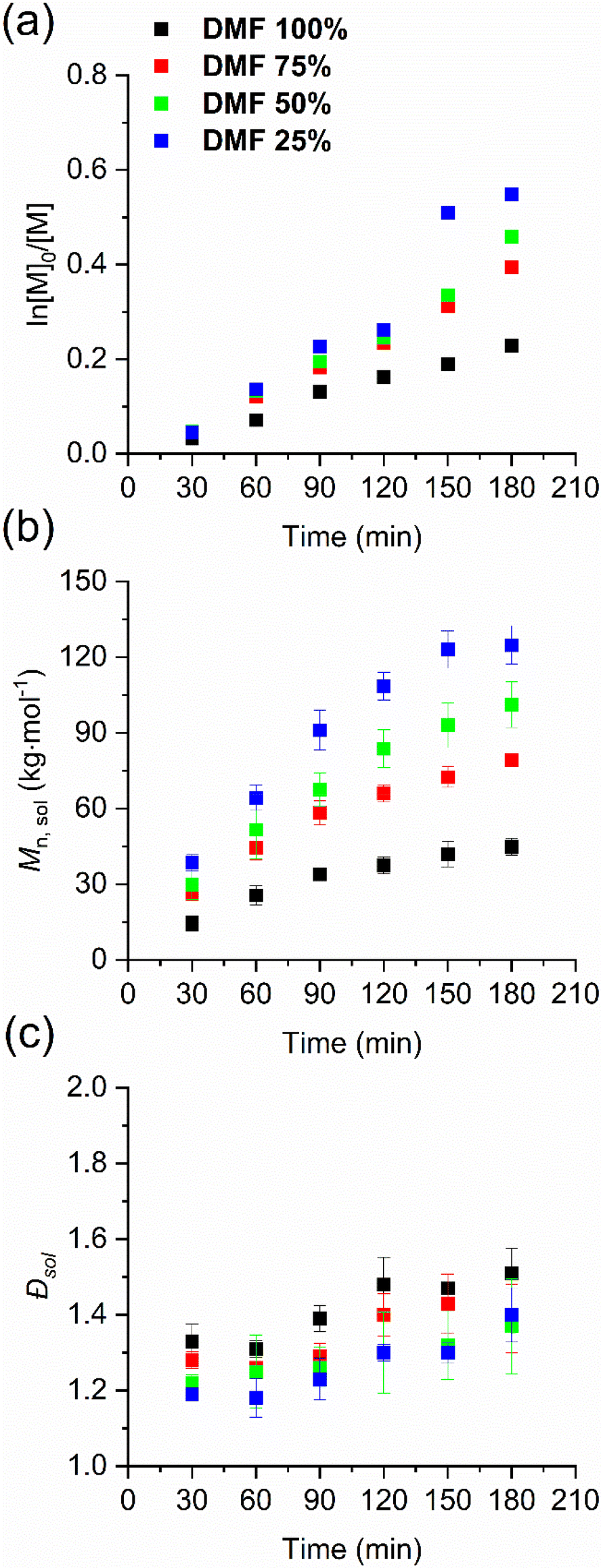 | ||
| Fig. 1 Kinetic plots of the solution-born p(HPMA) via SI-RAFT polymerization in the DMF/water system: (a) conversion, (b) Mn, sol, and (c) dispersity against time. Individual values are reported in Table S1, ESI.† | ||
Their work also presented temperature-dependent NMR experiments and computational simulation as evidence that in aprotic solvent, the intra- and intermolecular hydrogen bonding between polymer chains and monomer leads to retardation due to hindered fragmentation of the RAFT adduct. Increasing amounts of water in the solution mitigates the retardation. Notably, a control experiment was performed, where no free soluble CTA was added in the polymerization mixture (i.e. the only CTA present was the one immobilized on the surface). The obtained polymer demonstrated the expected uncontrolled polymerization process (Fig. S12 and S13, ESI†) as the surface-attached CTA does not affect the solution polymerization, probably owing to its minute amount and limited diffusion.
To analyse the surface-grafted polymers, the course of the polymerization on the surface was monitored by measuring the dry thickness of the polymer layer via spectroscopic ellipsometry (see Fig. 2a). As the thickness of the layers increases with time, the surface-grafted polymer chains appear to be growing during the 3 h of polymerization, suggesting that termination reactions are not significant on the surface. In the control experiment carried out without addition of free CTA in the solution, the thickness growth slows down and stops after 2 h (see Fig. S12, ESI†). This points to termination due to gradual loss of the CTA end groups and emphasizes the importance of the added free CTA to enable controlled SI-RAFT.
 | ||
| Fig. 2 Kinetic plots of the surface-grafted p(HPMA) via SI-RAFT polymerization in the DMF/water system: (a) dry thickness against time, (b) grafting density against solvent composition, and (c) the relation between Mn, sol and Mn, sur. Individual values are reported in Table S3, ESI.† | ||
Interestingly, the thickness of the controlled SI-RAFT did not follow the trend observed for the conversion and molar mass in solution. Indeed, the thicknesses obtained after 180 min under conditions DMF 75% (32.2 nm) and DMF 50% (25.6 nm) were 1.3 and 1.1 times higher than for DMF 100% (24.2 nm), respectively (see Table S3, ESI†). However, at the highest water content DMF 25% (14.8 nm) the thickness was thinner than that of DMF 100% by 0.6 times. This indicates that there is less polymer formed on the surface at the highest solvent water content than in pure DMF, in spite of the enhanced propagation rate observed in solution.
To characterize the molar mass of the surface-grafted polymers, we employed SMFS to obtain the contour length distribution from each solvent condition. The force–distance curves showing rupture events during retraction of the AFM tips were fitted according to eqn (S3)† to obtain the contour length, lc for each sampled polymer chain (see Fig. S3, ESI,† for a representative force curve fit) and calculate the number-average molar mass, Mn, sur (the obtained molar mass distributions are presented in Fig. S4–S7, ESI†).10,33,65 Increasing water content in the polymerization mixture leads to increased Mn, sur, in agreement with the faster propagation observed in solution (see Table S3, ESI†), however, Mn, sur showed a decreased propagation rate at condition DMF 25% compared to DMF 50%. This could be caused by mass transfer limitations: the propagation rate increases both in solution and on the surface, but the high local concentration of growing polymer chains on the surface rapidly depletes the monomer concentration in the interface region. At increasing propagation rates, the diffusion of reactants to the surface presumably creates a bottleneck for the continued growth of polymer on the surface, which lags the solution propagation when the solvent proticity increases. Moreover, increasing water content in the polymerization leads to increased conversion in solution, which could be associated with an increase in solution viscosity.
We hypothesized that an increase in viscosity could further limit mass transfer to the surface, as the rate of diffusion has an inverse dependence with viscosity, which poses a frictional resistance to diffusion. To assess this possibility, we measured the dynamic viscosity of the DMF/water polymerization system. Fig. 3 reports the shear rate dependence of the dynamic viscosity in various p(HPMA) DMF/water solutions at various polymerization times. We observed an increase in dynamic viscosity with increased reaction time and water content progressively, suggesting that the higher conversion and associated larger viscosity could play a role in increased mass transfer limitations at high water contents. Moreover, the shear-thinning behaviour of the solutions at high DMF/low water content may indicate interactions such as hydrogen bonding between the polymer, monomer, and solvent molecules. At higher water contents, these interactions are disrupted as observed by a shift in the dependence of dynamic viscosity on shear rate, when the shear-thinning behaviour (DMF 100% and DMF 75%) changes to a Newtonian fluid behaviour (DMF 50% and DMF 25%).66 This observation aligns with the conclusions from Thang and co-workers regarding solution RAFT polymerization of HPMA, where the disruption of these hydrogen-bonding interactions was found to be the primary mechanism by which water alleviates retardation and promotes faster propagation. Nevertheless, this does not explain the paradoxical tendency of the thickness, which reaches a maximum for condition DMF 75%, but decreases for conditions DMF 50% and DMF 25% in spite of the faster propagation in solution.
 | ||
| Fig. 3 Shear rate dependence of dynamic viscosity in various p(HPMA) DMF/water solutions at 23 °C. | ||
The amount of polymer formed on the surface is determined not only by Mn, sur but also by the number of chains on a given surface area, i.e. the polymer grafting density.20,21 We calculated the grafting density in each condition by plotting Mn, survs. the dry thickness and performed a linear regression fit using eqn (S6) (see Fig. S11a, ESI†).41 Interestingly, conditions DMF 100% and DMF 75% provided a comparable grafting density of 0.45 chain per nm2, but increasing water content led to lower grafting densities of 0.26 chain per nm2 and 0.16 chain per nm2 for DMF 50% and DMF 25%, respectively (see Fig. 2b). For the control experiment without free CTA in the polymerization mixture, SMFS measurements were attempted but did not yield reliable results, probably due to loss of the polymer CTA end groups during SI-RAFT polymerization (see Fig. S14, ESI†). Besides its influence on the propagation rate, increased solvent proticity may also affect the polymerization on the surface by a distinct mechanism. By disrupting polymer–polymer and polymer–monomer hydrogen bonding interactions on the surface, increased water content promotes solvent interactions with the grafted polymer. Accordingly, the grafted p(HPMA) chains would swell and stretch more due to increased uptake of solvent molecules, creating a larger steric hindrance with increasing water content (Scheme S1, ESI†). This limits the ability of the p(HPMA) chains to grow in close proximity to each other, lowering the grafting density. On the other hand, in pure DMF the p(HPMA) chains would tend to form hydrogen bonds between the side chains and adopt a more compact conformation. Thus, the polymer chains can grow closely together and reach a high grafting density. The lower grafting density in conditions DMF 50% and DMF 25% explains the decreasing thickness in spite of the faster polymerization kinetics in solution.
To further study the solvent effects on the SI-RAFT of HPMA, we used mixtures of 1,4-dioxane and water at varying ratios (Table 1, entries 5–7). As 1,4-dioxane is less polar than DMF, we expect a clear effect of the solvent mixture composition on the grafting density. The conversion plot showed a near-linear pseudo-first order kinetic (see Fig. 4a and detailed values at Table S2, ESI†). However, the molar mass reaches a plateau after about 120 minutes for 1,4-dioxane 75% and 1,4-dioxane 50% (see Fig. 4b and detailed values at Table S2, ESI†), with slightly increased dispersity in comparison to the DMF/water systems (see Fig. 4c). Compared with the DMF/water system, we observed that the 1,4-dioxane/water system had an overall faster propagation rate but a wider dispersity. Looking at the thickness of the concomitantly formed polymer layers (see Fig. 5a), we found that 1,4-dioxane 75% and 1,4-dioxane 50% reached a higher thickness than those of DMF/water mixtures. Importantly, increasing the water content to 75% (1,4-dioxane 25%) led to a much lower thickness, comparable to DMF 25%. This agrees with the observations in the DMF mixtures, but in the 1,4-dioxane/water system the difference in thickness was even more marked. To assess the grafting density, we employ SMFS to determine the Mn, sur (the obtained molar mass distributions are presented in Fig. S8–S10 and detailed values at Table S4, ESI†). We also plotted Mn, survs. the dry thickness and performed a linear regression fit using eqn (S6)† to obtain the grafting density for 1,4-dioxane/water systems (see Fig. S11b, ESI†). The grafting density of the p(HPMA) layer in 1,4-dioxane/water systems greatly decreased with increasing content of water in the polymerization mixture, in agreement with the results obtained for the DMF/water system (see Fig. 5b and Table S4, ESI†), supporting our hypothesis.
 | ||
| Fig. 4 Kinetic plots of the solution-born p(HPMA) via SI-RAFT polymerization in the 1,4-dioxane/water system: (a) conversion, (b) Mn, sol, and (c) dispersity against time. Individual values are reported in Table S2, ESI.† | ||
 | ||
| Fig. 5 Kinetic plots of the surface-grafted p(HPMA) via SI-RAFT polymerization in the 1,4-dioxane/water system: (a) dry thickness against time, (b) grafting density against solvent composition, and (c) the relation between Mn, sol and Mn, sur. Individual values are reported in Table S4, ESI.† | ||
Finally, we compared Mn, sol and Mn, sur in each condition, starting with the DMF/water system, and verified their discrepancy. In Fig. 2c, the position of each point represents Mn, sol and Mn, sur at a given polymerization for a given solvent condition. While both Mn, sol and Mn, sur increased with increasing the water percentage, the absolute values show a growing discrepancy, which is seen by Mn, sol increasing faster than Mn, sur. At the highest water content (condition DMF 25%) the largest discrepancy between the Mn, sol and Mn, sur was observed in comparison with the other three conditions, with the ratio Mn, sol/Mn, sur reaching as high as 2.4. Similarly, in the case of ATRP polymerizations the enhanced rate of propagation was found to play an important role in the discrepancy between the Mn of the polymer obtained in solution and on the surface.40 Solvent polarity and water content are critical in ATRP, as they shift the value of KATRP. The enhanced propagation observed in solution RAFT with increasing water content is caused the facilitated fragmentation of the RAFT adduct through hydrogen bonding with the solvent. Thus, both in ATRP and RAFT polymerization, the mechanism is of central importance for the effect of the solvent and for the observed differences in Mn between surface and solution. The comparison reported in our study experimentally demonstrated that higher propagation rate led to a larger discrepancy of molar masses between the surface-grafted and solution-born polymers. We hypothesized the larger discrepancy between the surface-grafted and the solution-born polymers was induced by the mass transfer limitation which accompanies the higher solution propagation rate. The increased viscosity caused by higher conversion and Mn, sol for the highest water content slows down diffusion to the surface further limiting surface propagation and causing it to lag propagation in solution. Similarly as for DMF/water mixtures, also in the case of the 1,4-dioxane/water systems the largest discrepancy between the molar masses of the polymer formed in solution and on the surface was observed for the highest water content, i.e. condition 1,4-dioxane 25%. This can be visualized in Fig. 5c by the larger Mn, sol in comparison to Mn, sur for each point for that condition.
It is interesting to compare these results with our previous observations on the SI-RAFT polymerization of HPMA in methanol using a low-temperature initiator.10 At 45 °C and with a low-temperature initiator, the polymerization proceeded much more slowly. After 24 h of reaction, the molar mass of the solution-grown polymer Mn, sol reached 44.6 kg mol−1 and the thickness of the grafted p(HPMA) layer was 13.5 nm with Mn, sur reached 49 kg mol−1. Notably and in contrast to the results of our current study, the molar masses of the p(HPMA) formed in solution and grafted from the surface were found to be comparable. This can be explained by the much slower polymerization kinetics, which negated the effect of mass transport limitations between surface and solution. Furthermore, the grafting density of the p(HPMA) brushes obtained under those conditions was 0.18 chain per nm2, which is comparable to the polymer layers prepared in the current study in solvent mixtures with 75% of water, both of DMF and 1,4-dioxane. This is consistent with the methanol being able to solvate the HPMA monomer and surface-grafted p(HPMA) thanks to its hydrogen bonding capacity. Taken together, these results confirm the decisive effect of polymerization kinetics on the discrepancy between the molar masses of the polymer formed in solution and grafted from the surface. They also support the role of the solvent in determining the grafting density of the polymer grafted during SI-RAFT.
Conclusions
In conclusion, enhancing the solution propagation rate by having a higher water content in the solvent leads to a larger discrepancy between the molar masses of the polymer grown on the surface and in solution. Importantly, we discover that the proticity and polarity of the solvent also directly affect the achievable grafting density of p(HPMA), which could be elucidated through the use of SMFS. This is probably due to the chains on the surface adopting a more extended conformation as the polymer–polymer interactions are reduced. As a consequence, in spite of the increased propagation rate, the thickness of the p(HPMA) layer is decreased under higher solvent proticity and polarity. Therefore, an optimized synthesis of p(HPMA) brushes via the SI-RAFT polymerization may be achieved in an aprotic solvent, under the precondition that a well-controlled CRP in the solution is maintained, in this case through the addition of a small fraction of water. Importantly, while in solution RAFT polymerization high conversions are generally targeted, the associated increase in viscosity contributes to mass transfer limitations in the surface polymerization and a discrepancy between solution and surface polymerization conditions. The present study highlights the importance of the solvent effects on SI-RAFT polymerization and how they act by multiple mechanisms simultaneously, exerting a complex influence on the polymer grafting process. It must be noted that the solvent effects discussed in the present report are specific to the HPMA monomer and its SI-RAFT polymerization in aprotic and water-containing solvent mixtures. Thus, further studies on other monomers and their interactions with different solvents during SI-RAFT and other SI-polymerizations present an interesting potential research direction to tune the molar mass and the grafting density of surface-grafted polymers. Such considerations are especially important for coating applications where the grafting density plays a decisive role, such as biomaterials.Author contributions
Y. W. – conceptualization, investigation, methodology, formal analysis, writing – original draft, writing – review & editing; A. K., Y. H., and H. B. – investigation, methodology, formal analysis, writing – review & editing; A. d. l. S. P. and O. P.-G. – conceptualization, supervision, writing – review & editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support from the Czech Science Foundation (project no. 22-02836S). A. d. l. S. P. acknowledges funding from the Czech Science Foundation (project no. 22-27329S). The work was supported by the project “National Institute for Cancer Research (Programme EXCELES, ID project no. LX22NPO5102) – funded by the European Union – Next Generation EU”. The experimental work of SMFS was performed during the implementation of the project Building-up Centre for advanced materials application of the Slovak Academy of Sciences, ITMS project code 313021T081 supported by the Integrated Infrastructure Operational Programme funded by ERDFW.References
- V. Šubr, L. Kostka, J. Strohalm, T. Etrych and K. Ulbrich, Macromolecules, 2013, 46, 2100–2108 CrossRef.
- J. Kopecek, P. Kopeckova, T. Minko and Z. Lu, Eur. J. Pharm. Biopharm., 2000, 50, 61–81 CrossRef CAS PubMed.
- O. Hovorka, T. Etrych, V. Šubr, J. Strohalm, K. Ulbrich and B. Říhová, J. Drug Targeting, 2006, 14, 391–403 CrossRef CAS PubMed.
- P. Chytil, T. Etrych, J. Kříž, V. Šubr and K. Ulbrich, Eur. J. Pharm. Sci., 2010, 41, 473–482 CrossRef CAS PubMed.
- M. Vorobii, A. de los Santos Pereira, O. Pop-Georgievski, N. Y. Kostina, C. Rodriguez-Emmenegger and V. Percec, Polym. Chem., 2015, 6, 4210–4220 RSC.
- A. de los Santos Pereira, S. Sheikh, C. Blaszykowski, O. Pop-Georgievski, K. Fedorov, M. Thompson and C. Rodriguez-Emmenegger, Biomacromolecules, 2016, 17, 1179–1185 CrossRef CAS PubMed.
- E. Roeven, A. R. Kuzmyn, L. Scheres, J. Baggerman, M. M. J. Smulders and H. Zuilhof, Langmuir, 2020, 36, 10187–10199 CrossRef CAS PubMed.
- P. Alves, L. C. Gomes, M. Vorobii, C. Rodriguez-Emmenegger and F. J. Mergulhão, Colloids Surf., B, 2020, 19, 110976 CrossRef PubMed.
- Z. Riedelová, A. d. l. S. Pereira, J. Svoboda, O. Pop-Georgievski, P. L. Májek, K. Pečánková, F. Dyčka, C. Rodriguez-Emmenegger and T. Riedel, Macromol. Biosci., 2022, 22, 2200247 CrossRef PubMed.
- Y. Wang, A. Kálosi, Y. Halahovets, I. Romanenko, J. Slabý, J. Homola, J. Svoboda, A. de los Santos Pereira and O. Pop-Georgievski, Polym. Chem., 2022, 13, 3815–3826 RSC.
- R. Wang, Q. Wei, W. Sheng, B. Yu, F. Zhou and B. Li, Angew. Chem., Int. Ed., 2023, 62, e202219312 CrossRef CAS PubMed.
- M. Badoux, M. Billing and H.-A. Klok, Polym. Chem., 2019, 10, 2925–2951 RSC.
- O. Roling, A. Mardyukov, J. A. Krings, A. Studer and B. J. Ravoo, Macromolecules, 2014, 47, 2411–2419 CrossRef CAS.
- W. Wei, A. Balamurugan, J. H. Dwyer and P. Gopalan, ACS Macro Lett., 2018, 7, 100–104 CrossRef CAS PubMed.
- A. C. Schmidt, H. Turgut, D. Le, A. Beloqui and G. Delaittre, Polym. Chem., 2019, 11, 593–604 RSC.
- S. Yamago, Y. Yahata, K. Nakanishi, S. Konishi, E. Kayahara, A. Nomura, A. Goto and Y. Tsujii, Macromolecules, 2013, 46, 6777–6785 CrossRef CAS.
- A. Layadi, B. Kessel, W. Yan, M. Romio, N. D. Spencer, M. Zenobi-Wong, K. Matyjaszewski and E. M. Benetti, J. Am. Chem. Soc., 2020, 142, 3158–3164 CrossRef CAS PubMed.
- R. Sivkova, J. Svoboda, J. Pánek, D. Appelhans and O. Pop-Georgievski, Prog. Org. Coat., 2023, 178, 107447 CrossRef CAS.
- Y. Ko, V. K. Truong, S. Y. Woo, M. D. Dickey, L. Hsiao and J. Genzer, Biomacromolecules, 2022, 23, 424–430 CrossRef CAS PubMed.
- S. Yamamoto, M. Ejaz, Y. Tsujii, M. Matsumoto and T. Fukada, Macromolecules, 2000, 33, 5602–5607 CrossRef CAS.
- S. Yamamoto, M. Ejaz, Y. Tsujii and T. Fukada, Macromolecules, 2000, 33, 5608–5612 CrossRef CAS.
- C. Rodriguez-Emmenegger, B. V. K. J. Schmidt, Z. Sedlakova, V. Šubr, A. B. Alles, E. Brynda and C. Barner-Kowollik, Macromol. Rapid Commun., 2011, 32, 958–965 CrossRef CAS PubMed.
- M. Zamfir, C. Rodriguez-Emmenegger, S. Bauer, L. Barner, A. Rosenhahn and C. Barner-Kowollik, J. Mater. Chem. B, 2013, 1, 6027–6034 RSC.
- X. Xu, X. Huang, Y. Chang, Y. Yu, J. Zhao, N. Isahak, J. Teng, R. Qiao, H. Peng, C. Zhao, T. P. Davis, C. Fu and A. K. Whittaker, Biomacromolecules, 2021, 22, 330–339 CrossRef CAS PubMed.
- A. R. Kuzmyn, A. T. Nguyen, L. W. Teunissen, H. Zuilhof and J. Baggerman, Langmuir, 2020, 36, 4439–4446 CrossRef CAS PubMed.
- A. R. Kuzmyn, M. van Galen, B. van Lagen and H. Zuilhof, Polym. Chem., 2023, 14, 3357–3363 RSC.
- J. Yuan, X. Huang, P. Li, L. Li and J. Shen, Polym. Chem., 2013, 4, 5074–5085 RSC.
- M. K. Cho, H. J. Seo, J. H. Lee, W. K. Cho and K. Son, Polym. Chem., 2021, 12, 7023–7030 RSC.
- Y. Zhao and S. Perrier, in Controlled Radical Polymerization at and from Solid Surfaces, ed. P. Vana, Springer, Cham: Switzerland, 2015, vol. 270, pp. 77–106 Search PubMed.
- C. Chen, F. Richter, C. Guerrero-Sanchez, A. Traeger, U. S. Schubert, A. Feng and S. H. Thang, ACS Macro Lett., 2020, 9, 260–265 CrossRef CAS PubMed.
- M. Li, M. Fromel, D. Ranaweera, S. Rocha, C. Boyer and C. W. Pester, ACS Macro Lett., 2019, 8, 374–380 CrossRef CAS PubMed.
- A. Halperin, Langmuir, 1999, 15, 2525–2533 CrossRef CAS.
- T. Tischer, R. Gralla-Koser, V. Trouillet, L. Barner, C. Barner-Kowollik and C. Lee-Thedieck, ACS Macro Lett., 2016, 5, 498–503 CrossRef CAS PubMed.
- L. Michalek, L. Barner and C. Barner-Kowollik, Adv. Mater., 2018, 30, 1706321 CrossRef PubMed.
- D. Goodman, J. N. Kizhakkedathu and D. E. Brooks, Langmuir, 2004, 20, 6238–6245 CrossRef CAS PubMed.
- M. Fromel, E. M. Benetti and C. W. Pester, ACS Macro Lett., 2022, 11, 415–421 CrossRef CAS PubMed.
- J. O. Zoppe, N. C. Ataman, P. Mocny, J. Wang, J. Moraes and H.-A. Klok, Chem. Rev., 2017, 117, 1105–1318 CrossRef CAS PubMed.
- E. Turan, S. Demirci and T. Caykara, Thin Solid Films, 2010, 518, 5950–5954 CrossRef CAS.
- E. H. Min, S. R. S. Ting, L. Billon and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3440–3455 CrossRef CAS.
- C. Kang, R. Crockett and N. D. Spencer, Polym. Chem., 2016, 7, 302–309 RSC.
- R. R. Patil, S. Turgman-Cohen, J. Šrogl, D. Kiserow and J. Genzer, Langmuir, 2015, 31, 2372–2381 CrossRef PubMed.
- A. Al-Baradi, M. R. Tomlinson, Z. J. Zhang and M. Geoghegan, Polym. Chem., 2015, 67, 111–117 CAS.
- A. R. Kuzmyn, T. G. Ypma and H. Zuihof, Langmuir, 2024, 40, 3354–3359 CAS.
- S. Yamamoto, Y. Tsujii and T. Fukuda, Macromolecules, 2000, 33, 5995–5998 CrossRef CAS.
- S. Al-Maawali, J. E. Bemis, B. B. Akhremitchev, R. Leecharoen, B. G. Janesko and G. C. Walker, J. Phys. Chem. B, 2001, 105, 3965–3971 CrossRef CAS.
- S. S. Sheiko, M. d. Silva, D. Shirvaniants, S. LaRue, S. Prokhorova, M. Moeller, K. Beers and K. Matyaszewski, J. Am. Chem. Soc., 2003, 125, 6725–6728 CrossRef CAS PubMed.
- S. S. Sheiko, O. V. Borisov, S. A. Prokhorova and M. Möller, Eur. Phys. J. E, 2004, 13, 125–131 CrossRef CAS PubMed.
- S. S. Sheiko, in New Developments in Polymer Analytics II, ed. M. Schmidt, Springer Berlin Heidelberg, 2000, vol. 151, pp. 61–174 Search PubMed.
- G. M. Scheutz, J. I. Bowman, S. Mondal, J. Y. Rho, J. B. Garrison, J. Korpanty, N. C. Gianneschi and B. S. Sumerlin, ACS Macro Lett., 2023, 12, 454–461 CrossRef CAS PubMed.
- Y. Terayama, M. Kikuchi, M. Kobayashi and A. Takahara, Macromolecules, 2011, 44, 104–111 CrossRef CAS.
- M. Kobayashi, H. Yamaguchi, Y. Terayama, Z. Wang, K. Ishihara, M. Hino and A. Takahara, Macromol. Symp., 2009, 279, 79–87 CrossRef CAS.
- W. A. Braunecker, N. V. Tsarevsky, A. Gennaro and K. Matyjaszewski, Macromolecules, 2009, 42, 6348–6360 CrossRef CAS.
- N. V. Tsarevsky, T. Pintauer and K. Matyjaszewski, Macromolecules, 2024, 37, 9768–9778 CrossRef.
- X. Pan, F. Zhang, B. Choi, Y. Luo, X. Guo, A. Feng and S. H. Thang, Eur. Polym. J., 2019, 115, 166–172 CrossRef CAS.
- S. Pirrier, Macromolecules, 2017, 50, 7433–7447 CrossRef.
- C. Rodriguez-Emmenegger, S. Janel, A. de los Santos Pereira, M. Bruns and F. Lafont, Polym. Chem., 2015, 6, 5740–5751 RSC.
- K. Ulbrich, V. Šubr, J. Strohalm, D. Plocová, M. Jelínková and B. Říhová, J. Controlled Release, 2000, 64, 63–70 CrossRef CAS PubMed.
- O. R. Courtney, S. M. Clouthier, S. Perrier, J. Tanaka and W. You, ACS Macro Lett., 2023, 12, 1306–1310 CrossRef CAS PubMed.
- D. K. Schneiderman, J. M. Ting, A. A. Purchel, R. Miranda Jr., M. W. Tirrell, T. M. Reineke and S. J. Rowan, ACS Macro Lett., 2018, 7, 406–411 CrossRef CAS PubMed.
- T. Nwoko, K. Nguyen, N. K. Saha, C. Barner-Kowollik and D. Konkolewicz, Polym. Chem., 2024, 15, 1052–1061 RSC.
- B. A. Abel and C. L. McCormick, Macromolecules, 2016, 49, 465–474 CrossRef CAS.
- M. S. Donovan, A. B. Lowe, B. S. Sumerlin and C. L. McCormick, Macromolecules, 2002, 35, 4123–4132 CrossRef CAS.
- M. S. Donovan, T. A. Sanford, A. B. Lowe, B. S. Sumerlin, Y. Mitsukami and C. L. McCormick, Macromolecules, 2002, 35, 4570–4572 CrossRef CAS.
- Z. Zheng, J. Ling and A. H. E. Müller, Macromol. Rapid Commun., 2014, 35, 234–241 CrossRef CAS PubMed.
- J. McClements and V. Koutsos, ACS Macro Lett., 2020, 9, 152–157 CrossRef CAS PubMed.
- R. G. Larson, The structure and rheology of complex fluids, Oxford University Press New York, New York, 1999 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00177j |
| This journal is © The Royal Society of Chemistry 2024 |